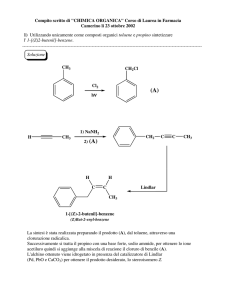
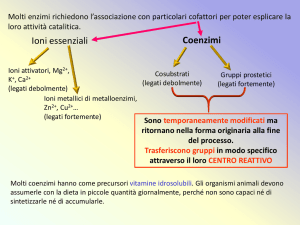
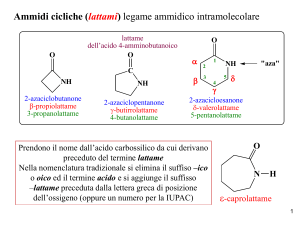
16/05/2011
PRINCIPI DELLA SPETTROMETRIA DI MASSA
A differenza delle altre tecniche analitiche (IR, UV-vis, NMR) in cui il campione
rimane intatto dopo l’analisi, la spettrometria di massa, che forma ed utilizza
ioni invece di molecole, è un metodo di analisi distruttivo.
In uno spettrometro di massa usato per analisi organica, di solito abbiamo a che
fare con l’analisi di ioni positivi derivati da molecole. La ionizzazione semplice di
una molecola si rappresenta con l’equazione:
M:
M+. + e
Lo ione M+. si chiama ione molecolare. La massa (m) e la carica (z) dello ione
vengono misurate dallo spettrometro di massa e quello che è importante è il
loro rapporto.
Spesso si formano ioni con una sola carica positiva è quindi il rapporto è
uguale alla massa dello ione che, a sua volta, è legata alla massa della
molecola (dato che l’elettrone ha una massa piccolissima, 5.49 x 10-4
rispetto alla massa di un protone).
La risoluzione di un buon spettrometro di massa è 1 amu (atomic mass
number) su104 amu.
Lo ione molecolare ha una configurazione elettronica con un numero
dispari di elettroni.
Frammentazione
Se stiamo formando uno ione molecolare con una carica positiva, significa
che dobbiamo raggiungere il potenziale di ionizzazione della molecola.
Questo valore dipende dalla natura dell’HOMO, dato che è da questo
orbitale molecolare che l’elettrone sarà rimosso.
I primi potenziali di ionizzazione della maggior parte delle molecole
organiche cadono nell’intervallo 8-15 eV
Il processo di ionizzazione spesso usa energie molto maggiori di quella
necessaria per la ionizzazione. Questo eccesso di energia può essere
trasferito allo ione molecolare quando si forma, sotto forma di energia
rotazionale, vibrazionale o elettronica.
Le ultime due sono le più importanti per la spettrometria di massa,
perché la frammentazione di questi stati eccitati produce un nuovo ione
ed una particella neutra.
1
16/05/2011
Lo ione molecolare può seguire due percorsi di frammentazione:
M+.
M+.
A+ + N .
B+. + N
Nel primo caso si ha la frammentazione a un catione con numero pari di
elettroni ed una specie neutra con un elettrone dispari.
Nel secondo caso si ha frammentazione a uno ione con numero dispari di
elettroni ed una specie neutra con numero pari di elettroni. In questo caso la
specie neutra può essere una molecola stabile
CH2
Prendiamo
una
molecola
semplice
come
C2H6
e
consideriamo possibili percorsi
di frammentazione dello ione
molecolare che comportino solo
scissione del legame C-H, ma
con formazione di un atomo
neutro, di un radicale neutro e
di una molecola neutra
H2C
H2C
H2C
H
+
+
H.
H
CH2
+.
H2C
.
+
H+
H
H
CH2
CH2
CH2 +.
CH2
+
H H +.
+ H H
La frammentazione non si ferma qui, perché gli ioni frammento possono a loro volta
frammentarsi, subendo anch’essi perdita di specie neutre con numero pari o dispari di
elettroni.
M +.
step Ia
.
A+ + N1
step IIa
C+ + N3
step Ib
B+. + N2
step IIb
D+ + N4.
Tutte queste frammentazioni formano un insieme di ioni che a loro volta
danno luogo allo spettro di massa del composto in questione.
La conoscenza dei percorsi tipici di frammentazione permette la ricostruzione
di un andamento di frammentazione (fragmentation pattern) caratteristico.
2
16/05/2011
Il cammino di frammentazione dipende da quattro fattori principali:
- la forza dei legami che si devono rompere
- la stabilità dei prodotti di frammentazione, sia ioni che specie neutre
- le energie interne degli ioni che si frammentano
- l’intervallo di tempo tra la formazione degli ioni e la loro rivelazione.
LO SPETTRO DI MASSA SEMPLICE
Analizziamo uno spettro di massa semplice. Un modo comune di mostrare uno spettro
di massa è quello di una traccia che si può ottenere direttamente dal registratore della
maggior parte degli spettrometri.
Consideriamo il metanolo: CH3OH, Peso Molecolare = 32
Ci si aspetta che il valore più grande m/z sia 32. Invece, osservando bene lo
spettro, si osserva un segnale di intensità piccola (abbondanza relativa
bassa), a M++1.
isotopi
Attorno ai picchi grandi ci possono essere picchi di abbondanza relativa
bassa. Per esempio, una piccola parte di molecole di metanolo avrà 13C
(1.08% di abbondanza naturale) invece di 12C e qualche atomo di idrogeno
sarà 2H (D) (0.015%) invece di 1H.
due tracce dello spettro di massa
del metanolo, una registrata con
una sensibilità dieci volte maggiore dell’altra. Questo permette di
vedere anche i picchi meno abbondanti.
m/z
attribuzione
32
CH3OH+.
31
CH2OH+
29
CHO+
15
CH3+
Lo ione con m/z 32 è lo ione molecolare: a parte un picolissimo picco a m/z 33, è lo
ione di massa più elevata nello spettro
Il picco più intenso viene chiamato picco base dello spettro.
Ad esso si assegna l’abbondanza relativa 100%: le abbondanze relative degli
altri picchi si misurano rispetto a questo.
3
16/05/2011
La frammentazione sarà tanto più facile quanto più stabile è il
catione che si forma.
Se sono possibili più frammentazioni che formano cationi di stabilità
analoga, sarà preferita la frammentazione che dà il radicale più stabile.
e
CH3 CH2 CH2 CH2 CH3
+
.
CH3 CH2 CH2 CH2 CH3
m/z = 72
2e
+
CH3 CH2 CH2 CH2
+
.
CH3
CH3 CH2 CH CH3
CH3
e
CH3 CH2 CH CH3
2e
CH3
CH3 CH2 CH + +
m/z = 57
.
+ CH3 CH2
m/z = 43
m/z = 57
CH3
CH3 CH2 CH2+
.
CH3
+
.
m/z = 72
CH3
+ CH CH +
3
.
CH3 CH2
m/z = 43
4
16/05/2011
M+ - 29
M+ - 15
M+
M+ - 29
M+ - 15
M+
ALCOOLI
O-H legato a C sp3
Caratteristica strutturale degli alcooli
Caratteristiche Spettrali
Spettrometria di massa
Lo ione molecolare ha intensità piuttosto bassa per gli alcooli primari e
secondari, è di solito assente negli spettri degli alcooli terziari
La frammentazione più importante avviene tra il C α all’OH ed il C β
(carbocatione stabile)
+.
R''
R
C
R'
OH
.
R
+
R'' +
C OH
R'
R''
C
+
OH
R'
Il gruppo alchilico più grande viene perso più facilmente (radicale
più stabile).
5
16/05/2011
1-esanolo, P.M. 102
C4H9+
C3H7+
CH2OH+
M+-H2O
2-esanolo, P.M. 102
M+-C4H9
CH3 CH2 CH2 CH2 CH CH3
OH
M+-H2O
M+-CH3
M+-C3H7
2-metil-2-pentanolo
P.M. 102
M+-CH3
CH3
CH3 C CH2 CH2 CH3
OH
6
16/05/2011
Solo legami σ: le transizioni n → σ*
richiedono λ = 185-195 nm (lontano
UV): fuori dal comune intervallo
UV-visibile
IR
stretching O-H
picco stretto, 3650-3600 cm-1 (OH libero)
picco largo, 3500-3200 cm-1 (OH in legame H)
stretching C-O
1250-1000 cm-1
Negli spettri di film liquido si vede solo la banda dell’OH impegnato in legame
idrogeno; in soluzione si vedono entrambe.
CH3 CH2 CH2 OH
(liquido puro)
CH3 CH2 CH2 OH
(in CCl4)
1-esanolo in CCl4
1-esanolo film liquido
7
16/05/2011
13C
NMR
1H
NMR
L’effetto -I dell’O diminuisce la densità elettronica
attorno al nucleo del 13C: deschermato
L’effetto -I dell’O diminuisce la densità elettronica
attorno al nucleo del 1H: deschermato a 3.5δ
Il nucleo 1H dell’OH può risuonare in un intervallo ampio di chemical shift,
tra 0.5 e 5.0 δ; la posizione dipende da vari fattori (concentrazione, temperatura,
solvente, legame idrogeno, ecc.)
H
R O
H
R O +
O
+
R OH2
R
L’H dell’OH è sufficientemente acido (H mobile) per dare equilibrio
acido-base veloce rispetto al tempo di risposta dell’NMR
Questo significa che, di solito, l’H non si ferma sull’O abbastanza a
lungo per influenzare il campo magnetico di un H su un C adiacente,
né per esserne influenzato.
l’H dell’OH è un singoletto, un po’ allargato, a meno di rallentare lo scambio,
per esempio abbassando la temperatura, eliminando le impurezze acide,
usando soluzioni diluite.
es.: metanolo
CH3
+ 37°C
+ 10°C
OH
- 10°C
- 65°C
8
16/05/2011
Se si vuole confermare che un segnale nello spettro 1H NMR sia dovuto a OH, si
aggiunge alla soluzione qualche goccia di D2O
R
OH + D O D
R
OD + D O H
Il protone mobile scambia con D ed il segnale diminuisce o scompare del tutto
esempio:
1-propanolo
CH3 CH2 CH2 OH
C
B
A
A
B
C
C
A
OH B
..
R-O-H
..
C H
BASE
alcossido
H C O-
a
C H
c
b
C H
..
H
d
C O
.. H
b
ACIDO
H
C X
C H
H
H C O+
H
a
C
b,c
C H
a,d
ossidazione
C
H
alcheni
C
O
aldeidi e chetoni
9
16/05/2011
Rottura del legame C-O
SOSTITUZIONE NUCLEOFILA
CH3 CH2 Br + H2O
OH + HBr
CH3 CH2
+
CH3 CH2 OH2 Br
H
H2SO4
CH2 CH2 OH
Δ
ELIMINAZIONE
CH2
CH2 + H2O
-OH è un cattivo gruppo uscente: può essere trasformato in un buon
gruppo uscente per protonazione
reazioni in ambiente acido
..
+
R OH
.. + H
H
R O+
Nu:-
R Nu + H2O
H
buon gruppo
uscente
esempi:
CH3CH2CH2OH + HBr
(CH3)2CHOH + HI
(CH3)3COH + HCl
Nu:- = I-, Br-, Cl-, F-
CH3CH2CH2Br + H2O
(CH3)2CH-I + H2O
(CH3)3C-Cl + H2O
A parità di alcool, la reattività dipende dalla forza dell'acido
HF
HCl
HBr
HI
pKa
3.2
-7
-9
-9.5
forza acida crescente
reattività crescente verso gli alcooli
A parità di acido, la reattività dipende dal tipo dell'alcool
metile < primario < secondario < terziario
reattività crescente degli alcooli con HX
Su questa diversa reattività si basa il saggio di riconoscimento degli alcooli
(saggio di Lucas)
alcool terziario
(CH3)3C-OH + HCl
25°C
(CH3)3C-Cl + H2O
10
16/05/2011
(CH3)2CH-OH + HCl
alcool secondario
ZnCl2
(CH3)2CH-Cl + H2O
ZnCl2
CH3CH2CH2-Cl + H2O
Δ
Il cloruro che si forma è immiscibile con la fase acquosa e si deposita
sul fondo (ha densità maggiore dell'acqua).
alcool primario
CH3CH2CH2-OH + HCl
Con H2SO4
stesso ordine di reattività
(CH3)3C-OH
terziario
secondario
(CH3)2CH-OH
primario
CH3CH2-OH
H2SO4
60°C
H2SO4
100°C
H2SO4
180°C
H3 C
C CH2 + H2O
H3 C
H3C CH CH2 + H2O
CH2 CH2 + H2O
Con H2SO4 si riesce ad isolare il prodotto di sostituzione solo a
temperatura molto bassa
+
R-CH2CH2-OH2 + HSO 4
R-CH2CH2-OH + H2SO4
H+, H2O
- H2O
nucleofilo debole
H2O
O
R CH2 CH CH2
H2SO4
R CH2 CH2 O S OH
O
ottimo gruppo uscente
Meccanismo:
+
OH2
OH
lento
+
veloce
CH3 CH CH3 + OH2
CH
CH3
CH3 CH CH3 + H2SO4
3 CH
H
+
CH3 CH
CH2
veloce
CH3 CH CH2
H+
11
16/05/2011
Orientamento:
OH
H2SO4
CH3 CH
CH3 CH2 CH CH3
CH CH3 + CH3 CH2 CH CH2
prevalente
H2O
Saytzeff
OH
CH2 CH CH3 H2SO4
CH CH CH3
H2O
unico prodotto
Come sempre, quando si formano carbocationi, c’è possibilità di trasposizione
H+
CH3 OH
CH3 C CH CH3
CH3
H2SO4
H3C
95°C
H3C
C
CH3
+
CH3 OH2
CH3 C CH CH3
CH3
CH3
C
CH3
H+
CH3
+
CH3 C CH CH3
CH3
+
CH3 C CH CH3
H2O
CH3
Se con H2SO4 si trattano 1,2-dioli:
H+
OH OH
CH3 C
CH3 O
CH3 C
C CH3
C CH3
+ H2 O
CH3
CH3 CH3
2,3-dimetil-2,3-butandiolo (pinacolo)
3,3-dimetilbutanone (pinacolone)
TRASPOSIZIONE PINACOLICA
H+
OH OH
CH3 C
C CH3
CH3 CH3
CH3
CH3 C
CH3
: OH
C+ CH3
+
OH2 OH
CH3 C
+
C CH3
CH3 CH3
+
CH3
C
- H+
CH3
OH
C CH3
CH3 CH3
H2O
CH3 OH
CH3 C
CH3 C
CH3 O
CH3 C
C
CH3
CH3
12
16/05/2011
Può migrare anche un H:
H+
OH OH
CH3 C
H
+
H+
OH2 OH
H
H
H
H
: OH
CH3 C
H
C CH3
CH3 C
+
H
OH
+
C CH3
CH3 C
C CH3
H
+ H2O
CH3 CH2 C CH3
H
OH OH
CH3 C
O
C CH3
CH3 C
H
H2O
+
C CH3
H
- H+
OH
C CH3
O
CH3 CH2 C CH3
H
Se i sostituenti del diolo sono diversi:
viene protonato l'OH più basico
migra il gruppo che lascia il carbocatione più stabile
MECCANISMO della SOSTITUZIONE degli alcooli in ambiente acido
SN1 o SN2 ?
Sostituzione di -OH2+
SN2
con metanolo ed alcooli primari
H+
CH3 CH2 OH
+
X
CH3 CH2 OH2
-
δX
δ+
OH2
CH2
X-CH2CH3
+ H2O
CH3
stato di transizione
SN1
con alcooli secondari e terziari
CH3
CH3 CH OH
H+
CH3
CH3
+
CH3 CH
CH3 CH OH2
H2O
+
X
-
CH3
CH3 CH X
13
16/05/2011
La sostituzione dell'OH può avvenire anche in ambiente non acido
però…..
Il gruppo -OH può essere sostituito SOLO DOPO TRASFORMAZIONE
IN UN BUON GRUPPO USCENTE
Gli alcooli si possono trasformare in alogenuri alchilici usando cloruri di P e S
PCl3
PCl5
tricloruro di fosforo
SOCl2
pentacloruro di fosforo
cloruro di tionile
3 R-OH + PCl3
R-OH + PCl5
3 R-Cl + H3PO3
R-Cl + POCl3 + HCl
R-OH + SOCl2
R-Cl + SO2 + HCl
Questi composti trasformano l’OH in un buon gruppo uscente, che viene
sostituito con un meccanismo SN2 (e quindi gli alcooli terziari non reagiscono)
P e S hanno un buon gruppo uscente (Cl), che
viene sostituito dall’alcool (O nucleofilo)
Con PCl5
R
..
O
.. H + Cl
PCl4
R
Cl-
.. +
O H
PCl4
HCl + R
O PCl4
buon gruppo uscente
buon gruppo uscente
R Cl +
Con SOCl2
R
O
..
O
.. H + Cl S Cl
R
Cl-
Cl
HOPCl4
.. +
O H
S
O
POCl3 + HCl
O
HCl + R O S Cl
buon gruppo uscente
buon gruppo uscente
R-Cl + SO2 + HCl
14
16/05/2011
Con PCl3
O H + Cl
R
HCl + R
PCl2
Cl + HO
R
O PCl2
PCl2
buon gruppo uscente
O H + Cl
R
HCl + R
P Cl
O P Cl
R Cl +
HO P Cl
OH
OH
OH
buon gruppo uscente
O H + Cl P OH
R
HCl + R
OH
O P OH
R Cl + HO P OH
OH
OH
buon gruppo uscente
ANDAMENTO STEREOCHIMICO
Con PCl3
1.
CH3
H
C
CH3 H
Cl
OH
+
CH2 CH3
P
H
Cl
C
+
Cl
(S)-2-butanolo
CH3
- H+
O P
CH2 CH3
Cl
Cl
C
H
Cl
O P
CH2 CH3 Cl
clorofosfito di (S)-2-butile
Nessuno dei legami del C* è
stato interessato dalla reazione
CH3
2.
H
Cl
C
Cl
O
CH2 CH3
P
Cl
S N2
CH3
Cl
C
H
+
CH2 CH3
-OPCl
2
(R)-2-clorobutano
Inversione di configurazione
analogamente con PCl5
15
16/05/2011
Con SOCl2
1.
L’andamento stereochimico dipende dalle condizioni di reazione
O
CH3
C
H
OH + Cl S
H
Cl
CH2 CH3
(S)-2-butanolo
CH3 H
O
C
S
O
+
- H+
Cl
CH2 CH3
CH3
C
H
O
O S Cl + HCl
CH2 CH3
clorosolfito di (S)-2-butile
In presenza di base (ammina terziaria):
R3NH+ +
R3N: + HCl
Cl
Nu-
O
CH3
2.
H
Cl
-
C
O
S
Cl
S N2
CH3
C
Cl
H + O
S
O + Cl-
CH2 CH3
CH2 CH3
(R)-2-clorobutano
Inversione di configurazione
In assenza di base, nel solvente poco polare (dietil etere) HCl è indissociato
manca il nucleofilo forte
gabbia del solvente
2.
