C.d.L. in Biotecnologie del farmaco
Corso di Chimica Farmaceutica Biotecnologica
Malattie Autoimmuni
Quando il sistema immunitario attacca i tessuti propri
dell’organismo invece degli elementi di aggressione
esterni, quali batteri o virus, si parla di malattia
autoimmune. Dal 5 all’8% della popolazione ne è
affetta.
Il sistema immunitario presenta un elevatissimo
numero di diverse specificità e poiché il repertorio
di queste specificità, espresse dalle generazioni
di linfociti B e T, viene generato in maniera casuale,
alcune di queste sono rivolte verso Ag costitutivamente
espressi dall’organismo. In questo modo si creano nel corpo umano meccanismi di
tolleranza che consentono di distinguere gli Ag propri dell’organismo (Self) da quelli
estranei (Non Self) allo scopo di evitare reazioni autoimmunitarie.
Tuttavia, esiste il rischio per tutti i sistemi di andare incontro a fallimento, e cosi
anche i meccanismi che permettono di riconoscere gli Ag Self non fanno eccezione
ed infatti è stato riconosciuto un certo numero di malattie autoimmuni causate
dall’abbondante produzione di autoanticorpi e di cellule T autoreattive.
I processi autoimmuni sono spesso causa di patologia; nei casi in cui si ritrovano
degli autoanticorpi associati con una particolare malattia vi sono tre diverse
possibilità:
1.
il processo autoimmunitario è responsabile delle lesioni della malattia
2. esiste un processo patologico che, attraverso la produzione di un danno
tissutale, conduce allo sviluppo di autoanticorpi
3. nel corpo è presente un fattore che costituisce l’Ag responsabile sia delle
lesioni che del processo autoimmunitario
In alcuni casi sono stati identificati autoanticorpi prodotti in seguito ad un danno,
quindi risulterebbe valida la seconda possibilità (ad esempio autoanticorpi diretti
verso Ag cardiaci possono essere prodotti dopo un infarto). Tuttavia, è raro che
vengano sintetizzati autoanticorpi in seguito al rilascio di autoantigeni durante
un semplice trauma.
Nella maggior parte delle associate ad autommunità ci sono prove che sostengono
l’ipotesi che il processo autoimmunitario sia l’agente causale delle lesioni.
Le malattie autoimmuni possono presentare un ampio spettro di gravità e possono
colpire qualsiasi organo e culminare con il decesso. Quasi tutte queste malattie
colpiscono più le donne che gli uomini e possono manifestarsi a qualsiasi età.
Molte malattie autoimmuni condividono dei meccanismi fisiologici simili, ma la loro
causa originaria è tuttora sconosciuta e non esiste possibilità di guarigione.
Le malattie autoimmuni possono essere classificate come organo-specifiche o non
organo-specifiche in base alla risposta, che può essere primitivamente diretta verso
Ag localizzati a livello di particolari organi oppure verso Ag ubiquitari:
Organo-specifiche
• tiroidite di Hashimoto
• Anemia perniciosa
• malattia di Addison
• Diabete Mellito insulino-dipendente
• Miastenia grave
• sclerosi multipla
• Anemia emolitica autoimmune
• Cirrosi biliare primitiva
• Epatite cronica attiva
• Artrite Reumatoide
Non organospecifiche
• Lupus Eritematoso Sistemico (LES)
Organo-specifiche
Nonostante le malattie
non organo-specifiche
producano
caratteristiche dei
sintomi a livello della
cute, delle articolazioni,
dei reni e dei muscoli;
alcuni organi sono
maggiormente colpiti da
queste malattie: come
ad esempio il rene dal
Lupus Eritematoso
oppure le articolazioni
dall’Artrite Reumatoide.
non organo-specifiche
Muscoli
DERMATOMIOSITE
SCLERODERMA
Solitamente i pazienti si lamentano per il dolore alle ossa ,in il dolore è delle
articolazioni; l’osso infatti di per sé è ben scarsamente capace di provocare dolore,
essendo privo o quasi dei nocicettori, cioè le terminazioni nervose all’origine delle
sensazioni dolorose.
Si parla di dolori articolari quindi, e non delle ossa, dolori che possono essere dovuti
a molte e svariate cause, ma che sono anche il principale sintomo di due importanti
malattie: artrite reumatoide (AR) ed artrosi.
Le stime del numero di pazienti affetti da AR
sono abbastanza difficili: si va dall’1 al 2-3% della
popolazione generale, ma vi sono anche stime molto
più elevate. Quello che è certo è che colpisce più
spesso le donne che non gli uomini (2-3 volte di più)
e che la comparsa di questa patologia si concentra
tra i 20 e i 40 anni ed in particolare nella quarta decade.
L’AR, come abbiamo già accennato in precedenza, è una malattia infiammatoria
cronica su base immunitaria, cioè costituita da una serie di reazioni immunitarie
rivolte verso l’organismo stesso (tessuti, cellule ma anche semplici proteine); è
come se il sistema immunitario si “sbaglia” e scatena l’attacco delle sue truppe
d’assalto contro l’amico e non il nemico. Quindi è un po’ come una reazione di
rigetto che, anzichè riguardare un organo estraneo trapiantato, riguarda un organo
proprio.
l’AR è una malattia progressiva ed
invalidante che coinvolge la membrana
sinoviale delle articolazioni diartrodiali
e distrugge le loro componenti
cartilaginee ed ossee.
Nonostante l’attività di ricerca scientifica, le cause della malattia, come abbiamo
detto prima, rimangono sconosciute. La patogenesi è probabilmente legata ai
seguenti fattori:
• Fattori genetici =
studi genetici familiari mostrano che l’AR ha una
componente
genetica; il Complesso Maggiore di Istocompatibilità (HLA,
sistema che permette di riconoscere i tessuti di un organismo da tutto ciò che
gli è estraneo) è un importante fattore genetico, e si ritiene che i rischi di
sviluppare AR possa essere associato a questo sistema.
•
Autoimmunità = sembra che le citochine prodotte dai macrofagi siano
coinvolte nel processo di induzione e mantenimento dell’infiammazione cronica
delle articolazioni tipica dell’AR. Elevati livelli di fattore reumatoide nel siero
(autoanticorpi diretti contro la porzione Fc delle IgG) sono associati con
sintomi più acuti dell’artrite alle articolazioni e in altre parti del corpo.
•
Agenti patogeni = alcuni virus, come quello della rosolia ed i parvovirus, sono
associati alla comparsa di poliartriti acute. Infezioni virali dovute ad esempio
all’Epstein-Barr virus o al Cytomegalovirus possono favorire la patologia; e a
conferma di questi studi i ricercatori hanno trovato segni di infezione dei
linfociti T presenti nel liquido sinoviale da parte dei virus in questione. Tuttavia
un legame di tipo causa-effetto non è stato ancora trovato.
Anche se il meccanismo di distruzione ossea e delle cartilagini che si osserva
nell’AR non è stato ancora del tutto chiarito; le citochine IL-1 e TNF-alfa
sembrano avere un ruolo certamente importante. Queste citochine infatti:
•
Sono presenti in gran quantità nelle articolazioni infiammate e promuovono
l’afflusso dei neutrofili dell’infiammazione acuta e dei monociti-macrofagi
all’interno delle articolazioni.
•
Stimolano la produzione di enzimi proteolitici da parte delle cellule della
sinovia, comprese le collagenasi e la stromalisina, che sono in grado di degradare
i tessuti
•
Causano sintomi quali senso generale di malessere e affaticamento
Pertanto, anche se la causa scatenante rimane sconosciuta, il perdurare e la
diffusione della malattia sembra siano da mettere in relazione con i processi
infiammatori mediati dal sistema immunitario (ne parleremo meglio nel dettaglio
successivamente). Quindi è probabile che interferendo con i passaggi chiave del
processo di infiammazione si possa ottenere un alleviamento dei sintomi ed un
rallentamento nella progressione della malattia.
