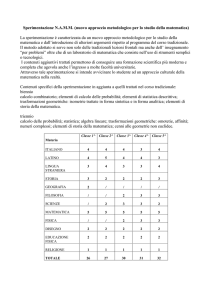
Doc. 14
VISITA DI MONITORAGGIO DEL CERS
Protocollo:
Codice CERS:
Sperimentatore Principale:
Data
approvazione:
Centro n°:
U.O.:
Sede:
Sponsor:
Farmaco/i:
Tipologia di
sperimentazione:
Dosaggio:
Data della Visita:
Forma farmaceutica:
Dispositivo Medico:
Integratore:
Sperimentatori dello staff medico presenti:
Componenti del CERS presenti come monitor:
Verifiche effettuate:
Data dell’ultimo paziente completato:
Data del primo paziente arruolato:
Stato del centro:
Numero dei soggetti screenati ___________
Arruolati ___________
Completati ___________
Discontinuati ___________
Pag 1/9
Compendio sullo Stato di avanzamento dello studio
Soggetto numero
Soggetto
iniziali
Data di
ottenimento del
consenso
informato
Data di
screening
Data di
randomizzazione
Pag 2/9
Stato del soggetto:
ongoing / discontinuato/
completato
Visita Data di conclusione
in
o di
corso
discontinuazione
Compendio sullo Stato di avanzamento dello studio
Soggetto numero
Soggetto
iniziali
Data di
ottenimento del
consenso
informato
Data di
screening
Data di
randomizzazione
Pag 3/9
Stato del soggetto:
ongoing / discontinuato/
completato
Visita Data di conclusione
in
o di
corso
discontinuazione
Lista dei soggetti discontinuati
Numero
paziente
Iniziali
paziente
Data
interruzione
Motivo dell’interruzione (AEs, violazioni al protocollo, ritiro del
consenso, perso al follow up, inefficacia del trattamento, morte)
Si sono verificati Eventi Avversi Seri?
Se si, compilare la lista degli Eventi Avversi Seri verificatisi:
Numero
paziente
Iniziali
paziente
Data
evento
Data
morte
Evento avverso
Pag 4/9
SI
NO
Criterio di serietà
Rispondere a tutte le domande scegliendo una delle opzioni. La scelta delle opzioni con (*) o con ‘N/A’ (non applicabile)
necessitano di un commento nella sezione “Relazione di monitoraggio”
Controllo dei documenti originali (cartelle cliniche o ambulatoriali, referti medici,
questionari e diari compilati dal paziente) e delle Case Report Form (CRF)
1.
Le cartelle cliniche o le cartelle ambulatoriali cartacee sono complete e leggibili?
2.
Sono disponibili tutti i referti medici e altri documenti originali inerenti lo studio?
3.
I documenti originali sono conservati in luogo sicuro?
4.
Sono scritti con penna biro?
5.
Le correzioni sono state siglate e datate, il dato errato è leggibile?
6.
Le cartelle cliniche o le cartelle ambulatoriali elettroniche sono complete?
7.
Le cartelle cliniche o le cartelle ambulatoriali elettroniche sono validate e protette?
-Hanno un sistema di memorizzazione/visualizzazione del dato errato e sostituito?
8.
I documenti originali e le CRF sono stati confrontati?
9.
I dati controllati sono speculari?
10.
Si sono verificati eventi avversi ed eventi avversi seri AEs/SAEs al centro?
a.
Gli AEs/SAEs sono stati documentati e controllati?
b.
Gli eventi avversi seri SAEs sono stati comunicati al CERS?
c.
Le nuove informazioni riguardanti gli AEs/SAEs noti sono state
documentate e controllate?
11.
I pazienti arruolati hanno firmato e datato il consenso informato approvato dal CERS?
12.
Le CRF sono state compilate in tempi ragionevoli?
Protocollo e Reclutamento pazienti
13.
Il ritmo di reclutamento dei pazienti rispecchia gli obiettivi dello studio?
14.
Sono state individuate significative deviazioni/violazioni al protocollo?
15.
Lo sperimentatore applica correttamente i criteri di inclusione/esclusione dei pazienti?
16.
Lo sperimentatore applica correttamente i criteri di discontinuazione dei pazienti?
17.
Lo sperimentatore aggiorna periodicamente il CERS sullo stato di avanzamento dello
studio?