H
CH3
C
O
O
CH2 CH3
S
Cl
SN i
H
O S O
CH3
C+
Cl
-
CH2 CH3
coppia ionica
+
CH3
H
C
Cl
CH2 CH3
(S)-2-clorobutano
SNi: il nucleofilo entra dalla stessa parte: “ritenzione” della configurazione
16
16/05/2011
ALTRE REAZIONI DEGLI ALCOOLI
ALCOOLI COME ACIDI
CH3 O -
CH3 OH + H2O
CH3 OH + OH
CH3 O
+ H3O +
+ H2O
Per avere formazione completa di alcossido:
CH3 O H
CH3 O - Na+
+ Na°
+ 1/2 H2
REAZIONI CON GLI ACIDI
Formazione di esteri
a) Acidi carbossilici
O
O
CH3 C
OH + H
O CH2 CH3
O CH2 CH3 + H2O
O
O
C
CH3 C
OH + H
O CH2 CH3
C
O CH2 CH3 + H2O
b) Acidi inorganici
CH2 O
CH
O
CH2 O
HO
NO2
CH2 O NO2
H + HO
NO2
CH O NO2 + 3 H2O
H
NO2
CH2 O NO2
H
HO
17
16/05/2011
Meccanismo:
1. formazione di NO2+
HO
+
H2O
+
H2O NO2 +
NO2
-O
+
ione NITRONIO (elettrofilo)
H2O + NO2
NO2
2. reazione di NO2+
H
H
+
+ NO
2
CH3 O
..:
+
nitrato di metile
S OH + H2O
CH3 O
O
O
H + HO S O CH3
solfato acido di metile
O
CH3 O
O
S O CH3 + H2O
O solfato dimetilico
ROSO2OH + ROSO2OR + H2O
0°C
ROH + H2SO4
CH3 O NO2
O
O
CH3 O
- H+
CH3 O NO2
O
+
H
HO S OH
CH3 O
(reazione acido-base)
NO2
Δ
alcheni + H2O
c) Acidi solfonici
O
O
R
O
H
+ HO S
R O S
CH3
H2O
O
acido p-toluensolfonico
CH3
O
p-toluensolfonato di R
(tosilato)
Per avere completamente il solfonato, l’alcool si fa reagire con il cloruro di p-metil
benzensolfonile
O
O
R
H + Cl
O
S
O
O
R O S
CH3
CH3
O
HCl
tosilato di R
S
O
CH3
Ts
Formare il tosilato è un altro modo di trasformare l’OH in un buon gruppo uscente
Cl
CH3
+ CH2 O Ts
..
CH3OH +
S N2
CH2 O Ts
CH3
Cl
S N2
CH2 CH3 +
-
O Ts
H
CH3 O CH2 CH3 + O Ts
+
CH3
O CH2 CH3
+
-H
18
16/05/2011
REAZIONI DI OSSIDAZIONE
Ossidazione biologica dell'etanolo
O
CH3 CH2 OH
O
CH3
O
[O]
C H
CH3
C
O-
alcool deidrogenasi
O
HSCoA
CH3 C
trigliceridi, ecc.
SCoA
CO2 + H2O
acetilcoenzima A
Ossidazione chimica
alcool 1o
O
[- 2H]
RCH2OH
R C
R C OH
R
X
alcool 3o
ossidanti tipici
[O]
X
H2CrO4
KMnO4, OH-
Na2CrO4 + H+
HNO3 conc.
ATTENZIONE!
O
[O]
R C H
O
[- 2H]
R2CHOH
[- 2H]
R3COH
alcool 2o
CH3 C H
K2Cr2O7 + H+
Gli alcooli terziari non vengono ossidati; però alcuni
ossidanti sono acidi e come tali possono reagire con
gli alcooli terziari
OH-
NESSUNA REAZIONE
R3COH + [O]
H+
prodotti di ossidazione dell'alchene
alchene
Meccanismo (semplificato)
..
R2CHOH
+ HCrO4-
- H2O
H
R
R
C
O
O
Cr
HO
O
O
R C R + Cr O
O
O
Cr(VI)
ESEMPI
KMnO4, H2O, tampone basico
-
Cr(IV)
CH3
CH
CH3
OH
C
O
72%
19
16/05/2011
O
HNO3
Br (CH2)5 C
Br (CH2)5 CH2 OH
OH
75%
Per ossidare gli alcooli primari ad aldeide è necessario usare reagenti
speciali, particolarmente blandi (mild) in condizioni controllate.
Reattivo di Jones
K2Cr2O7, H2SO4 diluito, (CH3)2C=O
O
RCH2OH
O
eccesso di reattivo
R C OH
R C H
Reattivo di Collins
CrO3 . 2
N
O
CH3 (CH2)5CH2 OH
CH3 (CH2)5 C H
. CrO . HCl
clorocromato di piridinio (PCC)
3
N
O
OH
H
87%
2-etilesanale
2-etil-1-esanolo
Ossidazione dei DIOLI
reagente caratteristico
scissione
R CH OH
R CH OH
HIO4
O
R
acido periodico
O
C H + H C
R + HIO3
perché la reazione avvenga è necessario che i due -OH siano su C adiacenti
20
16/05/2011
La scissione del legame C-C avviene anche se sono adiacenti un -OH
ed un C=O
O
O
O OH
HIO4
R C CH R
R
+
C OH
H C
R + HIO3
La scissione del legame C-C non avviene se sono adiacenti un -OH ed
una funzione -OR
OH
OH
HIO4
nessuna reazione
R CH CH2 CH R
OH
O
R C
HIO4
nessuna reazione
CH2 CH R
OH OCH3
HIO4
nessuna reazione
R CH CH R
Se ci sono più di due -OH su C adiacenti, TUTTI i legami C-C si rompono
OH
CH2
OH OH O
CH CH C H
HIO4
O
O
H
C H
+ 3 H
C OH
Meccanismo:
R
R C
R C
R
OH
..
OH
I
O
OH
..
R C
O
OH
O
I
O
R C
H 2O
R
O
O
R
R
R
C O
R
O
+
C O
+
HIO3
R
21
16/05/2011
ETERI
-O-
Caratteristica strutturale degli eteri
Caratteristiche Spettrali
Spettrometria di massa
La frammentazione più importante avviene tra il C α all’OH ed il C β
(carbocatione stabile)
+.
+
+
.
R + CH2 OH
H2C OH
R CH2 O R'
M+-C3H7
diisopropil etere, P.M. 102
CH3
M+-CH3
CH3 CH O CH CH3
Solo legami σ: le transizioni n → σ*
richiedono λ = 185-195 nm (lontano
UV): fuori dal comune intervallo
UV-visibile
IR
CH3
stretching C-O
CH3
1300-1000 cm-1
CH3
CH3 CH O CH CH3
22
16/05/2011
dipropil etere
13C
NMR
1H
NMR
L’effetto -I dell’O diminuisce la densità elettronica
attorno al nucleo del 13C: deschermato
L’effetto -I dell’O diminuisce la densità elettronica
attorno al nucleo del 1H: deschermato a 3.5δ
CH3
CH3
CH3
CH3 CH O CH
CH3
CH3
CH
CH
CH3 CH2 CH2 O CH2 CH2 CH3
A
C
B
B
A
C
A
C
B
23
16/05/2011
POCO REATTIVI
..
R-O-R'
..
USATI SPESSO COME SOLVENTI
O
Fra gli eteri, quelli più usati come
solvente, oltre al dietil etere, sono
quelli ciclici
O
O
ossaciclopentano
(tetraidrofurano)
1,4-diossacicloesano
(diossano)
Danno reazione solo con HI, a caldo
..
R O
.. R' + HI
..+
R O R'
I-
+
Δ
R OH + R'I + R' OH + RI
H
sostituzione nucleofila
reazione acido-base
esempio:
CH3CH2OCH2CH2CH2CH3 +
HI
Δ
CH3CH2OH + I-CH2CH2CH2CH3 +
CH3CH2-I + HOCH2CH2CH2CH3
se c'è un eccesso di HI e si lascia il tempo, anche gli alcooli reagiscono con HI
CH3CH2OH + CH3CH2CH2CH2OH
però
O CH3
+
HI
HI
Δ
CH2CH2I + CH3CH2CH2CH2I
+ CH2=CH2 + CH2=CHCH2CH3
OH
+
CH3I
Viene attaccato da I- SOLO il C sp3
+
O CH3
O CH3
-
ecc.
legame C(aromatico-O più
forte di C(alifatico)-O
Gli eteri ciclici con anelli tesi sono più reattivi perché, con l'apertura
dell'anello, hanno sollievo dalla tensione d'anello
OSSACICLOBUTANI
reagiscono anche con HBr, a caldo
APERTURA DELL'ANELLO
24
16/05/2011
Δ
O
HOCH2CH2CH2Br
HBr
H
meccanismo:
OSSACICLOPROPANI
MOLTO REATTIVI
O
O-
Nu:-
CH2 +
CH2 CH2
+
-:SCH
H+
CH2 CH2 Nu
esempio:
O
APERTURA DELL'ANELLO
SN2
a) con NUCLEOFILI FORTI
CH2
Br-
O
+
O HBr
OCH2 CH2 SCH3
3
O H
CH2 CH2 Nu
H+
HO CH2 CH2 SCH3
Se l'ossaciclopropano è sostituito, la reazione ha un orientamento dovuto al
meccanismo (SN2)
REGIOSPECIFICITA'
Di due "regioni" del substrato possibili e simili
una sola viene attaccata
REGIOSELETTIVITA'
Di due "regioni" del substrato possibili e simili
una viene attaccata di preferenza
S
O
H C
H
C
H
CH3
-OCH
3
CH3OH
H
H
O H
C
H3C O
C
H
CH3
S
Reazione regiospecifica; reazione stereospecifica
SN2: più reattivo C
meno sostituito
SN2: attacco di Nu da parte
opposta al gruppo uscente
25
16/05/2011
b) in assenza di NUCLEOFILI FORTI
Bisogna rendere -O- un miglior gruppo uscente
apertura dell'anello acido-catalizzata
..
O
..
H2C
H
O+
H+
..
H O:
H2C
CH2
..
H OCH
..
3
CH2
OH
H2C
H2C CH2
CH2
O+
H .. CH3
O CH3
H+
Il processo è SN1, ma non si forma un vero carbocatione, per l'interazione
con il doppietto elettronico dell'ossigeno
H C
H
H
S
O
O
H+
C
H C
H
H
CH3
δ+
+
C
H
H
O
O δ+
δ+
H C
H
H
CH3
C
H
H C
CH3 H
parziale
carbocatione
primario
δ+
H
H
O
O δ+
δ+
H C
H
C
H
H C
CH3 H
..
H O
CH3
(se ne forma di più)
R
H
H
CH3
H
H+
OH
C
H3C O
H
CH3
parziale carbocatione
secondario
S
δ+
C
δ+
C
C
H
CH3
prodotto principale
HO
+
H C
H
H
CH3
C
O CH3
prodotto secondario
Reazione regioselettiva; reazione stereospecifica
Si forma più (ma non solo)
“parziale” carbocatione
secondario
attacco di Nu da parte
opposta al gruppo uscente
POLIETERI CICLICI (ETERI CORONA)
O
O
O
O
O
O
18-Corona-6
18-Crown-6
26
16/05/2011
O
O
O
+
K
O
O
O
O
O
O
O
O
O
O
O
O
O
O
O
O
O
O
12-Crown-4
15-Crown-5
O HN
O
O
O
O
NH
HN
O
NH
HN
O
NH O
O
n+
O
N
N
O
O
O
O
CRIPTANDO
n+
+
O
O
O
O
O
O
N
catione
N
complesso CRIPTATO
27
16/05/2011
AMMINE
N:
Caratteristica strutturale delle ammine
Caratteristiche Spettrali
Spettrometria di massa
Lo ione molecolare è quasi sempre presente
La frammentazione più importante avviene tra il C α all’N ed il C β
(carbocatione stabile)
+.
R''
R
C
N
.
R
+
R'
R'' +
C N
R'
R''
C
+
N
R'
Per le ammine vale la “regola dell’azoto”
La regola dell’azoto
E’ una regola universale che riguarda sia gli ioni molecolari che i frammenti
con una singola carica.
“tutti gli ioni con numero dispari di elettroni hanno valori m/z pari,
tranne quando contengono un numero dispari di atomi di azoto”
Questa regola viene dal fatto che l’azoto ha massa atomica pari e valenza dispari.
1-butanammina, P.M. 73
30 (CH2=NH2+)
M+
1
16/05/2011
dietilammina, P.M. 73
M+-CH3
M+
Solo legami σ: le transizioni n → σ*
richiedono λ = 185-195 nm (lontano
UV): fuori dal comune intervallo
UV-visibile
IR
stretching N-H
3500-3300 cm-1
DUE bande per le ammine primarie; una banda per le
ammine secondarie (debole se alifatiche, forte se
aromatiche). Le amine terziarie non hanno assorbimento
bending N-H
stretching C-N
nelle ammine primarie, banda larga 1640-1560 cm-1
nelle ammine secondarie verso 1500 cm-1
1350-1000 cm-1
1-butanammina
2
%T
16/05/2011
2-butanammina
cm-1
dietilammina
13C
NMR
1-butanammina
L’effetto -I dell’N diminuisce la densità elettronica
attorno al nucleo del 13C: deschermato
AB
C
CH3 CH2 CH2 CH2 NH2
D
C
B
A
D
1H
NMR
L’effetto -I dell’N diminuisce la densità elettronica
attorno al nucleo del 1H: deschermato a 2.5-3 δ
Anche l’H sull’N è mobile: scambia abbastanza rapidamente da non dare
accoppiamento con nuclei adiacenti
3
16/05/2011
terz-butilammina
atomo basico, atomo nucleofilo
N:
cattivo gruppo uscente, anche in seguito a
protonazione
RNH2 + H+
R
+
NH3
NESSUNA REAZIONE
1. N come atomo basico o nucleofilo
con O come elettrofilo
(CH3)3N
H2O2
H2O
+
RNH2 + H+
con acidi
R
NH3
SOLO con ammine terziarie
(CH3)3N O +
CH3
H O
CH ..
CH3 2 2
N
CH3
H2O
CH3
CH CH3
+N O 98%
CH3
4
16/05/2011
O
R C
invece di H2O2 si possono usare peracidi
O
O O
R C
:N
H
R C
O
O OH
-O
+
OH
+
N
con alogenuri alchilici
La reazione dà miscele di prodotti, perché anche il prodotto della sostituzione
è nucleofilo
R
R
..
NH2 + CH3 CH2 Br
+
R NH CH2 CH3
R NH2 CH2 CH3 + Br+ HBr
CH2 CH3
+
R NH
R N CH2 CH3 + HBr
Br
..
NH + CH3 CH2 Br
CH2 CH3
CH2 CH3
R
CH2 CH3
CH2 CH3
..
N CH2 CH3 + CH3 CH2 Br
CH2 CH3
R
+
N CH2 CH3
Br-
CH2 CH3
Quando l’alchilazione dell’ammina si esegue fino al sale di ammonio si parla di
METILAZIONE ESAURIENTE
con acido nitroso
HNO2
La reazione con HNO2 è la reazione più caratteristica delle ammine, che
permette di distinguere tra ammine primarie, secondarie e terziarie.
Fatti sperimentali:
HNO2
H+
ammine primarie RNH2
ammine secondarie R2NH
ammine terziarie
R3N
alcooli, alcheni, N2
HNO2
H+
R2N-NO
nitrosammina
HNO2
H+
sviluppo di gas
precipitato giallo
NESSUNA REAZIONE
L'acido nitroso si genera in situ
NaNO2 + HCl
HNO2 + NaCl
5
16/05/2011
MECCANISMO
H
+
HO NO + H
+O
H2O +
NO
H
E+
+NO
catione
nitrosonio
ammine primarie
..
R NH2 + +NO
alcool
H2O
+
R
R NH2 NO
+
N
R+
N
alchene
N2
H2O
H+
i sali di diazonio si possono isolare solo con le ammine aromatiche
NH2
+
N
NaNO2
N
isolabile
H+
0°-5°C
+
N
+
N
N
N
+
-
+
N
N
-
+
+
N
N
-
+
N
N
+
ammine secondarie
..
R NH + +NO
R
H
+
R N NO
R
R
N NO
R
H+
ammine terziarie
R
R
R N:
+ +NO
R N
+
NO
R
R
i sali di ammonio quaternari possono essere usati come
catalizzatori di trasferimento di fase
-
CH3 CH2 CH2 CH2 CH2 CH2 CH2 CH2 Cl + Na+ C N
solubile in solvente idrocarburico
(solvente non polare)
NESSUNA REAZIONE
(neanche a caldo)
solubile in acqua
6
16/05/2011
se R contiene almeno 4 atomi di C, è solubile sia in
acqua (per l'N positivo) che in solventi non polari (per R)
R4N+
R4N+ Cl- + Na+ C N
R4N+ -C N +
Na+ Cl-
porta in fase organica il reagente nucleofilo
R4N+ -C N
+
R' Cl
+ R4N+ C N + Na Cl
+
R3N-
R4N+ Cl-
+
R4N+ Cl- +
R' C N
Na+ -C N
pessimo gruppo uscente
Metilazione esauriente - Degradazione di Hofmann
Reazione utilizzata per la determinazione della struttura di ammine
Esempi:
..
CH3 CH2 CH2 N H
H
CH3 CH2 CH2 N CH3 ICH3
Δ
+
CH3 CH2 CH2 N CH3 I-
metilazione esauriente
CH3
2 HI
CH3
+
CH3
3 CH3I
AgOH
AgI
β
CH3
+
CH3 CH CH2 N CH3 HO:H
CH3
β-eliminazione
CH3
CH3 CH CH2 + : N CH3 + H O
2
CH3
7
16/05/2011
2 CH3I
+
N
H
βH
H
HI
H3C
+
H3C
CH3I
H3C
N
H3C CH CH3
3
N
2) AgOH
CH3
Δ
+N
N
CH3
-OH
N
2) AgOH
H3C + CH3
2) AgOH
H3C CH CH3
3
+ H2O
+
H3C CH CH3
3
-OH
-OH
AgI
HO-
N
..
N
1) CH3I
1) CH I
3
+
H3C CH CH3
3
CH3
H3 C
AgOH
I-
+
Δ
1) CH I
3
CH3
N
N
Hβ
H
+
H3C
CH3
H2C
CH3
N
AgI
H
Δ
HO:-
+
N
N
HO-
AgOH
I-
Δ
N
H3 C
CH3
CH3
Δ
N
+ CH3
-OH
CH3
+
H3 C
N
CH3
+ H2O
8
16/05/2011
Applicazione: sintesi del cicloottatetraene (Wilstätter)
O
N
H3C
OH
Na, CH3CH2OH
H2SO4
N
H3C
84%
Composto naturale estratto
dalla corteccia del melograno
H 3C
CH3I
H3 C
H3 C
N+
Ag2O/H2O
Δ
H3C
CH3
N
CH3
CH3I
H3C
N
Br
(CH3)2N
HBr
N(CH3)2
68% (per i due passaggi)
Ag2O/H2O
H3C I-
94,5%
CH3
non isolato
10-20%
Δ
CH3
I-
I-
90%
72%
97,5%
3
H
H3 C +
H 3C N
CH3I
H3C CH3
HC N+
N
Br
Br2
84%
CH3
Δ
96%
N
H2O
Ag2O/H2O
I-
H3C
+ N CH
3
CH3
+ 2 (CH3)2N + 2 H2O + 2 AgI
Resa complessiva: 3%
Gli N-ossidi delle ammine terziarie danno β-eliminazione
Eliminazione di Cope
-O
N(CH3)2
R CH2CH R'
[O]
+
N(CH3)2
R CH2CH R'
([O] = reagente ossidante, per esempio H2O2)
meccanismo:
- ..
:O
.. +
N(CH3)2
H
R C
H
C
H
R'
reazione stereospecifica
δ-
H
O δ+
N(CH3)2
R C ------ C
H
H
=/
R
R'
C
R'
H
C
H
+ HO-N(CH3)3
eliminazione sin
9
ALCHENI
CICLOALCHENI
CARATTERISTICA FUNZIONALE:
legame π
C
C
Caratteristiche Spettrali
Spettrometria di massa
Una frammentazione importante dà un valore m/z = 41, che
corrisponde al carbocatione allilico
R
CH2 CH
CH2
+.
