Approfondimenti 07-11
A7: La struttura elicoidale
In una catena polimerica é possibile individuare l'eventuale presenza di una struttura
elicoidale prendendo in considerazione la posizione, in ognuno dei monomeri, di uno degli atomi
(scelto in modo arbitrario, ma sempre lo stesso sui vari monomeri) nella parte costante del
polimero (nel caso degli acidi nucleici, per esempio, uno qualsiasi degli atomi dello zucchero o
del fosfato che costituiscono lo scheletro).
Figura A7.1
La posizione del jesimo atomo (e quindi del jesimo residuo monomerico) é determinata, in coordinate
cilindriche e con l'asse z coincidente con l'asse di avvitamento dell'elica, dalle seguenti equazioni
parametriche
zj = hzj + z0
xj = r cos (j2π hz /P + δ0)
yj = r sin (j2π hz /P + δ0)
dove j = 0,1,2, ..., N, con N = numero di residui presenti nella struttura elicoidale.
Come mostrato nella Figura A7.1, r é la distanza dall'asse z della proiezione di un qualsiasi
residuo sul piano x-y, ovvero il raggio dell'elica; hz é la distanza, sull'asse z, tra due residui
adiacenti; P, o passo dell'elica, é definito come la distanza in z tra i due residui più vicini lungo
l'elica che hanno la stessa proiezione sul piano x-y, o, in altre parole, P é la distanza in z che
l'elica deve percorrere per fare un giro intero. z0 e δ0 individuano la posizione dello 0esimo residuo
(il primo residuo dell'elica). Poiché nell'elica di cui ci stiamo occupando ciascun residuo consiste
di molti atomi, é importante notare che il valore di P e hz non dipendono dalla scelta dell'atomo
che é stato preso per caratterizzare l'elica, mentre r, z0 e δ0 hanno valori diversi per i diversi
atomi dei residui. La quantità P/ hz é il numero di residui per giro dell'elica, che può anche non
essere un intero. Un'elica con M residui per giro é detta di molteplicità M. Spesso, in contesto
biologico, si parla di passo dell'elica intendendo però indicare la molteplicità. Così quando si
parla di α-elica nelle proteine si dice che questa ha un passo di 3.6, intendendo con ciò che ci
sono 3.6 residui ogni giro completo dell'elica. Doppie e triple eliche sono abbastanza comuni in
natura. In questo tipo di eliche le singole catene possono essere avvolte in direzioni parallele o
antiparallele.
18
A8: Le funzioni di stato termodinamiche
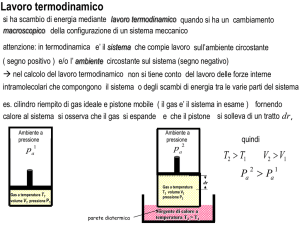
Con la dizione "sistema termodinamico" si intende genericamente indicare quella parte
dell'universo a cui siamo interessati. In pratica si stabilisce un confine arbitrario tra la regione di
spazio che ci interessa studiare e il resto dell'universo. Con "sistema termodinamico" si può
quindi indicare il sistema solare, la Terra, una singola cellula o una mole di acqua a 15 gradi e
alla pressione di un atmosfera.
Si possono definire tre tipi di "sistemi termodinamici":
1. Il sistema termodinamico isolato: che non scambia con l'esterno nè materia nè
energia;
2. il sistema termodinamico chiuso: che scambia con l'esterno energia, ma non
materia;
3. il sistema termodinamico aperto: che scambia con l'esterno sia materia che
energia.
Due cose occorre innanzitutto chiarire:
1. La scelta di dove porre il confine tra il sistema e l'esterno é una scelta arbitraria, ma va
operata in modo intelligente e opportuno.
Per fare un esempio supponiamo di avere un contenitore con del perossido di idrogeno in
soluzione e aggiungiamo alla soluzione l'enzima catalasi. La presenza dell'enzima come é noto
accelererà la reazione
H2O2→ H2O + 1/2 O2
e si produrrà ossigeno gassoso che uscirà dal contenitore. Se scegliamo come sistema il liquido
nel contenitore il sistema termodinamico andrà trattato come un sistema aperto, ma se scegliamo
il liquido più il gas prodotto nella reazione il sistema termodinamico é un sistema chiuso. Mentre
sappiamo trattare in modo relativamente semplice la termodinamica del secondo sistema, il primo
é di trattazione molto più difficile.
