MEDIATORI
DELL’INFIAMMAZIONE
Nabissi15
MEDIATORI DELL’INFIAMMAZIONE
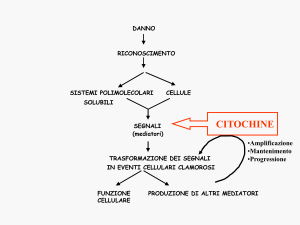
DANNO RICONOSCIMENTO
SISTEMIPOLIMOLECOLARICELLULE
SOLUBILI
SEGNALI
(mediatori)
TRASFORMAZIONEDEISEGNALI
INEVENTICELLULARICLAMOROSI
FUNZIONEPRODUZIONEDIALTRIMEDIATORI
CELLULARI
Nabissi15
• Amplificazione
• Mantenimento
• Progressione
MEDIATORI CHIMICI DELL’INFIAMMAZIONE
MEDIATORI
Tutte quelle molecole che determinano l’insorgere e partecipano allo sviluppo dei
fenomeni che si osservano durante l’evento infiammatorio.
I mediatori di derivazione plasmatica sono presenti nel plasma in forma inattiva mentre i
mediatori di origine cellulare sono sequestrati nei granuli intracellulari o sono sintetizzati de
novo in risposta ad uno stimolo.
REQUISITI
Assenti nello stato stazionario o presenti in forma inattiva o sequestrati all’interno delle
cellule
Presenti nello stato attivo (attivati o rilasciati dai depositi intracellulari o neosintetizzati)
Hanno vita media breve
In grado di mimare fenomeni infiammatori se iniettati
La loro inibizione determina la scomparsa dei fenomeni infiammatori osservati
Nabissi15
ANGIOFLOGOSI
MEDIATORI
EVENTI
1.
2.
1.
RISPOSTA VASCOLARE:
modificazioni del calibro e del flusso
sanguigno
MEDIATORI VASOATTIVI:
agiscono sul tono delle cellule muscolari
liscie (vasodilatazione)
fuoriuscita di molecole per alterazione
della barriera sangue-interstizio
(essudato)
promuovono la contrazione degli endoteli
(aumento permeabilità)
FASE CELLULARE:
- migrazione cellulare
- attivazione funzioni effettrici
- risposta immunitaria
2.
MEDIATORI CHE AGISCONO SUI
DIVERSI TIPI CELLULARI COINVOLTI
NELL’INFIAMMAZIONE:
- promuovono l’adesione e la chemiotassi
- modulano le funzioni dei fagociti
- modulano le risposte dei linfociti
Nabissi15
Nabissi15
MEDIATORI DERIVATI DAI SISTEMI POLIMOLECOLARI SOLUBILI DEL
PLASMA, DELL’INTERSTIZIO
1.
SISTEMA DELLA COAGULAZIONE (plasmina, fibrinopeptidi)
2.
SISTEMA DELLE CHININE (bradichinina, callicreina)
3.
SISTEMA FIBRINOLITICO (fibrinopeptidi)
4.
CASCATA DEL COMPLEMENTO (anafilotossine)
MEDIATORI CHIMICI DI FASE FLUIDA
Nabissi15
IL COMPLEMENTO
Il sistema del complemento consta di 20 componenti proteici che si trovano concentrati nel
plasma.
Questo sistema è coinvolto sia nella immunità innata che in quella acquisita nella difesa dai
microorganismi.
Nel processo d’attivazione del complemento vengono generati diversi fattori che causano un
aumento della permeabilità vascolare, chemiotassi e opsonizzazione, rilascio di
citochine ed attivazione della NADPH ossidasi.
Le proteine del complemento sono presenti in forma inattiva e classificate come C1-C9 e
molte di queste vengono attivate in modo da diventare enzimi proteolitici che degradano e
attivano altre proteine del complemento cosi’ da creare una cascata capace di notevole
amplificazione enzimatica.
Nabissi15
• La tappa critica ovvero l’attivazione (degradazione) della terza (C3)
componente puo’ avvenire attraverso tre vie:
- Classica: che è scatenata dal legame del componente C1 con anticorpi (IgM
o IgG) combinati con l’antigene, portando alla proteolisi di C2 e C4, con
formazione della C3 convertasi (C4bC2b)
- Via alternativa: stimolata da proteine di superficie dei microbi
(endotossine), amplifica la scissione del C3 in C3b che si fonde con il
fattore plasmatico Bb, formando il complesso C3bBb (C3 convertasi)
- Via della lectina: in cui la lectina plasmatica che lega il mannosio si lega ai
carboidrati presenti sui microbi e attiva direttamente il C1, che segue poi
l’attivazione della C3 convertasi (C4bC2b) come nella via classica.
Nabissi15
Nabissi15
IL COMPLEMENTO
• Qualunque sia la via, il risultato finale è la formazione dell’enzima attivo C3
convertasi che taglia il C3 in due frammenti funzionalmente distinti (C3a e
C3b).
• C3a è rilasciato mentre C3b si lega alla cellula o alla molecola che ha
scatenato l’attivazione del complemento.
• C3b forma anche la C5 convertasi (con altri frammenti precedentemente
generati) che scinde C5 generando C5a e C5b. C5b si lega ai componenti
C6/9 formando il COMPLESSO DI ATTACCO ALLA MEMBRANA
(MAC), il quale lisa le cellule.
Nabissi15
Nabissi15
Nabissi15
• I frammenti del complemento mediano vari fenomeni
dell’infiammazione.
– Fenomeni vascolari: C3a, C4a e C5a (anafilotossine) stimolano la liberazione di istamina
da parte dei mastociti, aumentando la permeabilità vascolare e causando vasodilatazione.
– C5a nei monociti e neutrofili attiva la via lipossigenasica del metabolismo dell’AA
rilasciando altri fattori dell’infiammazione.
– Adesione leucocitaria, chemiotassi e attivazione: C5a agisce come potente agente
chemiotattico per neutrofili, monociti, eosinofili e basofili.
– Fagocitosi. C3b quando si lega alla parete del batterio agisce come opsonina e favorisce la
fagocitosi da parte dei macrofagi, che presentano i recettori per C3b attivato.
– Enzimi proteolitici presenti nell’essudato infiammatorio possono attivare C3 e C5, che poi
fungono da componenti chemiotattili per i leucociti stessi.
L’attivazione del complemento è strettamente controllata da proteine regolatrici
circolanti associate alle cellule e queste molecole proteggono le cellule dell’ospite da
danni durante le reazioni di difesa da microbi.
Nabissi15
Nabissi15
Regolazione dell’attività del complemento
Regolazione della C3 e C5 convertasi. La formazione di C3 convertasi e la generazione di
C3b sono le fasi principali dell’attivazione del complemento. I regolatori di questi fattori
agiscono potenziando la dissociazione (degradazione) del complesso della convertasi
(Decay Accelerating Factor; DAF) o scindendo proteoliticamente il C3b.
Legame delle componenti attive del complemento. La via classica che inizia con il legame
del C1 ad un immunocomplesso, che viene bloccato da una proteina plasmatica chiamata
inibitore-C1 (C1NH), che interferisce con l’attività enzimatica di due delle proteine del
complesso C1.
Inoltre l’attività del complemento è impedita anche da proteine che inibiscono la
formazione del MAC.
Nabissi15
Deficienze del sistema del complemento
Deficienze nelle proteine del complemento possono dar luogo ad un aumento della
suscettibilità ad infezioni batteriche e virali (SEPSI).
I deficit di C2 e C4 sono associati a malattie autoimmunitarie (lupus eritematoso)
probabilmente a causa dell’impossibilità di eliminare immunocomplessi.
Edema angioneurotico ereditario: l’attivazione del C1 da parte degli immunocomplessi
non è controllata e si verifica aumento della degradazione di C2 e C4, formazione di un
frammento del C2 che agisce come la bradichinina.
Il C1NH (che è mutato in questa patologia) influisce anche sul sistema della coagulazione
in quanto inibisce anche la callicreina ed il fattore XII. Questa patologia genetica
comporta un aumento di bradichinina nel plasma con conseguente stato infiammatorio
cronico.
Nabissi15
RISPOSTA INFIAMMATORIA
Nabissi15
SISTEMA DELLA COAGULAZIONE
Il sistema della coagulazione è diviso in due vie convergenti che
culminano con l’attivazione della trombina e nella formazione di
fibrina.
La via estrinseca della coagulazione è composta da una serie di
proteine plasmatiche che possono essere attivate dal FATTORE
DI HAGEMAN (fattore XII), che è prodotto dal fegato e circola
in forma inattiva finchè non incontra il collagene, proteine della
membrana basale o le piastrine attivate.
Attivato, il Fattore XII va incontro a delle modificazioni
conformazionali (diventanto Fattore XIIa), esponendo il sito
attivo di serina e cosi’ acquisendo la capacità di tagliare substrati
proteici e attivare altri mediatori.