.
R +
CH2 CH
stretching =C-H
stretching C=C
CH2
CH2 CH CH2
Un solo legami π: le transizioni π → π*
richiede λ = 190 nm (lontano UV): fuori dal
comune intervallo. Però è una transizione
permessa dalla simmetria (ε= 15 000 per
l’etene)
UV-visibile
IR
+
+
a sinistra di 3000 cm-1
1660-1600 cm-1
bending =C-H (fuori dal piano)
1000-650 cm-1
C=C
C=C
=C-H
=C-H
2-metil-1-butene
Se lo stretching del doppio legame C=C non modifica il momento di dipolo, non si
ha assorbimento
1
1-pentene
cis-2-pentene
trans-2-pentene
13C
NMR
L’anisotropia diamagnetica descherma il nucleo
1-pentene
C2
C1
2
1H
NMR
L’anisotropia diamagnetica descherma il nucleo
Spesso gli spettri non sono del primo ordine
1-pentene
H vinilici
{
2-pentene
H vinilici
{
ALCHENI
CARATTERISTICA FUNZIONALE:
legame π
C
C
Più debole del legame σ, è il “punto
debole” degli alcheni.
la rottura del legame π porta a due valenze libere sui due atomi di C,
che vanno soddisfatte.
ADDIZIONE
la nuvola elettronica del legame π ATTIRA reagenti poveri di
elettroni, RESPINGE reagenti ricchi di elettroni.
ADDIZIONE ELETTROFILA
3
I reagenti elettrofili possono venire da una molecola di tipo X-Y o da una
molecola di tipo X-X
δ−
δ+
X Y
HCl, HBr, HI; H2O, H+; ICl; HOCl, H+ (HOBr, H+); BH3
HCl, HBr, HI; H2O, H+
E+ = H+
δ+
BH3
E+ = I+
E+ = atomo di B
(orbitale vuoto)
+
H O Cl
H
HO Cl + H+
+
H O Cl
δ−
I Cl
H2O +
E+ = Cl+
+
H O Br
Cl+
H2O + Br+
H
H
E+ = Br+
C
C
MECCANISMO
δ+
δ−
+ X Y
C
C
X
Y
Processo in due stadi
1.
C
+
H O Br
H
HO Br + H+
2.
+
C +X
lento
C
X
C C
+
X
δ+ X
X
δ+
C- - - -C
C
C
+
+
Y
-
C
C
X
Y
δ+
C
Y δ-
E
C
X
C
+
C C
X Y
C C
coordinata di reazione
4
REATTIVITA’
I fattori che stabilizzano il carbocatione:
abbassano l'energia dello stato di transizione
diminuiscono l'energia di attivazione
FAVORISCONO LA REAZIONE
E' PIU' REATTIVO L'ALCHENE che dà il CARBOCATIONE PIU’ STABILE
ORIENTAMENTO
Se da un alchene si possono formare due carbocationi di diversa stabilità,
si forma SOLO IL CARBOCATIONE PIU' STABILE (orientamento secondo
Markownikov)
REAZIONE REGIOSPECIFICA
ESEMPI
con HCl
equazione
Cl
CH3 CH CH3
CH2 CH CH3 + H Cl
2-cloropropano
meccanismo:
lento
CH2
+
CH3 CH CH3
CH CH3 + H Cl
..
- :Cl:
..
Cl
CH3 CH CH3
con HBr
CH3
equazione
CH2 C
Br
CH3 C CH3
CH3 + H Br
CH3
2-bromo-2-metilpropano
meccanismo:
CH2 C CH3 + H Br
CH3
lento
+
CH3 C CH3
..
- :Br:
..
CH3
Br
CH CH3
CH3
5
con HI
CH3
equazione
CH2 C
I
CH3 C CH3
CH3 + H I
CH3
2-iodo-2-metilpropano
..
-:I :
..
meccanismo:
lento
CH2 C CH3 + H
CH3
+
CH3
CH3 C CH3
I
Br
CH CH3
CH3
con H2O, H+
CH3
CH3 CH2 C
equazione
CH CH3 + H OH
OH
H+
CH3 CH2 C CH2 CH3
CH3
3-metil-3-pentanolo
meccanismo:
CH3 CH2 C
CH CH3 + H+
H+ H
..
O
H O
.. H
CH3 CH2 C CH2 CH3
CH3 CH2 C CH2 CH3
CH3
CH3
OH
lento
CH3
H+
+
CH3 CH2 C CH2 CH3
CH3
con ΙCl
Cl
CH3 CH2 CH CH2 I
CH3 CH2 CH CH2 + I Cl
equazione
meccanismo:
CH3 CH2 CH CH2 + I Cl
lento
+
CH3 CH2 CH CH2 I
- ..
:Cl:
..
Cl
CH3 CH2 CH CH2 I
con HOCl, H+
CH3 CH
equazione
OH
H+
CH2 + HOCl
CH3 CH CH2 Cl
1-cloro-2-propanolo
meccanismo:
H
H
HO Cl + H+
H
H
CH3 CH
CH2 + Cl+
lento
O + Cl+
+ O Cl
+
CH3 CH CH2 Cl
..
H O
.. H
CH3 CH CH2 Cl
O
CH3 CH CH2 Cl
OH
H + H
6
con BH3
IDROBORAZIONE
3 R CH CH2 + BH3
equazione
BH3
(RCH2CH2)3B
ottetto incompleto
B2H6
diborano
B2H6
H
H
B
H
H
B
H
H
B
H
H
B
H
H
orbitale vuoto
H
2 BH3
H
H
B
H
H
l’atomo di B è E+
meccanismo:
H
H
C
C
H
H
H
=
C
C
.
H
H
H
.
H
H
H
C
H
B
H
H
H
H
H
C
δ+
C
H2B-----H
δ-
=/
H
H
B
H
H
..
C
H
H
H
C
H
C
C
H2B
H
=/
H
H
H2B-----H
δ-
complesso tra
alchene e borano
H
δ+
C
H
H
anche gli altri legami B-H reagiscono allo
stesso modo
La reazione avviene in un unico stadio
H
H
C
C
H
H
H2B-----H
Reazione REGIOSPECIFICA
E
H
H
Anche se non si forma un intermedio
carbocationico, lo stato di transizione
ha un carattere di parziale
carbocatione (sul C dove è più stabile)
C
C
H
H
H
H
C
C
H2B
H
H
H
coordinata di reazione
7
I trialchilborani sono utili non come tali, ma perché il B viene sostituito facilmente da
un gruppo OH
HO OH
-OH
C B
H2O2, OH-
(RCH2CH2)3B
C OH
3 RCH2CH2OH
In questo modo è possibile ottenere da un alchene un alcool isomero rispetto a quello
della reazione con H2O, H+.
H+
+ OH
H2O:
2
CH2 CH CH3
H
CH CH3
CH2
BH3
+
CH2 CH CH3
CH2 CH CH3
BH2 H
H
H+
HO OH
OH
CH2 CH CH3
CH2 CH2 CH3
OH
H
1-propanolo
2-propanolo
MERCURIAZIONE-SOLVODEMERCURIAZIONE
Hg(OCOCH3)2 in solvente polare protico (H2O, ROH, CH3CO2H)
+
CH3 C O Hg
Hg(OCOCH3)2
O
O
C
+
CH3 C O Hg
+
O
Hg
lento
+
CH3 C O -
E+
O
O
C
CH3
--- +
O
+Hg
CH3
oppure
O
carbocatione
O
C
O
+Hg
O
C
O
C
O
Hg
CH3
+
ione mercurinio
O
Hg
CH3
SolvOH
H
O
+
Solv
H+
CH3
O Solv
8
ORIENTAMENTO
si forma il (parziale) carbocatione più stabile
I composti mercurati si trasformano in composti organici per trattamento
con idruro di boro e sodio (NaBH4) in ambiente basico
DEMERCURIAZIONE RIDUTTIVA
O
C
O
Hg
CH3
H
+ Hg°
H:-
precipita come mercurio
metallico (grigio)
O Solv
O Solv
SolvOH = H-OH
mercuriazione
OH
H2O
CH3 CH CH2 + Hg(OCOCH3)2
CH3 CH CH2 HgOCOCH3
H+
demercuriazione
OH
OH
H-
CH3 CH CH2 HgOCOCH3
alcool
CH3 CH CH3
Hg°
rispetto all'addizione di acqua acido-catalizzata, questo metodo ha il
vantaggio di non passare attraverso un carbocatione libero e quindi
di non correre il rischio della trasposizione.
SolvOH = R-OH
O
mercuriazione
ROH
CH3 CH CH2 + Hg(OCOCH3)2
H+
R
CH3 CH CH2 HgOCOCH3
demercuriazione
O
R
O
H-
CH3 CH CH2 HgOCOCH3
R
CH3 CH CH3
etere
Hg°
esempi:
CH3 CH CH2 + Hg(OCOCH3)2
CH3 OH
H+
O
CH3
CH3 CH CH2 HgOCOCH3
1-acetossimercurio-2-metossipropano
9
NaBH4
O CH3
CH3 CH CH3
isopropil metil etere
Hg°
CH3 CH CH CH2 CH3 + Hg(OCOCH3)2
CH3 CH2 OH
H+
CH3 CH2 O
HgOCOCH3
CH3 CH CH CH2 CH3
CH3 CH2 O
CH3 CH CH CH2 CH3
HgOCOCH3
+
3-acetossimercurio-2-etossipentano
2-acetossimercurio-3-etossipentano
NaBH4
Hg°
CH3 CH CH2 CH2 CH3 + CH3 CH2 CH CH2 CH3
O CH2 CH3
O CH2 CH3
2-etossipentano
3-etossipentano
SolvOH = CH3C(O)-OH
CH3
C
O
O
mercuriazione
CH3 CH CH2 + Hg(OCOCH )
32
CH3CO2H
CH3 CH CH2 HgOCOCH3
H+
demercuriazione
CH3
C
O
O
H-
CH3
C
O
O
CH3 CH CH3
CH3 CH CH2 HgOCOCH3
Hg°
estere
acetato di isopropile
10
Reazione con alogeni (Br2, Cl2)
Br CH2 CH2 Br
H2C CH2 + Br2
Il legame Br-Br (e Cl-Cl) è covalente, con gli elettroni tra due atomi con la
stessa elettronegatività (legame non polare).
Il legame Br-Br (e Cl-Cl) può essere polarizzato per interazione donatoreaccettore con un legame π. La polarizzazione è favorita da solventi polari.
dopo la polarizzazione
SCISSIONE ETEROLITICA
vuoto
C
C
C
C
C
.. C
Br
+
Br
Br : δ+
..
..
Br
Br δ-
(+)
ione BROMONIO
..
ione
bromuro
(-)
Br
complesso π
lento
C
C
+ Br Br
C
C
C
Br δ+
C
Br δ-
C
Br δ+
Br
+
----C
C
Br
C
Br
C
Br
C
+
Br-
Br
+
Br δ-
Br-
C
Br
Br
C
δ+
C
C
Br
C C
E
C
δ−
C
Br
+
Br
Br
π
REAZIONE
STEREOSPECIFICA
C C
Br
C C
C C
Br
Br
+ Br2
Br
coordinata di reazione
11
ANDAMENTO STEREOCHIMICO
Br δBr δ+
+ Br2
Br δ-
#
+ Br
Br δ+
Br
Br
+
-----
Br
π
Br
Br-
partendo da un isomero geometrico:
H
Br2
H
CH3
H
+ Br
CH3
H
H
(A)
CH3
H
Br
CH3
H
=
CH3
(A)
H
Br
CH3
H
CH3 Br-
Con Cl2
H
CH3
=
=
(A)
Br
CH3
H
CH3
CH3
S
Br
CH3
treo
S
CH3
Br
H
Br
(B)
H
Br
CH3
Br-
H
(A)
BrH
+ Br
CH3
(A)
CH3
=
Br
CH3
(B)
H
Br
H
H
CH3
CH3
CH3
+ Br
H
CH3
Br
H
#
H
CH3
π
CH3
δ- Br
δ+ Br
δ- Br
δ+ Br H
R
Br
H
H
Br
(A)
CH3
treo
R
Cl, più piccolo di Br, sovrappone meno bene l'orbitale pieno
con l'orbitale vuoto del C e quindi non forma un vero legame,
ma dà solo un'interazione accettore-donatore
Cl δCl δ+
+ Cl2
Cl δCl
-----
π
#
Cl:
δ+
:Cl
+
Cl-
+
+
Cl-
12
Cl
Cl
Cl
+
Cl
Cl
+
REAZIONE
STEREOSELETTIVA
Cl
minore
quantità
prevalente
REAZIONE con HBr in condizioni radicaliche
Fatto sperimentale:
l'addizione di HBr agli alcheni dà il regioisomero diverso
da quello atteso (orientamento anti-Markownikov)
meccanismo:
Δ
.RO-OR
RO + H-Br
C
C
+ Br.
lento
.
Vladimir Vasilevich
Markonikov
1838 - 1904
2 RO
.
R-H + Br
Br
.
C
C
H Br
Br H
C
C
+ Br
.
continuala reazione a catena
Si forma il radicale
più stabile
REAZIONE REGIOSPECIFICA
CH3 CH
CH2 +
Br
.
.
CH3 CH CH2 Br
.
CH3 CH CH2 Br + H Br
CH3 CH2 CH2 Br + Br
.
Orientamento anti-Markownikov
Quella con HBr in presenza di perossidi è la SOLA addizione radicalica
degli alcheni.
Con Br2 o Cl2 e hν o riscaldamento, hn alchene dà il RADICALE PIU’
STABILE, che è quello in posizione ALLILICA!!!!!
CH3 CH
CH2 + Br2
hν
CH2
Br
CH
CH2
13
RIDUZIONE
C
C
H
H
catalizzatore
C
+ H2
C
catalizzatore
HH H H
H H
H2C
CH2
Pt, Pd, Ni
H
H
H
H
C
C
H
H
H2C CH2
H H
superficie del
catalizzatore
H
H C
H
H
H
H
H
C
C
C
H
H
H
H
H
insormontabile senza
catalizzatore
E
---------
superabile con il catalizzatore
--------------
H
C C
C C H
enegia di attivazione
abbassata
REAZIONE STEREOSPECIFICA
coordinata di reazione
CH2 CH3
CH3
H2
H
H
CH2 CH3
CH2 CH3
+
PtO
H
CH3
enantiomeri cis H
CH3
14
H3C
CH3
D2
H
H
Pd-C
H3C
H
C
C
H
D2
CH3
D
Pd-C
CH3
D
C H = HC C
3
D
H
D
C
CH3
H
D
H3C
H C
C
C
H3C
H C
D
H
C
CH3
C
+ HC
3
H
D
D
(R,R)
meso
C
(S,S)
H
CH3
In soluzione (catalisi omogenea) si riesce ad avere reazione stereospecifica
con catalizzatori che hanno un legante chirale:
HO
HO
C
H2
H
HO
rodio.DIOP.Cl2
C
H2N
CO2H
enzimi
cerebrali
CH2
C
HO
HO
CO2H
H
H2N
(S)-(-)-DOPA
CH2 CH2
NH2
HO
dopammina
(neurotrasmettitore)
cura del morbo di Parkinson
OSSIDAZIONE DEGLI ALCHENI
senza scissione del legame C-C
C
OH OH
O
[O]
C
C
C
oppure
C
C
con scissione del legame C-C
[O]
C
C
C
O
O
O
oppure
REAGENTE
C
oppure
OH
PRODOTTO
Ossidazione senza scissione
KMnO4, OH-
1,2-Dioli
OsO4 e poi Na2SO3
1,2-Dioli
RCO3H
Ossidazione con scissione
KMnO4, OH-, Δ
O3 e poi H2O2, H+
O3 e poi Zn, H+
C
H
Ossaciclopropani
Acidi e/o Chetoni
Acidi e/o Chetoni
Aldeidi e/o Chetoni
15
Con KMnO4, -OH
KMnO4
CH2
CH2
25°C
CH2
CH2
O
O
-OH
Mn
O O-
CH2
CH2
OH
OH
REAZIONE STEREOSPECIFICA
O
O
-
H
H3C
C
C
CH3
H
C
H3C
C
C
-OH H3C
C
H
H3C
C
C
H
O
O
Mn
HO
O-
C
C
O
O
CH3
=
O
OH H
25°C
O
Os
O
O
H
H
C
H3C
C
CH3
H
OH
HO
H
=
CH3 (S,S)
H3 C
H C
CH3
H
CH3 HO
=
H
OH OH
OH
CH3
CH2
CH2
O
O
Os
O O
Na2SO3
H2O
CH2
CH2
OH
OH
C
OH2
H
CH3
O
+ Os°
Na2SO3
Os (riducente) Os°
OH OH
O
δ+ Os
δ- O δ- O
H C
H3C
H
C
O
O
CH3
OH
(R,R)
REAZIONE STEREOSPECIFICA
O
H
CH3
CH3
-OH
OsO4
CH2
C
CH3
Mn
-
Con OsO4
C
H
O
CH2
CH3
H
H
H3C
H3 C
CH3
H
O
H
CH3
H
H
OH
=
O-
H
OH
OH OH
-O
H3C
H
CH3
H
H2O
MnO2 + H O
2
O
O
Mn
OH OH
-OH
δ+ Mn
δ- O δ- O
O
O
O
O
Mn
+ MnO2
OH OH
H
H3C
C
C
H
CH3
16
OH
H
OH
H
H
CH3
=
C
H
OH
H
OH
CH3
H3C
O
C
C
O
O
Os
H2O
Os
O
O
meso
(R,S)
H
H
CH3
H3C
O
=
CH3
H
C
CH3
CH3
CH3
H
O
H OH
OH
H
CH3
H3C
OH2
CH3
H
CH3 HO
=
HO
OH OH
C
C
H
H
CH3
(R,S)
meso
O
Con peracidi
O
CH2
CH2 + RCO2H
CH2
CH2 + RCO3H
meccanismo:
C
C
CH3CO3H
acido
peracetico
O
C
C
H
O
C
O
HCO3H
acido
performico
(permetanoico)
R
O
R
O
C
+
O
H
CO3H
Cl
acido
m-cloroperbenzoico
17
con scissione del legame C-C
KMnO4, -OH, a caldo
KMnO4, -OH
CH3
H3C
C C
H
C O
Δ
CH3
CH3
H3C
O-
H+
O C
+
CH3
H3C
C O
HO
OH OH
C C
[O]
C C
O O
C C
[O]
[O]
con gli alcheni terminali si forma CO2
CH2
R
O
R'
R'
: :
CO2 + H2O
HO C OH
acido carbonico
acido metanoico
OZONO (O3)
..
O
..
..
-O
O
[O]
O + HO C H
C
CH2
O + CO2 + H2O
Δ
R
[O]
C
KMnO4, -OH
O
..:+
O
CH3
H3C
H3C
C C
H
CH3
O
O
O
C C
H3C
CH3
H
CH3
1,2,3-triossaciclopentano
O
C
O
H
CH3
C
CH3
1,2,4-triossaciclopentano
(3,3,5-trimetil)
(4,4,5-trimetil)
meccanismo probabile:
O
O
H3C
H
H3C
O
C C
CH3
CH3
δO
C
δ+
H
-O
O
O
+
C
CH3
H 3C
H
CH3
O
C
C
O
CH3
CH3
Instabile, non si isola
18
TRATTAMENTO RIDUTTIVO
O
H3C
O
C
H
C
O
+ O
C
H3 C
H2O
CH3
ad aldeide
CH3
O
Zn,H+
CH3
C
CH3
H
a chetone
ad H2O2
TRATTAMENTO OSSIDATIVO
O
H3C
C
H
ad acido
carbossilico
O
C
O
H 2O 2
CH3
H+
CH3
ad H2O2
O
H 3C
C
+ O
OH
CH3
C
CH3
a chetone
Addizione di carbeni
Molecole contenenti un C BIVALENTE sono rare in Chimica Organica.