La Cartilagine è un tipo specializzato
di tessuto connettivo in cui le cellule,
chiamate condrociti, sono immerse in
una sostanza fondamentale gel-simile
consistente. A differenza di altri tessuti
connettivi essa non è vascolarizzata.
Le sue cellule sono isolate in piccole
cavità nella sostanza fondamentale, chiamate lacune, e sono nutrite per diffusione di
piccole molecole attraverso la fase acquosa della matrice da capillari localizzati nel
tessuto connettivo intorno alla cartilagine.
Le singolari proprietà viscoelastiche della sua sostanza fondamentale conferiscono alla
cartilagine grande consistenza e tale proprietà è mantenuta durante la crescita. Ciò
rende la cartilagine un materiale scheletrico ideale durante lo sviluppo embrionale. La
quantità di C. è ridotta durante la vita post-natale ma continua a svolgere un ruolo
importante nella crescita in lunghezza delle ossa lunghe. Quando è raggiunta l’età
adulta tutta la cartilagine è sostituita da osso eccetto che sulla superficie delle
articolazioni, le estremità ventrali delle coste, i dischi intervertebrali della colonna
vertebrale e nel naso, nella laringe e nella trachea.
Cartilagine ialina
Cartilagine elastica
Nella parte superiore della cartilagine ialina si notano le cellule appena formate singole
ed allungate; mentre quelle più in basso sono più grandi, sono comprese in gruppi e
sono circondate dalla matrice che si colora più intensamente con coloranti basici.
La cartilagine elastica presa dall’epiglottide umana è invece formata da fasci scuri di
fibre elastiche nella matrice tra gruppi di condrociti
L’osso è un tessuto connettivo in cui
le cellule e le fibre sono incluse in
una matrice (sostanza fondamentale)
che contiene cristalli di un sale
complesso di calcio, idrossiapatite.
La calcificazione della sua matrice lo
rende particolarmente duro, ideale
per la sua funzione di supporto e protettiva nello scheletro. Le ossa forniscono siti di
inserzione e leve per i muscoli della locomozione; essi proteggono organi vitali delle
cavità craniche e toraciche e costituiscono un deposito di calcio che può essere
prelevato quando la sua concentrazione ematica è bassa. Uno strato di tessuto
connettivo dell’osso, non calcificato, il periostio, ricopre la superficie esterna delle ossa,
ed uno strato simile, più sottile, l’endostio, le riveste internamente. Oltre ai fibrobalsti,
ambedue questi strati contengono cellule osteoprogenitrici, precursori inattivi delle
cellule destinate a formare l’osso. Le cellule dell’osso comprendono gli osteoblasti, che
secernono il collagene, proteoglicani ed idrossiapatite della matrice ossea; gli osteoiciti,
che sono localizzati in lacune della sostanza fondamentale; gli osteoclasti, che
rimuovono tessuto osseo in un continuo processo di riassorbimento e rimodellamento
dell’osso che si verifica nel corso della vita.
Gli osteoblasti sono cellule cuboidi allineate sulle superfici dell’osso durante la
deposizione della nuova matrice ossea. Essi non formano tuttavia un epitelio tipico.
Infatti sono lievemente distanziati tra di loro anche se mantengono il contatto mediante
processi laterali. Il nucleo si trova all’estremità apicale arrotondata della cellula ed il
citoplasma circostante è basofilo. Il loro ruolo e quello di sintetizzare collagene di tipo I,
glicoproteine e proteoglicani della sostanza fondamentale, comprese molte componenti
proteiche minori (osteocalcina, osteonectina, osteopontina ecc..). La membrana
plasmatica contiene recettori per vari ormoni, vitamine e citochine che controllano
l’attività cellulare. Quando gli osteoblasti diventano inattivi, si appiattiscono contro
l’osso circostante e diminuisce la loro basofilia. Essi sono localizzati verso la superficie
che è in contatto con l’osso sottostante, ma il rilascio di componenti della matrice ossea
non è limitato solo a questa superficie. Alcuni di essi restano intrappolati dalle proprie
secrezioni e quindi imprigionati come osteociti in lacune all’interno della matrice ossea
neoformata.
Durante la vita dell’adulto, l’osso va incontro ad un continuo processo di
rimodellamento interno e rinnovo che comporta la rimozione della matrice ossea
calcificata e la sua sostituzione con un nuovo tessuto osseo. Gli osteoclasti sono infatti
responsabili della rimozione di osso in questo processo. Al di sotto di ciascun
osteoclasto vi è una piccola depressione sulla superficie ossea ( lacuna di Howship) che
viene provocata dalla digestione enzimatica della sottostante matrice ossea da parte di
enzimi lisosomiali rilasciati dagli osteoclasti.
La artrologia studia le modalità
con cui i segmenti ossei si
mettono in rapporto tra di loro a formare le articolazioni o giunture. Le articolazioni si
distinguono in fisse (sinartrosi), semimobili (anfiartrosi) o mobili (diartrosi).
Diartrosi = sono caratterizzate da una cavità articolare che separa i due capi ossei e
consente loro movimenti reciproci più o meno ampi. Gli elementi che caratterizzano
tutte le diartrosi sono:
1. Cavità articolare: si può considerare virtuale in quanto i capi ossei sono a contatto tra
di loro con le superfici ricoperte di cartilagine articolare
2. Capsula articolare: distinta in capsula fibrosa (manicotto fibroso che si inserisce sui
due capi ossei connettendoli tra di loro ed impedendo il distacco; racchiude
l’articolazione e la isola dal contesto) e membrana sinoviale (strato più interno della
capsula che passa a rivestire i capi ossei arrestandoli ai bordi della cartilagine
articolare. Può essere molto sottile o complessa).
3. Cartilagine articolare: si tratta di cartilagine ialina molto liscia, adatta ad
assorbire e resistente alle sollecitazioni meccaniche
4. Liquido sinoviale o sinovia: è un trasudato plasmatico prodotto dalla
membrana sinoviale. Esso è denso, filante, ricco di acido ialuronico. Svolge il
compito di lubrificare e nutrire la cartilagine articolare
5. Legamenti articolari: fanno parte dei dispositivi di unione che servono ad
impedire il distacco assicurando nel contempo la stabilità ed il grado più
ampio possibile di movimento.
Le diartrosi, come affermato nella definizione, consentono il movimento reciproco dei
capi ossei:
• Adduzione / Abduzione = l’arto superiore si avvicina (adduzione) o si allontana
(abduzione) dal corpo ruotando su un asse sagittale.
• Flessione = l’articolazione del gomito consente all’estremità dell’avambraccio di
avvicinarsi o di allontanarsi dalla spalla.
• Rotazione = l’arto ruota sul proprio asse.
• Circonduzione = l’arto descrive nello spazio un cono.
enartrosi
condilartrosi
sella
artrodia
trocoide
troclea
……….ritornando all’AR…………..
Nell’AR l’elemento fondamentale è rappresentato dai linfociti T, linfociti B, neutrolfili,
macrofagi, la cui attivazione determina poi a cascata la secrezione di sostanze
infiammatorie (interleuchine, TNF-alfa ecc…) . Queste sostanze hanno il compito di
erodere i tessuti articolari, a cominciare dalla sinovia, e poi l’osso stesso che vengono a
poco a poco erosi, più o meno rapidamente a seconda dell’andamento della malattia e
della sua aggressività.
Secondo i dati clinici, nell’arco dei primi tre
anni di malattia il 70% dei pazienti presenta
lesioni delle articolazioni evidenti anche alla
radiografia e la metà di questi ha sviluppato
erosioni e riduzione dello spazio all’interno
dell’articolazione già nel primo anno. Dopo
il sesto anno, però, le lesioni progrediscono
molto più lentamente.