18.
In caso di chiusura dello studio, lo sperimentatore ha fornito al CERS un rapporto di
fine studio completo?
Farmaco/Dispositivo/Integratore in sperimentazione e altro materiale utilizzato per la
sperimentazione
19.
Il farmaco/dispositivo/integratore in sperimentazione ha una data di scadenza
compatibile con i tempi (arruolamento, trattamento) della sperimentazione in corso?
20.
Il farmaco/dispositivo/integratore è stoccato in condizioni appropriate?
21.
La scorta di farmaco/dispositivo/integratore è sufficiente?
22.
Sono stati aperti dei codici di randomizzazione?
a.
Se si l’evento è opportunamente documentato?
23.
I documenti originali, le CRFs, i referti, le cartelle cliniche, le cartelle ambulatoriali, il file
della sperimentazione sono archiviati in modo appropriato?
24.
Il materiale della sperimentazione è accessibile solo agli sperimentatori?
25.
Ci sono procedure validate di raccolta, etichettatura, stoccaggio, e trasporto dei
campioni biologici?
Pag 5/9
SI
NO
N/A
*
*
*
*
*
*
*
*
*
*
*
*
*
*
SI
NO
N/A
*
*
*
*
*
*
SI
NO
*
*
*
*
*
*
*
*
*
N/A
File della sperimentazione e documenti regolatori
Rispondere alla check list (verificare la presenza di tutti i documenti)
Protocollo
Emendamenti
Lettera di intenti
Polizza assicurativa in toto
Elenco dei centri partecipanti allo studio
Sinossi al protocollo
Investigator Brochure
Modello di CRFs
Lista degli sperimentatori
Curricula degli sperimentatori
Parere del CERS del centro coordinatore
Attestazione di versamento al CERS
Pareri e comunicazioni del CERS
Delibera autorizzativa
Convenzione Economica
SI
Versione finale
Tutti
In corso
In corso
Versione finale
In corso
In corso
Aggiornata
Aggiornati
Tutti
Modello di foglio informativo e modulo del consenso informato
Consensi firmati dai pazienti
Lista dei pazienti arruolati/randomizzati
Lista dei pazienti screenati
Lista di identificazione del codice paziente
CIOMS, SUSARS
SAEs
Rapporti periodici al CERS
Rapporto finale di chiusura del centro al CERS
Moduli di invio farmaco e relativi certificati di qualità
Lista di movimentazione del farmaco
Lista del farmaco ritirato dal centro
Lista dei campioni biologici
I range di laboratorio, le certificazioni/accreditamento di laboratorio
Commenti sui documenti mancanti:
Pag 6/9
Tutte le versioni
approvate
Tutti
Aggiornata
Aggiornata
Aggiornata
Tutti
Tutti
Tutti
Tutti
Aggiornata
Aggiornata
Aggiornata
Aggiornati
NO
N/A
Relazione di monitoraggio
(Riportare in questa sezione i commenti relativi a tutti i punti che necessitano di chiarimenti (punti critici, non conformità,
azioni correttive), indicando tra parentesi il punto cui ci si riferisce, riportare informazioni e appunti vari, concludere con una
relazione finale sulla qualità del lavoro svolto dal centro, chiarire se il centro lavora nel rispetto delle GCP.)
Nome del/dei componenti il CERS
monitoratore/i:
Firma/e:
Data di lettura in seduta di CERS:
Firma di approvazione:
Pag 7/9
GLOSSARIO
Sperimentatore Principale: il responsabile di tutta la sperimentazione in corso.
Co-sperimentatore: il collaboratore che si occupa della sperimentazione.
Centro sperimentale: reparto presso cui si svolge la sperimentazione.
Monitoratori: coloro che ispezionano il centro sperimentale, garantendo il rispetto delle GCP.
GCP: Good Clinical Practice.
Pazienti Screenati: pazienti valutati ma non arruolati in sperimentazione, la fase di screening
prevede la firma del consenso e la valutazione del paziente sotto il profilo dei criteri di inclusione ed
esclusione. I pazienti che non rientrano in tutti i criteri di inclusione e che presentano anche un solo
criterio di esclusione non verranno arruolati.
Pazienti randomizzati: pazienti arruolati in sperimentazione e sottoposti al sistema di assegnazione
casuale di un trattamento farmacologico/placebo/trattamento non farmacologico.