Nabissi15
Fattori della coagulazione
•
•
•
•
•
•
I – Fibrinogeno
II – Protrombina
III – Fattore tissutale
IV – Calcio
V – Proaccelerina
VI – unassigned
(prev Factor Va)
• VII – Proconvertina
•
•
•
•
VIII – fattore antiemolitico
IX – Christmas Factor
X – Stuart-Prower Factor
XI – Plasma
Tromboplastina
Antecedente
• XII – Hageman Factor
• XIII – fattore stabilizzante la
fibrina
Nabissi15
La via intrinseca comporta l’attivazione della via delle chinine che
attraverso la formazione di callicreina attiva il Fattore XII.
Il fattore XII attivato (XIIa) porta all’attivazione di trombina ed alla
formazione di fibrina mediante processazione del fibrinogeno (azione
coagulante).
L’enzima callicreina attiva un altro processo enzimatico:
-formazione di plasmina che svolge un ruolo litico a livello del coagulo
(azione anticoagulante)
Nabissi15
SISTEMA DELLE CHININE
• Il sistema delle chinine genera peptidi vasoattivi a partire da proteine
plasmatiche, detti CHININOGENI, per azione di proteasi specifiche dette
callicreine.
• L’attivazione del sistema porta alla formazione di Bradichinina.
• La bradichinina aumenta la permeabilità vascolare a causa la contrazione
del muscolo liscio, dilatazione dei vasi e dolore quando iniettato.
• L’azione della bradichinina è molto rapida e viene inattivata dall’enzima
chininasi
Nabissi15
Nabissi15
SISTEMA DELLA COAGULAZIONE
• La plasmina taglia anche il fattore del complemento C3 producendo
C3a e C3b e scinde la fibrina (che forma dei prodotti coinvolti nella
permeabilità vascolare).
• La plasmina circola sotto forma di precursore (plasminogeno) legata al
suo inibitore tPA (plasminogen tissue activator) che quando si lega alla
fibrina si attiva ed attiva la plasmina.
• UROCHINASI: attivatore del plasminogeno (formazione di plasmina) con
un ruolo molto importante nel dissolvere trombi ed emboli trombotici
Nabissi15
ATTIVAZIONE XII
• Una varietà di sostanze organiche ed inorganiche cariche
negativamente attivano il fattore XII. Fra le sostanze
organiche ci sono collagene, eparina e fra le sostanze
inorganiche il vetro, silicio, cristalli di pirofosfato.
Nabissi15
Sistemi anticoagulanti naturali
Antitrombina III: prodotta dal fegato e dalle cellule endoteliali è in grado
d’inibire la trombina
Proteina C: inibisce i fattori V e VIII limitando la produzione di trombina
Proteina S: prodotta dalle cellule endoteliali e modula l’intero processo
della coagulazione
Nabissi15
Funzione:
lisi del
coagulo,
permeabilità
vascolare
Funzione:
Vasodilatazione
liberazione
d’istamina
Nabissi15
Funzione:
Funzione:
vasodilatazione formazione
attivazione
di fibrina
chemiotassi
vasodilatazione
ISTAMINA
Il primo mediatore chimico ad entrare in funzione nell’infiammazione acuta è
l’istamina e se l’alterazione è di modesta entità rimane anche l’unico. Infatti
l’istamina è responsabile “soltanto” delle modificazioni vascolari che si attuano nei
primi 15-30 minuti e per cio’ la sua azione è abbastanza fugace anche se la sua
azione è di fondamentale importanza.
L’istamina quindi si degrada velocemente o viene disattivata dalle istaminasi
presenti negli eosinofili
Nabissi15
ISTAMINA
L’istamina modula a diversi livelli la risposta immune antigene-specifica
ed è coinvolta nella regolazione del rilascio di mediatori e neurotrasmettitori.
L’istamina deriva prevalentemente dalla degranulazione di mastociti e basofili,
ma può anche essere sintetizzata de novo da altre cellule ematopoietiche e da
queste immediatamente rilasciata. Essa è depositata all’interno di granuli
citoplasmatici specifici, presenti in mastociti e basofili, complessata con
eparina e condroitin-solfato.
Nabissi15
L’istamina provoca effetti sulla muscolatura liscia, inducendo la contrazione delle vie
aeree, intestino e vasi sanguigni e la vasodilatazione (rilassamento) delle arteriole.
La sua liberazione avviene mediante degranulazione in risposta a diversi stimoli
infiammatori : lesioni fisiche, reazioni immunitarie che comportano il legame di anticorpi
ai mastociti (IgE con
il recettore ad alta affinità FcεRI) presenti sui mastociti,
anafilotossine (frammenti del complemento C3a, C5a), proteine di derivazione leucocitaria,
neuropeptidi (Sostanza P), citochine infiammatorie e chemochine (IL-1, IL-8).
Nabissi15
I mastociti elaborano e rilasciano
eparina, istamina e numerosi altri fattori
Nabissi15
Nabissi15
Nabissi15
EFFETTI FARMACOLOGICI DELL’ISTAMINA
Nabissi15
ISTAMINA
L’istamina è un costituente naturale del corpo ed è un’ammina a basso peso molecolare
sintetizzata a partire dall’aminoacido essenziale L-istidina mediante una reazione
enzimatica esclusiva della istidina decarbossilasi.
L’istidina decarbossilasi è espresso in quasi tutte le cellule del corpo, inclusi i neuroni del
sistema nervoso centrale, mucosa gastrica, mastociti e basofili.
L’azione dell’istamina è svolta grazie al suo legame a quattro tipi di recettori (H1, H2, H3,
H4), recettori a 7 domini transmembrana accoppiati a G-protein (Gs, Gq, Gi/o).
Nabissi15
Caratteristiche
H1
H2
H3
H4
Sequenza aa
487
359
445
390
Loc.cromosomica
3p25
5q35
20q13
18q11
Espressione
Largamente
distribuito,
neuroni compresi
Largamente
distribuito
compresa la
mucosa gastrica
Poco distribuito,
presente nei
neuroni
istaminergici
Presente in tessuti
ematopoietici
periferici, midollo
osseo
Proteina G
accoppiata
Pathways attivati
Fosfolipasi A,
NfkB, cAMP,
NOS
Fosfolipasi C,
Protein chinasio
C, c-fos
MAP chinasi,
inbizione cAMP
MAP chinasi,
inbizione cAMP
Azione
dell’istamina
Prurito, dolore,
vasodilatazione,
ipotensione,
tachicardia
Secrezione
gastrica acida,
permeabilità
vascolare
Previene
broncocostrizione
media prurito
Differenziamento
di mieloblasti e
promielociti
Nabissi15
GHIANDOLE ESOCRINE
Ghiandole gastriche: aumento della secrezione di H+, di pepsina e del fattore
intrinseco, mediati da recettori H2 (localizzati sulle cellule parietali)
Ghiandole salivari, pancreatiche, intestinali, lacrimali e bronchiali: aumento
della secrezione, mediata da recettori H1
SISTEMA IMMUNITARIO
Mastociti e granulociti basofili: diminuzione della secrezione di autacoidi
(ormoni ad azione locale) mediata da recettori H2 (meccanismo a feed-back
negativo)
Granulociti neurotrofili: diminuzione della secrezione di enzimi lisosomiali
mediata da recettori H2
Linfociti: diminuzione della produzione di anticorpi e linfochine mediata da
recettori H2
Nabissi15
Metaboliti dell’acido arachidonico
Quando le cellule sono attivate da stimoli di varia natura, i lipidi presenti sulla membrana
plasmatica sono rimodellati per generare mediatori lipidici che fungono da segnali
intracellulari o extracellulari influenzando diversi processi biologici , fra cui
l’infiammazione.
Questi lipidi formati, definiti AUTACOIDI, sono ormoni che svolgono la loro funzione a
livello locale (autocrina o paracrina).
Nabissi15
Metaboliti dell’acido arachidonico
L’acido arachidonico (AA) è un grasso polinsaturo a 20 atomi di carbonio che
deriva da fonti alimentari o dalla conversione dell’acido linoleico.
L’acido arachidonico non si trova libero nelle cellule, ma è normalmente
esterificato nei fosfolipidi di membrana.
Viene rilasciato da questi ultimi, attraverso l’azione di fosfolipasi cellulari, le
quali possono essere attivate da stimoli chimici, fisici, meccanici o altri
mediatori (C5a).
Metaboliti dell’acido arachidonico
•
•
I mediatori dell’acido arachidonico vengono denominati EICOSANOIDI, si legano a
recettori accoppiati alle proteine G e mediano ogni fase della risposta infiammatoria.
Sono sintetizzati da due importanti classi di enzimi:
– 1) ciclossigenasi: da cui si ottengono prostaglandine e trombossani
– 2) lipossigenasi da cui si ottengono leucotrieni e lipossine
Nabissi15
Ciclossigenasi
La via ciclossigenasica è mediata da due diversi enzimi (COX-1 e COX-2).
La COX-1 è espressa costitutivamente, la COX-2 è inducibile.