Possibili cause
Se il meccanismo dell’AR è in buona parte noto, meno chiaro è quale sia l’evento che
fa scattare la reazione autoimmune. Nell’introduzione abbiamo accennato quali
possono essere i possibili fattori scatenanti questa malattia come fattori ereditari,
autoimmunitari e virali. Si può parlare quindi di una malattia multifattoriale, alla cui
origine ci sono caratteristiche delle persone e, probabilmente, fattori ambientali.
L’AR è una malattia ad andamento cronico
e se non interviene una guarigione completa
nell’arco del primo anno, è quasi escluso che
possa presentarsi in seguito. Nella maggior
parte dei casi l’andamento è pulsante: a fasi
di attenuazione dei sintomi si alternano fasi
di aggravamento.
Sintomi e manifestazioni
Ovviamente, il primo e più importante segno / sintomo dell’AR è l’infiammazione
dolorosa delle articolazioni. Si usa ovviamente il plurale perché uno dei tratti distintivi
della malattia è di interesse sempre più di una articolazione, cosi come l’andamento
simmentrico: le articolazioni delle due mani, dei due polsi, quelle dei gomiti e cosi via.
Di solito le prime ad essere colpite sono proprio le piccole articolazioni citate prima. Un
altro elemento importante è la rigidità dell’articolazione, che si presenta dopo un
periodo di riposo. Difatti uno dei criteri per la diagnosi è proprio la rigidità mattutina, cioè
la difficoltà a muovere l’articolazione dopo il riposo notturno.
Anche se ora il criterio non è più accettato unanimamente, una rigidità articolare che
dura per almeno un’ora al mattino deve far pensare all’AR.
Trattandosi di un processo infiammatorio, l’articolazione colpita può risultare calda e
gonfia (calor, tumor). Quest’ultima caratteristica è dovuta all’accumulo di liquido
sinoviale, che è una delle risposte dell’articolazione a tutti gli insulti (anche traumi). Altri
sintomi di esordio possono essere oltre che articolari anche sistemici, come febbre,
astenia, perdita di peso, mialgie ecc…
In teoria, l’AR può colpire qualsiasi articolazione, tuttavia è frequente l’interessamento
delle mani, piedi, gomiti, anche e ginocchia. Per quanto riguarda la colonna vertebrale,
di solito l’artrite si manifesta quasi sempre alle vertebre cervicali.
La diagnosi
Purtroppo è difficile poter distinguere tra l’AR e le altre malattie che interessano le
articolazioni basandosi semplicemente su uno o più test.
Esistono dei test che provano l’esistenza generica dell’infiammazione (VES, la proteina
C reattiva, il fibrinogeno, l’amiloide sierica A) ma non sono, appunto, specifici. Esiste poi
il cosiddetto fattore reumatiode che altro non è che un autoanticorpo, come detto in
precedenza, però malgrado il nome non è un indicatore certo della presenza di AR:
infatti 1/3 dei malati non è positivo a questo test, mentre questo fattore viene riscontrato
nel 5% della popolazione sana; inoltre con l’età il fattore reumatoide tende a presentarsi
più spesso anche in assenza della malattia (nel 10-20% degli over 65).
Nemmeno le indagini radiologiche sono di grande aiuto nella prima fase della malattia:
al massimo si possono riscontrare alterazioni della sinovia tipiche di un po’ di tutti i
disturbi articolari. Risulta chiaro che se si riscontrano erosioni di cartilagine ed osso
significa che la malattia è attiva da mesi.
La diagnosi va posta, quindi, sulla base dell’esame dello specialista (il reumatologo)
che terrà presente tutti gli aspetti citati finora.
Dal 1987 l’American College of Rheumatology ha messo a punto un insieme di
criteri utili sia per la diagnosi sia per la valutazione della gravità della malattia.
Per affermare che un paziente soffre di AR devono essere presenti almeno 4 di questi 7
requisiti:
• rigidità mattutina che si protrae per almeno un’ora
• infiammazione / dolore di tre o più aree articolari (falange, metacarpo, polso, gomito,
ginocchio ecc…)
• infiammazione / dolore delle articolazioni della mano
• simmetria dell’artrite
• presenza di noduli reumatoidi (presenza di noduli sottocutenei nelle aree vicine alle
articolazioni o in punti anatomici soggetti a pressione, come la pleura)
• presenza di livelli ematici elevati di fattore reumatoide
• segni di erosione delle articolazioni della mano o del polso visibili alle radiografie
Principali forme di artrite
Ci sono cosi tante forme di artrite che non sarebbe possibile elencarle tutte qui.
Secondo l’Arthitis Foundation, il termine artrite si riferisce a più di 100 differenti
malattie che provocano dolori, gonfiori e limitazioni al movimento delle articolazioni e
del tessuto connettivo in tutto il corpo.
Le tre forme più diffuse di artrite sono:
1. Osteoartrite = conosciuta anche come artrosi,
è una malattia degenerativa delle articolazioni.
Essa è provocata dall’usura e dal logoramento
dovuto all’invecchiamento che provocano un
deterioramento della cartilagine nella parte finale
delle ossa. Questo provoca un indebolimento dei
muscoli, dei tendini e dei legamenti che sostengono
l’articolazione. Il tentativo dell’organismo di
stabilizzare la giuntura ha come effetto la formazione
di speroni ossei osteofiti (nuove formazioni ossee).
Il processo può essere accelerato dal sovrappeso,
da lesioni articolari, da una dieta inadeguata e da
scarso o inadatto esercizio fisico.
2. Fibromialgia = essa è una condizione di origine
sconosciuta,caratterizzata da dolore cronico e
generalizzato alle muscolature, dolori articolari,
rigidità, astenia, parestesie, insonnia, ansia,
cefalea e sindrome dell’intestino irritabile.
Colpisce più frequentemente le donne di età
variabile fra i 20 e i 50 anni.
3. Gotta = essa è un tipo di artrite causata da un
aumento dei livelli di acido urico nei liquidi del corpo.
L’acido urico in eccesso si deposita sotto forma di
cristalli i quali, a loro volta, si depositano nelle articolazioni (specialmente la I
metatarso falangea) ed in altri tessuti, inclusi i reni,
causando notevoli danni ed infiammazione. La Gotta
era denominata come “la malattia dell’uomo ricco”
(perché solo il ricco poteva permettersi di mangiare
tanto e tanta carne) e storicamente è stata associata
all’abbondanza. Questa immagine tradizionale ha
delle basi reali in quanto la degradazione degli aa
(carne e proteine in genere), in carenza di un
determinato enzima, causa l’aumento dell’acido
urico e l’assunzione da alcool diminuisce la capacità dei reni di eliminarlo
Manifestazioni extra articolari
L’AR presenta varie manifestazioni al di fuori dell’articolazione (extra articolari)
piuttosto frequenti. Di solito tali manifestazioni si presentano in individui con titoli elevati
di autoanticorpi contro il fattore reumatoide.
• noduli reumatiodi: si sviluppano nel 20-30% dei pazienti con AR. Sono in genere
localizzati a livello delle superfici estensorie o di altre aree soggette a pressione
meccanica, ma possono presentarsi anche in altri distretti come le pleure e meningi. Le
dimensioni e la consistenza sono variabili. Sono raramente asintomatici, ma talvolta
possono rompersi per effetto di traumi o infettarsi. Essi sono costituiti da materiale
necrotico che comprende fibre di collagene e detriti cellulari, macrofagi e un tessuto di
granulazione. In alcuni pazienti la terapia con Metotrexato può aumentare il numero di
noduli.
• senso di debolezza e l’atrofia muscolare: l’atrofia appare poche settimane dopo
l’esordio ed interessa in genere i muscoli adiacenti all’articolazione colpita.
• vasculite reumatoide:
essa può interessare qualsiasi organo ed è più comune nei
pazienti con AR grave e titoli elevati di fattore reumatoide. Può causare ulcere cutanee
con necrosi del derma, gangrena ed infarto viscerale (tali complicanze sono però rare).
Essa si manifesta con macchie brunastre raggruppate nei polpastrelli delle dita.
