")
1
2
Edmund Vincent Cowdry li identificò nel 1918
come organelli intracellulari deputati alla
produzione di energia.
3
4
Invaginazioni della
membrana interna
Consistenza gelatinosa
proteine idrosolubili; contiene
ribosomi 70s, enzimi e DNA
circolare
Costituita da lipidi ed enzimi; molto
permeabile, grazie alle “porine”
Costituita da proteine e lipidi; poco
permeabile, grazie alla “cardiolipina”
Composizione simile al
citoplasma
5
6
•Ciclo dell’acido
citrico (di Krebs o
anche degli “acidi
tricarbossilici”);
•Catena respiratoria;
•Fosforilazione
ossidativa.
matrice
membrana interna
7
8
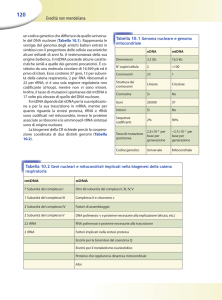
DNA circolare, doppia membrana, dimensioni, struttura
ribosomi, metodi di replicazione e divisione, tipi di
rRNA…
9
“
” di
Umberto Pierantoni (1876-1958).
10
11
I mitocondri possiedono un
proprio genoma
•Si trova nella matrice in copie multiple;
DNA “NUDO” NUCLEOIDI;
•Organizzato in plasmidi, simili a quelli dei
batteri;
•Costituito da 16600 bp,(meno dell’1% del
genoma totale nei mammiferi);
•Codifica per alcune proteine utili al mitocondrio
stesso;
•Scarsa presenza di tratti di regolazione;
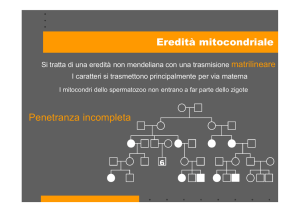
•Eredità matrilineare;
•Più grande nei vegetali.
12
•I geni contenuti nell’mtDNA sono
in tutto 37.
•Le proteine codificate dall’mtDNA sono
solo 13. La maggior parte delle proteine
necessarie per le funzioni del mitocondrio
vengono tradotte a partire dal genoma
nucleare.
13
Durante la fecondazione è l’oocita a
garantire la presenza di numerosi mitocondri
(e di tutti gli altri organuli) per lo sviluppo
dello zigote.
Essendo la prima DONNA individuata da
studi antropologici l'AFRICANA (LUCKI),
l’origine di ognuno di noi, maschio o
femmina che sia, ha una eredità genetica
AFRICANA.
14
15
Ogni essere umano pertanto eredita
il suo DNA mitocondriale
solo ed esclusivamente dalla madre.
16
17
•Raddoppia la massa del mitocondrio;
•A livello della membrana interna si forma un
solco, per la scissione del vecchio mitocondrio
in due nuovi.
18
Nella traduzione sono coinvolti 22 tRNA diversi.
Il primo porta la “formil-metionina”, come nei
batteri.
19
AUA
AUG
UGA
AGA
AGG
nDNA
mtDNA
Isoleucina (Ile)
Metionina (Met)
Metionina (Met)
Metionina (Met)
STOP
Triptofano (Trp)
Arginina (Arg)
STOP
Arginina (Arg)
STOP
20
21
Le malattie mitocondriali sono
molto variabili e perciò difficili
da diagnosticare.
22
N.°
casi
20
Incidenza patologie mitocondriali
18
16
14
12
10
8
6
4
2
0
<5
6-10
11-20
21-30
31-40
41-50
51-60
>60
23
Età
Organi ed apparati coinvolti:
•Apparato respiratorio: scompensi respiratori;
•Muscoli scheletrici: stanchezza, debolezza, crampi;
•Cervello: epilessia, demenza, emicrania;
•Occhi: impossibilità nel muovere gli occhi da un senso all’altro
(Oftalmoplegia esterna), cecità (retinitis pigmentosa), palpebre
afflosciate (Ptosis);
•Cuore: cardiomiopatia;
•Fegato: ipoglicemia;
•Pancreas: diabete;
•Sistema uditivo: diminuzione dell’udito;
•Rene: Sindrome di Toni-Fanconi-Debre (perdita di metaboliti
essenziali nelle urine);
•Tratto digestivo: reflusso acido, vomito, diarrea
cronica, ostruzione intestinale.
24
25
MANIFESTAZIONI CLINICHE
PRINCIPALI
•Nell’infanzia: rallentamento o arresto della
crescita, mioglobinuria ricorrente, danni
renali, nanismo, diabete, atrofia ottica e
sordità, od encefalopatia progressiva;
•Nell’età adulta: intolleranza allo sforzo e
facile affaticamento.
26
Quando sospettare una malattia
mitocondriale?
In genere, una patologia mitocondriale
deve essere sospettata in tutti i pazienti
che presentino sintomi a carico di più
organi non correlati tra loro, con
prevalente compromissione
neuromuscolare e con un decorso
progressivo.
27
Aggressività della mutazione è in
relazione con diversi fattori:
•Numero di mutazioni;
•Localizzazione delle mutazioni;
•Percentuale di mitocondri mutati;
•Tipo di mutazione.