Entrambe le vie portano alla produzione di PROSTAGLANDINE (PG).
Le prostaglandine sono codificate con il suffisso PG, una lettera (PGD, PGE,..)
e da un numero che indica il numero di doppi legame del composto (PGE2).
Le prostaglandine sono coinvolte anche nel meccanismo del dolore e della febbre
durante il processo d’infezione.
La prostaglandina PGE2 è iperalgesica in quanto rende la cute ipersensibile agli
stimoli dolorosi.
La prostaglandina PGI2 è il principale metabolita della via ciclossigenasica nei
mastociti, causando vasodilatazione ed aumento della permeabilità delle venule
potenziando cosi’ la formazione dell’edema.
Nabissi15
TERAPIA ANTINFIAMMATORIA
Inibitori ad ampio spettro: Comprendono i glucocorticoidi che sono potenti antinfiammatori
che inibiscono la trascrizione della COX-2, fosfolipasi A2, citochine (IL-1, TNF) e NOS.
I glucocorticoidi inoltre stimolano la trascrizione di geni che codificano per proteine
antinfiammatorie, come la LIPOCORTINA 1 che inibisce il rilascio di AA dai fosfolipidi di
membrana.
Nabissi15
VIA LIPOSSIGENASICA
I metaboliti iniziali sono prodotti da tre diversi lipossigenasi:
1) La 5-lipossigenasi, presente nei neutrofili che porta alla produzione di 5-HETE, che è
un’agente chemiotattico per i neutrofili e viene convertito in una famiglia di composti detti
LEUCOTRIENI.
La loro funzione è quella di facilitare l’adesione e l’aggregazione dei leucociti, la generazione
di radiacali liberi dell’O2, il rilascio di enzimi lisosomiali vasocostrizione e broncospasmo.
Nabissi15
Legandosi a specifici recettori i LT
promuovono l’accumolo e le funzioni di tutte
le classi di leucociti.
Queste risposte sono importanti in diversi
tipi di patologie (asma,
cardiovascolari, tumori, ecc..).
Il LT stimolano la crescita delle CD34+
pluripotent hematopoietic stem-cell
progenitors e la loro migrazione nel plasma.
LT incrementano l’espressione delle
molecole d’adesione e promuovono la
motilità cellulare.
Mediante LTB-receptor 1 (BLT1), LTB4
recluta i mastociti, neutrofili, e cellule T
Nabissi15
Nabissi15
LIPOSSINE
Le lipossine derivano da meccanismi di sintesi transcellulare, cioe’ la loro
produzione coinvolge due tipi celluari.
I neutrofili producono intermedi della sintesi di lipossine che vengono poi
convertite in lipossine dalle piastrine.
Le lipossine, inibiscono il reclutamento dei leucociti e delle componenti
cellulari dell’infiammazione, inibendo la chemiotassi dei leucociti e
l’adesione. Questo suggerisce che le lipossine siano dei regolatori negativi
dell’azione dei leucotrieni e quindi coinvolti nel processo di risoluzione
dell’infiammazione.
Nabissi15
OSSIDO NITRICO
L’ossido nitrico (NO) è un mediatore pleiotropico dell’infiammazione e fu
inizialmente scoperto come un fattore rilasciato dalle cellule endoteliali che
causa vasodilatazione, rilassando la muscolatura liscia dei vasi e vene.
NO è un gas solubile prodotto dall’enzima nitrico sintetasi (NOS) a partire da
L-arginina. Vi sono tre tipi di NOS: endoteliale (eNOS), neuronale (nNOS) e
inducibile (iNOS). I primi due sono espressi costitutivamente a bassi livelli e
possono essere attivati rapidamente da un aumento degli ioni Ca2+ intracellulari,
provocando una rapida produzione di NO, iNOS viene invece indotto quando i
macrofagi e altre cellule sono attivate da citochine (TNF).
NO agisce a livello paracrino sulle cellule bersaglio attraverso l’induzione di
GMP (guanosin-monofosfato) ciclico che a sua volta da inizio a una serie di
eventi intracellulari che generano rilassamento muscolare a livello dei vasi.
Nabissi15
OSSIDO NITRICO
• Inoltre NO riduce l’aggregazione e l’adesione inibendo alcune caratteristiche
dell’infiammazione indotta dai mastociti e funge da regolatore endogeno del
reclutamento dei leucociti.
• Quindi NO rappresenta un meccanismo endogeno compensatorio che riduce la
risposta infiammatoria.
• Inoltre NO ed i suoi derivati sono microbicidi, quindi NO è anche un mediatore
nella risposta dell’ospite alle infezioni.
• Infatti:
– Le specie reattive che derivano da NO hanno attività antimicrobiche
– Interazioni fra NO e specie reattive dell’O2 producono metaboliti
antimicrobici
– La produzione di NO aumenta durante la risposta dell’ospite alle infezioni
– L’inattivazione del gene per la nitrico sintetasi facilita la replicazione
microbica
Nabissi15
Nitric Oxide (NO)
Nabissi15
CITOCHINE INFIAMMATORIE
Con il termine citochine s’intendono mediatori polipeptidici non antigene-specifici, che
funzionano come segnali di comunicazione intercellulari (cellule del sistema immunitario
vs. organi e tessuti).
Le citochine sono prodotte da diversi tipi di cellule e svolgono la loro azione a breve
distanza, con azioni in parte simile fra citochine diverse (es. IL-1 e TNF), con effetto
pleiotropico.
Le citochine possono essere suddivise in base al loro ruolo funzionale in:
emopoietiche
dell’immunità specifica
infiammatorie primarie
anti-infiammatorie ed immunosoppressive
infiammatorie secondarie (le chemochine)
LE CITOCHINE INFIAMMATORIE PRIMARIE SONO : IL-1, TNF e IL-6
IL-1 e TNF attivano l’intera cascata dei mediatori dell’infiammazione, mentre IL-6 si
definisce anche secondaria perché agisce soprattutto nello stimolare la produzione di
proteine di fase acuta, responsabili dell’amplificazione dei meccanismi dell’immunità
innata.
Nabissi15
Proprietà generali delle citochine
Le citochine, non sono generalmente immagazzinate come molecole preformate all’interno
delle cellule, ma la loro sintesi è attivata dalla trascrizione dei loro geni. Le citochine
svolgono un’azione biologica sia autocrina che paracrina, legandosi, con alta affinità, a
specifici recettori di membrana.
L’espressione dei recettori per le
citochine è regolata da segnali
esterni, inducendo un maggiore
risposta delle cellule che li
esprimono, alle citochine. Ad
esempio la stimolazione dei linfociti
B e T agli antigeni induce un
aumento dell’espressione dei
recettori per le citochine.
Nabissi15
CITOCHINE INFIAMMATORIE
Le due citochine principalmente studiate nel processo infiammatorio sono il TNF (tumor
necrosis factor) e IL-1 (interleuchina-1), che sono prodotte nei macrofagi attivati.
La secrezione di TNF e IL-1 puo’ essere stimolata da lesioni, endotossine e vari stimoli
infiammatori.
La loro azione nell’infiammazione riguarda gli effetti sull’endotelio (inducendo la
produzione di molecole d’adesione), sui leucociti e fibroblasti (stimolando la sintesi di
mediatori dell’infiammazione (PG e NO), chemochine) e l’induzione della reazione
sistemica di fase acuta.
La risposta sistemica di IL-1 e TNF agisce attraverso l’induzione di IL-6, che induce la
sintesi di proteine di fase acuta, amplificando a livello sistemico l’immunità innata e la
rigenerazione (o riparazione) tissutale.
Nabissi15
Classificazione delle citochine in base al ruolo funzionale
Mediatori che regolano l’immunità innata: sono prodotti principalmente dai
fagociti mononucleati in risposta ad agenti infettivi come prodotti virali,
molecole batteriche, RNA a doppio filamento o dai macrofagi attivati dai
linfociti T antigene-stimolati. Molte di queste citochine agiscono sulle cellule
endoteliali e sui leucociti inducendo le prime risposte infiammatorie.
Mediatori e regolatori dell’immunità adattativa: sono citochine prodotte
principalmente dai linfociti T in risposta a specifici antigeni e regolano la
crescita e differenziamento di varie popolazioni linfocitarie o reclutano e
attivano altre cellule effettrici specializzate come neutrofili e eosinofili che
possono eliminare gli antigeni.
Stimolatori dell’ematopoiesi: stimolano la crescita ed il differenziamento di
leucociti immaturi e sono prodotte dalle cellule stromali del midollo osseo.
Nabissi15
Nabissi15
La regolazione negativa dell’azione delle
citochine è svolta da citochine antiinfiammatorie, come IL-10 e TGF-β, prodotti
dai monociti-macrofagi .
Un’altra regolazione negativa è indotta dalla
produzione di ACTH da parte
dell’ipofisi, indotta da fattori ipotalamici
stimolati da IL-1 e TGF (feedback
negativo). ACTH stimola il rilascio di
glucocorticoidi da parte del surrene, i
quali inibiscono la produzione di IL-1 .