• manifestazioni pleuropolmonari: comprendono pleurite, fibrosi interstiziale, noduli
pleuropolmonari (descritti in precedenza), polmonite ed arterite. Si può verificare anche
un’ostruzione delle alte vie respiratorie
• complicanze cardiache: sono rare, tuttavia nel 50% dei pazienti si rileva, in sede
autoptica, una pericardite.
• manifestazione neurologiche: esse possono derivare anche da lussazioni dei tratti
atlanto-assiale e medio-cervicale. Si avranno neuropatie a carico dei nervi del braccio e
della gamba dovute a lesioni dei nervi conseguenti a sinoviti o deformazioni articolari.
• l’occhio viene colpito meno dell’1% dei casi.
• osteoporosi: essa è secondaria all’interessamento reumatoide e può essere
aggravata dalla terapia cortisonica.
……........Numerose evidenze indicano
che l’interleuchina-1 (IL-1) e il fattore di
necrosi tumorale- alfa (TNF-alfa) sono
mediatori chiave della degradazione dei
tessuti nell’AR. Entrambe le citochine
possono influenzare le manifestazioni
locali e sistemiche della malattia,
nonché i meccanismi che provocano la
distruzione della cartilagine. In particolare,
gli effetti di IL-1 sull’induzione di enzimi
proteolitici, quali proteasi della matrice,
sembrano essere critici per la
fisiopatologia del danno strutturale a
livello articolare…………………
Modelli Sperimentali
Secondo alcuni studi sperimentali condotti sul sangue periferico umano nonché su
modelli animali, il TNF-alfa induce la produzione di IL-1, a sua volta responsabile della
sintesi di TNF-alfa nonché dell’ulteriore rilascio di IL-1.
Malgrado l’apparente sovrapposizione della azioni esercitate da queste citochine, i
risultati emersi da modelli animali indicano che IL-1 e TNF-alfa possono produrre effetti
diversi e distinti. In un modello di AR murina indotta dalla parete cellulare streptococcica,
per esempio, sono stati osservati aumenti considerevoli della produzione di IL-1
indipendenti da TNF-alfa. In questo modello sperimentale, nonostante il trattamento con
anti-TNF, le erosioni hanno continuato a progredire fino a raggiungere un’estensione
superiore a quella evidenziata nei topi che presentavano carenza di TNF.
L’AR quindi si sviluppa anche in assenza di TNF, ma non nei topi che non sono in
grado di produrre IL-1.
In un altro studio che ha valutato l’effetto di anti-TNF sulla produzione di IL-1, non è
stata osservata alcuna riduzione significativa dei livelli di IL-1 circolante, rispetto al
placebo.
La produzione di IL-1 è quindi indipendente dalla presenza di TNF.
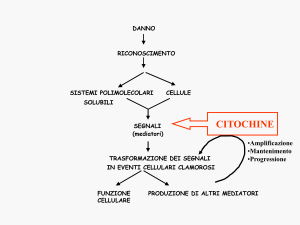
Citochine
Le citochine sono una classe eterogenea di glicoproteine secretorie prodotte da diversi
tipi di cellule, ed hanno funzione di condizionare il comportamento di altre cellule. Sono
quindi dei mediatori tra le cellule ed agiscono come segnali inter-cellulari. Sotto il nome
di citochine sono raggruppate molecole prodotte da linfociti ( Linfochine) o da monociti
(Monochine), hanno le più svariate funzioni; soprattutto possono comportarsi in modo
diverso, in una stessa cellula, a seconda della presenza o meno di altre citochine
(sinergia).
Esse possono agire a livello della stessa cellula che le ha prodotte (azione Autocrina),
oppure agire su altre cellule ma vicine a quella che le ha prodotte (azione Paracrina) o
ancora sulle cellule lontane (azione Endocrina) utilizzando il sistema ematico come
mezzo di trasporto. Molte citochine presentano gli stessi effetti una volta che agiscono,
l’affinità verso i loro recettori è molto alta se comparata ad altri ormoni. Diverse
citochine possono sinergizzare od antagonizzarsi, diviene quindi difficile capirne l’effetto
se in presenza di diverse specie.
Le azioni delle citochine possono essere riepilogate in 5 aree:
• attivazione della risposta immunitaria cellulare ed umorale
• induzione dell’infiammazione
• regolazione dell’emopoiesi
• controllo sulla differenziazione e proliferazione cellulare
• modulazione della guarigione
IL-1: struttura e funzione
L’IL-1 è un monomero di 159 aa del PM di 17,5 KDa, è una glicoproteina responsabile
dello stimolo della risposta della fase acuta, dell’indebolimento muscolare e di malattie
infettive. Essa è un mediatore endogeno della febbre.
Sono state descritte due isoforme di IL-1 (IL-1 alfa e beta) entrambe con attività
biologica sovrapponibile ed agiscono sullo stesso recettore. Esistono due tipi di recettori
per IL-1: RI (espresso su tutte le cellule) ed RII (espresso sui linfociti B).
L’IL-1 viene prodotta da una grandissima varietà di cellule, ma prevalentemente da
macrofagi in seguito a stimoli immunologici ed infiammatori.
Questa citochina è una delle più pleiotropiche intervenendo, oltre che in vari aspetti della
reattività immunitaria, nei processi di riparazione tissutale, nel favorire il rilascio di fattori
di crescita e di differenziazione dei precursori midollari; inoltre si comporta da potente
pirogeno ed ha una spiccata azione pro-infiammatoria. Eccone le principali azioni:
• effetto pirogeno a livello del SNC
• espansione di cellule T CD4+, rilascio di citochine ed incremento dell’espansione
recettoriale di alcune di esse (come IL-2, IL-3)
• proliferazione e differenziazione di cellule B
• induzione della chemiotassi della funzione dei macrofagi
• ecc…....ecc……ecc……ecc……
Ruolo dell’IL-1 nella patogenesi dell’AR
L’IL-1 svolge un ruolo fondamentale nella
fisiopatologia dell’AR in quanto agisce su
diverse cellule presenti nello spazio articolare
amplificando e perpetuando il processo
patologico che sta alla base della malattia.
In particolare IL-1 è un potente stimolatore dei
sinoviociti, dei condrociti e degli osteoclasti.
IL-1 attiva le cellule legandosi al recettore
di tipo1 (IL-1RI) situato sulla membrana. Il
legame IL-1 / IL-1RI forma quindi un complesso
eterotrimerico con la proteina accessoria del
recettore per IL-1 (IL-1RAcP) attivando poi i
meccanismi di trasduzione del segnale.
Proteasoma 26S
Effetti infiammatori dell’IL-1
I segni e i sintomi propri dell’AR sono in gran parte causati dal processo infiammatorio.
Nella fase iniziale, IL-1 media l’infiammazione reclutando i neutrofili all’interno
dell’infiammazione (funge quindi da sostanza chemiotattica), successivamente attiva i
macrofagi e stimola la proliferazione e differenziazione dei linfociti B e T. I sinoviociti,
esposti all’azione dell’IL-1, proliferano e producono IL-6, prostaglandine (come PGE2)
e metalloproteasi della matrice (MMP).
Attraverso tutte questi prodotti l’IL-1 contribuisce al dolore ed alla tumefazione,
tipicamente presenti nell’AR, ed è in grado di alterare la riparazione dell’osso e della
cartilagine.
Degradazione della cartilagine
La produzione di MMP da parte dei sinoviociti attivati da IL-1 scatena un processo di
degradazione dei proteoglicani che a sua volta determina la distruzione della cartilagine.
L’enzima stromalisina sembra svolgere un ruolo fondamentale nell’attivazione della
collagenasi necessaria per la degradazione della cartilagine. L’IL-1 agisce sui condrociti
inibendo la sintesi dei proteoglicani e stimolando la degradazione del collagene
ostacolando cosi il normale processo di conservazione della cartilagine. L’IL-1, inoltre,
determina la produzione di ossidi di azoto che distruggono i condrociti (cellule coinvolte
nel rimodellamento della cartilagine).