2. L'altra cosa da chiarire é che siamo interessati alle variazioni di energia che caratterizzano
l'evolversi del sistema e molto meno al suo valore assoluto.
Per fare un esempio concreto pensiamo ad una reazione chimica innescata dall'aggiunta di una
certa quantità di un dato composto. L'informazione sulla quantità di materia aggiunta é
determinante per definire l'evoluzione della reazione, ma sebbene l'aggiunta di materia abbia
come conseguenza anche una variazione dell'energia potenziale del sistema (pari a E = mc2),
questa non é di nessun interessa se non ha effetto sulla reazione chimica in studio.
Ciò premesso, consideriamo un sistema termodinamico chiuso in assenza di campi esterni (é
chiaro che l'assenza di campi esterni é solo una ragionevole approssimazione) costituito, per
esempio, da un liquido puro di cui conosco P, V e T. Conoscere il valore di queste tre variabili é
19
sufficiente per derivarne altre, caratteristiche dello stato del sistema, quali per esempio: densità,
energia etc.
P, V e T , insieme ad altre proprietà del sistema sono dette variabili di stato se dipendono
solo dallo stato del sistema e non da come lo stato é stato raggiunto. Esse hanno l'importante
caratteristica che, date alcune variabili di stato sufficienti a descrivere lo stato del sistema, le altre
sono da queste determinate implicitamente e da esse derivabili. Si definisce inoltre funzione di
stato una funzione delle variabili di stato la cui variazione dipende solo dallo stato iniziale e
finale del sistema.
Il primo principio della termodinamica
dU = δQ - δL
Eq.A8.1
ci dice che U (la funzione energia interna) é una funzione di stato, mentre lavoro e calore non lo
sono (d.significa differenziale, δ significa variazione)
Il secondo principio della termodinamica definisce un'altra funzione di stato, la funzione
entropia
δQ = TdS
Eq.A8.2
Per un sistema idrostatico (fluido perfetto) il primo principio può essere espresso
dU = TdS - pdV
Eq.A8.3
dove si vede che U ≡ U(S,V), ovvero, la funzione di stato energia interna é esprimibile come
funzione di S e di V, e che
∂U
=T
∂S
∂U
= −p
∂V
Eq.A8.4
che ci dice che nota U ≡ U(S,V) é possibile ricavare T e p che sono quindi variabili dipendenti.
Per specifici problemi é a volte conveniente avere funzioni di stato che sono funzione di
variabili di stato diverse da S e V.
Vogliamo per esempio costruire la funzione di stato entalpia che sarà una funzione delle
variabili S e p in modo tale che rimangano valide le (A8.4). A questo scopo utilizziamo un
procedimento che va sotto il nome di trasformata di Legendre. Sia f(x, y) la funzione di partenza
delle variabili (x,y) , si definisce differenziale la quantità:
20
df =
∂f
∂f
dx +
dy
∂x
∂y
Eq.A8.6
Definiamo la nuova funzione g(u, y) attraverso la formula
g (u, y ) = Min [ f ( x, y ) − xu]
Eq.A8.9
x
dalla quale si ha
∂f ( x , y )
−u=0
∂x
Eq.A8.10
Una volta ricavata da questa equazione x come funzione di u e y, x = x(u,y), questa espressione va
sostituita nella A8.9. In concreto per costruire l'entalpia, utilizzando la A8.9 poniamo
H ( S , p) = Min [U ( S ,V ) + pV ]
Eq.A8.11
V
La condizione
∂U ( S ,V )
+ p=0
∂V
Eq.A8.12
risolta rispetto a V fornisce V = f(S,p) ovvero la vecchia variabile come funzione delle nuove
variabili, che consente di eliminare V esprimendolo in funzione di S e p.
Quindi il differenziale della nuova funzione di stato entalpia si può scrivere
dH =
∂H
∂H
dS +
dp
∂S
∂p
Eq.A8.13
confrontando con
dH = dU + pdV + Vdp = TdS - pdV + pdV + Vdp = TdS + Vdp
si ottiene
∂H
=T
∂S
e
∂H
=V
∂p
Eq.A8.14
Eq.A8.15
In modo analogo si procede per definire la funzione energia libera di Helmhotz, F = F (T,V)
δU
F (T ,V ) = Min [U ( S ,V ) − TS ] usando la condizione
= T per eliminare la dipendenza da S.