Si chiaano CARBENI e possono essere di due tipi, a seconda della
configurazione elettronica
E+
orbitale p vuoto
C
orbitale sp2
pieno
metilene singoletto
:CH2 (C sp2)
orbitale p
(un e)
H
orbitale p
(un e)
H
R.
metilene tripletto
.
HCH (C sp)
.
19
Si formano per decomposizione fotochimica del diazometano:
:CH2
+
N N
+
N N
CH2
diazometano
:CH2
+
N
N
hν
:CH2 + N N
Il metilene attacca un legame π C=C, sia come singoletto che come tripletto
:CH2
R
con metilene
singoletto
R
R
H
cis
CH2 R
H
H
cis
H
REAZIONE STEREOSPECIFICA
.
.
H-C-H
.
R
R
con metilene
tripletto
H
R
CH2
.
R
R
CH2 H
+
H
H
possibilità
di rotazione
H
cis
CH2 R R
H
H
H
cis
trans
R
Carbeni sostituiti con alogeni sono più stabili e si preparano più facilmente
:CCl2
(CH3)3CO
-
+
Cl
: C Cl
diclorocarbene
(CH3)3COH
H CCl3
:CCl2
+
-CCl
3
-
+ Cl
Cl
Esempio:
Cl
+ :CCl2
Cl
Reazione di Simmons-Smith
CH2I2 + Zn(Cu)
(C2H5)2O
I
CH2 ZnI
CH2
CH2I2 + Zn(Cu)
Esempio:
H 3C
CH CH2
carbenoide
H 3C
CH CH2
(C2H5)2O
20
ALCHINI
CARATTERISTICA FUNZIONALE:
legame π
Caratteristiche Spettrali
Spettrometria di massa
Una frammentazione importante dà un valore m/z = 39, che
corrisponde al carbocatione propargilico
+.
+
. +
R + CH2 C CH
R CH2 C CH
CH2 C CH2
4-ottino
1-ottino
M+
IR
stretching
stretching =C-H
C C circa 2150 cm-1
a circa 3300 cm-1
manca negli alchini
simmetricamente disostituiti
1-ottino
21
2-ottino
4-ottino
13C
NMR
L’anisotropia diamagnetica scherma
il nucleo
C sp δ 70-90 ppm
4-ottino
C C
1-ottino
68.11
84.66
C2
C1
22
1H
L’anisotropia diamagnetica scherma
il nucleo
NMR
C C H
δ 1.9 – 2.5 ppm
C C H
Danno le stesse reazioni degli alcheni (legame π), con lo stesso meccanismo
L'addizione del primo equivalente lascia ancora un legame π,
che perciò può dare ulteriore addizione
X
C
C
+ X Y
C
C
Y
X
Y
C
C
X
Y
il prodotto della reazione dipende perciò dal numero di equivalenti del
reagente elettrofilo
esempi:
H
H3 C C
C CH3 + H Cl
CH3
C
H3 C
C
Cl
trans
REAZIONE STEREOSPECIFICA
23
con HBr
H3 C C
(addizione elettrofila)
..
-:Br:
+
H3C C
C H + H Br
C H
(carbocatione
più stabile)
Br
H3C C
H Br
C H
..
C H
H
..
CH2
Br
H3C C
Br H
H
H3 C C
H
..
-:Br:
+
H3C C
Br
CH3
Br
(carbocatione
più stabile)
con HBr
(addizione radicalica)
RO
Δ
OR
RO. + H Br
H3C C
C H +
Br.
RO-H + Br.
.
H3C C
(radicale
più stabile)
H
H3 C C
C H +
H3C C
Br
H3C CH CH Br
(radicale
più stabile)
H
H Br
C H
.
Br.
Br
con H2O, H+
2 RO.
C H
+ Br.
continua
la catena
Br
H
H Br
H3 C C
Br
H
Br
.
C H + Br
Br
continua
la catena
servono anche sali di Hg2+ come catalizzatori
O
H+
H3C C CH3
H3C C C H + H2O
Hg2+
meccanismo:
HC
C CH3 + H+
H
H C
+
C CH3
carbocatione più stabile
H
H
:OH2
H
C
C CH3
+ OH2
24
H
H
H C C CH3
H O
C CH3
H C
enolo OH
TAUTOMERIA
C
C
O
C
C
O
H
H
anche a temperatura ambiente si ha
scissione del legame C-C
Ossidazione con KMnO4
R C
C R'
KMnO4
-OH, 25°C
H+
O
R C
O
O-
+
-O
O
O
R C
C R'
+ R' C
OH
OH
La riduzione catalitica arriva ad ALCANO, se
non si usano catalizzatori speciali
RIDUZIONE
H2
C
C
Pt
C
H
C
H
H2
Pt
H
C
H
C
H
H
Per fermarsi ad alchene è necessario che il catalizzatore sia meno attivo
C
C
H2
C C
Pd "avvelenato"
H
disattivato H
cis
Per ridurre alchini interni ad alcheni trans
C
C
Na
NH3
H
C C
H
trans
REAZIONE STEREOSPECIFICA
con Na° (K°, Li°)
in NH3 liquida
REAZIONE STEREOSPECIFICA
25
ATTENZIONE!!
C
GLI ALCHINI TERMINALI DANNO GLI ALCHINURI
C H + Na°
NH3
C C: Na+ + 1/2 H2
meccanismo:
.-.
Na+ R C
.-.
R C
Na+ +
C R + Na°
R C
.
.
R
.
Na+ -NH2 +
C R + NH3
C
.
R
C
+ Na°
C
C
anione vinilico
R
..
R
Na+
C
H
R
H
..
R
Na+ +
radicale vinilico
C
H
R
C
H
R
Na+ -NH2 +
+ NH3
C
anione radicale
C R
H
C
C
R
H
R
trans
L'andamento stereochimico viene spiegato con la maggiore stabilità del
radicale vinilico con i gruppi R in trans, rispetto a quello con i gruppi R in
cis (repulsione per ingombro sterico).
.
R
C
H
R
R
C
C
R
C
H
.
ACIDITA' del legame Csp-H
I sali degli alchini terminali (alchinuri) si possono ottenere, oltre che con Na°,
con idrossidi di metalli di transizione
R C C H
+ Ag(NH3)2OH
R C
"Cu+ -OH"
+ H2O + 2 NH3
legame covalente polare
"Ag+ -OH"
R C C H + Cu(NH3)2OH
C Ag
R C
C Cu
+ H2O + 2 NH3
legame covalente polare
26
DIENI
legame π
CARATTERISTICA FUNZIONALE:
Caratteristiche Spettrali
UV-visibile
Analoghe a quelle dgli alcheni
La coniugazione abbassa la differenza di
energia HOMO-LUMO (shift batocromico)
A
B
C
3-metil-1,4-pentadiene
=CH2
=C-H
142.45
ppm
113.13
ppm
27
1,4-esadiene
H vinilici
{
{
C sp2
Stesse reazioni degli alcheni; il prodotto dipende dal numero di equivalenti
del reagente (elettrofilo)
Cl
HCl
CH2 CH CH2 CH2 CH
CH3 CH CH2 CH2 CH CH2
CH2
HCl
Cl
CH3 CH CH2 CH2 CH CH3
Cl
DIENI CONIUGATI
Anche con un solo equivalente si ottengono più prodotti:
H
H2C CH CH CH2 + HCl
Cl
H
Cl
CH2 CH CH CH2 + CH2 CH
addizione
1,2
a bassa temperatura
prevale addizione 1,2
a temperatura elevata
prevale addizione 1,4
CH CH2
addizione
1,4
28
addizione 1,2
Cl δ-
+
H3C CH CH CH2
H2C CH CH CH2
+ H
+
H3C
CH
+
CH CH2
Cl-
#
H3C CH CH CH2
δ+
H3C
CH
δ+ #
CH CH
unico carbocatione
DUE siti positivi
δ-Cl
addizione 1,4
E
CH
H3 C
Ea
H3C CH
+
CH
CH2
1,2
+
CH CH2
H3 C
Cl
CH CH CH2
1,4
H2 C
Cl
CH CH CH2
CH2 CH CH CH3
coordinata di reazione
1,21,4-
controllo cinetico
controllo termodinamico
Reazione di Diels-Alder
Δ
π3*
LUMO
HOMO
π1
Addotto di Diels-Alder
+
diene
dienofilo
nuovi legami σ
nuovo
legame π
29
Kurt Alder
1902-1958
Otto Paul Hermann Diels
1876-1954
La geometria del dienofilo si mantiene nell’addotto
O
O
H C CH3
C CH3
O
O
+
O
O
H C CH3
C CH3
O
O
dienofilo cis
H
+
H3C
O
C
O
O
C
addotto cis
O
O
C
CH3
H
C
O
dienofilo trans
O
O
CH3
CH3
addotto trans
Se il diene ed il dienofilo sono sostituiti non simmetricamente, si ottengono
due addotti
CH3
H
O
C
H
H
+
CH3
CH3 O
C
CH3
CH3
+
C
O
CH3
30
Regola endo
Quando il dienofilo contiene un legame π nel gruppo elettron attrattore, gli
orbitali π in quel gruppo si avvicinano agli atomi centrali del diene. Questa
sovrapposizione secondaria aiuta a stabilizzare lo stato di transizione della
formazione dell’addotto.
Fenomeno osservato formando i
prodotti biciclici dal ciclopentadiene
H
H
+
O
O
O
H
O
O
H
CHO
e non
H
O
OCH3
OCH3
+
H
H
O
H
H
CO2CH3
OCH3
CO2CH3
e non
H
CO2CH3
H
H
H
H
H
H
H
H
H
H
H
H
H
O
eso
eso
endo
endo
H
H
O
H
C
=
==
=
C
=
==
C
=
==
C
C
H
C
H
Sovrapposizione
secondaria
C
=
==
=
=
O
H
CH2
CH2 CH2
C
O C H
H
31
16/05/2011
COMPOSTI AROMATICI
L’aromaticità è l’aspetto strutturale determinante per le caratteristiche spettrali e di
reattività di questi composti.
Caratteristiche Spettrali
La maggior parte dei composti aromatici
presenta un picco dello ione molecolare
molto intenso.
Spettrometria di massa
Gli alchilbenzeni hanno una frammentazione molto facile con m/z 91.
E’ stato dimostrato che il carbocatione benzilico traspone a catione eptatrienilio.
CH2
R
+.
+
CH2
.
m/z 91
M+
m/z 91
M+
(128)
(78)
CH3
+
R +
91
92
CH2 CH3
91
108
1
16/05/2011
Il benzene ha legami π coniugati
UV-visibile
Lo spettro di assorbimento elettronico è caratterizzato da due bande dette
“primarie” e una banda detta “secondaria” o della struttura fine.
λ = 184 nm
(ε = 47 000)
A
λ = 202 nm
(ε = 7 000)
struttura fine
λ = 255 nm
(ε = 230)
È dovuta alle interazioni
dei livelli elettronici con i
modi vibrazionali
stretching =C-H
IR
bending =C-H (fuori dal piano)
stretching C=C
nm
a sinistra di 3000 cm-1
900-690 cm-1
spesso in coppia a 1600 e 1475 cm-1
2
16/05/2011
CH2 CH3
13C
NMR
L’anisotropia diamagnetica descherma il nucleo
δ 120-140 ppm
1H
NMR
L’anisotropia diamagnetica descherma il nucleo
δ 7-8 ppm
3
16/05/2011
COMPOSTI AROMATICI
I composti aromatici reagiscono MANTENENDO l’aromaticità
ADDIZIONE
H
+
E+
E
+
SOSTITUZIONE
H+
possibili meccanismi?
1) meccanismo sincrono
H δ+
E
δ+
E
+ E+
2) meccanismo in due stadi
H
H
E
E
+
+
+
+E
+
E
+ H
+
H
E
Evidenze sperimentali
La reazione è stata eseguita sul deuterobenzene
D
D
D
D
D
k / kH = 1
D
D
il legame C-D è piu forte del legame C-H:
significa che nello stadio lento non c'e rottura del legame C-H
4
16/05/2011
CH3
In alcuni casi si è riusciti
ad isolare l'intermedio
H
H
+
H3C
H
CH3
+
BF4
H
SbF6
MECCANISMO IN DUE STADI
MECCANISMO DELLA SOSTITUZIONE ELETTROFILA AROMATICA
primo stadio
=/
E δ+
H
lento
+
E
E+
+
H
δ+
addotto sigma
primo stato di transizione
strutture di
risonanza
dell'intermedio
secondo stadio
H
E
+
+
H
E
H δ+
E
H
+
H
E
=/
E
veloce
E
+
δ+
secondo stato di transizione
Energia in funzione della coordinata di reazione:
1° ST
2° ST
E
+
H
E
---------------
E
- 10.8 kcal/mole
---------------------------------coordinata di reazione
5
16/05/2011
Tutte le variazioni strutturali che abbassano l'energia
dell'intermedio abbassano anche l'energia dello stato di
transizione che porta all'addotto σ e quindi rendono più
facile la reazione.
SOSTITUZIONI ELETTROFILE AROMATICHE
principali reazioni di Sostituzione Elettrofila
NO2
+
NITRAZIONE
HNO3
H2SO4
+ H2O
SO3H
+
SOLFONAZIONE
SO3
H2SO4
X
FeX
ALOGENAZIONE
+ X2
+ HX
Cl
(X = Cl)
+ Cl2
FeCl3
+ HCl
clorurazione
+ HBr
bromurazione
Br
(X = Br)
FeBt3
+ Br2
R
+ RX
ALCHILAZIONE
AlCl3
+ HX
Alchilazione di Friedel-Crafts
O
AlCl3
O
ACILAZIONE
+
R
X
R
+ HX
Acilazione di Friedel-Crafts
Queste reazioni permettono di introdurre nel benzene gruppi funzionali
diversi, ma avvengono con lo STESSO MECCANISMO. E' perciò importante
capire quale è il reagente elettrofilo e come si forma.
6
16/05/2011
Nitrazione
+
HNO3 + H2SO4
+ HSO4
H O NO2
-
H
+
+NO
2
H 2O +
H O NO2
E+
H
catione NITRONIO
H3O+ + HSO4=
H2O + H2SO4
NO2+ + H3O+ + 2 HSO4=
HNO3 + 2 H2SO4
H
+NO
2
+
NO2
NO2
+
H+
Solfonazione
O
E+
S O
O
+
O
HO
S O
+
S O
SO3H
HO
O
O
+
SO3H
S OH
H+
O
Alogenazione
Cl Cl + FeCl3
FeCl4
Cl+
+
"Cl+"
+
-
H
+
-
Cl
FeCl4
Cl
E+
H+ ClFeCl3
Br Br + FeBr3
H
+
Br+
+
Br
-
FeBr4
FeBr4
-
+
"Br+"
E+
Br
H+ BrFeBr3
7
16/05/2011
Alchilazione (di Friedel-Crafts)
δ+
δR Cl + AlCl3
δδ+
R ... Cl ... AlCl3
-
H
R+
+
"R+
AlCl4
+ R
E+
R
H+
AlCl3
Acilazione (di Friedel-Crafts)
R
Cl
δ+ C δO
R
+ AlCl3
+
C O:
R
R
+
C
+
C
O
R
+
+
O
C O+
R
-
AlCl4
+
R C O
E+
H
+
+
O
H
C
AlCl4
H+ ClAlCl3
C
R
O O
C
R
+
AlCl3
O
C
AlCl3
R
AlCl3 si deve
usare in quantità
stechiometrica
perché dà
complesso di
Lewis con il
prodotto
Charles FRIEDEL
(1832 - 1899)
James Mason CRAFTS
(1839 – 1917)
8
16/05/2011
Sostituzioni elettrofile aromatiche dei Benzeni SOSTITUITI
Tutte le posizioni del benzene sono equivalenti.
Quando c'è un sostituente, le posizioni sono diverse: 2 orto, 2 meta e 1para
se i prodotti si formassero solo su basi statistiche, si dovrebbe avere 40% di
orto, 40% di meta e 20% di para
a distribuzione dei prodotti osservata sperimentalmente non è
MAI quella statistica
questo significa che il sostituente presente sull'anello del benzene indirizza
l'attacco dell'elettrofilo
ORIENTAMENTO
per esempio:
CH3 E+
H+
CH3
+
E
E
CH2Cl
CH3
CH2Cl
E+
H+
E
Non è un effetto del sostituente sul substrato!
Stadio lento: formazione dell’addotto σ: più stabile è
l'addotto sigma, minore è l'energia dello stato di
transizione che porta all'addotto.
ORIENTAMENTO
1. Si scrivono tutti i possibili addotti e le rispettive strutture di risonanza.
2. Si esamina se c'è una struttura con la carica positiva sul C che lega il
sostituente.
3. Si guarda se il sostituente stabilizza (o destabilizza) la carica positiva.
CH3
+ E+
CH3
H
+ E
CH3
+
H
E
CH3
H
E
+
CH3
+
H
E
CH3
H
E
+
E
CH3
+
CH3
CH3
CH3
H
E
CH3
CH3
+
+
+
H E
H
E
H
E
9
16/05/2011
CH3
E
benzene
H
+
meta
E
H
orto,para
CH3
+
+
E
CH3
H
+
E
H
E
CH3
+ E+
+ E+
E
coordinata di reazione
La reattività del benzene sostituito rispetto a quella del benzene
dipende dall'energia di attivazione dello stadio lento e si vede dalla
stabilità dell'addotto σ più stabile (del benzene sostituito) rispetto a
quella dell'addottodel benzene.
+
CH2Cl
H
CH2Cl
H
+
E
CH2Cl
E+
+
+
+
+
H
H
+
E
H
E
CH2Cl
E
CH2Cl
E
CH2Cl
E
CH2Cl
CH2Cl
CH2Cl
CH2Cl
H
+
+
+
H
E
H
E
H
E
10
16/05/2011
CH2Cl
+
orto,para
CH2Cl
H
+
E
E
H
meta
CH2Cl
E
benzene
H
E
+
+
H
E
CH2Cl
+ E+
o
coordinata di reazione
OH
OH
OH
+
+
E+
OH
H
H
E
+
+
H
E
E
+
OH
H
E
OH
OH
OH
+
+
H
H
+
E
E
E
OH
OH
H
OH
+
+
+
H
E
H
+
E
OH
H
H
E
E
11
16/05/2011
OH
meta
H
E
+
benzene
E
H
E
+
orto,para
+
OH
+
OH
H
E
E
H
OH
+ E+
o
coordinata di reazione
NO2
NO2
NO2
+
+
E+
E
H
E
+
E
+
NO2
NO2
NO2
+
+
H
H
+
NO2
H
H
E
NO2
H
E
NO2
E
NO2
+
+
+
H
E
H
E
H
E
12
16/05/2011
NO2
NO2
H
+
E
E
H
orto,para
+
meta
NO2
E
benzene
H
+
E
H
+
E
NO2
+ E+
o
coordinata di reazione
Cl
Cl
Cl
Cl
H
H
+
+
E+
E
H
E
+
E
+
+ Cl
H
E
Cl
Cl
Cl
+
+
H
H
+
E
Cl
Cl
H
E
E
Cl
+
+
+
H
E
H
Cl +
E
H
H
E
E
13
16/05/2011
Cl
meta
+
H
E
orto,para
Cl+
E
Cl+
H
E
benzene
H
E
+
H
E
Cl
+ E+
o
coordinata di reazione
reagisce meglio con centri
RICCHI di elettroni
E+ è specie a difetto elettronico
REATTIVITA’
Si deve considerare la densità elettronica
delle POSIZIONI REATTIVE (orientamento)
Esempio:
mettere i seguenti composti in ordine decrescente di reattività, nei confronti di
un reagente elettrofilo
NO2
OCH3
CH3
Cl
1. Si individua la posizione (o le posizioni) di attacco, cioè l'orientamento
(sulla base della stabilità degli addotti σ).