Degradazione dell’osso
Gli effetti dell’IL-1 sul riassorbimento osseo sono mediati indirettamente dall’interazione
tra l’antagonista osteoprotegerina (OPG) e il suo ligando (RANKL = ligando
dell’attivatore recettoriale di nf-kb, noto anche come fattore di differenziazione degli
osteoclasti) nonché dagli effetti che il complesso esercita sugli osteoclasti.
OPG è un fattore solubile che si lega a RANKL inibendo la maturazione e l’attivazione
degli osteoclasti e riducendo di conseguenza il riassorbimento osseo da parte degli
osteoclasti stessi. L’equilibrio tra OPG ed il suo ligando determina il grado di erosione
ossea che si verifica nell’articolazione. Nell’AR la concentrazione di RANKL è maggiore
di quella di OPG
IL-1 agisce direttamente sui linfociti T e sugli osteoblasti, potenziando l’espressione di
RANKL, che a sua volta stimola la differenziazione dei precursori degli osteoclasti in
osteoclasti maturi. RANKL , a sua volta, induce la produzione di citochine proinfiammatorie quali IL-1 e di altre citochine che stimolano i linfociti T e ne inducono la
differenziazione (come IL-12 e 15 prodotte dalle cellule APC). Inoltre RANKL agisce
direttamente sugli osteoclasti maturi aumentando la loro attività di riassorbimento,
favorito anche da IL-6 e PGE2.
Inoltre è stato dimostrato che IL-1 induce l’apoptosi degli osteoblasti impedendo la
formazione del nuovo tessuto osseo, effetto particolarmente importante per l’eventuale
processo di riparazione.
Antagonista del recettore dell’IL-1
Già negli anni 1983/1984, prima del clonaggio di IL-1, si sospettava l’esistenza di un
potenziale inibitore di IL-1. Infatti mentre si cercava di isolare grandi quantità di IL-1
usando il saggio biologico della stimolazione delle collagenasi e PGE2 nelle cellule della
sinovia, venne rivolta l’attenzione verso patologie associate alla presenza di grandi
quantità di monociti (leucemia monocitaria) o a patologie associate a temperatura
elevata o a patologie croniche come l’AR.
Con sorpresa si notò che non si rilevava alcuna attività biologica della IL-1 nel siero o
nell’urina di pazienti gravemente affetti dalle citate patologie. Questo indusse ad
ipotizzare che la presenza di IL-1 potesse essere mascherata da molecole inibitrici: la
purificazione chimica condusse all’isolamento di un fattore di circa 17 KDa dall’urina di
pazienti affetti da leucemia monocitaria.
Questo fattore bloccava specificamente le attività biologiche di IL-1, senza influire su
TNF-alfa. Si trattava della prima purificazione dell’antagonista endogeno di IL-1.
L’antagonista naturale di IL-1, IL-1Ra, interferiva con il legame di IL-1 ai linfociti.
Successivamente si notò che in pazienti con AR giovanile, elevati livelli di IL-1Ra erano
associati a fasi afebbrili; mentre bassi livelli di IL-1Ra erano associati alla fase febbrile
della malattia. In seguito all’osservazione che IL-1Ra naturale bloccava il legame di IL-1
alle cellule, l’IL-1Ra fu clonato. I glucocorticoidi stimolano la espressione di questo
antagonista
Cosi potendo disporre dell’antagonista IL-1Ra ricombinante, si stabili che l’antagonista
naturale e quello ricombinante erano simili, in quanto entrambi inibivano il
riassorbimento osseo e la produzione di PGE2, mediati dall’IL-1. Nel 1991 la forma
ricombinante di IL-1Ra (Anakira, del cui farmaco ne parleremo in seguito) fu introdotta in
studi clinici in pazienti con AR.
TNF: struttura e funzione
Il fattore di necrosi tumorale (Tumor Necrosis Factor) è una citochina coinvolta
nell’infiammazione sistemica ed è membro di un gruppo di citochine che stimolano la
reazione della fase acuta. Il TNF è coinvolto in numerosissimi processi come la morte
apoptotica delle cellule , la proliferazione, il differenziamento, la cancerogenesi e la
replicazione virale. Il TNF viene detto talvolta cachessina, cachectina o TNF-alfa.
Il principale ruolo del TNF sta nella regolazione delle cellule del sistema immunitario.
Difetti nella regolazione, in particolare la sovrapproduzione di TNF, sono implicati in
numerose malattie dell’uomo, come il cancro.
Il TNF fu isolato come fattore solubile rilasciato dalle cellule dell’ospite di un tumore
trapiantato (un sarcoma), questo fattore causava la necrosi del tumore. Sebbene il TNFalfa causasse la necrosi di alcuni tumori, può stimolarne la crescita di altri tipi. In questo
senso il nome della citochina può essere frainteso.
Nel 1984, fu sintetizzato il primo cDNA di TNF e ne fu individuata l’omologia di funzione
e struttura con la linfotossina (LT).
Il TNF è prodotto, dapprima, come una proteina transmembrana di tipo II lunga 212 aa
ed arrangiata in uno stabile omotrimero. Da questa forma legata alla membrana, diventa
una citochina solubile omotrimerica (sTNF) grazie al taglio proteolitico di una
metalloproteasi (TACE = enzima convertitore il TNF-alfa). La forma trimerica solubile,
del PM di 51 KDa, tende a dissociarsi per concentrazioni inferiori all’ordine delle
nanomoli, perdendo la sua bioattività.
Meccanismo di azione
Il TNF, come IL-1, è predominante nel processo infiammatorio dell’AR. Esso è presente
nella sinovia, stimola la proliferazione dei sinoviociti e la produzione di mediatori
infiammatori, portando al reclutamento di cellule infiammatorie, alla neo-angiogenesi ed
alla distruzione articolare. A basse concentrazioni esso agisce a livello locale, ad alte
concentrazioni agisce a livello sistemico (causando shock endotossico).
Il TNF deve legarsi a 2-3 molecole di recettori di superficie perché avvenga la
trasmissione del segnale e si produca l’effetto biologico. Il legame con un singolo
recettore di membrana non trasduce nella trasmissione del segnale e la cascata non si
innesca.
La sua iperproduzione non è causa solo di un danno locale, ma anche della
sintomatologia sistemica (ne parleremo più in dettaglio dopo).
Studi mirati hanno quindi dimostrato che la sua attività è modulata non solo dall’azione
delle cellule immunitarie e delle altre citochine, ma anche dalla presenza di mediatori
solubili (sTNF) specifici che si legano ad esso inattivandolo. Però questi recettori solubili
non sono sufficientemente prodotti e non riescono a controbilanciare la produzione di
TNF.
Recettori del TNF
Alcuni anni dopo furono scoperti due recettori di membrana in grado di legare il TNF. I
due tipi di recettori a singolo dominio transmembrana sono:
• TNF R1= p55, è fondamentalmente un recettore di morte, cioè attiva un programma di
morte apoptotica centrato sull’attivazione di una cascata di enzimi proteolitici detti
caspasi;
• TNF R2 = p75, recettore che attiva nella cellula un profilo trascrizionale di tipo
infiammatorio ed in parte inibisce il programma di morte cellulare attivata da TNF R1.
Pur condividendo il ligando, l’attivazione delle due specie recettoriali, determinata dalla
multimerizzazione delle catene recettoriali per interazione con il ligando omotrimerico,
attiva differenti vie di trasduzione del segnale.
Trasduzione del segnale
La cascata di trasduzione del segnale attivata da TNF R2 dipende dal reclutamento di
molecole adattatrici (TRAF1 e TRAF2) che attivano le cascate chinasische dipendenti
da JNK e p38. TRAF2 media anche il reclutamento della chinasi RIP1 (ReceptorInteracting Protein 1), che attivando IKK-beta la fosforilazione di IKB-alfa che
degradandosi, rilascia nfkb libero ed attivo nel citosol, il quale entra nel nucleo e si ha
l’induzione di un programma genetico pro-infiammatorio analogo a quello indotto da
IL-1.
