Ritardo mentale:
quando il cariotipo è diagnostico
Casi clinici del mercoledì: 07/11/2012
Tutors
AIF
Dott. A. Romano/N. Brunetti-Pierri
Dott.ssa M. Cerbone
Giunge a ricovero Mohamed…
Motivo del ricovero:
ritardo nel linguaggio
difficoltà nell’apprendimento
All’ingresso:
Età: 8 anni
Peso: 25,500 Kg (50-75° ct)
Altezza: 130 cm (50-75° ct)
Circ. cranica: 53 cm (media / +2DS)
Etnia: marocchina
EO: numerose macchie caffè-latte diffuse su tutto il corpo
note dismorfiche lievi con ponte nasale ampio e depresso
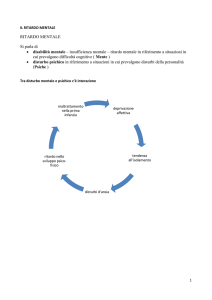
Ritardo mentale
DEFINIZIONE DSM IV
CRITERIO A:
Significativa limitazione delle funzioni cognitive:
QI < 70
CRITERIO B:
Significativa limitazione funzioni adattive in almeno 2 aree:
comunicazione
cura della propria persona
vita in famiglia
capacità sociali/interpersonali
uso risorse di comunità
autodeterminazione
CRITERIO C:
Esordio < 18 anni
capacità di funzionamento
QUOZIENTE
RITARDO MENTALE
INTELLETTIVO
scolastico
lavoro
Lieve
50/55 – 70/75
tempo libero
Moderato
30/40 - 50/55
salute
Grave
20/25 – 35/40
sicurezza
Profondo
< 20/25
Eziologia
RITARDO
SEVERO
RITARDO
LIEVE-MODERATO
Eziologia
Vasto campo di
indagine
Alta frequenza
insuccessi diagnostici
Grandi progressi negli ultimi 20 anni nel rendimento diagnosico:
dismorfologia
citogenetica
genetica
molecolare
neurofisiologia
neuroimaging
Diagnosi
Identificazione del ritardo: utilizzo di test specifici
Storia familiare con albero a 3 generazioni e storia perinatale
EO accurato: alterazioni cutanee, descrizione e misurazione di
eventuali anomalie, foto e video
Test genetici: cariotipo standard, cariotipo ad alta risoluzione, FISH,
analisi dei subtelomeri, indagine genetica per X-fragile e sindrome di
Rett, array-CGH
Test metabolici: resa diagnostica bassa (1%) nei casi non selezionati
(maggiore per ricerca di difetti della glicosilazione e della sintesi e del
trasporto della creatina)
Indagini neuroradiologiche: RM preferita alla TC
Indagini neurofisiologiche: EEG (epilessia, grave compromissione del
linguaggio, s. Angelman...)
Valutazione sensoriale visiva ed uditiva: screening visivo, esame
oftalmologico ed audiometrico
La storia clinica
ANAMNESI FAMILIARE
Genitori non consanguinei originari dello stesso villaggio in Marocco
Tre sorelle in buona salute
Alcuni membri familiari deceduti per cause sconosciute
Un cugino di I grado con ritardo mentale
ANAMNESI PERSONALE
Fenomeni perinatali normoevoluti
Numerose macchie caffè-latte presenti dalla nascita
SPM ritardato: deambulazione autonoma a 20 mesi, prime parole a 12
mesi
Trasferimento in Italia all’età di 4 anni
Difficoltà nell’apprendimento e ritardo nel linguaggio
14 y
13 y
7y
6y
Work-up diagnostico
a) Identificazione del ritardo:
• Leiter-R (intelligenza non verbale): QI = 65 (ritardo mentale lieve)
b) Indagini di laboratorio:
• Profilo tiroideo: nella norma
• Indagini metaboliche su sangue ed urine: nella norma
c) Valutazione sensoriale:
• Esame audiometrico: nella norma
• Valutazione oftalmologica: nella norma
d) Indagini neuroradiologiche:
• RM encefalo: cisti aracnoidea di grandi dimensioni nella regione
temporale di destra comprimente il lobo temporale ed il ventricolo
laterale
Roid GMM, L.J. International Performance Scale-Revised: Examiners Manual. Wood Dale, IL: Stoelting Co., 1997
Work-up diagnostico
e) Indagini genetiche:
• X-fragile:
non evidenza di espansione della tripletta CGG nell’esone 1 del gene FMR-1
• Cariotipo standard:
L’analisi ha evidenziato una linea cellulare, presente per il 50%, con cariotipo normale maschile 46,XY.
L’altro 50% delle cellule esaminate presentano elevata instabilità della eterocromatina pericentromerica
dei cromosomi 1 e 16. In particolare si riscontrano delezioni, associazioni, interscambi e duplicazioni di
parti o interi bracci di tali cromosomi che formano delle strutture a bracci multipli il cui centro è sempre
rappresentato dall’eterocromatina presente sui cromosomi 1 e 16.