2. Si considera l'effetto del sostituente SU QUELLA POSIZIONE.
3. Maggiore è la densità elettronica, maggiore è la reattività (a parità di
elettrofilo).
14
16/05/2011
Tutte le posizioni sono equivalenti. Non ci sono
sostituenti, la densità elettronica è quella di
riferimento.
Orientamento in META.
In questa posizione il gruppo NO2 esercita solo
l'effetto induttivo.
NO2
attrazione elettronica
-I
Le posizioni reattive del nitrobenzene sono più povere di elettroni (e
quindi MENO reattive) delle posizioni reattive del benzene
Orientamento in ORTO + PARA.
In queste posizioni il gruppo OCH3 esercita sia
l'effetto induttivo che quello coniugativo.
OCH3
rilascio elettronico
-I , + R
Le posizioni reattive del metossibenzene sono più ricche di elettroni
(e quindi PIU’ reattive) delle posizioni reattive del benzene
CH3
Orientamento in ORTO + PARA.
In queste posizioni il gruppo CH3 esercita solo
l'effetto induttivo.
rilascio elettronico
+I
Le posizioni reattive del metilibenzene sono più ricche di elettroni (e
quindi PIU’ reattive) delle posizioni reattive del benzene
Orientamento in ORTO + PARA.
In queste posizioni il gruppo OCH3 esercita sia
l'effetto induttivo che quello coniugativo.
Cl
-I , + R
attrazione elettronica
Le posizioni reattive del clorobenzene sono più povere di elettroni (e
quindi MENO reattive) delle posizioni reattive del benzene
Il rilascio elettronico dovuto a risonanza è maggiore di quello induttivo (alchili)
Reattività:
CH3
OCH3
>
>
Cl
>
>
NO2
15
16/05/2011
Sostituzioni Elettrofile sul NAFTALENE
A differenza del benzene, le posizioni del naftalene non sono tutte equivalenti:
ORIENTAMENTO
α
α
β
β
β
per conoscere l’orientamento,
basta confrontare gli addotti
sigma nelle posizioni α e β
+ E+
β
α
α
E
H
E
H
E
H
+
+
+
H
H
+
H
E
E
E
+
+
L’altro anello mantiene la completa aromaticità 2 volte su 3 nella risonanza dell’addotto
in α, solo 1 nella risonanza dell’addotto in β e quindi è più stabile
Le sostituzioni elettrofile aromatiche sul naftalene avvengono in α
NO2
H2SO4
+ HNO3
+
+H
Un caso a parte è la solfonazione, in cui l’orientamento dipende
SO3H
dalla temperatura
T<100oC
H2SO4
H2SO4 , Δ
+ SO3
SO3H
T>150oC
E
a T<100°C: controllo cinetico,
a T>150°C: controllo termodinamico
α
β
O
H
O
H
S
O
H
O
S
O
OH
coordinata di reazione
H
16
16/05/2011
REAZIONI CON ELETTROFILI DEBOLI
Avvengono solo con aromatici MOLTO reattivi
1. Reazione con i sali di benzendiazonio
:OH
+ OH
OH
H N
N
N
N+
X
-
+ HX
N
N
4-idrossiazobenzene
L’elettrofilo (voluminoso) va in in orto solo se la posizione para è occupata
Reagiscono solo: benzenolo (=fenolo) e benzenammina (=anilina)
(e analoghi del naftalene)
2. Reazione di carbossilazione (Reazione di Kolbe)
O - Na+
CO2
OH O
C
Δ
O
+
O
δO
C δ+
+
H
O
O
C
-
O
O
OH O
C
-
+
solo con fenolo
O - Na+
-
O
H
O
C
+
-
O
OH O
C
O
OH
H
C
O
O
O
H
O
C
O
acido salicilico
(acido 2-idrossibenzencarbossilico)
O
C
OH
O
aspirina
C
O
CH3
(acido acetilsalicilico)
acido 2-acetossibenzencarbossilico
17
16/05/2011
3. Reazione di formilazione (reazione di Reimer-Tiemann)
O - Na+
Na+ - O
solo con fenolo
O
C
CHCl3
H
KOH
-
CHCl3 + -OH
O-
O:CCl2
+
Cl- + :CCl2
:CCl3
O
H
CCl2
+
OH
O-
H
CCl2
-
-
OH O
C
OH OH
C OH
H
CHCl2 H2O
HO-
O
H
+
CCl2
+
-
Elettrofilo
H
H
CCl2
-
aldeide salicilica
(2-idrossibenzencarbaldeide)
4. Reazione di idrossimetilazione
solo con fenolo
O-
O
O
H
O
+ C
H
H
+
H
CH2 O
-
CH2 OOH
OH
CH2 OH
+
CH2 OH
O
OH
CH2 OH
OH
CH2 OH
CH2
CH2 OH
O
H2O
O
H
H2O
o-chinometano
p-chinometano
CH2
18
16/05/2011
PREPARAZIONE E REATTIVITA' DEI SALI DI BENZENDIAZONIO
[H]
NH2
NO2 (Fe, HCl)
HNO3
+
N N
NaNO2, HCl, O°C
[O]
H2SO4
(CF3CO3H)
+
OH
N N
H2O, Δ
Cl
-
H
H3PO2
CuCl
Cl
KI
CuCN
CuBr
Br
AgBF4
I
C N
NaNO2,
+N N
Cu
--
F
BF4
(solido)
Δ
NO2
USO DEI SALI DI BENZENDIAZONIO
1. Introduzione di gruppi che non si possono introdurre diversamente
OH
-OH è nucleofilo
il benzene reagisce con gli elettrofili
da
HNO3
NH2 NaNO
2
HCl, 0°C
NO2 [H]
H2SO4
H2O
-OH
OH
+
+
N N
Cl-
N2
2. Introduzione di un secondo sostituente in una posizione diversa da quella
possibile con una sostituzione elettrofila aromatica, sulla base dell'orientamento provocato dal primo sostituente
N
C
esempi
H3 C
?
19
16/05/2011
PROBLEMI
difficile introdurre CN
CN orienta un elettrofilo in meta
sali di benzendiazonio
SOLUZIONE
CH3
CH3
(CH3CO)2O
NH2
NH C
CH3Br
NH C
O
AlCl3
CH3CO2H
O
H3C
serve per ingombrare stericamente le posizioni orto
ed avere solo la sostituzione in para e per diminuire
la reattività altrimenti si avrebbero prodotti di polimetilazione
H2O, H+
NH2 NaNO
2
HCl, 0°C
H 3C
CH3
+
N N
ClH3C
PROBLEMA
N2
sali di diazonio
CH3
Br
CH3
Br2
NH C
H3C
sia -CH3 che -Br orientano in orto, para
SOLUZIONE
Br
C N
CuCN
-NHCOCH3
NH C
(|+R|>|-I|)
O
O
gruppo a rilascio
elettronico più forte
di -CH3
FeBr3 H C
3
H3C
(preparato come prima)
CH3
Br
NH C
Br
H2O, H+
NH2
O
H3C
H3C
Br
H3C
+
N N
HSO4-
NaNO2
H2SO4, 0°÷ 5°C
Br
H3PO2
H
H3C
20
16/05/2011
Br
?
Br
Br
- Br orienta in orto, para
PROBLEMA
Br2
Br Br2
Br
Br
+
FeBr3
FeBr3
Br
Br
Br2
FeBr3
Br
Br
+
Br
Br
SOLUZIONE
NH2
SALI DI DIAZONIO
Br2
NH2
Br
Br
gli atomi di Br sono nella
posizione relativa voluta:
basta togliere il gruppo
-NH2
H2O
in queste condizioni reagiscono
tutte le posizioni influenzate da
-NH2
Br
N
NH2
Br
Br
Br
Br
X-
N+
NaNO2
Br
Br
H3PO2
Br
Br
HX, 0°÷ 5°C
Br
Br
Br
21
16/05/2011
REATTIVITA' DI ANTRACENE E FENANTRENE CON I REAGENTI ELETTROFILI
L'aumento del numero di anelli condensati diminuisce l'aromaticità
Fenantrene ed antracene (meno aromatici di benzene e fenantrene)
danno ANCHE REAZIONI DI ADDIZIONE
il legame C9-C10 ha caratteristiche di un
doppio legame poco delocalizzato
FENANTRENE
Br2
+
(FeCl3)
Br
Br
Br
9-bromofenantrene 9,10-dibromo-9,10-diidrofenantrene
nel nome del prodotto di addizione va indicata la presenza
dei due H : 9,10-dibromofenantrene indica che in 9 e 10 ci
sono due atomi di Br AL POSTO DI DUE H
ATTENZIONE!!
Br
Br
9,10-dibromofenantrene
il fenantrene si comporta con HX come un alchene
HCl
9-cloro-9,10-diidrofenantrene
Cl
L'addizione avviene al legame C9-C10, perché in questo modo rimane intatta
l'aromaticità degli altri due anelli benzenici
Si comporta anche come un diene coniugato, dando
prodotti di addizione 1,4, oltre ai prodotti di sostituzione
elettrofila aromatica (in posizione 9)
ANTRACENE
Br
Br
Br2
+
(FeCl3)
Br
9-bromoantracene
9,10-dibromo-9,10-diidroantracene
L’antracene dà le caratteristiche reazioni dei dieni coniugati e perciò reagisce con
elettrofili del tipo HX e con i dienofili
22
16/05/2011
HX
9-X-9,10-diidroantracene
X
O
O
O
O
O
O
addotto di Diels-Alder
O
O
O
Reazioni in catena laterale
I benzeni sostituiti possono reagire anche in catena laterale (cioè sul sostituente), se
si usano condizioni opportune.
CH2 Cl
hν
CH3
+ Cl2
CH3
CH3
Cl
FeCl3
Br
CH CH3
CH CH2
+ HBr
+
Cl
REAZIONI DI OSSIDAZIONE
Si ossidano a acido benzencarbossilico
Monoalchilbenzeni
CH3
KMnO4, OH-
OC O
H+
OH
C
O
Δ
23
16/05/2011
CH2 CH3
KMnO4, OH-
Δ
CH2 (CH2) CH3
n
KMnO4, OH-
Δ
Anello benzenico
OC O
CO2 +
OH
H+
C
OC O
(n + 1) CO2 +
O
OH
H+
C
O
molto resistente all'ossidazione
O
O2, V2O5
O
400-440°C
O
O
O2, V2O5
Il naftalene è un po’ più facile da ossidare
O
350-400°C
O
Un’ossidazione meno drastica si può avere con CrO3 e gli aromatici con più anelli
condensati
O
CrO3
1,4-naftalenchinone
(1,4-naftochinone)
O
O
CrO3
CrO3
O
O
9,10-fenantrenchinone
benzendioli
OH
OH
O
9,10-antracenchinone
Si ossidano molto facilmente a chinoni
Ag2O
O
O
o-benzochinone
OH
HO
O
Ag2O
O
p-benzochinone
24
16/05/2011
REAZIONI DI RIDUZIONE
- Con H2
La riduzione catalitica richiede pressioni e temperature elevate per la
prima addizione (perdita dell’aromaticità), mentre le addizioni successive
sono più facili.
3 H2, P, Δ
cat. (Pt, Pd, Ni, Ru, o Rh)
- Con Na°
Riduzione di Birch
Na (o Li)
NH3 liquida, CH3CH2OH
meccanismo:
.
+
Na°
CH3CH2OH
H
..
-
H
H
CH3CH2OH
H
H
CH3CH2O-
H
carbanione
H
Se la reazione si effettua su un benzene sostituito
Si formano carbanioni
Na°
H
radicale
anione radicale CH3CH2O
..
-
.
ORIENTAMENTO
stabilizzati da gruppi ad attrazione elettronica,
destabilizzati da gruppi a rilascio elettronico
Un carbonio che porta un gruppo ad attrazione elettronica viene ridotto
CO2H
CO2H
CO2H
Na (o Li)
H
H
H
NH3 liquida, CH3CH2OH
H
H
intermedio stabilizzato
Un carbonio che porta un gruppo a rilascio elettronico non viene ridotto
H H
CH3
CH3
Na (o Li)
NH3 liquida, CH3CH2OH
H H
-
H
H
intermedio destabilizzato
H H
O
CH3
O
Na (o Li)
NH3 liquida, CH3CH2OH
H H
CH3
CH3
H
H
-
O
CH3
intermedio destabilizzato
25
16/05/2011
Il benzene non reagisce con i nucleofili
Però …..
+ Nu-
la sintesi industriale del fenolo parte dal clorobenzene
Cl
OH
KOH, Δ, P
…..e allora ????
KCl
SOSTITUZIONE NUCLEOFILA AROMATICA
Perché i composti aromatici possano reagire con NUCLEOFILI, devono
possedere i seguenti requisiti
1. BUON GRUPPO USCENTE
2. SOSTITUENTE/I A FORTE ATTRAZIONE ELETTRONICA (NO2)
In questo caso si ha:
Meccanismo di ADDIZIONE-ELIMINAZIONE
La reazione avviene in due stadi, con formazione nello stadio lento di un
intermedio (addotto σ negativo o complesso di Meisenheimer), stabilizzato
per risonanza dal sostituente
CH
Cl O CH3
Cl
+ - O CH
3
+
- N
O
O
δ- OCH
δ- Cl
3
Cl
δ-
-
E
Cl
Cl
N+
O
OCH3
-
N+
O
Cl OCH
O
3
-
+
- N
O
O
X
+
- N
O
O
N+
O- O
X
OCH3
X
OCH3
O
-
δCl OCH3
X
-
Cl
OCH3
3
O
lento
=/
ΔH
Cl OCH3
X
N
-O + O
coordinata di reazione
26
16/05/2011
Evidenze sperimentali:
qualche complesso di Meisenheimer è stato isolato
F
Cl
è più reattivo di
il legame C-F è più forte di C-Cl, ma
NO2
NO2
il legame con il gruppo uscente non si rompe nello stadio lento e F (più elettronegativo)
favorisce l'attacco del nucleofilo, rendendo più positivo il C a cui è legato
Però …..
la sintesi industriale del fenolo parte dal clorobenzene
Cl
OH
KOH, Δ, P
…..e allora ????
KCl
Se manca il sostituente a forte attrazione elettronica, si può avere reazione
con i nucleofili, purché vengano soddisfatti i seguenti requisiti:
1. BUON GRUPPO USCENTE
KOH (solida)
NaNH2 in NH3 liquida
2. il nucleofilo è UNA BASE FORTE
In questo caso si ha:
Meccanismo di ELIMINAZIONE-ADDIZIONE
La base forte strappa un protone acido (in orto al gruppo uscente) e la base
coniugata forma il benzino, che, essendo molto reattivo, addiziona l'acido
coniugato della base
Cl
H
Cl
H
OH
H
..
+ -OH
-
+ H2O
H2O
Cl-
BENZINO
OH
H
l'addizione può avvenire in entrambi i modi
H
HO
Evidenze sperimentali:
Cl
*
OH
OH
-OH
*
+
*
* = 13C
Cl-
con un clorobenzene sostituito si hanno miscele di prodotti
27
16/05/2011
p-metilclorobenzene
NH2
Cl
H
NH2
+ NH2-
+
CH3
CH3
CH3
Cl
- CH
H
H
Cl
2
H 2N
..
-
+ NH3
H
NH2
ClCH3
CH3
o-metilclorobenzene
Cl
H
CH3
H NH
2
H2N H
NH2
CH3
CH3
H2N
CH3
+
Cl-
m-metilclorobenzene
Cl
H
H
H3 C
H2 N
Cl-
H2N
H
+
H3 C
H3 C
H
+
H3C
H3 C
NH2
NH2
NH2
+
+
CH3
CH3
CH3
28
16/05/2011
ALDEIDI e CHETONI
Caratteristica strutturale di
aldeidi e chetoni
O sp2 legato a C sp2
C=O
Caratteristiche Spettrali
Spettrometria di massa
Lo ione molecolare di solito è osservabile, sia con le aldeidi che con i chetoni.
Una scissione molto facile è la “rottura α” (α-cleavage), che lascia
un carbocatione acilio (stabilizzato per risonanza):
R
.
H
+.
H
C O
.
R + H
+
C
O
M+ - H
R
O
H
R'
C O
H
C
C
+.
R'
R
+
R
+
.
+
+
C
C
R
O
+
O
+
C
O
+
.
R + R' C O
R
+
C
R' C
O
+
O
M+
O
P.M = 106
CH3
C
m/z 105
m/z 105
O
P.M = 120
+
C O
+
C O
M+
1
16/05/2011
UV-visibile
due transizioni:
π → π* a λ = 190 nm (lontano UV) permessa dalla simmetria (ε> 10 000)
n → π* a λ = 280-290 nm proibita dalla simmetria (ε 15-20)
La transizione n → π* può non vedersi se la soluzione è diluita
La coniugazione con doppi legami sposta I massimi di assorbimento verso
lunghezze d’onda maggiori (shift batocromico)
O
IR
π → π* a λ = 231 nm (ε = 12 600 M-1cm-1)
n → π* a λ = 321 nm (ε =
40 M-1cm-1)
ALDEIDI
stretching C=O a circa 1725 cm-1, spostato verso valori più bassi
dalla coniugazione
stretching C-H (del –CHO)
deboli bande a 2750 e 2850 cm-1.
3-metilbutanale
benzencarbaldeide
2
16/05/2011
IR
CHETONI
stretching C=O
a circa 1715 cm-1, spostato verso valori più bassi
dalla coniugazione
3-pentanone
fenil metil chetone
13C
NMR
L’effetto -I dell’O ed il campo magnetico generato
dagli elettroni π diminuiscono la densità elettronica
attorno al nucleo del 13C: molto deschermato
δCO 205-185 ppm (aldeidi) 220-190 ppm (chetoni)
3-metilbutanale
CO
3-pentanone
CO
3
16/05/2011
1H
NMR
ALDEIDI
L’effetto -I dell’O ed il campo magnetico generato dagli elettroni π
diminuiscono la densità elettronica attorno al nucleo del 1H del CHO: molto
deschermato (9,5-10,5 δ)
CHO
benzencarbaldeide
3-metilbutanale
CHO
1H
NMR
CHETONI
L’effetto -I dell’O diminuisce la densità elettronica attorno al nucleo del 1H
sul C adiacente: deschermato da 2,0-2,5 (CH3) fino a 3 (CH2, CH) δ
3-pentanone
fenil metil chetone
4
16/05/2011
ALDEIDI e CHETONI
δ+
sp2
Nu:
δ-
C O
H
E+
C
H acido
ADDIZIONE NUCLEOFILA
Reazione caratteristica
Meccanismo:
1.
Nu:-
lento
C
O
sp2 (trigonale
planare)
C
Nu
E+
Nu
O-
C
O
E
sp3
(tetraedrico)
E+ = H+
generalmente
2.