Nabissi15
IL-1 and TNF
Nabissi15
IL-1 α –IL-1β sono molecole,
codificate da geni distinti, con il 20%
di omologia di sequenza
aminoacidica ma con attività
funzionale simile. La famiglia IL-1
comprende anche la IL-1a, che è
prodotta dalle stesse cellule che
producono IL-1 ma ha un ruolo
inibitorio. IL-1a si lega allo stesso
recettore di IL-1 ma non attiva la
trasduzione del segnale.
Un altro meccanismio inibitorio è
dato dalla presenza di un falso
recettore (decoy receptor) che ha
affinità di legame per IL-1 ma non
trasduce per segnali intracellulari.
IL-1(α /β/a)
• Interleuchina1
• La fonte cellulare principale di IL-1 sono i fagociK mononucleaK che la
rilasciano dopo sKmolazione da parte di LPS (LIPOPOLISSACARIDI DELLA
PARETEBATTERICA)odialtrecitochine(comeilTNF).InoltreIL-1èprodoPa
da altre cellule come neutrofili, cellule epiteliali (cheraKnociK) e cellule
endoteliali.
IL-1 viene secreta come molecola di 33kD (IL-1α) e può agire come forma
intera o come soPoprodoPo di 18 kD, agendo tuPe e due le forme sullo
stessorecePore.IL-1β,lasecondaformadiIL-1diventaaXvadopotaglioda
partedell’enzimaIL-1β-converKngenzyme(ICE).
Nabissi15
TNF
IlTNFèilprincipalemediatoredellarispostainfiammatoriaaibaPerigram-
negaKviedadaltrimicrobi,sopraPuPorilasciatoinrispostaaendotossine
baPeriche(LPS)edèresponsabiledellecomplicazionialivellosistemico
causatedadiversiKpid’infezioni.
IlTNF(denominatoTNFα)èprincipalmenteprodoPodaifagociKmononucleaK
aXvaK,anchesevienesecretoanchedacelluleNK,linfociKTAnKgene-sKmolaK
emastociK.
IlTNFvieneprodoPocomeproteinadimembrananonglicosilataconun
dominioaminoterminaleintracellulareeunlargodominiocarbossi-terminale
extracellulare.
Nabissi15
Laformadimembranavienepoitagliatadauna
metalloproteasidimembranaerilasciatocome
polipepKdedi17kDcheformanopoiunomotrimero
di51kDchecos=tuiscelaformaa?vadelTNF.Il
TNFsecretoassumeunaformaapiramidelacui
basehaaffinitàperilrecePore,inmanierataleche
ognimolecoladiTNFaXvopossalegaretre
recePoricontemporaneamente.
Nabissi15
• Funzionibiologiche
• La funzione principale del TNF è quella di sKmolare il reclutamento di
neutrofili e monociK nel sito infiammatorio, ma il TNF media anche diversi
effeXsiasuileucociKchesullecelluleendoteliali.
• A livello delle cellule endoteliali il TNF sKmola l’espressione delle molecole
d’adesione, come le selecKne E e le immunoglobuline, inoltre sKmola il
rilasciodichemochinesianelleendotelialicheneimacrofagi.
• Induce la secrezione di IL-1 nei fagociK mononucleaK, la quale agisce poi in
modosimilealTNF.
Nabissi15
IrecePoriperilTNFsonodidueKpi,TNFRIop55(55kD)ilTNFRIIop75(75kD),presenKin
quasituPelecellule.IlTNFRIIaXvatorecluta,alivellocitoplasmaKcounaproteina
adaPatricedenominataTRAF,conconseguenteaXvazionedelpathwayNF-kBedella
proteinad’aXvazione1(AP-1),mentrel’aXvazionedelTNFRIportaalreclutamentodi
proteineadaPatricicheaXvanoilpathwayestrinseco
Nabissi15
AncheilTNFRIcheaXvato,induceapoptosi,puo’ ancheindurreuna
rispostaan=-infiammatoriaedan=apopto=caquandoTRADDlegaTRAF2e
conseguentementeaXvailpathwayNF-kB,medianteaXvazionedellaIkB
chinasidapartediTRAF2.TRAF2(comealtriTRAF)aXvaanchelacascata
delleMAPchinasi,checomportal’aXvazionediJNK,fosforilazionedic-june
formazionedelfaPoretrascrizionaleAP-1compostodac-junec-fos.Questa
aXvazionecomportalatrascrizionedigenicoinvolKnell’infiammazione
(molecoled’adesioneendoteliale,citochine,chemochine)edlatrascrizione
(dapartediNf-kB)digenicoinvolKnelprocessoanKapoptoKco(IAPs).
FENOTIPO PROLIFERATIVO E
RESISTENTE ALL’APOPTOSI
Nabissi15
• RILASCIOECCESSIVODITNF
• induzionedelmeccanismodellafebbrealivelloipotalamico,mediante
sKmolazionedellaproduzionediprostaglandine
• agiscealivellodegliepatociK,sKmolandolasintesiedilrilasciodiproteine
sieriche(proteinaamiloideAefibrinogeno),inducendolarisposta
infiammatoriadifaseacuta.
• ProduzioneprolungatadiTNFinducecachessia,mediantesoppressionedella
sensazioned’appeKtoeriduzionedellasintesidilipasi(enzimiresponsabilidel
rilasciodilipoproteine,necessariealmetabolismoKssutale).
• TNFcausatrombosiintravascolari,riducendoleproprietàanKcoagulanK
dell’endoteliomediantelosKmolodifaPoricoagulanKedinibizionedifaPori
anKcoagulanK.
Nabissi15
IL-6
IL-6 interagisce con un recettore costituito da due catene, la catena gp130 che è
comune ad altre citochine ed una catena IL-6R che è specifica per IL-6.
IL-6R è in grado di creare un complesso attivo (in forma solubile o legato alla
membrana), dopo legame a IL-6, con il gp130, permettendo anche alle cellule che
non possiedono Il-6R di rispondere a IL-6.
Questo sistema permette, ad esempio, alle cellule endoteliali che non esprimono
IL-6R di rispondere a IL-6, che stimola la produzione di molecole d’adesione e
chemochine.
Il complesso IL-6R/IL-6/gp130 attiva una cascata intracellulare che attiva una
tirosin-chinasi (JAK) che fosfoforila il recettore. In forma fosforilata,
IL-6R/
IL-6/gp130 attira dei fattori trascrizionali (STAT) che vengono a loro volta
fosforilati e sotto forma di eterodimeri migrano nel nucleo.
Nabissi15
Nabissi15
Interferone di tipo I (IFN)
Media la risposta immunitaria innata contro infezioni virali ed in particolare
viene sintetizzato in presenza di RNA a doppio filamento, ed è composto da due
proteine IFN-α IFN-β prodotti da due geni distinti.
Legano lo stesso tipo di recettore, attivando una serie di risposte (attraverso il
pathway JAK/STAT) che inducono:
blocco della replicazione virale, mediante stimolazione della sintesi di enzimi
(oligoadenilato sintetasi) che disturbano la replicazione virale incrementano
l’espressione di molecole MHC (complesso maggiore d’istocompatibilità) di tipo
I, che riconoscono antigeni virali e li presentano ai linfociti CD8+ che uccidono le
cellule infettate
Nabissi15
Interferone-γ (IFN-γ)
E’ prodotto dalle cellule NK, dai linfociti CD4+Th1 e dai CD8+ e svolge la sua
attività biologica legandosi a recettori che inducono l’attivazione di STAT1, il
quale induce la trascrizione di geni delle molecole MHC e di enzimi responsabili
della produzione di sostanze ad azione antimicrobica.
INF-γ attiva i macrofagi
Promuove il differenziamento dei linfociti Th1 e inibisce la proliferazione delle
cellule Th2
Stimola la produzione di IgG da parte delle plasmacellule
Attiva i neutrofili
Nabissi15
Nabissi15
Interleuchina 12 (IL-12)
E’ uno dei primi mediatori della risposta precoce dell’immunità innata ed induce
la risposta immunitaria cellula-mediata. La sua azione principale sulle cellule del
sistema immunitario riguarda l’induzione della produzione di IFN-γ da parte
delle cellule T.
IL-12 è presente in due forme di 35 e 40 kD (forma attiva) ed agisce attraverso un
recettore di membrana composto da catene β1 e β2, in cui la subunità β2 è
responsabile della trasmissione del segnale mediante il pathway JAK/STAT.