Trasduzione del segnale (continuo)
Diversamente da TNF R2, TNF R1 non esprime significativa affinità per le proteine
TRAF, ma recluta TRADD (TNF R1-Associated Death Domain), un adattatore
intracellulare a sua volta capace di interagire con TNFR2. il dominio DD di TRADD
interagisce con l’omologo dominio DD di un secondo adattatore, la molecola FADD
(Fas-Associated Death Domain), che infine recluta ed attiva la caspasi 8, portando
all’avvio del processo di morte cellulare programmata.
Oltre a TNF-alfa, questa famiglia include altre 10 citochine tra cui : linfotossina alfa e
beta; i ligandi Fas; CD27: CD30: CD40; TRAIL (TNF-related apoptosis-inducing ligand);
RANKL). Diversamente di TNF-alfa, la gran parte di queste molecole non un ruolo
significativo nella risposta infiammatoria, ma hanno in comune la capacità di regolare la
vitalità cellulare. Queste molecole interagiscono con un crescente numero di recettori di
membrana strutturalmente analoghi a TNF-alfa.
TNFRII
Effetti sistemici del TNF
Il TNF è prodotto in massima parte dai macrofagi, ma anche da una serie di altri tipi
cellulari inclusi cellule linfoidi, mastociti, cellule endoteliali, fibroblasti e cellule nervose.
Una grande quantità di sTNF è rilasciata dopo il contatto del macrofago con un
Lipopolisaccaride (LPS = endotossina che compone la membrana esterna della parete
cellulare dei batteri Gram- e che viene rilasciata dopo la lisi del batterio).
Il TNF agisce su organi e sistemi, generalmente in associazione con IL-1 ed IL-6:
• sull’ipotalamo (stimola l’asse ipotalamo-ipofisi-surrene aumentando il rilascio
dell’ormone di liberazione della corticotropina, ACTH; sopprime l’appetito ed induce
febbre)
• sul fegato (stimola la risposta della fase acuta portando l’aumento della proteina C
reattiva e di altri mediatori; induce una insulino-resistenza promuovendo la fosforilazione
della serina del substrato recettore dell’insulina-1, il quale blocca il sistema di
segnalazione dell’insulina)
• attrae potentemente i neutrofili e li aiuta ad agganciarsi alle cellule endoteliali per
extravasare)
• sui macrofagi (stimola la fagocitosi, la produzione di NO, dell’IL-1, di PGE2)
• sul sistema cardiocircolatorio (diminuisce la pressione in seguito alla vasodilatazione;
diminuisce la contrattilità del miocardio; induce la formazione di placche aterosclerotiche
e di trombi)
L’aumento locale della concentrazione del TNF causa i segni tipici dell’infiammazione:
calore, rossore, bruciore e gonfiore. Sebbene alte concentrazioni di TNF possano
indurre sintomi shock-simili, la prolungata esposizione a basse concentrazioni di TNF
può portare a chachessia (sindrome che porte ad una deplezione del patrimonio
proteico e lipidico dei tessuti, in particolare quello muscolare ed adiposo). Questo tipo di
risposta si riscontra nei pazienti affetti d tumore.
Inibitori del TNF
Un’analisi di studi che hanno impiegato Ab anti-TNF nel trattamento dell’AR, ha
confermato l’aumento del rischio di gravi infezioni e di tumori associati a questi farmaci.
I farmaci anti-TNF non sono solo utilizzati nell’AR ma anche nella psoriasi e nel morbo
di Crohn
Un gruppo di ricercatori ha compiuto una ricerca ed ha trovato 9 casi clinici che hanno
incontrato criteri di inconclusione: è stato osservato che i pazienti trattati con anti-TNF
presentavano un rischio 3 volte superiore di sviluppare tumore rispetto ai pazienti trattati
con placebo, ed un rischio 2 volte superiore di presentare gravi infezioni.
Secondo questi ricercatori, gli Ab anti-TNF sono associati ad un aumentato rischio di
infezioni e di tumori perché questi farmaci interferiscono con i meccanismi immunologici
che difendono l’organismo dalle infezioni e dai tumori.
I pazienti che assumono Ab anti-TNF dovrebbero valutare con attenzione i possibili
sintomi di infezioni e sottoporsi a periodici controlli per l’individuazione di tumori.
Riassumendo
IL-1 e il TNF-alfa sono mediatori chiave dell’infiammazione, sono prodotti
principalmente dai monociti-macrofagi in seguito all’attivazione di fattori solubili al
contatto con i linfociti Th1 stimolati. Il contatto tra linfociti e macrofagi è regolato da
ligandi e controligandi (integrine; CD40 ecc…) e dalle lipoproteine plasmatiche (apoA-1
associata alle HDL)
IL-1 e TNF sono potenti induttori delle metalloproteinasi della matrice (MMP),
eicosinoidi, dell’enzima iNOS, del ligando dell’attivatore recettoriale di nfkb (RANKL),
nonché dei prodotti coinvolti nella distruzione della matrice extracellulare e della
cartilagine e nel riassorbimento osseo.
IL-1 è particolarmente importante a livello locale, è più potente del TNF nella
stimolazione delle MMP e nel blocco della riparazione della cartilagene
Tuttavia IL-1 e TNF sinergizzano fortemente in molteplici funzioni biologiche.
Il blocco di IL-1 da parte del suo antagonista endogeno ( IL-1 Ra) e del recettore solubile
di IL-1 (s IL-1R2) in combinazione con la forma solubile della proteina accessoria del
recettore di IL-1 (IL-1R AcP), produce un beneficio a lungo termine nelle malattie
infiammatorie croniche.
Curare l’AR significa agire su due fronti: da una parte contrastare il dolore, a volte
fortissimo e comunque più forte di quello causato dall’artrosi; mentre dall’altra parte
impedire la progressione delle lesioni articolari.
Le terapie della medicina convenzionale comprendono la soppressione
dell’infiammazione con anti-infiammatori (come FAS e FANS) e antidolorifici e, come
ultima risorsa, la chirurgia. Questo approccio provoca seri problemi e complicanze.
I farmaci tipo il cortisone deprimono il sistema immunitario le cui conseguenze sono
probabili malattie da malfunzionamento immunitario ed infezioni, accanto a molti altri
effetti collaterali quali l’ipertensione, il diabete, la ritenzione idrica, degenerazione dei
tessuti, ulcera gastrica ed atrofia muscolare.
I farmaci non steroidei, d’altra parte, causano anch’essi ulcere, problemi renali, enteriti
ed in effetti contribuiscono alla distruzione delle articolazioni impedendo la sintesi della
cartilagine.
Le enteriti, che possono colpire circa il 70% delle persone che prendono questi farmaci
per almeno due settimane, provocano diarrea e dolori addominali che possono portare
ad altre reazioni allergiche e ad un ulteriore aggravamento della patologia autoimmune
per assorbimento di sostanze tossiche.
Evitare la progressione delle lesioni articolari
richiede l’impiego di farmaci capaci di interrompere
in qualche modo la reazione autoimmune.
Le sostanze impiegate a questo scopo sono chiame
farmaci di fondo o DMARD ( Disease Modifyng
Anti-Rheumatic Drugs = farmaci antireumatici capaci
di modificare il decorso della malattia). In questa
categoria rientrano sostanze note da tempo ed altre
recentissime, che molto spesso non sono neppure
nate per curare l’AR, ma per altre indicazioni.
Ad esempio:
• l’Idrossiclorochina (antimalarico)
• Il Metotrexate (antitumorale analogo strutturale dell’acido Folico)
• la Ciclosporina (antirigetto).
Molecole più recenti, come la Leflunomide, sono state messe a punto già in vista di
questa indicazione.
Sostanzialmente, tutti questi farmaci hanno l’effetto di
interrompere l’attivazione o la proliferazione delle cellule
del sistema immunitario, oppure, come accade con i
cosiddetti anti-TNF, di inibire la liberazione di una o più
sostanze responsabili del processo infiammatorio.