O + H+
C
C
+
O H + :Nu
sp2 (trigonale
planare)
C
lento
+
C
+
O H
O H
Nu
C
O
H
sp3
(tetraedrico)
5
16/05/2011
Sono reazioni di equilibrio
Risentono dell'effetto sterico
Risentono degli effetti elettronici dei sostituenti
Sostituenti ad attrazione elettronica RENDONO PIU' POSITIVO il C carbonilico
FAVORISCONO l'attacco del nucleofilo
REATTIVITA' CRESCENTE
CHETONI < METILCHETONI < ALDEIDI < METANALE
a. NUCLEOFILI ALL'OSSIGENO
H2O
OH
O
R
C
H +
R
H 2O
C
aldeide idrata
OH
O
R
C OH
H
R +
R
H 2O
C OH
R
chetone idrato
Meccanismo:
:O
C
O
+
H
H
+
lento
OH2
OH
C
C
O-
OH
6
16/05/2011
ROH
Meccanismo:
:O
C
R
H
+
+
lento R OH
C
O
OR
OH
O-
esempi:
H3C
H
C
H3C
+ CH3CH2OH
C
CH3
O CH2 CH3
C
H
O
H3C
EMIACETALE
C
OH
H3C
+ CH3CH2OH
C
H3C
O
O CH2 CH3
OH
La reazione può avvenire anche in modo intramolecolare:
O
O
H2C
HO CH2 CH2 CH2 C H
H
C
H2C
CH2
OH
Se la reazione con alcool si fa in ambiente ACIDO ANIDRO, prosegue:
O
C H + 2 R'OH
R
OR'
H+(*)
R C OR'
+ H2O
H
(*) HCl (gas)
O
R
C R" + 2 R'OH
ACETALE
OR'
H+(*)
R C OR' + H2O
R"
ACETALE (CHETALE)
(*) HCl (gas)
Meccanismo:
R
C
O
H
+ R' O H
OR'
R
C
H
OH
come già
descritto
7
16/05/2011
OR'
R
..
H+
OR'
C +
H
O H
H
R
C
H
R
OH
OR'
C +
H
O R'
H
C
H
..
HO
+
C OR'
R'
H
H
OR'
R
R
+
OR'
C
H2O +
R
+ H+
OR'
Con I chetoni (meno reattivi delle aldeidi) l’equilibrio è spostato verso sinistra
H3C
C
CH3
H+
H3 C
+ 2 CH3CH2OH
C
H3 C
O
O CH2 CH3
O CH2 CH3
I chetoni danno chetali con equilibrio spostato verso destra solo se il nucleofilo è un
diolo (1,2- o 1,3-): la reazione è intramolecolare e si forma un anello stabile
H3 C
C O
H3C
HO
H3C
C
H3C
C
H3C
CH2 OH
H 3C
H3C
C
O CH2
CH2
KMnO4
CH OH
CH2 OH
OH
C
H3 C
O CH2
CH2 OH
HO
H3C
O CH2
H3C
H3C
C+
H3C
CH2 OH
H
O
+ CH2
Esempio di applicazione:
CH
+
OH2
H3C
H+
O CH2
H3 C
O
C +
O CH2
H3 C
H
CH2 OH
CH2
OH
-
H3C
..
HO CH2
CH2
O CH2
O
CH2
C
+ H+
O CH2
H
O
O
C
C
come ottenere CH OH da
CH
CH2
CH2 OH
H
H
però, con KMnO4:
C
O KMnO4
HO
?
C
O
8
16/05/2011
H
O
+
C
HO CH2 H
CH +
HO CH2
CH2
O
CH2 CH CH
OH OH
O
O
CH2
CH2
CH CH
CH2
O
CH2 H2O, H+
CH2
KMnO4
OH-
O
CH2 OH
CH2 CH CH +
CH2 OH
OH OH
b. NUCLEOFILI ALLO ZOLFO
RSH
R
:S
C
H
+
lento
+
R SH
SR
C
C
O
EMITIOACETALE
OH
O-
esempi:
H3 C
H
C
+
H3 C
CH3CH2SH
H
O
H3 C
C
C
CH3
H 3C
+ CH3CH2SH
H3 C
O
S CH2 CH3
OH
C
S CH2 CH3
OH
Se la reazione con si fa in ambiente ACIDO ANIDRO, prosegue:
H+ anidro
C
C
+ 2 CH3CH2SH
S CH2 CH3
TIOACETALE
S CH2 CH3
O
Con ditioli la reazione è intramolecolare quando si possono formare anelli
stabili (5 e 6 termini).grado di
R
C O
..
HS
CH2
HS
CH2
CH2
R
R
-
O
C +
S CH2
R
H CH
2
CH2 SH
SH
R
OH
C
R
S
CH2
CH2 CH2
9
16/05/2011
H+
+
OH2
R
C
R
H
R
C
R
S
+
R
CH2
R
S
CH2
CH2
C
CH2
+ H+
S CH2
TIOACETALE (TIOCHETALE)
CH2
solo con aldeidi
-O
: S OH
C
-O
H
CH2
S CH2
Reazione di Bertagnini
+
C
C+
R
O
O
HS
R
CH2
CH2 CH2
CH2
S
NaHSO3
S
SH
O
OH O
S OH
C
S O-
H
O
H O
reazione acido-base
Serve per purificare le aldeidi: l’addotto precipita, si filtra, si torna ad aldeide
c. NUCLEOFILI ALL’AZOTO
con i nucleofili all'azoto i prodotti di addizione perdono una molecola diacqua
ammoniaca ed ammine primarie
O
R
C
+ :NH3
H
H+
NH3, RNH2
OH
R CH
NH + H2O
IMMINA
R CH
NH2
(base di Schiff)
O
R
+
C
H
..
R'NH2
H+
OH
R CH
NH R'
R CH N R' + H2O
IMMINA
(base di Schiff)
Meccanismo:
C O + H+
veloce
+
C OH
+
C OH
10
16/05/2011
..
+
C OH + H2N R
OH veloce
C +
NH2
lento
+
OH2
C
R
veloce
+
C NH R
..
NH
R
veloce
N
C
R
H+
H2O
5÷6
La velocità della reazione dipende dal pH
k
la velocità massima si ha a pH 5÷6
R
O
C
NH2 + H+
+ H+
R
+
NH3
+O H
O
C
C
+
H
pH
a pH troppo acido manca il nucleofilo
a pH non abbastanza acido, la concentrazione di composto carbonilico protonato è
troppo bassa
11
16/05/2011
R2NH
ammine secondarie
O
H2C
R'
H
OH
H+
..
R2NH
+
C
R' CH2 CH
R' CH CH NR2 + H2O
ENAMMINA
NR2
Meccanismo:
veloce
+ H+
C O
CH2
+
C OH
CH2
.. R
HN
R
+
C OH
OH
C +
NHR2
CH2
lento
+
OH2
veloce
C
H
CH
NH
R
CH CH N
R
R
H
CH2
Esempi:
H+
O
CH3 C
+
CH3
H N
H2O C
CH3
CH2 CH N
CH3
OH
+ (CH3)2NH
ammine terziarie
C
derivati dell'ammoniaca
O
H+
C
veloce
+ OH
N
CH3
CH2 CH N
H2O
CH3
CH3
NESSUNA REAZIONE
H2N-X
..
H2N OH
X = OH
C
H2O, H+
R3N
O
+
R3 N C
O
+
C
N
H
H
R3 N
CH2
CH3
O
CH3 C
+ H2 O + H+
+
C
OH
IDROSSILAMMINA
..
H2N OH
lento
H
+
H N OH
C
OH
12
16/05/2011
H OH
:N
C
+ OH
H + OH
N
OH
N
C
OSSIMA
C
H2 O
2
Esempio:
O
C
OH
H+
H
CH N OH
CH NH OH
+ H2N OH
H2O
H+
O
CH3 CH2 C CH3 + H2N OH
ossima della
benzencarbaldeide
OH
CH3 CH2 C NH OH
CH3
CH3 CH2 C N OH
CH3
H2O
ossima del
butanone
X = NH2
.. ..
H2N NH2
H+
C
O
veloce
+
C
OH
C
+ OH
H + NH
2
N
IDRAZINA
..
H2N NH2
lento
N
C
H+
H
+
H N NH2
H NH
2
:N
C
C
+ OH
OH
2
H2O
NH2
C
IDRAZONE
Esempio:
O
CH3 CH2 C H + H2N NH2
H+
OH
CH3 CH2 CH N NH2
CH3 CH2 CH NH NH2
H2O
idrazone del propanale
13
16/05/2011
Nu
X = NH-C6H5
.. ..
H2N NH
H+
+
C
C
C
veloce
O
+ OH
H NH
:N
C
+ OH
2
OH
H + NH
N
H2O
C
FENILIDRAZINA
..
H2N NH
lento
H
+
H N NH
C
OH
NH
N
H+
C
FENILIDRAZONE
Esempio:
.. ..
H2N NH
O
C
CH N NH
CH3 +
H 2O
fenilidrazone del
fenil metil chetone
H2N NH
O
CH3CH2CH2 C H +
CH3CH2 CH2 CH N NH
H 2O
fenilidrazone del
butanale
Nu
.. ..
H2N NH
NO2
2,4-DINITROFENILIDRAZINA
NO2
14
16/05/2011
NO2
.. ..
H2N NH
C
NO2
H+
NH
NO2
N
+
O
C
NO2
2,4-DINITROFENILIDRAZONE
Esempio:
O
H2N NH
NO2
CH3 C CH3 +
H+
H3 C
C
N
NH
H3 C
NO2
H2 O
NO2
NO2
2,4-difenilfenilidrazone
del del propanone
X = CONHNH2
O
..
H2N C
..
NH NH2
SEMICARBAZIDE
Nu
C
O
O
..
+ HN C
..
2
NH NH2 H O
2
O
NH C
N
NH2
C
SEMICARBAZONE
15
16/05/2011
d. NUCLEOFILI AL CARBONIO
HCN
OH
R C H
O
+ HCN
C
R
H
C N
cianidrina
OH
O
R
C
+ HCN
R'
R C R'
C N
cianidrina
meccanismo:
O
-: C
+
C
..
:O:-
lento
..
:OH
H+
C C N:
N:
C C N:
Addizione di carbanioni (composti organometallici)
RMgX
O
δ-
+
δ+ C
O - +MgX
C
δ- δ+
RMgX
OH
H2O
(H+)
R
C
+ Mg(OH)X
R
Esempi:
H
OMgBr
MgBr
O
C
H
O
H3C
OMgI
C
H
+ CH3MgI
H 3C
CH
OMgI
C
CH3
+ CH3CH2MgI
H3 C
OH
H2 O
CH3
O
H3 C
CH2 OH
H2 O
CH2
+
C CH2CH3
CH3
H 3C
CH
H 2O
CH3
OH
H3 C
C CH2CH3
CH3
16
16/05/2011
RLi
δ-
O
+
δ+ C
δ- δ+
OLi
RLi
C
R
esempio:
O
Li
OLi
CH3 CH C H +
CH3
OH
H2O
(H+)
C
R
OH
H2O
CH3 CH CH
CH3
+ Li(OH)
CH3 CH CH
CH3
REAZIONE DI WITTIG
FOSFORANO
alogenoalcano + trifenilfosfina +
base forte + composto carbonilico
ALCHENE
1. Sostituzione nucleofila
R CH2 Br +
SN 2
..
P
R CH2
+
P
Brsale di FOSFONIO
(bromuro di alchiltrifenilfosfonio)
2. Reazione acido-base
+
R CH2 P(C6H5)3
..- +
R CH P(C6H5)3
base
Nu:
BASE
R CH P(C6H5)3
FOSFORANO o ILIDE (yilide)
butillitio (CH3CH2CH2CH2Li) oppure idruro di sodio (NaH)
IL FOSFORO PUO' ESPANDERE L'OTTETTO: stabilizza il carbanione
3. Addizione nucleofila
..- +
R CH P(C6H5)3
O
+
C
R"
R'
R CH P(C6H5)3
R" C
R'
O
BETAINA
o
FOSFETANO
17
16/05/2011
R CH
R" C
+
P(C6H5)3
fosfinossido
O
R'
"driving force“
(forza trainante)
Il legame P-O è più forte del legame P-C
Esempio:
O
1. :PPh3
CH3 CH2 CH2 Br + CH3 CH
2. NaH
CH3 CH CH CH2 CH3
+
..NaH
+
CH3 CH2 CH PPh3
CH3 CH2 CH2 PPh3
H2
CH3 CH2 CH2 Br + :PPh3
O
+
..+
CH3 CH2 CH PPh3 CH3 CH
O
PPh3
CH3 CH CH CH2 CH3
CH3 CH CH CH2 CH3
O=PPh3
Esempio:
O
CH3 CH2 CH2Br +
..- +
CH3 CH2 CH PPh3 +
CH3
C
CH3
1. :PPh3
2. C4H9Li
O
O
C
CH3 C
CH3
CH3 CH2 CH C
PPh3
CH CH2 CH3
CH3 CH2 CH
CH3
C
CONDENSAZIONE ALDOLICA
2
H3C
O
C
10% NaOH in H2O
H
O
OH
H3C CH CH2 C
H
ALDOLO
18
16/05/2011
meccanismo:
genera il nucleofilo
1. reazione acido-base
O
α
H CH2 C
H
HO- +
HO H + -CH2 C
O-
O
CH2 C
H
H
2. addizione nucleofila
O
OH3C CH CH2 C
H
O
CH2 C
H
O
CH3 C
H
3. reazione acido-base
O
O+ HO H
H3C CH CH2 C
H
O
OH
CH
C
H3C CH
2
H
+ HOla sua
concentrazione
non cambia
l'aldolo, sottoposto a riscaldamento, perde una molecola di acqua
O
Δ
O
OH
H3C CH CH2 C
H
H3C CH CH C
H 2O
H
doppio legame coniugato
perché la condensazione aldolica possa avvenire, è necessario che
l'aldeide abbia almeno un H in α al carbonile
O
O
CH
per esempio:
H
CH
CH3 O
H3C C CH
non reagiscono
CH3
aldeidi con più C dell'etanale, comunque danno il carbanione
in α al carbonile
HO-
H
+
O
CH3 CH C
α
H
O
CH3CH C
H
-
OCH3 CH C
H
+ H2O
19
16/05/2011
O
δ-
-
OO
CH3 CH2 CH CH C
H
CH3
O
CH3 CH2 C δ+ + CH3 CH C
H
H
OO
+ H2O
CH3 CH2 CH CH C
H
CH3
OH
O
+ HOCH3 CH2 CH CH C
H
CH3
CONDENSAZIONE ALDOLICA MISTA
La reazione con due aldeidi, entrambe con H in α, dà una miscela di prodotti
O
CH3 C
H
HO-
+
O
CH3 CH C
H
O
CH3 CH2 C
H
HO H
O
CH2 C
H
+ H2O
O
OH
OH
O
OH
O
+ CH3 CH2 CH CH C
+ H C CH CH C
H3C CH CH2 C
3
H
H
H
CH3
CH3
OH
O
+ CH3 CH2 CH CH2 C
H
la condensazione aldolica mista (o incrociata) diventa utile quando
una sola delle aldeidi ha un H in α (precursore del nucleofilo).
O
C
O
C
H
+
O
CH3 C
H
HO-
O-
nucleofilo
O
CH CH2 C
H
O
CH2 C
H
H
O
CH2 C
H
H2O
OH
O
CH CH2 C
H
+ HO-
20
16/05/2011
H
C
OH
O
CH CH2 C
H
O
CH C
H
+ H2O
la coniugazione con l'anello
benzenico facilita la disidratazione
ATTENZIONE!
la condensazione aldolica in ambiente basico con i chetoni
non avviene
equilibrio spostato a sinistra
O
2
C
CH3
O
OH
H3C C CH2 C
CH3
CH3
CH3
i chetoni possono essere utilizzati come fonti di carbanioni in condensazioni
miste con aldeidi senza H in α
REAZIONE DI CLAISEN-SCHMIDT
O
C
O
C
+
H
CH3
O
-OH
C
CH3
CH3
CH2
nucleofilo
meccanismo:
OO
C
O
H + CH2 C
CH3
O
CH CH2 C
H
C
spontanea
OH
O
CH CH2 C
CH3
H2O
CH3
O
CH C
CH3
+ H2 O
la coniugazione con l'anello
benzenico facilita la disidratazione
esempio:
O
O
C
H
+
H
C
CH3
-OH
-O
O
CH2 CH2 C CH3
CH3
H2O
O
CH2 CH2 C CH3
OH
In questo caso, per avere disidratazione occorre SCALDARE (meglio se in ambiente acido)
O
CH2 CH2 C CH3
OH
Δ
O
CH2 CH C CH3
H+ + OH H
2
O
CH2 CH C CH3 + H2O
21
16/05/2011
REAZIONE DI CANNIZZARO
Le aldeidi SENZA H in α al carbonile possono reagire con NaOH, in condizioni
più drastiche di quelle della condensazione aldolica (concentrazione più
elevata di base, temperatura elevata)
O
2
-OH
C
H
H
Δ
O
CH3OH + C H
O
Reazione di disproporzione
(un -CHO si ossida e un altro di riduce)
meccanismo:
1. attacco nucleofilo al C carbonilico
O δC
H δ+ H
O+
-OH
Cannizzaro (1826-1910)
H C H
OH
Questo EQUILIBRIO si ha anche nel caso della condensazione aldolica, ma in
quel caso c’è un’altra strada (attraverso la base coniugata) che porta a
prodotti e quindi questo equiibrio è come se non ci fosse.
2. trasferimento di idruro (H con gli elettroni di legame)
C
H
δ-
O-
O
H
C
H
H
H C H
OH
3. reazione acido-base
OH
O
OC H
+
H C H
OH
H
O
C H
CH3OH + HCO2-
O
C H
H
OH C H
=/
δ-
O
+
OH
H
Danno la reazione di Cannizzaro tutte le aldeidi SENZA H ACIDI
Di solito gli H acidi sono in α al carbonile, ma NON NECESSARIAMENTE
esempio:
O
C
-OH
CH2OH
H
H
ma:
C
O
C
+
O-
O
-OH
?
H acido!
CH3
22
16/05/2011
H
C
H
O
H
O
C
-OH
C
H
O
C
O
H
C
O-
ecc.
- CH
CH3
CH2
2
CH2
Nucleofilo
H
C
H
C
+ H2O
CH2
condensazione aldolica
O
O
O
-
H
C
H C CH2
H2O
O
- CH2
CH3
CH3
OH
H C CH2
HC CH
C
O
CH3
H
H
C
O
H2O
CH3
23
16/05/2011
TAUTOMERIA
C
C
O
H
C
O
C
H
L'equilibrio tautomerico è catalizzato da acidi e da basi
meccanismo in ambiente basico:
O
C
H
C
+ OH
O
O
..C
C
HO
C
C
+ H2O
C
enolo
ione enolato
reazione acido-base
C
reazione acido-base
l'equilibrio è spostato verso la forma carbonilica
O
OH
C
C
CH2
H 3C
0.01%
CH3 CH3
99.99%
OH
O
C
CH3
H
99.90%
C
H
CH2
0.10%
OH
O
H
H
H
0.06%
99.4%
l'equilibrio è spostato verso la forma enolica se ci sono fattori particolari che la
stabilizzano
O
C
CH3
O
CH2
C
O
C
CH3
CH3
20%
..H
....
O
O
C
C
CH3
CH3 CH
OH
C
CH3
CH
80%
stabilizzazione per
legame idrogeno
meccanismo in ambiente acido:
O
C
H
C
+
H3O+ H O
reazione acido-base
C
H
C
H O
+C
H
C
HO
:OH2
C
C
reazione acido-base
24
16/05/2011
Un C chirale in α al carbonile in ambiente acido o basico racemizza
O
H
H
H
H
(S)
-OH
(S)
H
CH3 + H
*
oppure
O
H
H
CH3
CH3
*
O
OH
H+
CH3
*
H
(R)
RIDUZIONE
OH
R CH R
H2, cat.