L’azione biologica della IL-12 consiste in:
stimolare la produzione di IFN-γ nei linfociti e nelle NK
stimolare la differenziazione dei linfociti T-helper CD4+ in Th1 (produttori di
IFN-γ) induce l’attivazione dei linfociti T CD8+ citolitici
Nabissi15
Nabissi15
CITOCHINE ANTI-INFIAMMATORIE
Interleuchina 10 (IL-10)
Inibisce l’azione dei macrofagi
attivati, controllando cosi’ la
risposta infiammatoria cellulomediata
Inibisce la produzione di IL-12
Inibisce l’espressione delle
molecole MHC di tipo II, inibendo
cosi’ l’attivazione dei linfociti T e
inducendo la terminazione della
risposta cellula-mediata
Nabissi15
• TGF-β
• Inibisce la proliferazione e l’attivazione dei
linfociti e di altri leucociti ed è prodotto, dalle
cellule T-antigene stimolate e dai fagociti, come
precursore ed attivato da taglio proteolitico.
• Inoltre TGF-β stimola la produzione di IgA
(necessarie per l’immunità delle mucose),
stimola la sintesi di proteine della matrice
extracellulare (collagene), di metalloproteasi e
d’integrine
Nabissi15
RISPOSTA POLARIZZANTE
Quando l’agente lesivo comporta una risposta immunitaria specifica si ha la presenza di
linfociti che producono citochine con conseguente attivazione dei macrofagi.
L’interazione dei macrofagi con linfociti T Helper di tipo 1 o 2 comporta una
polarizzazione della risposta.
RISPOSTA POLARIZZANTE DI TIPO 1:
Presenza di linfociti Th1 che secernono INFγ che agisce su diversi tipi di popolazioni
cellulari stimolando il reclutamento, a livello citoplasmatico, di due protein chinasi JAK1 e
JAK2 che fosforilano il fattore trascrizionale STAT1.
STAT1 attiva la trascrizione di molti geni coinvolti nella :
- attivazione macrofagica (induzione di recettori di membrana di tipo opsoninico)
- induzione del gene della NADPH ossidasi, responsabile della produzione di ROI
- Induzione del gene della Nitrico sintasi (iNOS)
- Induzione del gene di IL-1, IL-6 e TNF
Nabissi15
RISPOSTA POLARIZZANTE DI TIPO 2
In particolare nelle risposte infiammatorie di tipo allergico, la presenza di linfociti di tipo
Th2 è dominante, con conseguente secrezione di IL-4,
IL-13, IL-5. Queste citochine attivano le protein chinasi JAK1 e JAK3 che fosforilano il
fattore trascrizionale STAT6.
STAT6 oltre ad attivare geni coinvolti con le proprietà delle IL-4 e IL-13 inducono i Th2 ,
inibiscono IFN-γ, determinano la produzione di IgE, la sintesi del decoy receptor per IL-1 e
l’induzione di recettori per il mannosio e scavenger (recettori non opsoninici)
Nabissi15
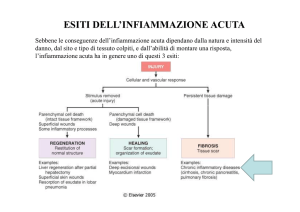
ESITI
DELL’INFIAMMAZIONE
ACUTA
Esiti dell’infiammazione acuta
1) Completa risoluzione: rappresenta l’esito normale quando il danno è limitato
2) Guarigione tramite sostituzione con tessuto connettivo (fibrosi)
3) Infiammazione cronica
Completa risoluzione: rappresenta l’esito normale quando il danno è limitato
La risoluzione consiste nella neutralizzazione o nella perdità dell’attività dei
mediatori, con il ripristino della normale permeabilità vascolare, la cessazione
dell’infiltrazione leucocitaria, la morte dei neutrofili e la rimozione del liquido e
proteine dalla sede del danno.
Guarigione tramite sostituzione con tessuto connettivo (fibrosi).
Avviene dopo un danno tissutale di notevole entità, quando la lesione
infiammatoria colpisce i tessuti che non sono in grado di rigenerare o quando vi è
un’essudato ricco in fibrina. In molte infezioni si ha la formazione di pus
(infiltrato di neutrofili e liquefazione dei tessuti). Il tessuto distrutto è poi
riassorbito e sostituito da fibrosi.
Progressione della risposta tissutale verso l’infiammazione cronica
L’infiammazione cronica puo’ seguire quella acuta, oppure la risposta
infiammatoria puo’ definirsi cronica fin dall’inizio. La transizione da acuta a
cronica avviene quando la risposta infiammatoria non puo’ essere risolta.
Tipi morfologici dell’infiammazione
acuta
Infiammazione sierosa: caratterizzata
dalla fuoriuscita di liquido a scarso
contenuto proteico che a seconda della
sede di lesione deriva dal plasma o
dalle secrezioni delle cellule
mesoteliali che rivestono le cavità
(pericardio, pleura o peritoneo). Il
liquido fuoriuscito viene denominato
versamento.
Infiammazione fibrinosa: in presenza di
lesioni piu’ gravi con fuoriuscita di proteine e
fibrinogeno, dalle lesioni dei vasi, si forma
fibrina che viene depositata nello spazio
extracellulare. Gli essudati fibrinosi possono
essere asportati per fibrinolisi ed asportazione
di altri detriti da parte dei macrofagi. Quando
la fibrina non puo’ essere rimossa essa puo’
stimolare l’accrescimento di fibroblasti e vasi
sanguigni, portando quindi alla formazione di
cicatrici.
Infiammazione suppurativa o purulenta:
produzione di grandi quantità di pus
costituito da neutrofili, cellule necrotiche
e liquido. Gli ascessi, ad esempio, sono
raccolte localizzate di tessuto
infiammatorio purulento.
Ulcere: lesione locale della superficie di
un organo o tessuto, prodotta
dall’eliminazione (distacco) di tessuto
infiammatorio necrotico. L’ulcerazione
si verifica quando la necrosi tissutale e
l’infiammazione ad essa conseguente
sono localizzate in prossimità o sulla
superficie del tessuto.
Infiammazione cronica
L’infiammazione cronica è considerata una infiammazione di durata prolungata in
cui procedono contemporeanamente l’infiammazione attiva, la distruzione del
tessuto ed i tentativi di riparazione.
Spesso ha un’esordio insidioso, in quanto si manifesta come una risposta debole,
persistente e spesso asintomatica (artrite reumatoide, aterosclerosi, tubercolosi e
patologie polmonari croniche).
Cause dell’infiammazione cronica
Infezioni persistenti: agenti virali, batterici o funginei che sono dotati di bassa
tossicità ed evocano una risposta immunitaria detta ipersensibiltà ritardata.
Prolungate esposizioni ad agenti potenzialmente tossici, esogeni ed endogeni:
inalazione di materiale inorganico che genera malattie infiammatorie, ad esempio
il silicio che provoca la silicosi (infiammazione cronica bronchiale)
Autoimmunità: reazioni autoimmunitarie contro i tessuti dell’individuo stesso, che
portano a malattie autoimmuni.
BRONCOPOLM. TUBERCOLARE
TUBERCOLOSI
micobatteri
T) granuloma tubercolare
B) bronchi invasi da macrofagi
Infiammazione cronica
L’accumolo di macrofagi nell’infiammazione cronica è mediato da:
Reclutamento di monociti dal circolo: che derivano dall’attivazione delle
molecole d’adesione e da fattori chemiotattici
Proliferazione locale di macrofagi: avviene dopo la loro migrazione dal torrente
sanguigno. E’ un processo predominante in alcune infiammazioni croniche.
Immobilizzazione dei macrofagi: causata da citochine e lipidi ossidati
I prodotti, dei macrofagi attivati, servono ad eliminare gli agenti lesivi ed a iniziare
i processi riparativi, ma sono anche responsabili del danno tissutale.
Infatti la distruzione del tessuto è uno dei segni caratteristici dell’infiammazione
cronica
Riparazione del tessuto attraverso sostituzione con tessuto connettivo che si realizza con
proliferazione di piccoli vasi e fibrosi.
DEPOSIZIONE DI COLLAGENE
all’inizio connettivo lasso
poi sempre più denso
Infiammazione granulomatosa
Formazione di granulomi quando l’agente lesivo agisce in modo localizzato
formando un nodulo di tessuto infiammatorio ben circoscritto. Il nodulo è formato
dal materiale inerte non eliminabile dal sistema dei fagociti e non immunogenico.
Quindi si ha assenza di risposta immunitaria specifica, ma una reazione fibrosa che
tende ad isolare l’agente lesivo.
Agenti antigenici sviluppano granulomi immunologici, spesso causati da agenti
lesivi virali, inglobati dai macrofagi ma non annientati. Solo una risposta
immunitaria specifica puo’ risolvere la causa, quindi si forma un infiltrato
linfocitario caratteristico di questa forma di granuloma.
Si ha reclutamento di linfociti ed attivazione mediata dalla presentazione
dell’antigene ad opera di fagociti, fino ad eradicazione dell’agente lesivo.
Processo Riparativo
La riparazione tissutale è un fenomeno complesso che comprende eventi cellulari
migratori, proliferativi, apoptotici e/o differenziativi oltre ad un ruolo diretto dei fenomeni
diretti dei mediatori dell’infiammazione. La riparazione comprende due processi:
rigenerazione e reintegrazione connettivale.
