I difetti della catena respiratoria possono quindi
essere causati da mutazioni nel nDNA (e quindi essere ereditati secondo le leggi della genetica mendeliana classica, AD, AR, XL) oppure da mutazioni del
mtDNA (in questo caso sono ereditati secondo regole proprie della genetica mitocondriale). Esistono
poi alcune alterazioni del mtDNA che sono secondarie a difetti del nDNA (vedi riquadro).
A livello del mtDNA possiamo ritrovare mutazioni puntiformi o riarrangiamenti su larga scala (delezioni o duplicazioni). Alcune mutazioni sono ricorrenti (le delezioni comuni e le sostituzioni a livello
dei nucleotidi 8993, 3243 e 8544 del mtDNA), mentre altre sono state identificate solo in uno o due pazienti. Esse possono essere trasmesse con delle modalità diverse da quelle della genetica mendeliana
classica.
Le caratteristiche della genetica mitocondriale
sono:
1) l’eredità materna;
2) l’eteroplasmia, cioè la presenza di una proporzione variabile di mtDNA normale e mutato in una
cellula;
3) l’effetto soglia;
4) la segregazione casuale.
Inoltre, le mutationi del mtDNA possono interessare anche i geni per i tRNA e gli rRNA, mentre non
esistono esempi analoghi nel DNA nucleare.
Alterazioni del mtDNA secondarie a
mutazioni nucleari
Alcune alterazioni del mtDNA non sono degli eventi
primitivi, ma sono determinate da mutazioni in alcuni geni nucleari che controllano la replicazione e la
stabilità del mtDNA. Per deplezione di mtDNA
si intende un’alterazione del numero di copie per
cellula di genoma mitocondriale (che per il resto
sono strutturalmente normali) al di sotto di un valore minimo necessario per assicurare una normale
biogenesi della CR. Le sindromi con deplezione di
mtDNA sono di solito ereditate come caratteri autosomici recessivi e sono causate da mutazioni nel
gene per la DNA polimerasi mitocondriale (PolG)
e nei geni che regolano il metabolismo del pool di
deossinucleotidi mitocondriali, ma possono essere anche secondarie a cause non genetiche, quali
l’assunzione di farmaci antiretrovirali che possono
interferire con PolG.
Alcuni pazienti presentano un particolare quadro di
anomalie del genoma mitocondriale detto “delezioni multiple”. Consiste nell’accumulo di molecole di mtDNA recanti delezioni di diversa ampiezza,
soprattutto a livello del muscolo, causate da mutazioni in geni che regolano il processo di replicazione
del mtDNA. Possono essere ereditate con modalità
dominante o recessiva e non vi è trasmissione alla
prole dei genomi mitocondriali anomali, ma le delezioni si formano nuovamente in ogni generazione di
individui affetti.
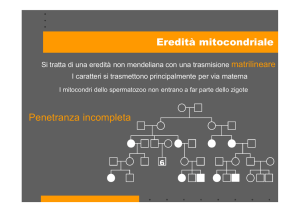
Eredità materna
Il mtDNA è ereditato solo dalla madre. Dopo la
fecondazione, infatti, i mitocondri dello spermatozoo che eventualmente sono penetrati nello zigote
vengono attivamente eliminati. Ciò fa sì che possono essere affetti da mutazioni del mtDNA sia i maschi che le femmine, ma solo le femmine possono
trasmettere la mutazione alla prole.
Eteroplasmia
Ogni cellula contiene 2 copie per ogni gene nucleare, mentre la situazione per i geni mitocondriali
è completamente diversa. Infatti, vi sono in media
circa 5-15 molecole di mtDNA per ogni mitocondrio
e vi è un numero variabile di mitocondri per cellula
(da >50 a >1000), per cui ogni cellula possiede da un
minimo di diverse centinaia fino ad alcune decine di
migliaia di copie di mtDNA. Ne consegue che le mutazioni possono interessare proporzioni variabili del-
le molecole di mtDNA in una cellula o in un tessuto
(da 0% a 100%). Per eteroplasmia si intende la coesistenza di due popolazioni di mtDNA (wild type e
mutato) all’interno di uno stessa cellula o di uno
stesso tessuto.
Il grado di eteroplasmia può variare tra cellula e
cellula in un dato tessuto e tra diversi tessuti in uno
stesso organismo.
Per omoplasmia si intende la presenza di molecole di mtDNA di un unico tipo (o wild type o mutato) in un dato tessuto o organismo.
Le mutazioni patologiche sono solitamente eteroplasmiche, mentre le varianti omoplasmiche sono
di solito dei polimorfismi. Le mutazioni omoplasmiche sono rare e per poter essere compatibili con la
vita devono essere necessariamente lievi o avere un
effetto tessuto-specifico (ad es. la mutazione A1555G
Malattie mitocondriali
121