OH
O
R
idruro
C
R CH R
(a) oppure (b)
R
R
idruro = NaBH4, LiAlH4
CH2 R
NH2
[H] NH3
R
(a) Zn(Hg), HCl
Clemmensen
(b) NH2NH2, OH-, Δ
Wolff-Kischner
CH R
meccanismi:
O
O
H BH3
C
BH3
..
+ H2 N
C
C
H
ADDIZIONE NUCLEOFILA
NH2
O
OH
H+
H2O
C
H
..
H2N
NH2
C N
NH2
H2O
..
NH3
O
+
..
NH3
C NH
H2O
H2
Ni
Nu: = H2N-NH2
-OH
Δ
ADDIZIONE NUCLEOFILA
C
Nu: = H-
ADDIZIONE NUCLEOFILA
NaBH4
N
N
+
CH2
Nu: = :NH3
CH
NH2
idrogenazione catalitica
25
16/05/2011
OSSIDAZIONE
O
R
C
R
H
O
R
O
[O]
C
[O]
C
tutti gli ossidanti già visti con gli
alcooli
OH
[O]
NESSUNA REAZIONE
R'
O
Saggio di Tollens O
R
C
+ Ag(NH3)2OH
C
R
H
O
+ Ag°
specchio
d'argento
gli α-idrossichetoni possono
dare un saggio positivo:
R CH C R'
OH O
[-2H]
Gottfried Tollens
R C
Ag(NH3)2OH
C R'
O
O
+ Ag°
REAZIONI IN CATENA LATERALE
α-ALOGENAZIONE
I chetoni vengono alogenati in α al carbonile. La reazione è promossa da
basi oppure acido-catalizzata (Le aldeidi vengono ossidate dagli alogeni).
promossa da basi:
O
H3C
C
CH3
O
O
+ Br2 + -OH
+ Br2 + -OH
H3C
C
CH2 Br
O
+ Br- + H2O
+ Br- + H2O
Br
la base si consuma
acido-catalizzata
O
O
H3C
C
+ Br2
CH3
H3C
C
CH2 Br
+ Br- + H+
26
16/05/2011
O
O
+ Br- + H+
+ Br2
Br
l'acido non si consuma
La velocità dell'α-alogenazione (sia in ambiente basico che in ambiente acido)
NON dipende dalla concentrazione di alogeno.
meccanismo della reazione promossa da basi:
O
H3C
C
CH3
+ -OH
O
C CH2
H3C
O-
O
lento
C CH2
H3C
H3C
C
CH2
+ H2O
O
Br Br
H3C
C
+ Br-
CH2 Br
meccanismo della reazione acido catalizzata:
O
H3C
C
+O
+
CH3 + H
H3C
C
H
CH3
+OH
H3C
C
OH
lento
CH2
H3C
+ H+
C
CH2
H
H3C
C
+ OH
:OH
OH
+
Br Br
CH2
H3C C CH2 Br
+
+ OH
H3C C CH2 Br
H3C C CH2 Br
O
H3C C CH2 Br
27
16/05/2011
Con i metilchetoni
reazione dell'aloformio
con eccesso di alogeno e NaOH in acqua
O
C
CH3
O
-OH, H O
2
+ I2
C
O-
+ CHI3
solido
giallo
iodoformio
meccanismo:
O
R
O
R
C
CH2 I
O
R
O
R
C
C
I I
C CH I
-OH
CHI2
CH2 I
Acidità maggiore di
quella del metile
CH I
I
O
O
C CI2
R
R
C
CI3
3 atomi di iodio
(elettronegativi)
O
R C CI3
CI3
C
R
O+ -OH
C
R
O
O
R
O
C CH2
R
CH3
-OH
C
O
-OH
R C
OH
+
OH
:CI3
O
R C
O-
+ CHI3
Si separa; giallo
ATTENZIONE!!! Ci possono essere “falsi positivi”
I2
ossidante
anche composti diversi dai metilchetoni possono dare lo iodoformio (falsi
positivi) se la molecola può essere ossidata dallo iodio a metilchetone
CH3 CH2 OH
CH3 CH CH3
OH
O
I2
CH3 C H
I2
CH3 C CH3
O
I2
-OH
I2
-OH
O
O C H
+ CHI3
O
H C O- + CHI3
28
16/05/2011
COMPOSTI CARBONILICO α,β-INSATURI
centro nucleofilo
O
CH2 CH CH
O-
O
+
CH2 CH CH
CH2 CH CH
+
centro elettrofilo
Con reagenti elettrofili
O
Addizione 1,2 e 1,4
+
OH
H+
CH2 CH CH
CH2
OH
CH CH
CH2 CH CH
+
Addizione 1,2
O
CH2 CH CH
CH2
OH
+
CH2 CH CH
Addizione 1,4
OH
Cl CH2 CH CH
OH
CH CH Cl
+ HCl
Con reagenti nucleofili
O
Cl CH2 CH2 CH
Addizione 1,2 e 1,4
O
HCN
CH C CH3 + C N
CH2
CH2
OH
CH C CH3
C N
O
+
CH2CH2 C CH3
C N
meccanismo:
1,4
O
CH C CH3
CH2
C N
HCN
1,2
CH2
CH2 CH C CH3
C N
O
CH2CH2 C CH3
C N
OH
+
C N
CH2 CH C CH3
C N
O
CH C CH3
C N
-O
O
CH2 CH C CH3
C N
OCH2 CH C CH3
C N
HCN
OH
CH2 CH C CH3 + -CN
C N
con nucleofili basi più forti è preferita l'addizione 1,2 (al C=O)
29
16/05/2011
O
CH2 CH C CH3 + CH3MgI
OH
OMgBr
CH2 CH C CH3
CH3
CH2 CH C CH3
CH3
con nucleofili basi più deboli è preferita l'addizione 1,4
O
N C CH2 CH2 C CH3
O
CH2 CH C CH3 + HCN
RIDUZIONE di composti carbonilici contenenti C=C
il prodotto dipende dal catalizzatore e dalle
condizioni di reazione
O
si riduce C=C
CH CH CH CH CH
H , Ni
CH3 CH
O
CH CH2 CH
2
25°C
H2, Ni, Δ
3
2
2
2
OH
CH3 CH2 CH2 CH2 CH2
si riducono entrambi
1. NaBH4
2. H2O
OH
CH3CH CH CH2 CH2
si riduce C=O
30
ACIDI CARBOSSILCI e DERIVATI
O
Caratteristica strutturale di acidi carbossilici e
derivati
C
Y
Caratteristiche Spettrali
Spettrometria di massa
Acidi carbossilici
Lo ione molecolare di solito è spesso debole.
La “rottura α” (α-cleavage), lascia il carbocatione acilio (stabilizzato
per risonanza):
+.
HO
R
.
HO +
C O
R
.
+
+
C
O
R
C
+
O
R+
Può essere perso il gruppo –CO2H
R CO2H +.
R
CO2H +
m/z 45
Anche con gli altri gruppi funzionali è favorita la scissione dello ione molecolare
che porta al carbocatione acilio
Le ammidi non sostituite sull’N hanno un picco caratteristico a m/z 44
+.
O
R C NH2
.
R +
+
O
C NH2
m/z 44
acido butanoico,
PM 88
CH3CO+
acetato di etile,
PM 88
M+
M+
1
+
propanammide,
PM 73
O
cloruro di butanoile,
PM 106
C NH2
m/z 44
M+ - Cl
M+
+
C
O
Analogo a quello di aldeidi e chetoni
UV-visibile
IR
stretching O-H
anidride
benzencarbossilica,
PM 226
ACIDI CARBOSSILICI
banda molto larga a 2400 cm-1, spesso sovrapposta
alle bande CH
O
HO
R C
C R
OH
O
stretching C=O a circa 1730-1700 cm-1, spostato verso valori più
bassi dalla coniugazione
stretching C-O
tra 1320 e 1210 cm-1
acido butanoico
2
IR
ESTERI
stretching C=O
a circa 1735 cm-1, spostato verso valori più bassi
dalla coniugazione con R. La coniugazione con OR’
sposta l’assorbimento verso valori più alti.
stretching C-O
due o più bande tra 1300 e 1000 cm-1
acetato di etile
IR
AMMIDI
stretching C=O
a circa 1670-1640 cm-1.
stretching N-H
3500-3100 cm-1 (CONH2 hanno due bande)
propanammide
O
H C
N CH3
H3C
3
IR
CLORURI ACILICI
stretching C=O
a circa 1800 cm-1, spostato verso valori più bassi dalla
coniugazione.
cloruro di butanoile
cloruro di benzoile
IR
stretching C=O
stretching C-O
ANIDRIDI
sempre due bande tra 1830 a 1800 cm-1 e tra 1775 e 1740,
spostato verso valori più bassi dalla coniugazione con R.
tra 1300 e 900 cm-1
anidride propanoica
4
IR
stretching
NITRILI
C N a circa 2250 cm-1, spostato verso valori più bassi dalla
coniugazione con R.
propanonitrile
benzenecarbonitrile
13C
NMR
L’effetto -I dell’O ed il campo magnetico generato
dagli elettroni π diminuiscono la densità elettronica
attorno al nucleo del 13C: molto deschermato
δCOX 180-160 ppm
CO
acetato di etile
acido butanoico
CO
COCO
anidride propanoica
CO
propanammide
5
1H
NMR
L’effetto -I dell’O diminuisce la densità elettronica attorno al nucleo del 1H
sul C adiacente: deschermato da 2,0-2,5 (CH3) fino a 3 (CH2, CH) δ
OH (negli acidi arbossilici) fortemente deschermato: 10-15 δ;
protone mobile, scambia con D2O
NH (nelle ammidi) deschermato e molto allargato: 6-7 δ; protone
mobile, scambia con D2O
acido butanoico
acido
benzencarbossilico
OH
OH
acetato di etile
propanammide
NH
N-metiletanammide
anidride benzoica
NH
6
ACIDI CARBOSSILCI e DERIVATI
H acido
H
O+ C O
δ
sp2
H
δ
H+
C
Nu:
H acido
Caratteristiche:
Acidità del legame O-H
Il C del carbossile subisce attacco da parte dei nucleofili
I protoni in α al carbossile sono relativamente acidi e
possono essere sostituiti
gruppo uscente
Y
C O
H
H acido
♦ sull'O del carbonile (o sull'N del
nitrile) si ha protonazione
δ-
δ+
sp2
Caratteristiche:
H+
C
♦ I protoni in α al carbonile sono
relativamente acidi e possono
essere strappati con basi forti
♦ Qualche reazione caratteristica
delle ammidi avviene sull'N
Nu:
Reazione caratteristica
δ+
sp
C N
H
SOSTITUZIONE NUCLEOFILA
δ-
(la sostituzione nucleofila può avvenire con
meccanismo base- o acido-catalizzato)
H acido
H+
C
Nu:
7
Meccanismo:
+
H Nu:
lento
R
C
Nu H
R
O
C
O
Nu
-
sp2
(trigonale planare)
C
O
R
Y
Y
sp2
(trigonale planare)
sp3
(tetraedrico)
SOSTITUZIONE NUCLEOFILA AL CARBONIO ACILICO
Principali nucleofili:
:NuH = H2O, ROH, NH3, RNH2, R2NH, RSH, RCO2H
Nu:- = RCO2-, R-, H-
O
<
C
R
O
R
NH2
<
C
OR'
O
O
C
C
R
O
O
<
<
C
R
R
O
C
R
SR'
Cl
reattività crescente
Questo ordine di reattività è il risultto di più fattori
meno basico: miglior gruppo uscente
Basicità del gruppo uscente
Basicità:
-NH2 < -OR < -O-C(O)R < Cl-
capacità come gruppo uscente
Risonanza
O-
O
C
R
R
C
Cl
R
Cl+
O
O-
R C
C
C +
R
SR'
R C
SR'
R C
O
C
OR'
R
O+
O+
R C
C
R
O-
O-
O
+
OR'
C
O
O-
C
O
R
R C
O
O
R
O-
O
C
NH2
R
+
NH2
8
il contributo della struttura di risonanza a separazione di carica diminuisce
la bontà del gruppo uscente ed è tanto maggiore quanto migliore è la
sovrapposizione degli orbitali (cioè con gli elementi della stessa riga).
Effetto induttivo
Maggiore è l’effetto ad attrazione elettronica, più positivo è il C carbonilico
più facile è l’attacco del nucleofilo
O
R C
Cl
REATTIVITA‘
CRESCENTE
O
R C
O
R C
H2O
O
O
O
R C
OH
H+
oppure
OH-
R C
OR'
O
R C
NH2
R C
N
O
R C
REATTIVITA‘
CRESCENTE
Cl
O
R C
O
R"OH
O
R C
OR"
R C
O
O
R C
OR'
O
R C
REATTIVITA‘
CRESCENTE
Cl
O
R C
O
R C
O
NH3
O
R C
NH2
O
R C
OR'
9
1.
Nu = H2O
REAZIONE CON H2O
a) Alogenuri acilici
lento
O
R C
+ :OH2
Cl
O-
O
+
R C OH2
R C
OH
HCl
Cl
Sostituenti ad attrazione elettronica su R FACILITANO l'attacco del nucleofilo
b) Esteri
Gli esteri sono meno reattivi degli alogenuri acilici: l’acqua da sola non reagisce)
-OH,
Nu = -OH
In ambiente basico
-:OH
+
OR'
R C
O
lento
O
H2O oppure H2O, H+
R
O
O
+ -OR'
R C
C OH
R C
OH
reazione acido-base
OR'
O
- + R'OH
L'ultimo passaggio rende IRREVERSIBILE l'intero processo
Sostituenti ad attrazione elettronica su R FACILITANO l'attacco del nucleofilo
In ambiente acido
O
Nu = H2O
H+
+O H
R C
R C
OR' H O
2
OH
OH
R C
OR'
+ OH
2
R C
OH
O H
R C
+
OR'
OH
lento
R C
OR'
:OH2
OR'
+OH2
+
H
OR'
+
R C +
OH
Processo REVERSIBILE
O
OH
OH
R C
+ R'OH
R C
OH
H+
OH
Questo significa che in ambiente acido si può
idrolizzare un estere ad acido carbossilico
L'idrolisi acida degli esteri è praticamente insensibile agli effetti elettronici
dei sostituenti; è invece sensibile agli effetti sterici
ATTENZIONE
Questa insensibilità agli effetti elettronici (induttivo
e coniugativo) dei sostituenti è una caratteristica
SOLO dell’IDROLISI ACIDA degli ESTERI
è il risultato di effetti opposti, praticamente uguali
10
Lo stadio lento è l’attacco del nucleofilo sull’estere protonato:
dipende quindi dalla sua concentrazione, che, a sua volta, dipende
dalla basicità dell’estere
K=
[RC(O)OHR'+]
[RCO2R'][H+]
[RC(O)OHR'+] = K [RCO2R'][H+]
v = k[RC(O)OHR'+] = kK [RCO2R'][H+]
La reattività dipende dalla costante di velocità dello stadio lento (k) E
dalla costante di equilibrio acido-base preliminare (K)
k
l'attacco del nucleofilo è favorito dai sostituenti ad attrazione elettronica
K
la protonazione di una base è sfavorita dai sostituenti ad attrazione elettronica
Solo nel caso dell'idrolisi acida degli esteri gli effetti hanno valori molto simili,
cioè la sensibilità della protonazione e dell'attacco nucleofilo all'effetto dei
sostituenti è praticamente la stessa.
Essendo di segno opposto, gli effetti si annullano.
c) Ammidi
Le ammidi sono ancora meno reattive: l’acqua da sola non reagisce)
-OH,
Nu = -OH
In ambiente basico
O
R C
+ -:OH
O-
lento
R
C OH
NH2
Ci sono solo CATTIVI gruppi uscenti, -OH e
-NH2, ma -OH è più ACIDO di –NH2
C OH
R
NH2
O-
H2O oppure H2O, H+
NH2
O-
-:OH
R
C O
NH2
-
O
R C
-
+ -NH2
H2O
O
R C
O
reazione acido-base
O-
+ NH3
L'ultimo passaggio rende IRREVERSIBILE l'intero processo
Sostituenti ad attrazione elettronica su R FACILITANO l'attacco del nucleofilo
11
In ambiente acido
H+
O
Nu = H2O
+O H
R C
R C
NH2 H O
2
OH
OH
R C NH2
+ OH
R C
O H
NH2
R C
+
O
R C
OH
:OH2
R C NH2
+ OH
2
+ OH
R C +
OH
OH
2
NH2
OH
H
NH2
+
OH
lento
+ NH3
R C
OH
+ NH4+
L'ultimo passaggio rende IRREVERSIBILE l'intero processo
Sostituenti ad attrazione elettronica su R FACILITANO l'attacco del nucleofilo
d) Anidridi
Nu = H2O
Le anidridi sono più reattive di esteri ed ammidi: basta l’acqua da sola
O-
O
C
R
O
R
R
+ :OH2
C
-
O
+
OH2
R
C
O
R
C
C
R
C
O
O
O
O-
O
+
R
OH
O H
+
C OH2 + R
OH
2
C
O
R
C
O
L'ultimo passaggio rende IRREVERSIBILE l'intero processo
Sostituenti ad attrazione elettronica su R FACILITANO l'attacco del nucleofilo
e) Nitrili
I nitrili sono poco reattivi (meno delle ammidi): l’acqua da sola non reagisce)
-OH,
H2O oppure H2O, H+
12
In ambiente acido
OH
tautomeria
+ R C N H
H+
O
:OH2
+
OH2
lento
+
R C N H
R C N H
+
R C N H
+
R C N: + H
H+
Nu = H2O
R C
NH2 H O
2
OH
+O H
R C NH2
+ OH
R C
R C
+
O
R C
OH
OH
R C NH2
+ OH
NH2
:OH2
+ OH
OH
H
NH2
+
R C +
OH
OH
2
PROSEGUE con l'idrolisi
acida delle ammidi
O H
R C NH2
OH
H+
O
R C NH2
2
+ NH3
R C
OH
+ NH4+
L'ultimo passaggio rende IRREVERSIBILE l'intero processo
Sostituenti ad attrazione elettronica su R FACILITANO l'attacco del nucleofilo
2.
REAZIONE CON ALCOOLI
a) Acidi carbossilici
In ambiente acido
+O H
O
+ H+
R C
OH
OH
R C
+ OH
O H
R C+
OH
R C
OH
OR'
H2O +
Nu = R’OH
OH
R C OR'
+
lento
+
: O R'
H
OH
R C
OH
H
O
R C OR'
H
OR'
+
+
H+ + R C
O
OR'
2
La reazione intramolecolare è facile, se si formano anelli stabili (5 e 6 termini)
R
HO:
O
R CH CH2 CH2 C
γ
OH
Δ
H+
CH
H2O + CH2 O
CH2 C
γ-lattone
O
13
HO:
O
R CH CH2 CH2 CH2 C
δ
OH
Δ
R
CH2 CH
H2O + CH2
O
CH2 C
δ-lattone
O
H+
se l'OH è più vicino al carbossile, ci sono altri percorsi di reazione, più facili della
chiusura di un anello teso
R
Δ
O
R CH CH2 C
+
OH H
β
HO
CH O
CH2 C
O
O
R CH CH C
OH β-eliminazione
α
OH
R CH C
O
OH
OH
HO
C CH R
O
R
+
Δ H
O
CH C
O
CH
R
O
lattide
O
C
+ 2 H2O
se l'OH è più lontano dal carbossile, il prodotto dipende dalle condizioni di reazione
in soluzione molto diluita:
OH
OH
CH2CH2 CH2 CH2 CH2 CH2 C
O
Δ
O
O
H+
in soluzione concentrata:
OH
OH
n CH2 CH2 CH2 CH2 CH2 CH2 C
O
Δ
H+
O
(
O CH2 CH2 CH2 CH2 CH2 CH2 C
)
n
+ n H2O
b) Alogenuri acilici
-
O
R C
Cl
..