La rigenerazione consiste nel ripristino delle cellule funzionali del tessuto leso,
la reintegrazione connettivale consiste nella sostituzione delle cellule danneggiate con
tessuto connettivale e perdita di funzionalità (fibrosi).
Nei processi riparativi sono coinvolte tutte le cellule presenti ed attive nel processo
infiammatorio
Rigenerazione
Restituzione del tessuto perso
Tessuti con alta capacità proliferativa = tessuti labili (cellule ematopoietiche,
cellule epiteliali della pelle e del tratto gastrointestinale che rigenerano da
cellule staminali)
Tessuti quiescenti = tessuti stabili hanno normalmente bassi livelli di
replicazione, ma possono andare incontro a rapida divisione cellulare quando
stimolati. ( cellule parenchimali del fegato, rene, pancreas, cellule di origine
parenchimale come linfociti, fibroblasti cellule muscolari liscie, endoteliali)
Reintegrazione connettivale
La cicatrizzazione è una risposta fibro-protettiva che risolve il danno piu’
che restituire la funzionalità tissuttale e coinvolge i seguenti processi:
Induzione di una risposta infiammatoria che rimuove il tessuto morto e
danneggiato
Proliferazione di cellule del tessuto parenchimale e connettivo
Angiogenesi e formazione di tessuto granulomatoso
Sintesi di proteine della ECM e deposito di collageno.
Rimodellamento tissutale
Contrazione della ferita
Formazione della cicatrice con perdita della funzionalità tissutale
ECCESSO DI RIPARAZIONE: GENESI DELLA FIBROSI
RIPARAZIONE DELLE FERITE DERMO-EPIDERMICHE
GUARIGINE DI PRIMA E SECONDA INTENZIONE
Guarigione di prima intenzione: ferite con margini giustapposti (incisione
chirurgica)
Guarigione di seconda intenzione: ferite con margini opposti irregolari
Tessuto granulare con consistente neo-vascolarizzazione, presenza di
magrofagi (causa presenza d’infezioni), fibriblasti e perdita della connessione
della ECM
Incremento dell’accumulo di collagene, cicatrizzazione, zone dense di
collagene e fibre di elastina.
Prolungarsi delle fasi di guarigione
Riparazione delle ferite dermo-epidermiche
Entro pochi minuti formazione di coagulo
contenente fibrina e fibronectina, con formazione di
crosta (escara) nella parte superficiale.
Proliferazione dei cheratinociti
Rilascio PDGF e TGF-β che attivano il reclutamento
di piastrine e neutrofili, con inizio del processo
infiammatorio.
Dopo 24/48 ore la rigenerazione epiteliale è
completa, con invasione del coagulo sottostante da
parte di
neutrofili e macrofagi che sono
responsabili dell’eliminazione dei detriti cellulari e
del rilascio di fattori di crescita.
A 72 ore la fase di proliferazione inizia con il
reclutamento di fibroblasti stimolati da TGF-β e FGF.
Sintesi di collagene, vascolarizzazione.
Entro una settimana si ha la regressione del
componente leucocitario e dell’angiogenesi e nel
periodo succesivo completa formazione della
cicatrice, che puo’ portare in casi di mal
rigenerazione alla formazione di cisti epitelioidi,
cicatrice ipertrofica o cheloidi (cicatrici deturpanti)
Difetti della
cicatrizzazione
ulcere
Deinescenza
cheloidi
Eccessiva
cicatrizzazione
contratture
Rigenerazione epatica
Le cellule coinvolte nella rigenerazione epatica sono di tre tipi:
– Epatociti maturi che vanno incontro a cicli proliferativi limitati
– Cellule progenitrici duttali che possono differenziarsi in epatociti o cellule
biliari
– Cellule staminali periduttali, molto rare ma pluripotenti con alto potenziale
differenziativo
– La rigenerazione epatica è attivata principalmente da TNF-α che induce
l’espressione di IL-6 che attiva STAT 3 seguendo la via di NfkB con
induzione di geni che codificano per prodotti coinvolti nell’attivazione del
ciclo cellulare (c-jun, c-fos, c-myc) e di geni anti-apoptotici (BclXL).
Altri fattori di crescita, come HGF (hepatocytes growth factor) inducono
l’espressione di geni che attivano il ciclo cellulare e blocco dell’apoptosi.
Nel casi di danno ridotto la riparazione è caratterizzata da ripresa della
funzionalità d’organo, altrimenti si ha reintegrazione connettivale con
FIBROSI EPATICA, che puo’ compromettere la funzionalità dell’organo
con evoluzione verso la CIRROSI EPATICA
CIRROSI EPATICA
CIRROSI A LIVELLO MACROSCOPICO E MICROSCOPICO
Fegato cirrotico
Fegato normale
Le cellule del Kupffer secernono citochine che attivano le cellule stellate le
quali possono differenziarsi in miofibroblasti deponendo i componenti della
matrice extracellulare (collagene, ecc..) diventando responsabili del processo
di fibrosi
Riparazione dell’ulcera peptica
• La mucosa gastrointestinale che è esposta continuamente all’azione lesiva della
secrezione cloridro-peptica, mantiene la sua integrità morfo-funzionale grazie
all’azione di fattori come il muco e bicarbonato, EGF, TGF-α, HGF e PDGF
che consentono di mantenere un’equilibrio fra riparazione e danno.
• Il danno della mucosa gastrica puo’ consistere in una desquamazione
superficiale o una lesione piu’ profonda (ulcera peptica) fino ad arrivare ad
interessare il peritoneo (ulcera perforata).
• Il danno profondo comporta lesione dei componenti tissutali sottoepiteliali come
la musculatura, i nervi ed i vasi sanguigni, quindi il processo riparativo risulta
piu’ complesso.
Ulcera
duodenale
Ulcera gastrica
con sangue
digerito sulla
superficie
tissutale
Helicobacter pylori
H. Pylori si adatta nell’ambiente acido dello stomaco, mostrando una serie di
aspetti caratteristici CHE GLI PERMETTONO di attraversare il muco, nuotare ed
orientarsi nel muco, aderire alle cellule epiteliali della mucosa, evadere dalla
risposta immunitaria, colonizzare e proliferare
Esprime proteine che codificano per geni variabili
come:
• enzimi che sono in grado di modificare la struttura
antigenica delle molecole della superficie batterica.
• Controllare l’entrata di DNA esterno
• Regolare la motilità dei flagelli
H.Pylori produce ureasi che idrolizzano l’urea in CO2 e ammoniaca creando un ambiente
basicointornoallacoloniaba:ericamaaumentandol’aciditàdellostomaco.
H.pyloriesprimela95-kD vacuolaKngcytotoxin
(VacA), che secreta nello stomaco ha come
bersaglio le membrane mitocondriali delle
cellule della mucosa, inducendo il rilascio di
citocromoCediconseguenzaapoptosi.
Un’altraproteinaprodoPadaH.pylorièCagAcheaXvaKrosinchinasinellecelluleospite
inducendorilasciodifaPoridicrescitaecitochine,inducendoproliferazioneestato
infiammatorio.
Riparazione del miocardio
• Il tessuto cardiaco è composto principalmente da cardiomiociti, cellule con
capacità contrattili differenziate e non in grado di dividersi.
• Quindi lesioni del tessuto cardiaco comportano una guarigione mediante
sostituzione con tessuto cicatrizzante.
• Questo comporta aumento del collagene e neovascolarizzazione che possono
coinvolgere zone non direttamente colpite dall’infarto, instaurando
un’insufficienza ventricolare sinistra cronica.
• La capacità contrattile dei cardiomiociti è indotta da angitensina II che deriva
dalla conversione dell’angiotensinogeno, nel cuore infartuato, attuata da renina
e catepsina D. L’angiotensina I prodotta è poi convertita in angiotensina II da
ACE (angiotensin-converting enzyme).
• I livelli di ACE ed angiotensina II aumentano nell’infarto.
• Recentemente sono state identificate cellule staminali adulte in grado di
originare cardiomiociti maturi, celllule muscolari liscie e cellule endoteliali.
Rappresentazione della progressione della necrosi da miocardio, dopo occlusione
della coronaria
REAZIONI SISTEMICHE DELL’INFIAMMAZIONE
Nabissi15
Ipersensibilità
Tipo I
• Ipersensibilità Immediata o Anafilassi
Tipo II
• Ipersensibilità mediata da anticorpi citotossici
Tipo III
• Ipersensibilità mediata da immunocomplessi
Tipo IV
• Ipersensibilità di tipo ritardato o cellulo-mediata
Le reazioni d’ipersensibilità sono definite come risposte del sistema
immunitario contro antigeni che sono di per se innocui ma che possono
generare dei danni attraverso le due vie del sistema immunitario
(anticorpi o linfociti T effettori).
Questa risposta immunitaria permette di classificare le risposte
d’ipersensibilità in ANTICORPO-MEDIATA o CELLULO-MEDIATA.