28
29
30
ALCUNE PATOLOGIE
31
Malattia
Acronimo
Sintomatologia
Gene affetto
Oftalmoplegia cronica
progressiva esterna
CPEO (Chronic Progressive
External Ophthlmoplegia)
Miopatia mitocondriale e paralisi
dei muscoli motori dell’occhio
Perdita di più geni per delezione
Sindrome di Kearns-Sayre
KSS (Kearns-Sayre Syndrome)
CPEO più atassia, deterioramento
retinico, cardiopatia, sordità,
diabete e insufficienza renale
Perdita di più geni per delezione
Neuropatia ottica ereditaria di
Leber
LHON (Leber’s Hereditary Optic
Neuropathy)
Cecità (completa o parziale) per
degenerazione del nervo ottico
Mutazioni in subunità della
NADH-deidrogenasi
Sindrome di Leigh
SNEM (Subacute Necrotizing
Encephalomyelopathy)
Degenerazione dei gangli della
base con conseguente perdita
delle capacità motorie e verbali
ATP-sintasi
Encefalopatia mitocondriale con
acidosi lattica ed episodi ictali
MELAS (Mithocondrial
Encephalomyopathy Lactic
Acidosis and Stroke-Like Episodes)
Disfunzione del tessuto encefalico
con conseguenti demenza e
accessi epilettici, miopatia
mitocondriale e acidosi lattica
tRNALeu
Ipostenia muscolare neurogena,
atassia e retinite pigmentosa
NARP (Neurogenic Muscle
Weakness, Ataxia and Retinitis
Pigmentosa)
Debolezza muscolare, atassia e
cecità
Subunità della ATP-sintasi
Disfunzione del midollo osseo
infantile con conseguenti
anomalie multiple del sangue e
insufficienza pancreatica
Perdita di più geni per delezione
Accessi epilettici, demenza,
debolezza muscolare associata
con la presenza di fibre “raggedred”(RRF)o rosso sfilacciato
tRNALeu
Sindrome di Pearson
Epilessia mioclonica con fibre a
brandelli
MERRF (Myoclonic Epilepsy and
Ragged-Red Fiber)
32
•Manifestazione di un insieme di mutazioni a carico
dell’mtDNA note come miopatie o citopatie che
causano disturbi nella motilità oculare;
•Degenerazione lenta;
•Causa genica: transizione A3243G, a livello di un
mt-tRNA;
•Diagnosi: attraverso biopsia muscolare, e analisi
attraverso la colorazione tricromica di Gomori grazie alla
quale è visibile un gran numero di mitocondri
ipertrofici;
•Nessuna terapia esistente; la ptosi è correggibile con
chirurgia che comunque non garantisce la chiusura delle
palpebre.
33
34
•Variante più aggressiva della CPEO;
•Causa genica: delezioni nel mtDNA variabili in dimensioni (1.3-8
Kbp) e posizioni. La più comune delezione (presente in 1/3 degli
individui affetti da KSS) è di 4.9 Kbp localizzata dalla posizione
8469 alla 13147 del genoma;
•Effetti: progressiva diminuzione delle forze fino alla paralisi
totale (nella maggior parte dei casi), causata dalla perdita della
piena funzionalità di occhi, muscoli e cuore exitus letalis.
•Eredità: mitocondriale o autosomica dominante/recessiva.
•Danni: i più gravi sono a carico del blocco atrio ventricolare
dove si blocca la conduzione elettrica da atrio a ventricolo.
•Sintomi: sincope e bradicardia.
•Diagnosi: mediante biopsia
35
(Lebher’s Hereditary Optic Neuropaty)
•Causa genica: transizioni
G11778A, G3460A, T14484C;
•Effetti: lenta degenerazione delle cellule gangliari
retiniche perdita della visione centrale.
•Evolve in atrofia ottica.
36
•Forma più grave e più rara della LHON
•Oltre a disfunzioni dell’occhio, include
l’incapacità del cervello di controllare i
muscoli, tremore e aritmia cardiaca.
37
•Causa genica: delezione sull’mtDNA
•Caratterisitche: Anemia sideroblastica e
disfunzione pancreas esocrino;
•Fatale nell’infanzia. Pazienti
sopravvissuti sviluppano sintomi
riconducibili alla KSS, meno di 100 casi
accertati nel mondo.
38
ETEROPLASMIA ED
EFFETTO SOGLIA
•Eteroplasmia: mtDNA mutato + mtDNA sano;
•Effetto soglia: rapporto limite tra mtDNA
mutato ed mtDNA inalterato, oltre il quale si
hanno disfunzioni patologiche.
È tanto più basso quanto più il tessuto o
l'organo ha elevato fabbisogno energetico.
39
E la ricerca?
40
Negli ultimi anni i ricercatori si sono uniti a
formare una vera e propria "task-force" per
chiarire molti aspetti ancora oscuri sulle patologie
mitocondriali.
In Europa si è costituito il consorzio MitEURO, che
raggruppa 51 laboratori di ricerca, per chiarire i
meccanismi molecolari, cellulari e fisiopatologici
delle malattie mitocondriali con lo scopo di
identificare terapie efficaci.
41
Il caso della scimmia con due
madri
Messa a punto di un metodo sperimentale di
fecondazione assistita:
Trasferimento del materiale genetico da un oocita
con mtDNA mutato, in un altro oocita anucleato
con mtDNA inalterato e fertilizzato dal seme di un
donatore.
Prole sana!
42
Questa tecnica non va in contro a problemi
di natura etica, in quanto questi risultati
hanno il potenziale di permettere ad una
coppia di avere un figlio biologicamente
proprio.
43
GFER (nDNA)
La mutazione di questo gene è alla base di tre
patologie differenti patologie:
1. Cataratta congenita;
2. Iposviluppo somatico;
3. Miopatie mitocondriali.
44
La proteina codificata da tale gene
offre un importante apporto al
mitocondrio in piccole proteine
essenziali per la catena
respiratoria
45
46
")