Pazienti arruolati: pazienti che partecipano alla sperimentazione.
Pazienti discontinuati: pazienti che per motivi di varia natura interrompono anticipatamente la loro
partecipazione allo studio clinico.
Pazienti completati: Pazienti che hanno portato a termine tutte le procedure della sperimentazione
sancite dal protocollo.
CRFs Case Report Form: sono le schede raccolta dati.
AE: Eventi avversi, eventi che peggiorano lo stato di salute del paziente ma non sono
necessariamente correlati al farmaco in sperimentazione.
SAE: Eventi avversi seri, eventi che peggiorano lo stato di salute del paziente ma non sono
necessariamente correlati al farmaco in sperimentazione, essi rientrano nei criteri di serietà
morte,
pericolo di vita,
ospedalizzazione o prolungamento di ospedalizzazione,
inabilità o disabilità,
difetti e malformazioni alla nascita.
CERS: Comitato Etico per la Ricerca e Sperimentazione.
Violazioni al protocollo: pazienti in sperimentazione benché non soddisfino tutti i criteri di
inclusione e rientrino in uno o più criteri di esclusione.
Deviazioni al protocollo: deviazioni di qualsiasi natura rispetto a quanto viene stabilito dal
protocollo sperimentale ed eventuali emendamenti.
Codici di randomizzazione: sono buste sigillate entro le quali sono riportati i trattamenti che il
paziente sta assumendo in cieco. Ogni paziente viene individuato da un codice numerico che
corrisponde ad un trattamento assegnato a random ossia casualmente, ad ogni codice corrisponde una
busta, se la busta viene aperta si conoscerà quale trattamento sta assumendo il paziente che possiede
quel numero. I codici di randomizzazione si aprono solo in caso di Evento Avverso Serio,
allorquando è necessario conoscere il trattamento per poter intervenire adeguatamente sul paziente.
Il File della sperimentazione: archivio contenente tutti i documenti inerenti la sperimentazione.
Investigator brochure: scheda tecnica del farmaco sperimentale.
CIOMS (The Council for International Organization of Medical Science): sono singoli SAE
report provenienti da tutto il mondo per tutti i tipi di sperimentazione clinica realizzata con il farmaco
in sperimentazione presso il centro.
SUSARs: Suspected Unaspected Serious Adverse Reactions: rapporti periodici di sicurezza del
farmaco in sperimentazione, dati ottenuti da studi clinici realizzati in tutto il mondo con il farmaco in
questione.
Anonimato del paziente
Per garantire l’anonimato del paziente in sperimentazione e per permettere la sicura
identificazione dello stesso, si applicano le seguenti regole:
il paziente che ha fornito il Consenso Informato viene identificato con le proprie iniziali seguite
dal numero di screening, ossia il numero sequenziale di valutazione del paziente al centro.
Successivamente, se il paziente verrà arruolato, potrà rimanere identificato con questo numero
oppure sostituito con un numero di arruolamento. Se è prevista la randomizzazione, il paziente
assumerà anche il numero di randomizzazione che identifica inequivocabilmente il trattamento
che il paziente sta assumendo in cieco.
Le CRFs (di proprietà dello sponsor) sono completamente anonime, identificate solo da questi
codici alfanumerici riportate nell’intestazione di ogni pagina e possono essere esibite e uscire
dal centro solo a coloro che, nell’ambito del trial, si occuperanno della parte statistica.
I documenti originali (di proprietà della struttura ospedaliera), invece, non essendo anonimi, si
possono esibire solo in caso di ispezione del monitoratore dello Sponsor, del CERS,
dell’Authority Sanitaria e della Segreteria di Stato alla Sanità della repubblica di San Marino,
della CRO, non devono mai uscire dal centro e non possono essere esibiti a persone non
autorizzate. La lista di identificazione del codice paziente è una lista che riporta: il nome e
cognome per esteso del paziente, le iniziali, tutti i numeri ad esso assegnati, il codice fiscale, la
data di nascita, il recapito del paziente, il sesso. Questi dati sono fondamentali per
l’identificazione del paziente così come lo sono le CRFs e gli altri documenti. Tale
documentazione deve essere conservata obbligatoriamente per almeno quindici anni nel centro
e resa ostensibile al solo personale autorizzato.
Pag 9/9