Ovviamente, poiché tutte queste sostanze hanno l’effetto
di ridurre le difese immunitarie, il paziente trattato può
facilmente andare incontro ad infezioni, come accennato
in precedenza, fenomeno del resto che accompagna anche
la somministrazione prolungata di cortisone & derivati.
Il paziente dunque deve tener presente che solo di rado un
solo farmaco è sufficiente. Infatti ai farmaci di fondo spesso
si affiancano gli analgesici, oppure si impiegano in
associazione diversi farmaci di fondo.
Però, in certi casi, gli effetti delle due classi di farmaci (farmaci di fondo ed analgesici)
possono sovrapporsi: anche i farmaci di fondo hanno un effetto analgesico cosi come, a
rigore, gli analgesici antinfiammatori dovrebbero tamponare la reazione autoimmune e,
almeno in parte, ridurre il danno. Questa ipotesi è stata avanzata per un farmaco noto
come il Prednisone
Da dove si comincia?
Tradizionalmente l’approccio all’AR era per cosi dire gradualista. Si cominciava con la
terapia analgesica e solo in un secondo momento si passava ai farmaci di fondo.
Attualmente si propende per un approccio più aggressivo sin dalle prime fasi, soprattutto
quando il paziente presenta i segni di una rapida progressione del danno: fattore
reumatoide, VES e proteina C reattiva elevati.
Trattamento non farmacologico
In primo luogo, è di estrema importanza l’informazione del paziente. Come per le altre
malattie croniche, anche per l’AR il paziente deve essere portato a conoscenza della
natura della sua malattia, della tendenza a perpetuarsi, della possibile insorgenza di
limitazioni funzionali delle articolazioni, oltre che delle possibilità terapeutiche, dei loro
limiti e degli eventuali effetti collaterali.
Oggi è dimostrato che un paziente con AR “informato”, che conosce da un lato i limiti
imposti dalla sua patologia e dall’altro come si articola la sua terapia, permane in una
migliore qualità di vita e ricorre meno spesso all’ospedalizzazione, o comunque meno
gg di ricovero.
Necessario è inoltre, per il mantenimento di una normale funzione articolare e di un
migliore stato psicologico, il coinvolgimento fin dal momento della diagnosi di specialisti
di fisioterapia.
I pazienti con danno articolare tale da limitarne seriamente la funzione, o con un livello
inaccettabile di dolore, devono ricorrere a procedure chirurgiche ortopediche.
Rimedi Naturali
Molti sono i rimedi proposti dalla medicina naturale. Il primo passo per un approccio
benefico all’AR è quello di ottimizzare il funzionamento del sistema immunitario
diminuendo al massimo gli effetti autoaggressivi.
In medicina naturale si dice “Dolori = Intossicazione”. Importantissimo sarà non
introdurre più tossine e quindi lo stile di vita (fumo, alcool), l’aria che respiriamo, i cibi
che mangiamo (cibi biologici), l’acqua che beviamo (acqua pura e salutare –
purificazione per osmosi inversa. Bisognerà quindi individuare le intolleranze alimentari
ed eliminare i cibi tossici.
Gli antiossidanti proteggono le articolazioni dal deterioramento. Le vitamine C ed E, il
betacarotene, il tè verde, ma in particolare gli estratti dei semi della vite sembrano
essere utili a questo scopo.
Fu scoperto che alti dosaggi di antiossidanti, in particolare la vitamina C, possono
ridurre il rischio di perdita di cartilagine ed il peggioramento dei pazienti affetti da
osteoartrite. Ma ciò non rispetta il concetto di naturale.
Da molto tempo si conoscono le proprietà degli acidi grassi essenziali ( omega-3 e
omega-6) nel trattamento dell’AR. Questi tipi di oli si possono trovare nei pesci ed in
alcune piante (olio di semi di lino, di enagra e di borragine). In teoria però, questi acidi
grassi essenziali possono generare problemi perché si ritiene possano favorire la
formazione di Radicali liberi nell’organismo, con conseguenti pericoli di malattie
cardiocircolatorie.
Quindi è consigliato assumere assieme agli acidi grassi anche sostanze antiossidanti.
Un antiossidanre completo, disintossicante e rigenerante è il DEGEN-X. Nell’AR i
dosaggi dovranno essere adattati al paziente, ma si consiglia di iniziare con poche
gocce (4 o 5) tre volte al die e salire gradualmente, diminuendo nuovamente in caso di
dolore, per poi riprendere gradatamente a salire.
Anche l’ossigeno-ozono terapia si è rivelata utile nell trattamento dell’AR mediante il
GAET ma, come per il DEGEN-X, le sedute vanno modulate in base alle risposta del
paziente.
Trattamento Farmacologico
Esso consiste principalmente nella combinazione di farmaci sintomatici (FAS e FANS,
che non alterano il decorso clinico dell’AR) e farmaci di fondo (DMARDs).
FANS
Queste sostanze, utili nel controllo della sintomatologia dolorosa, non sono in grado di
prevenire l’evoluzione del danno anatomico articolare se non in associazione con i
farmaci di fondo.
Tuttavia essi vengono impiegati spesso per la loro veloce, seppure fugace, azione sul
dolore. Una buona protezione della mucosa gastrica, nell’impiego di questi farmaci, si è
dimostrata efficace nella prevenzione dell’ulcera peptica; infatti gli inibitori selettivi della
COX2 (come ad esempio il Nimesulide) hanno una minore tossicità gastro-enterica.
Bisogna però ricordare che i malati di AR sono a rischio di un accelerato processo di
aterosclerosi che risulta meglio profilassato con inibitori selettivi della COX1 piastrinica.
Quindi è raccomandabile, nei pazienti maschi over 60 con persistenti indici di flogosi,
l’impiego di un anti-aggregante piastrinico (aspirina 100 mg al die)
Questi farmaci quindi non alterano il decorso della malattia, ne prevengono la comparsa
di erosioni articolari; per questo non possono costituire il solo trattamento farmacologico
dell’AR.
Il malato inoltre NON DEVE FUMARE, deve correggere il peso corporeo
mediante appropriata dieta e, nei limiti del possibile, condurre una vita non sedentaria
FAS
La capacità antinfiammatoria degli steroidi ha reso questa categoria di farmaci molto
usati nell’AR: in quanto essi determinano una rapida inibizione contemporaneamente
sia della produzione di citochine ad azione flogistica (TNF, IL-1) sia del loro meccanismo
di azione.
Essi vengono impiegati a basso dosaggio nell’AR (ad esempio il Prednisone, con un
dosaggio tra 7,5 e 10 mg al die, è in grado di interferire con la progressione dei processi
erosivi articolari oltre che con il ruolo antinfiammatorio, sempre in associazione con un
DMARDs.
Il paziente è invitato ad eseguire una dieta ipocalorica e di mantenere una buona
idratazione.
La posologia di questi farmaci viene ridotta o sospesa al raggiungimento della
remissione indotta dai farmaci di fondo.
Ricercare precocemente segni di osteoporosi (specie nelle donne in menopausa) ed
impostare una giusta terapia con calcio, vitamina D, sarebbe auspicabile
L’azione antinfiammatoria dei glucocorticoidi è dovuta a :
•
Attivazione dell’espressione genica:
lipocortina 1 (annessina 1)
IL-1RII, IL-1Ra
IkB-alfa
•
Inibizione dell’espressione genica:
citochine (IL-1, 2, 3, 4, 5, 6, 8, 11, 13, TNF-alfa, GM-CSF, IL-2R)
chemochine (RANTES, ecc…)
enzimi (iNOS, COX)
molecole di adesione (integrine, selettine, CAMs)
fosfolipasi A2
collagenasi
Il fattore di trascrizione nucleare che codifica per queste proteine, sia anti che pro
infiammatorie, è l’NFkB che viene bloccato dai glucocorticoidi.