Entrambe le risposte immunitarie non si verificano al primo contatto con
l’antigene, in quanto deve esserci una fase di sensibilizzazione che
precede il manifestarsi della reazione.
Un’altra caratteristica è che le reazioni anticorpo dipendenti tendono a
svilupparsi velocemente, in quanto gli anticorpi specifici (all’antigene)
sono molecole preformate ed immediatamente disponibili. Quella di tipo
IV, viene definita ritardata in quanto dipende dalla mobilizzazione delle
cellule che dopo essere state attivate richiedono circa 18-20 ore per
raggiungere il numero sufficiente per contrastare l’antigene
Quattro tipi di ipersensibilità
Allergie ed Ipersensibilità
Le allergie sono delle reazioni d’ipersensibilità. Una risposta di IgE
verso antigeni innocui.
L’Ipersensibilità è una reazione immunitaria verso un antigene innocuo
che provoca un danno cellulare
Un antigene che causa allergia è detto allergene
Reazione immediata che avviene in soggetti geneticamente
predisposti pochi minuti dopo la seconda esposizione ad un
antigene (allergene) e coinvolge le IgE
Patologie:
• Asma
• Rinite allergica
• Eczema
• Orticaria
• Anafilassi
Allergie: Ipersensibilità di tipo I mediata da IgE sui mastociti
Degranulazione dei Mastociti attivata dall’antigene (allergene):
legame tra FcεR ed IgE
Le IgE sono prodotte dal
tessuto linfoide al primo
contatto con un allergene
Quando interviene una
seconda esposizione
l’antigene si combina con le
IgE precedentemente prodotte
e legate ai mastociti tramite la
regione Fc, provocando il
rilascio dei mediatori chimici
contenuti.
Ipersensibilità di tipo II o Citotossica
E’ una reazione mediata da anticorpi (IgG o IgM) che
legano antigeni di superficie delle cellule ospiti.
Apteni - sostanze capaci di rendere antigenica una
proteina innocua.
Bersagli comuni sono i Globuli Rossi e le Piastrine.
L’anticorpo complessato all’antigene si lega sulla
superficie cellulare fissando ed attivando il complemento.
L’attivazione del complemento porta alla lisi cellulare con
conseguente anemia emolitica nel caso dei globuli rossi
ed emorragia nel caso delle piastrine.
Altro esempio è la trasfusione di sangue incompatibile.
Ipersensibilità di tipo III o da immunocomplessi
E’ caratterizzata dalla formazione di
immunocomplessi che si formano in circolo od a
livello delle membrane basali dei vasi.
Gli immunocomplessi sono costituiti da antigene,
IgG o IgM e complemento.
L’attivazione da parte degli IC del complemento comporta la formazione di
C3a e C5a che inducono il rilascio di sostanze vasoattive, aumento della
permeabilità vasale nelle prima fase (antigene in eccesso), nella fase
successiva (equilibrio fra antigene e IC) gli IC tendono a precipitare ed a
essere fagocitati.
La vasculite consiste in accumulo sottoendoteliale a livello dell’aorta e dei vasi
polmonari di cellule mononucleate, necrosi della tonaca media (causata
principalmente da neutrofili) e distruzione della membrana elastica interna dei
vasi a medio calibro (cuore). La glomerulonefrite è caratterizzata da
proliferazione di cellule endoteliali con conseguente restringimento del lume
capillare e depositi densi lungo la membrana basale. Le lesioni flogistiche
colpiscono i glomeruli con conseguente proteinuria, ematuria, ipertensione,
edema fino all’insufficienza renale.
Ipersensibilità di tipo IV ritardata o cellulo-mediata
Richiede la presenza di
Linfociti T sensibilizzati (da qui
il cellulo-mediata) ed un
antigene.
Si manifesta dopo 24-48 ore
dalla stimolazione (da qui il
ritardata).
Viene provocata da alcuni batteri:
• Bacillo tubercolare,
• Brucella,
• Virus (morbillo e parotite),
• Funghi
• Punture d’insetto,
• Sostanze chimiche e
farmacologiche.
Alla base della reazione di tipo IV c’è una reazione tra antigene,
linfociti T sensibilizzati all’antigene e macrofagi
L’antigene reagisce con un linfocita sensibilizzato stimolando la
produzione di citochine (linfochine) che:
• Richiamano monociti e macrofagi,
• Li trattengono nel sito interessato,
• Li attivano.
I Macrofagi attivati, rilasciano enzimi lisosomiali e (citochine)
che causano:
• la distruzione del tessuto,
• infiammazione
• ulteriore richiamo di macrofagi.
FEBBRE
Meccanismi di termoregolazione
La temperatura corporea dell’uomo è mantenuta in
condizioni fisiologiche entro livelli costanti (37 °C±1),
mediante un sistema di termoregolazione che
conprende diversi meccanismi fra cui i sensori di
temperatura centrali e periferici, un centro neuronale
regolatorio ed effettori che attraverso reazioni
chimico-fisiche sono in grado di variare i livelli della
temperatura corporea.
I sensori rilevano le informazioni derivanti dalla periferia
corporea (terminazione nervose) e dal sangue, le quali
vengono percepite dai neuroni ipotalamici (neuroni W) e da
altri tipi di cellule attivando risposta di termodispersione o
termoproduzione. La termodispersione avviene
principalmente mediante evaporazione (eliminazione di
vapore acqueo mediante la respirazione) o la sudorazione,
mentre se l’ambiente esterno è freddo, l’ipotalamo attiva una
risposta che tende prima a conservare e poi a produrre calore
nei tessuti periferici.
L’aumento di temperatura corporea si puo’ distinguere in
due processi: ipertermia e febbre che differiscono in quanto
nella febbre si ha una alterazione del centro di
termoregolazione, mentre nell’ipertermia non si ha
attivazione di questo centro. L’ipertermia si verifica in
condizioni di lavoro fisico eccessivo o nel colpo di calore
favorito da particolari condizioni climatiche (caldo e
umidità), farmaci (cocaina, LSD), che rendono difficile la
termodispersione, portando in alcune condizioni a
temperature corporee anche letali.
La febbre caratteristica nei processi infiammatori è causata
da una diminuita termodispersione e dall’attivazione
dell’espressione di specifici geni (che sono coinvolti nel
processo di neotermogenesi) ed è indotta da fattori pirogeni
endogeni (citochine pirogene) e prostaglandine (PG). La
diminuita termodispersione (termoconservazione) è attuata
mediante vasocostrizione superficiale con vasodilatazione
degli organi interni, mentre la neotermogenesi avviene
mediante aumento del metabolismo di alcuni tessuti,
fenomeni entrambi regolati dall’ipotalamo stimolato dai
pirogeni interni (IL-1, IL-6, TNF-α).
PATOGENESI DELLA FEBBRE
I pirogeni attivano la sintesi di PGE2,
all’attivazione del nucleo sopraottico e
paraventricolare che rilascia peptidi che
attivano l’ipofisi ed i centri vasomotori.
Ad esempio a livello dell’adenoipofisi si
ha il rilascio dell’ormone stimolante le
tireotropine (TSH), che agisce a livello
tiroideo stimolando la sintesi e rilascio di
T3 e T4. Quest’ultimi attivano le ATP-asi
i o n i c h e c h e c o n s u m a n o AT P
producendo calore, le termogenine I e II
che attivano produzione di calore e la
lipolisi e la glicolisi necessari a
riprodurre l’ATP.
I pirogeni si possono suddividere in esogeni ed endogeni, i
primi sono principalmente le endotossine batteriche in grado di
evocare la risposta febbrile, mentre i secondi sono
principalmente le citochine pirogene e la PGE2 che
direttamente o indirettamente agiscono a livello ipotalamico
mediante la stimolazione della sintesi di cAMP, che permette
un resettaggio del centro termoregolatore verso un livello di
temperatura maggiore. I pirogeni endogeni non sono solo
prodotti in presenza di endotossine, virus od altri parassiti, ma
anche quando si hanno danni endogeni come necrosi
cellulare, infarto, ictus, ecc..
La febbre viene caratterizzata da dei profili termici qualitativi e
quantitativi (curva termica) che possono essere di valido aiuto
nel diagnosticare la causa dello stato febbrile.
La curva termica si puo’ distinguere in tre fasi:
innalzamento, fastigio e defervescenza. La prima è
determinata dal rialzo termico con caratteristici brividi
e contrazioni muscolari, il fastigio si caratterizza per i
valori termici raggiunti (bassi, medi, alti, altissimi), la
durata (ore, giorni, anni) e per come vengono
mantenuti (continui o discontinui). La defervescenza
indica la scomparsa della febbre e puo’ essere
rapida (per crisi) o lenta (per lisi).
Fase del rialzo termico: sensazione di freddo, brividi, pallore
cutaneo con conseguente vasocostrizione (riduzione della
termodispersione).