S
δS
21
Confrontando
dF =
∂F
∂F
dT +
dV
∂T
∂V
Eq.A8.16
con
dF = dU - TdS - SdT = TdS - pdV - TdS - SdT = - pdV - SdT
∂F
= −p
∂V
si ottiene
e
∂F
= −S
∂T
Infine
si
definisce
l'energia
libera
di
Gibbs,
G (T , p) = Min [U ( S ,V ) − TS + pV ] ove utilizziamo le condizioni
Eq.A8.17
Eq.A8.18
G
=
G(T,p)
,
V ,S
∂U
=T
∂S
e
∂U
= −p
∂V
Eq.A8.19
per eliminare la dipendenza da S e da V. Al solito confrontando
dG =
∂G
∂G
dT +
dp
∂T
∂p
Eq.A8.20
con
dG = dU -TdS - SdT + pdV + Vdp = TdS - pdV - TdS - SdT + pdV + Vdp =
= Vdp - SdT
Eq.A8.21
si trova
∂G
=V
∂p
e
∂G
= −S
∂T
Eq.A8.22
Per chiarire meglio il concetto di Funzione di stato diamo un paio di semplici esempi della loro
utilizzazione pratica
a) Misura dell'entalpia di una reazione a p = costante
Dalle A8.14 e A8.2 (definizione di entropia dal secondo principio)
dH = TdS + Vdp = TdS = δQ
Eq.A8.22
per misurare l'entalpia di una reazione a pressione costante sarà sufficiente quindi misurare la
quantità di calore scambiata con l'esterno durante la reazione stessa.
b) Variazione di energia libera di Gibbs.
22
Si abbia una reazione che porta il sistema da uno stato 1 ad un stato 2. Per determinare se la
reazione può o no avvenire in modo spontaneo è sufficiente determinare il segno della variazione
di energia libera di Gibbs. Infatti a
∆Q
(II Principio)
S 2 − S1 ≥
T
∆Q = U 2 − U1 + p(V2 − V1 )
(I Principio)
ne segue che a per una reazione spontanea
− G 2 = TS 2 − U 2 − pV2 ≥ −G1 = TS1 − U1 − pV1 )
Dunque
a p, T = costanti
∆G < 0
∆G = 0
∆G > 0
la reazione procede spontaneamente
la reazione é all'equilibrio
la reazione non procede spontaneamente
23
A9: La distribuzione Poissoniana
Poichè la distribuzione Poissoniana può essere definita come una distribuzione limite della
Binomiale vediamo prima com'è fatta quest'ultima.
La distribuzione Binomiale misura la probabilità che un evento si presenti con una data modalità,
k volte su n prove.
Indichiamo con p la probabilità che, in una singola prova, l'evento si verifichi con una data
modalità, allora, q = 1 - p, sarà la probabilità che nella singola prova l'evento non si verifichi con
quella modalità.
Poiché si fa l'ipotesi che le singole prove siano tutte indipendenti una dall'altra1, la probabilità che
l'evento si verifichi per k volte con la modalità che ha probabilità p e non si presenti più con
questa modalità per le restanti n - k volte è data da
p k q n−k
Eq. A9.1
⎛ n⎞
Sappiamo però che ci sono ⎜⎜ ⎟⎟ modi indipendenti diversi in cui l'evento si presenta k volte su n
⎝k ⎠
⎛ n⎞
prove. Il simbolo ⎜⎜ ⎟⎟ indica il cosiddetto coefficiente binomiale
⎝k ⎠
⎛ n ⎞ n(n − 1) K (n − k + 1)
n!
2
⎜⎜ ⎟⎟ =
=
k
1
×
2
×
3
×
×
k
k
!
(
n
−
k
)!
K
⎝ ⎠
Eq. A9.2
La probabilità totale che l'evento con quella data modalità si presenti k volte su n prove é data da
⎛n⎞
Pn,k (k ) = ⎜⎜ ⎟⎟ p k q n−k
⎝k ⎠
Eq.A9.3
1
Ricordiamo che la probabilità del verificarsi di eventi indipendenti é pari al prodotto delle probabilità dei
singoli eventi. Quindi la probabilità che si verifichi k volte l'evento con probabilità p è pari a pxpxpx...xp,
k volte, ovvero pk
2
Avendo introdotto la notazione "fattoriale": n!=1×2×3×K×n
24
La distribuzione Poissoniana é la distribuzione limite della Binomiale quando n→ ∞ (n: numero
di eventi o prove) e p→ 0 (p: probabilità che l'evento si presenti con una data modalità) in modo
che np = m sia finito, fisso e ≠ 0. Poiché la Poissoniana é ottenuta nel limite p→ 0 viene anche
detta "Legge degli eventi rari".