DMARDs
Tutti i pazienti con AR sono candidati a terapia con questi farmaci, trattamento che
dovrebbe essere instaurato non appena posta la diagnosi.
Dal momento che il trattamento con un singolo DMARD spesso non controlla in modo
soddisfacente i sintomi, o non impedisce la progressione del danno articolare, sempre
più diffusa è la tendenza ad associare DMARDs diversi. Essi sono sostanze che
alterano il decorso clinico dell’AR e rallentano nel tempo l’evoluzione del danno
anatomico delle articolazioni
Le più comuni associazioni, che hanno dimostrato una migliore capacità nel controllare
la patologia rispetto ai DMARDs impiegati da soli sono:
• Metotrexato e Ciclosporina
• Metotrexato, Salazoprina, Idrossiclorochina
• Etanercept e Metotrexeto
Numerosi sono i fattori che influenzano la scelta delle combinazioni di questi farmaci.
Infatti il paziente ed il medico devono insieme valutare la gravità dell’artrite; l’efficacia
attesa dal singolo farmaco o dalla combinazione; le potenzialità tossiche e la frequenza
di manifestazione di questa, il numero e tipologia dei controlli di monitoraggio necessari
ed il costo complessivo inerente.
INFLIXIMAB (Remicade)
Esso è un anticorpo monoclonale di origine chimerica (75% della sequenza umana e
25% della sequenza murina). Questo mAb gioca un ruolo fondamentale nella
patogenesi dell’AR: infatti lega il TNF e non consente l’interazione della citochina con il
suo recettore (anti-TNF). Questo farmaco può essere somministrato solo in ospedale, in
ambiente specialistico reumatologico, per via dei costi molto elevati.
Sono previste infusioni endovenose lente (2 h almeno) di 3 mg/kg ogni due mesi. Viene
somministrato in associazione con il Metotrexato.
Esso si è dimostrato molto efficace: infatti oltre il 70% dei pazienti affetti da AR
refrattaria ad altri trattamenti, hanno risposto in modo significativo al farmaco.
Nei pazienti trattati con il Remicade è stato osservato l’arresto della progressione del
danno anatomico articolare; per questo motivo è in corso una sperimentazione nella AR
di recente insorgenza.
Gli effetti collaterali più frequenti sono: infezioni e reazioni vasomotorie durante
l’infusione; febbre, orticaria, ipotensione ed immunodepressione
ETANERCEPT (Enbrel)
Esso è un recettore solubile chimerico che riconosce e si lega al TNF e lo blocca.
Anch’esso può essere dispensato solo in ospedale.
Viene somministrato per via sottocutanea dove viene lentamente assorbito. Raggiunge
la massima concentrazione dopo circa 48 ore. La sua biodisponibilità e del 76%; viene
eliminato lentamente dall’organismo. Ha una lunga emivita di 70 ore circa. La presenza
di insufficienza renale o epatica non richiede la modifica del dosaggio.
Gli effetti collaterali più frequenti sono: infezioni e reazioni cutanee locali, nel sito di
iniezione
ADALIMUMAB (Humira)
Esso è un mAb umano anti-TNF; si somministra per via sottocutanea ogni 15 gg.
Genera, come effetto tossico infezioni e reazioni vasomotorie durante l’infusione
ANAKINRA (Kineret)
Il Kineret è una forma ricombinante dell’antagonista endogeno dell’IL-1 (IL-1Ra),
r-metHuIL-1ra, prodotta mediante la tecnologia del Dna ricombinante utilizzando
Eschlerichia Coli come sistema di espressione. Il Kineret è analogo a IL-1Ra endogeno
ad eccezione dell’aggiunta di un solo residuo di metionina in posizione N-terminale.
Da un punto di vista terapeutico il Kineret neutralizza l’attività biologica di IL-1 inibendo,
con un meccanismo competitivo, il suo, legame con IL-1R1. esso impedisce quindi la
trasduzione del segnale nonché la produzione di NO, di PGE2, dell’enzima collagenasi
da parte dei sinoviociti, dei fibroblasti e dei condrociti.
Il Kineret è ben assorbito per via sottocutanea con un dosaggio raccomandato di 100
mg da somministrare una sola volta al die; la somministrazione di questo farmaco deve
essere effettuata nella stessa ora, la sua biodisponibilità è del 95%, l’emivita è di 4-6
ore
Esso è utilizzato per il trattamento dei segni e sintomi dell’AR in associazione con il
Metotrexato.
Il kineret è stato somministrato ad oltre 2600 pazienti nel corso di studi clinici su larga
scala (fase III). L’evento avverso segnalato è stato una reazione nel sito di inezione
risultata di entità lieve o moderata nel 95% dei pazienti
RITUXIMAB (Mabthera)
Esso è il primo mAb utilizzato nell’AR mirato ai linfociti B. Grazie al legame specifico
con una molecola sulla superficie delle cellule B, CD20, l’anticorpo spezza la cascata
infiammatoria che provoca i sintomi della malattia. Le cellule B, grazie al legame con il
farmaco, vemgono inibite. Grazie alla eliminazione delle cellule B, non vengono più
prodotti Ab che attaccano i tessuti dell’organismo e che promuovono l’infiammazione.
Poiché né le cellule staminali nè le cellule plasmatiche subiscono l’azione del farmaco,
la popolazione di cellule B potrà essere ripristinata tramite le cellule staminali con la
conservazione di livelli normali di Ab. In questo modo il sistema immunitario rimane
integro.
L’azione selettiva sulle cellule B permette ai pazienti, che hanno avuto una risposta
insufficiente o che non tollerano la terapia con anti-TNF, di ricorrere ad una efficace
alternativa terapeutica.
TOCILIZUMAB (Actemra)
Esso è un mAb umanizzato in gradi di bloccare i recettori dell’IL-6 quindi impedire il
legame dell’interleuchina con il suo recettore. In questo modo il farmaco inibisce
un’importante mediatore dell’infiammazione dell’AR.
Studi nella fase III, condotti dalla Chugai in Giappone appartenente al gruppo Roche,
sulla monoterapia di questo farmaco, hanno dimostrato che Actemra è migliore dei
farmaci anti-reumatici modificatori della malattia (DMARDs) sia nel ridurre i segni e
sintomi dell’AR che nel ridurre in modo significativo l’entità della distruzione articolare.
Attualmente è in corso un vasto programma di fase III su Actemra nell’AR al di fuori del
Giappone, con l’arruolamento previsto di oltre 4000 pazienti in 41 paesi.
Actemra è un anticorpo dei recettori di IL-6 di qualità superiore ed è dotato di un nuovo
meccanismo di azione che può fornire un trattamento nuovo ed efficace contro l’AR.
Una proteina chiave per l’AR
Una nuova possibile arma contro l’AR è data dall’inibizione di un enzima chiamato
MKK3 che, all’interno delle cellule, svolge un ruolo fondamentale nello scatenamento
della risposta infiammatoria da parte delle cellule.
Si tratta di una scoperta importante dal momento che potrebbe portare a farmaci meno
costosi e, nelle speranze delle persone, più sicuri per trattare l’AR.
Come detto in precedenza, il TNF scatena l’infiammazione, la sua sovrapproduzione
porta all’AR. L’inibizione del TNF, tuttavia, ha un inconveniente di sopprimere anche la
risposta immunitaria normale.
Per molti anni si è cercato di trovare un’altra strada per la terapia puntando sui
meccanismi di azione degli enzimi p38 MAP chinasi, che hanno il compito di regolare
l’espressione di citochine. Però questa è un’azione troppo drastica, poiché inibisce
anche la risposta utile all’organismo.
Allora i ricercatori hanno puntato con successo su in altro regolatore, l’enzima MKK3.
Sperimentazioni in vitro hanno dimostrato che un deficit di questo enzima porta ad una
drastica diminuzione dell’infiammazione reumatica, lasciando intatte le altre funzioni
cellulari intatte.