SOSPETTO DI SINDROME ICF
Sindrome ICF
(Immunodeficienza, instabilità centromerica, anomalie facciali)
Rara malattia genetica autosomico recessiva (circa 50 casi
descritti)
Infezioni ricorrenti: secondarie ad ipo-agammaglobulinemia
Note dismorfiche: ipertelorismo, ponte nasale appiattito ed
ampio, epicanto, orecchie a basso impianto, macroglossia
Ritardo mentale
Aumentato rischio di patologie ematologiche e tumori
Prognosi: influenzata dalla severità dell’immunodeficienza
Blanco-Betancourt et al. Defective B-cell-negative selection and terminal differentiation in the ICF syndrome Blood 2004.
Ehrlich M et al Immunodeficiency, centromeric region instability, facial anomalies syndrome (ICF). Orphanet J Rare Dis 2006
Sindrome ICF
(Immunodeficienza, instabilità centromerica, anomalie facciali)
Marker diagnostico: instabilità centromerica derivante da
un’ipometilazione del DNA aspetto caratteristico dei
cromosomi
Tipi (clinicamente indistinguibili):
• ICF tipo 1 (60%): mutazioni nel gene della metiltransferasi di
tipo 3B (DNMT3B)
• ICF tipo 2 (40%): mutazioni diverse da DNMT3B tra cui
mutazioni nel gene ZBTB24
Blanco-Betancourt et al. Defective B-cell-negative selection and terminal differentiation in the ICF syndrome Blood 2004.
Ehrlich M et al Immunodeficiency, centromeric region instability, facial anomalies syndrome (ICF). Orphanet J Rare Dis 2006
Conferma diagnostica
A. Southern Blot per analisi
dell’ipometilazione di NBL2 e
degli α-satelliti
Ipometilazione di entrambi: ICF2
B. Sequenziamento della regione
di ZBTB24:
mutazione missenso in
omozigosi nel gene ZBTB24
(c.1222T>G)
Genitori: eterozigoti per la
mutazione
Immunodeficienza e sindrome ICF
Ipo-agammaglobulinemia nell’ 89% dei pazienti descritti
Infezioni: presentazione clinica più frequente
40.5% dei casi descritti deceduti per infezioni respiratorie severe o
sepsi
Patient
Control range
IgG (mg/dl)
10.1
6.73-17.34
IgM (mg/dl)
0.19
0.47-3.11
IgA (mg/dl)
1.47
0.41-3.68
IgG1 (mg/dl)
7.05
4.00-10.8
IgG2 (mg/dl)
1.30
0.85-4.10
IgG3 (mg/dl)
1.77
0.13-1.42
IgG4 (mg/dl)
0.03
0.00-1.89
White blood cells (x103/µL)
6.70
4.50-13.50
% lymphocytes
39.00
20.0-51.0
% granulocytes
51.00
40.0-75.0
CD22+ (cells/mm3)
360.0
200.0-1300.0
CD16+ (cells/mm3)
104.0
200.0-400.0
CD3+
1846.0
1000.0-3900.0
(cells/mm3)
CD4+ (cells/mm3)
CD8+ (cells/mm3)
a) Riduzione dei livelli di
IgM sieriche
b) Riduzione dei livelli delle
cellule NK
560.0-2700.0
ASSENZA 854.0
DI MANIFESTAZIONI
CLINICHE DI
728.0
330.0-1400.0
IMMUNODEFICIENZA!!
Hagleitner et al. Clinical spectrum of immunodeficiency, centromeric instability and facial dysmorfism. J Med Genet 2008
Peculiarità del caso
Assenza di immunodeficienza:
- presenza di casi senza immunodeficienza non diagnosticati?
Presenza di una malformazione cerebrale:
- mai descritta prima (casi non investigati con RM?)
- mutazione ZBTB24 ipometilazione del gene DNMT3B
cambiamenti di espressione di geni critici nella neurogenesi
- RM indicata in pazienti con ICF?
Interesse pediatrico
Cariotipo nella diagnostica del ritardo mentale: quale ruolo?
Raccomandazioni recenti sulla diagnostica del ritardo mentale
suggeriscono di utilizzare l’array-CGH come indagini genetica
di prima linea
Test genetici e ritardo mentale
Miller DT. Am J Hum Genet 2010
Liang J-S. Pediatr. Neonatol 2008
Test genetici e ritardo mentale
TEST GENETICO
ARRAY-CGH
RESA DIAGNOSTICA
NON SINDROMICI
SINDROMICI
7.8%
10.6%
CARIOTIPO STANDARD
4%
FISH
3.5%
3%(18.6%)*
4.2%
*
s. Down inclusa
Array-CGH potere diagnostico maggiore del cariotipo standard fatta
eccezione per i casi in cui ci sia un forte sospetto di cromosomopatia
Riarrangiamenti bilanciati e mosaicismi di basso grado non sono
individuabili con l’array-CGH, anche se queste alterazioni
rappresentano < 1% dei casi di ritardo mentale.
Michelson et al et al. Evidence report: genetic and metabolic testing on children with global developmental delay. Neurology 2011
Conclusioni
RITARDO MENTALE:
SINDROME
ICF:
Attenta valutazione
di anomalie cutanee e tratti dismorfici lievi
utile
Sospettarla
anche
in assenza
immunodeficienza
a definire
l’eziologia
del di
ritardo
mentale
Esecuzione di una RM encefalo nei pazienti con ICF?
Alcune malattie genetiche responsabili di ritardo mentale
possono essere individuate con il cariotipo e restare
misconosciute effettuando la sola analisi con l’array-CGH
GRAZIE
DELL’ ATTENZIONE