Fase del fastigio: quando i centri termoregolatori si posiziona
a temperature maggiori di 37 °C, scompare la sensazione di
freddo e compare quella di calore. Si attiva con l’aumento di
PGE2 e si mantiene per tutto il periodo in cui si ha produzione
di PGE2 in eccesso.
Fase di defervescenza: sensazione di caldo ed abbassamento
della temperatura, con riduzione delle citochine
infiammatorie e di conseguenza di PGE 2 . I centri
termoregolatori riportano lentamente (per crisi) o
velocemente (per lisi) il corpo a livelli di temperatura normale
attivando la sudorazione per facilitare l’abbassamento della
temperatura.
Per quanto riguarda il trattamento dello stato febbrile
si utilizzano, quando necessario, antipiretici che
agiscono inibendo le cicloossigenasi (COX) che sono
responsabili della produzione di PGE2 come
l’aspirina o altri farmaci non steroidei, oppure farmaci
steroidei (glucocorticoidi) che inibiscono la fosfolipasi
A2 responsabile del rilascio di acido arachidonico
necessario per la sintesi di PGE2.
TIPI DI FEBBRE
Febbre continua: rialzo termico che si mantiene costante
durante i periodo di fastigio, classica nelle malattie infettive.
Febbre remittente: oscillazioni di temperatura durante il
periodo di fastigio, con variazioni non superiori ad 1 °C,
senza mai raggiungere la defervescenza. Tipica del tifo.
Febbre continua-remittente: aumenti di temperatura di 1°C
durante il periodo di fastigio senza mai raggiungere la
defervescenza.
Febbre intermittente: rialzi di temperatura seguiti da
temperatura normale con alternanza spesso regolare. Si
indica quotidiana quando il rialzo è mattutino per poi ridursi
durante la giornata.
Il significato patologico della febbre
Aspetti positivi
Febbre = sintomo = indice di evolutività della malattia causale
Aumenta le difese dell’organismo, in particolare la produzione di heat
shock proteins (HSPs) dette anche chaperons, che proteggono proteine
cellulari dal danno
Aspetti negativi
L’ipertermia stessa è problemi di tolleranza (astenia + intensa,
compl. neurologiche, disidratazione…)
I pirogeni stessi possono indurre gravi complicanze generali = la
sindrome maligna
FEBBRE EMORRAGICA
Nabissi15
Classificazione del virus Ebola
Ordine: Mononegavirales
Filoviridae
• Ebolavirus – 5 viruses/species
– Ebola (Zaire)
– Sudan
– Bundibugyo
– Tai Forest
– Reston
• Marburgvirus
• Cuevavirus
Struttura del virus
§ Il virione ha una struttura tubulare
variabile
§ I virioni misurano
complessivamente 80 nm di
diametro e circa 1000/1400 nm di
lunghezza
§ Al centro del virione è presente
il”nucleocapside” composto da
RNA genomico e da un complesso
proteico NP, VP35, VP30 ed L
§ E’ presente una glicoproteina
(GP) virale derivante dalla
membrana della cellula ospite
§ Nella matrice situata tra
membrana e nucleocapside si
trovano le proteine virali VP40 e
VP24
Genoma
•
Ciascun virione contiene una molecola anti-senso di RNA (sssRNA)
composta da due estremità: 3’ e 5’
•
Il genoma codifica per 7 proteine strutturali disposte in linea:
- 4 strutturali (VP 30, VP35, NP e una polimerasi [ L] )
- 3 associate alla membrana ( GP, VP40 e VP24)
•
La regione codificante è:
3’-LEADER-NP-VP35-VP40-GP/sGP-VP30-VP24-TRAILER-5’
Replicazione citoplasmatica
La replicazione citoplasmatica avviene
attraverso:
• Aggancio ai recettori tramite GP ed
endocitosi del virus in vescicole
della cellula ospite
• Il virione entra negli endosomi
mediante macropinocitosi
• Fusione della membrana dei virus
con membrana vescicolare e
rilascio del ribonucleotide nel
citoplasma
• Trascrizione sequenziale,
maturazione degli mRNA virali
tramite aggiunta di basi e
poliadenilazione con polimerasi
• Replicazione
• Il ribonucleotide interagisce con le
proteine della matrice e attraverso
gemmazione rilascia il virione
Serbatoio e trasmissione all’uomo
q
Probabilmente i pipistrelli della frutta sono il serbatoio naturale del virus Ebola
q I pipistrelli possono infettare altri animali tra cui scimpanzé, gorilla e antilopi
q Gli esseri umani maneggiano e consumano carne cruda di questi animali
selvatici
q Infezione umana passa da persona a persona
Patogenesi - Come il virus causa la malattia
q Il virus entra nel corpo umano tramite sangue e fluidi corporei infetti
(urina, feci, vomito, saliva,sudore, sperma e latte materno) che
entrano in contatto diretto con mucose, ferite aperte e via
parenterale (punture)
q Virus replica preferenzialmente in monociti / macrofagi e cellule
dendritiche che facilitano la diffusione del virus in tutto il corpo
attraverso il sistema linfatico.
q Altre cellule vengono infettate in via secondaria, provocando una
rapida crescita virale in epatociti, cellule endoteliali e tessuti epiteliali
q Nella fase progressiva della malattia si verifica una forte cascata
infiammatoria con rilascio di citochine pro-infiammatorie tra cui
interferone, interleuchine (IL-2, IL-6, IL-8, IL-10) e fattori di necrosi
tumorale (TNF-α)
Patogenesi – La risposta infiammatoria
q Provoca danno endoteliale, aumento della permeabilità vascolare e
shock.
q A sua volta ciò comporta disfunzioni a livello di un singolo organo o
multi-organo
q Diffusa coagulazione intravascolare disseminata(CID) con consumo
di piastrine e fattori della coagulazione che portano a emorragia.
q Presenza di IgM a due giorni e IgG a 5-8 giorni dall'infezione.
Risposta immunitaria si correla con la sopravvivenza del paziente
infetto
q Aumento della probabilità di guarigione in coloro che attivano tale
risposta immunitaria e sopravvivono per un periodo > 1 settimana
Manifestazioni cliniche
q Il periodo di incubazione del virus è solitamente di 8-10 giorni (range
2-21)
Sintomi precoci: (da 0 a 3° giorno)
q comparsa improvvisa di febbre> 38.6°C
q sintomi simil-influenzali: mialgia, artralgia, malessere e brividi, mal di
gola,difficoltà di deglutizione,astenia, cefalea
Sintomi successivi (da 3° a 12° giorno)
q nausea, vomito, dolore addominale, diarrea
q dolore toracico, difficoltà respiratorie e tosse
q sintomi SNC: mal di testa, confusione e coma
Manifestazioni cliniche
Sintomi gravi (intorno al 5° giorno)
q ipotensione, edema periferico
q manifestazioni emorragiche (interne/esterne) si sviluppano nel 40- 50% dei
pazienti malati
q rush cutanei, ematomi, petecchie ed ecchimosi, epistassi, emottisi,
ematuria, ematemesi, e massiccia perdita di sangue a livello GI
q disidratazione e perdita di peso
q disfunzioni a livello di reni e fegato
Anomalie in esami di laboratorio confermano:
q trombocitopenia e leucopenia
q transaminasi elevate (AST> ALT), amilasi, D-dimeri
q Riduzione di albumina
Sintomi terminali
q Tachipnea, anuria, esaurimento fisico, shock ipovolemico ed insufficienza
multi-organo
Fattori di rischio a confronto
Sono fattori di rischio
Non sono fattori di rischio
§ Esposizione a oggetti e/o ambienti
contaminati con secrezioni infette
§ Contatti frequenti tra malati e parenti
§ Precarie condizioni igienico-sanitarie
§ Allattare al seno bambini dopo aver
contratto la malattia
§ Partecipare alla cerimonia funebre
toccando il cadavere di una persona
deceduta a causa dell’infezione
§ Fare sesso con una persona infetta
o con persona guarita dalla malattia
da poco tempo
§ Riutilizzare siringhe con aghi non
sterili
§ Entrare in contatto con persone
che non presentano sintomi
§ Viaggiare in aereo con persone
che hanno sviluppato i sintomi solo
successivamente
§ Puntura di zanzare
§ Trasmissione aerea (non possibile)
Prevenzione e Gestione dei pazienti
Il personale sanitario deve
Ø Indossare dispositivi di protezione individuali(DPI)
Ø Utilizzare cautela nella vestizione e rimozione
Ø Utilizzare corrette misure di controllo e
sterilizzazione
Ø Isolare i pazienti con malattia sospetta e quelli con
malattia accertata
Le popolazioni delle aree endemiche devono
Evitare contatto con fluidi corporei infetti
Non maneggiare oggetti probabilmente contaminati
Evitare funerali e cerimonie di sepoltura
Evitare contatti con pipistrelli e di mangiare carne di
scimmia (bush-meat)
Ø Praticare un’attenta igiene delle mani e del corpo
Ø
Ø
Ø
Ø