Si può dimostrare che si ha
lim Pn, p (k ) = Pm (k ) =
n →∞
p →0
m k e −m
k!
Eq.A9.4
Si noti che la distribuzione dipende solo dal parametro m, che k é un numero intero ≥0, e che σ2
= m. Si ricorda che la varianza o momento secondo, σ2 , della distribuzione di probabilità ,
Pm (k ) ,
∞
é data da ∑ ( k − m ) 2 Pm ( k ) .
k =0
25
A10:Catena di Markov (o Processo di Mrkov)
Si definisce Catena (o Processo) di Markov, un processo (o catena di stati) in cui la probabilità di
occorrenza di uno stato dipende dalla precedente occorrenza di un numero finito, r, di stati
immediatamente precedenti. Parleremo allora di processo di Markov di ordine r.
In modo più formale poniamo la seguente definizione. Partiamo dal caso r =1 (processo di
Markov di ordine 1)
Definizione:
Sia
{X (t ), t ≥ 0}
un processo stocastico (t non é necessariamente la variabile temporale). Il
processo é detto di Markov se ∀ n ≥ 0 , detti 0 ≤ t 0 < t1 < K < t n < t n+1 i passi del processo e
i 0 , i1 ,K i n , i n+1 i corrispondenti stati si ha che
P[ X (t n+1 ) = i n+1 X (t n ) = i n , X (t n−1 ) = i n−1 ,K X (t 0 ) = i 0 ] = P[ X (t n+1 ) = i n+1 X (t n ) = i n ]
Questa definizione stabilisce che solo lo stato attuale, i n , dà informazioni sul futuro
comportamento del processo. La conoscenza della storia precedente del processo, (i n−1 K i 0 ) , non
aggiunge nessuna nuova informazione.
Se r > 1 , l'ordine di una catena di Markov corrisponde al numero di stati (precedenti) che
concorrono a definire la probabilità dello stato attuale.
26
A11: Teorema di Bayes
Il Teorema di Bayes si ottiene come segue.
Siano A e B due eventi le cui probabilità sono date rispettivamente da P(A) e P(B).
Sia P ( A ∩ B ) la probabilità dell'evento ( A ∩ B ) (che si legge: A intersezione B), ovvero la
probabilità dell'evento : A.AND.B. Sia P ( A B) ⋅ P ( B) la probabilità condizionale, ovvero la
probabilità che dato A (o B), segua B (o A), allora
P ( A ∩ B) = P ( A B) ⋅ P ( B)
Eq.A11.1
P ( B ∩ A) = P ( B A) ⋅ P ( A)
Eq.A11.2
Poiché
P ( A ∩ B ) = P ( B ∩ A)
Eq.A11.3
segue
P ( A B) ⋅ P ( B) = P ( B A) ⋅ P ( A)
Eq.A11.4
e dunque
P ( A B ) = P ( B A)
P ( A)
P ( B)
Eq.A11.5
Supponiamo che A1 A2 K An sia un insieme completo di eventi incompatibili, ovvero
⎧ Incompatib ilità : Ai ∩ A j = 0, ∀i ≠ j per cui
⎪
⎪ P ( Ai ∪ A j ) = P ( Ai ) + P ( A j )
⎪
A1 A2 K An ⎨
⎪Completezz a : ∀B → B = ( B ∩ A )
U
i
⎪
i
⎪
⎩
Eq.A11.6
27
Si noti che ( B ∩ Ai ) ∩ ( B ∩ A j ) = 0 e quindi
⎛
⎞
P ( B ) = P ⎜⎜ U ( B ∩ Ai ) ⎟⎟ = ∑ P ( B ∩ Ai ) = ∑ P ( B Ai ) ⋅ P ( Ai )
i
⎝ i
⎠ i
Eq.A11.7
e sostituendo nella A11.5 per ogni j si ha
P ( A j B) =
P(B A j ) ⋅ P( A j )
∑i P ( B Ai ) ⋅ P ( Ai )
= P (B A j ) ⋅
P( Aj )
P ( B)
Eq.A11.8
che é la formulazione più generale del Teorema di Bayes, nella quale P ( A j B) é la cosiddetta
probabilità "a posteriori" mentre P ( A j ) é quella "a priori".
28