+ HOR'
lento
O +
R C OR'
Cl
H
O
R C
HCl
OR'
E’ opportuno effettuare la reazione in presenza di una base (per HCl)
14
es.:
O
O
C
C
N
Cl + CH3CH2CH2OH
OCH2CH2CH3 +
N + ClH
c) Anidridi
-
O
R
C
R
C
O
..
O + HOR'
C
R
O
R
R
C
O H
+
O
O
O
C
R
O
+
OR'
R
+
+
O
C
R
OR'
Nu = -OR’’
+ -:OR"
R
H+
OR' R"OH
O
C OR"
+ -OR'
R C
OR"
OR'
In ambiente acido
R C
O-
lento
OR'
H
R C OR'
+
OR"
C
TRANSESTERIFICAZIONE
In ambiente basico
R C
O
OH
R
O
d) Esteri
O
-
C
H
OH
-
C OR'
R
C
O
O
O
H
OR'
+
Nu = R’’OH
+O H
R C
OR'
OH
R C +
OR"
O H
R C
+
OH
lento
OR'
: O R"
H
R C OR'
+ OR"
+ OH
+ R'OH
R C
OR"
H
O
R C
OR"
H+
15
e) Ammidi
3.
Essendo meno reattive, richiedono condizioni di reazione
troppo energiche per essere utili
REAZIONE CON AMMONIACA ED AMMINE
a) Acidi carbossilici
con NH3
O
O
R C
R C
+ :NH3
OH
O-
NH4+
reazione acido-base
Scaldando il carbossilato di ammonio solido:
O
R C
Δ
NH4+ (solido)
O-
NH2
Meccanismo:
R C
O-
O
O
O- NH4
R C
+
O
R C
OH
+ :NH3
R
+ H2 O
O-
+
C NH3
C NH2
R
OH
O
R C
NH2
OH2
+
+ H2O
con ammine primarie
O
OH
O
R C
O-
O
+ RNH2
R C
RNH3+
R C
Δ
(solido)
O-
RNH3+
O
+ H2 O
R C
NH R
Meccanismo:
-
O
R C
RNH3+
O-
-
O
O
R C
OH
+ RNH2
C NH R
OH
+ 2
+
NH2 R
OH
O
R
R C
O
R C
NH R
+ H2O
16
con ammine secondarie
O
R C
OH
O
R C
O-
O
+ :NHR2
R C
Δ
R2NH2+ (solido)
R2NH2+
O-
O
R C
NR2
+ H2 O
Meccanismo:
O
R C
O-
O
R
O-
O
R2NH2+
R C
+ :NHR2
OH
R
+
C NHR2
OH
O
C NR2
R C
+ H2 O
NR2
OH2
+
Se si ha un amminoacido con il gruppo amminico in γ o δ al carbossile, la
reazione è intramolecolare (si formano anelli a 5 o a 6 termini)
R
NH2
R
Δ
OH
CH CH2 CH2 C
γ
CH
CH2
H+
O
+ H2O
NH
γ-lattame
CH2 C
O
NH2
R CH CH2 CH2 CH2 C
δ
OH
Δ
O
H+
R
CH2 CH
CH2
NH
+ H2O
CH2 C
δ-lattame
O
se l‘NH2 è più vicino al carbossile, ci sono altri percorsi di reazione, più facili della
chiusura di un anello teso
R
CH NH
β
OH
H+
O
Δ
R CH CH2 C
NH2
CH2 C
O
OH
+ NH3
R CH CH C
β-eliminazione
O
17
b) Alogenuri acilici
Meccanismo:
R C
O
-
+
R C NH3
+ :NH3
Cl
R C
O
lento
O
O
+ 2 :NH3
O
R C
NH2
O
+ 2 R'NH2
O
+ NH4+Cl+ R'NH3+Cl-
R C
NH R'
Cl
R C
NH2
HCl
Cl
Cl
R C
O
R C
O
+ 2 R'R"NH
+ R'R"NH2+Cl-
R C
N R'
R"
Cl
c) Anidridi
O-
O
C
R
R
R
+ :NH
3
O
O
+
NH3
C
R
O
R
C
R
C
R
C
-
+
NH3 + R
C
NH2
O H
+
O
O
O
O
C
-
O
O
R
C
C
OH
NH2 + R
O
O
R
O
O
R
R
O
+ NH3
NH2
O
C
R
OH
+
O
O +NH
4
NH3 R
O
18
R
R
O
O
O
O
R
+ R'R"NH
O
R
R
+ R'NH2
N
O
R' R O
+ O
R"
N R' R
+
R
O
O
R'NH2
-+
R
O
R'NH3
O
-
R
O R'R''NH
O
O
O
+
R'R''NH2
Le anidridi cicliche danno immidi
O
O
O
..
O + 2 NH3
NH
.. 2
+ NH
3
OH
NH2
O+
NH4
O
anidride ftalica
O
O
O
Δ
NH
H2O
ftalimmide
O
O
:B-
O
NH
base debole, H acido
N
BH
O
-
O
d) Esteri
lento
O
R C
OR'
+ :NH3
-
O-
+
R C NH3
O
R
C
O
NH2
R C
+ R'OH
NH2
+ OR'
H
OR'
Gli esteri sono meno reattivi di alogenuri acilici e anidridi e perciòbisogna scaldare
O
+ H2N R"
R C
Δ
O
R C
NH R"
OR'
O
R C
+
OR'
HN R"
R'''
+ R'OH
Δ
O
+ R'OH
R C
N R"
R'''
19
4.
REAZIONE CON ACIDI CARBOSSILICI E CARBOSSILATI
Con questi nucleofili possono reagire solo I derivati acilici più reattivi
-
a) Alogenuri acilici
O
O
R C
R
O
O-
+ R C
Cl
R
O
C Cl
R C
O
C
R C
O
Cl-
O
O
OO
O
Cl
+O H
C
R
O
+ R C
R C
OH
O
R
R C
C Cl
O
R C
O
HCl
neutralizzato con una base
man mano che si forma
O
es:
O
CH3 CH2 CH2 C
Cl
O
+ CH CH CH C
3
2
2
O
C
N
CH3 CH2 CH2
OH
N
H
C
O
CH2 CH2 CH3
+ Cl-
b) Acidi carbossilici
(SOLO con acidi bicarbossilici in grado di formare
O
anelli stabili) O
C
C
Δ
CH2
OH
CH2
O
OH
CH2
CH2
C
C
H2 O
O
O
2.
REAZIONE CON TIOLI
a) Alogenuri acilici
O-
O
R C Cl
R' +SH
+ R C
R' SH
Cl
O
R C
HCl
SR'
es:
O
O
CH3 CH2 C
+ CH3 CH2 SH
N
CH3 CH2 C
Cl
N+
H
S CH2 CH3
Cl-
20
6. REAZIONE CON COMPOSTI ORGANOMETALLICI
con reattivi di Grignard e litioorganici
a) Esteri
O
δ - δ+
R' C OR" + R MgX
O- +MgX
O
R' C
R' C OR"
R
+
R"OMgX
R
da qui prosegue la reazione dei chetoni
O
R' C
δ- δ+
R + R MgX
O +MgX H O, H+
2
R' C R
OH
R' C R
R
R
i chetoni sono più reattivi degli esteri e perciò si arriva all'alcool terziario
es.:
MgBr
O
CH3 CH2 O C CH3 +
2
CH3 C
O- +Li
O
δ- δ +
+
R
Li
R' C OR"
OMgBr
R' C OR"
H2O, H+
CH3 C
OH
O
R' C
R + R"OLi
R
da qui prosegue la reazione dei chetoni
O
R' C
δ- δ+
R + R Li
O +Li
R' C R
H2O, H+
OH
R' C R
R
b) Cloruri acilici
O
R' C Cl
δ- δ+
+ R MgX
R
O- +MgX
R' C Cl
O
R' C
R + MgXCl
R
con accorgimenti sperimentali, è possibile fermare la reazione alla
formazione del chetone, altrimenti, si arriva ad alcool terziario
O
R' C
δ- δ +
R + R MgX
O +MgX H O, H+
2
R' C R
R
OH
R' C R
R
21
O
O
δ - δ+
+ R Li
R' C Cl
+Li
O
R' C Cl
R' C
+ LiCl
R
R
O
O +Li
δ- δ+
R + R Li
R' C
H2O, H+
R' C R
OH
R' C R
R
R
con cadmioorganici e cuprati organici
Per fermare la reazione a chetone si devono usare:
composti organometallici meno reattivi
solo con i cloruri acilici
2 RMgX + CdCl2
R2Cd + MgCl2 + MgX2
(R = CH3, alchile primario, fenile)
esempio:
MgBr CdCl
2
Cd
-
O
O
+ CH3 CH2 C
Cd
O
CH3 CH2 C
Cl
Cl
δ- δ+
+ R CuLi
2
R' C Cl
O
CH3 CH2 C
O-
O
R' C
R' C Cl
R
R
es.:
O
O
C
C
Cl
CH3
+ (CH3)2CuLi
O
O
C
H
C C
CH3 CH
Cl
(CH3CH2)2CuLi
CH3
-78°C
H
C
C
CH3 CH
CH2
CH2
CH3
CH3
CH2 CH3
C
CH3
(feromone delle formiche)
92%
22
RIDUZIONI
a) Con idruro
LiAlH4
O
R C
R CH2 OH
OH
R C
O LiAlH4
Cl
R CH2 OH
LiAlH4
O
R CH2 OH
R C
OR'
LiAlH4
O
R CH2 NH2
R C
NH2
R C
LiAlH4
N
R CH2 NH2
Meccanismo:
+
AlH3 (Li )
+ H
H
O
O
-
O
R C
H2
+ R C
(Li+)
-
AlH3
O
-
Y = Cl, OR', OAlH3
O
O
R C
-:H
+
O
R C H
R C H
Y
+ Y-
Y
O
R C H
-
+
O-
-:H
R C H
H2 O
H
Y = NH2
O
R C
+
Y
-:H
OR C H
NH2
OH
R C H
H
OH
R C H
- NH
R C H + OH
NH
23
H
-:H
R C H +
R C H
H
N
+
R C H
- NH
NH
R C
H
H2 O
-:H
R C
-
N
NH2
H
-:H
+
R C
N
H
2 - 2 H2 O
R C
NH2
H
H
Diminuendo la reattività dell'idruro è possibile fermare ad aldeide la riduzione
dei CLORURI ACILICI
-
O
+ [(CH3)3CO]3AlH
R C
O
Li+
R
Cl
es.:
-
O
CH3 CH2 CH2 C
+ [(CH3)3CO]3AlH
C
H
O
Li+
CH3 CH2 CH2 C H
Cl
b) Con idrogeno e catalizzatore
H2, Pt
N
R C
R CH2 NH2
Con catalizzatore disattivato è possibile fermare ad aldeide la riduzione
degli ALOGENURI ACILICI
Pd, BaSO4
Pd, chinolina
N
O
R C
Cl
es.:
O
+ H2
R
C
H
O
O
C
Pd
C
Cl
H
+ H2
N
24
REAZIONI SPECIFICHE DELLE SINGOLE CLASSI DI GRUPPI FUNZIONALI
1.
Acidi carbossilici
(a) trasformazione del gruppo OH in buon gruppo uscente
OH
cattivo gruppo uscente
O
Per trasformarlo in buon gruppo uscente si possono utilizzare i
reagenti che trasformano -OH in -Cl
R C
Con agenti alogenanti
OH
R C
O
+ HCl + SO2
R C
O
Cl
OH
+ PCl5
R C
Cl
+ SOCl2
+ HCl + POCl3
R C
O
O
Cl
OH
+ PCl3
3R C
O
O
O
O
C
OH
+
CH3
Cl S Cl
esempi:
O
C
O
CH3 C Cl + HCl + SO2
O
C
OH + PCl
5
+ P(OH)3
3 R C
Cl + HCl + POCl
3
O
CH3 CH2 CH3 C Cl
O
CH3 CH2 CH3 C OH + PCl3
+ P(OH)3
Meccanismo con SOCl2:
O
O
CH3 C O S Cl
O
O
CH3 C OH + Cl S Cl
O
O
S Cl
C
O
CH3
+
CH3 C O
+
CH3 C O
+ HCl
-
+
O
O S Cl
O
CH3 C Cl + O S O
Con diazometano: CH2N2
O
R C
+
OH
-:
CH2
+
N
+
CH2 N
.. N:
-:
O
N
reazione acido-base
R C
Nu
O
+
CH3
sp3
+
N
+
CH2 N
N
N
ottimo gruppo uscente
25
O
N2
Con diazometano si formano esteri metilici
+ R C
O CH3
Con dicicloesilcarbodiimmide, DCC
O
+
R C
O H
N
O
C
C
R
C N
..
H2N R'
H
N
O
N
N
H
-
O
R
C
C
N
O
C
R' NH2
+
N
N
Nu
Addizione nucleofila
buon gruppo uscente
H
H
O
R
+
NH2 R' + O
C
O
N
C
R
N
C NH R' + HO
C
N
N
reazione acido-base
H
N
La DCC permette di ottenere ammidi dagli
acidi carbossilici in condizioni molto blande
O C
NH
dicicloesilurea
b) reazione in posizione α
OH
PBr3, Br2
R CH2 C
oppure
P + Br2
O
OH
Reazione HVZ
(Hell-Volhard-Zelinski)
R CH C
Br
O
α-alogenazione
Meccanismo:
..
OH
O
Br
Br
PBr3
R CH2 C
R CH C
R CH2 C
O
OH
gli alogenuri acilici enolizzano più facilmente degli acidi carbossilici
Il C=C addiziona Br2
26
OH
R CH C
Br
+
OH
R CH C +
Br
Br-
Br
Br
OH
H+
R CH C
O
R CH C
Br
Br
Br
Br
Br
Quando si lavora la reazione (aggiunta di acqua), il bromuro acilico è più reattivo di
quello alchilico. Oppure si possono aggiungere nucleofili
O
R CH C
NH3
O
2.
R CH C
H+
Br
O
O
ROH, H+
R CH C
Br
Br
Br
OH
H2O
R CH C
NH2
Br
OR
Esteri
Con alcossido (condensazione di Claisen)
CH3CH2O-Na+
O
2 CH3 C
OCH2 CH3
O
O
CH3 C CH2 C O CH2 CH3 + CH3CH2OH
L’alcossido DEVE essere identico al gruppo -OR dell’estere
Meccanismo:
(Na+)
O
CH3 C
+ CH3CH2O-Na+
O CH2 CH3
E
reazione acido-base
O
-
CH2 C
+ CH3CH2OH
O CH2 CH3
(equilibrio spostato a sinistra)
Nu
OO
CH3 C
O
-
CH3 C O CH2 CH3
O
O CH2 CH3
H2C C
(equilibrio spostato a sinistra)
O CH2 CH3
:
+ CH
2
C
O CH2 CH3
27
O
O
CH3
O
C
O CH2 CH3
CH2
+ CH3CH2O-
C
CH3
C
O
CH
-
C
O CH2 CH3
+ CH3CH2OH
(equilibrio spostato a destra)
H più acidi di quelli dell’estere di partenza
CH3
O
O
C
C
CH
(Na+)
O
O CH2 CH3
H2O
OH-
CH3
C
O
C
CH2 O CH2 CH3
E' possibile la condensazione incrociata, quando un solo estere ha H in α:
O
O
O
O CH CH
C
2
3
O
1. CH CH O-Na
C
3 2
CH2C O CH2CH3
+ CH3 C O CH2CH3 2. H+
qualsiasi carbanione è in grado di dare condensazione
O
NaNH2
H3C C C(CH3)3
O
(Na+) H2C C C(CH3)3
NH3
O
O
O
O
CH3 C CH
C(CH3)3
C
CH
+
CH
CH
CH
C
O
CH
C
2C
3
2
2
3
3
CH3
CH3
CH3CH2O-
3.
Ammidi
Con anidride fosforica, P2O5
O
R
C
R C N + H2O
NH2
Con con Br2 + NaOH (Trasposizione di Hofmann)
O
R C NH2 + Br2 + 4 NaOH
R-NH2 + 2 NaBr + Na2CO3 + 2 H2O
(oppure NaOBr, OH-)
meccanismo
O
R C NH2 +
O
R C
NH
-OH
O
-
R C NH + H2O
O
+ Br Br
R C NH Br +
N-bromoammide
Br-
28
O
+
R C NH Br
O
-OH
O
O
..
R C N
..
nitrene
..
N
..
C
H2O
+
Br-
R N C O
isocianato
OH H O
2
R N C O
R N C O + -OH
OH
- OH
R NH C O
OH
OH
R NH C O +
+
O
..R C N
.. Br
R
-
R C N Br
-OH
R NH C O
OH
R NH C O + H2O
O
O
+
R NH2 C O
O
OH
R NH C O
O
OH
O C O
O
R NH2 +
DECARBOSSILAZIONE
a) da carbossilati di metalli pesanti (reazione di Hunsdiecker)
O
O
R
C
+ Ag2O
C
OH
R
- +
O Ag
O
O
C
R
+ HgO
R
-)
O 2
Hg2+
O
O
C
C
R
OH
+ Pb(OCOCH3)4
R
OH
R
O
C
C
O )
Pb4+
4
Δ
+ Br2
O Ag+
(o I2)
R Br + CO2
(R I )
29
meccanismo:
O
R C O
R
inizio
O
R C O Br
acil ipobromito
Br Br
:O:
O .. ..
C O
.. :
... Br
.. .
.. .
+ : Br
R C O
..
..
radicale carbossilato
:O:
.. .
R C O
..
propagazione
.
R
O
+
O
Br O C
R Br + R
R
b) da β-chetoacidi
C
R
O
C
CH2
Δ
R
O
R
C
O
O
O
C
CH2
C
H
R
O
O
R
C
H
O
+
CH2
C
O
O
R
CH2
in ambiente basico:
+ CO2
CH3
#
H
H
..
.
O
..
C
O
OH
R
R
Δ
O
O
O
.
R + O C O
O
C
C
CH3
O
O
CH2
C
O-
R
O
R
-
C
C
CH2
+ CO2
CH2
30
b) da acidi β-dicarbossilici
O
HO
H
Δ
O
C
C
O
CH2
HO
O
HO
C
C
#
O
O
H
O
C
CH2
+ CO2
CH3
HO
OH
H
O
HO
O
O
O
H
HO
O
C
+
C
CH2
O
O
C
HO
CH2
CH3
la decarbossilazione è favorita da sostituenti ad attrazione elettronica
SINTESI "ACETOACETICA"
O
OCH2CH3
C
+ CH3CH2OCH2
Na+
C
O
R"
Sfrutta l'acidità degli H in mezzo a due CO e la facile
decarbossilazione dei β-chetoacidi
O
C
C
-:C
O
OCH2CH3
R
R"
OCH2CH3
R
H2O, OHidrolisi
basica
R"
R"
R1 X
R"
O
OC CH C
O
R
H+
O
OH
R" C CH C
O
R
(K+)
OCH2CH3
1. H+
C
O R1 OH2O, OH2. Δ
R1 C R
R" C C C
R"
idrolisi
C
O
R
CO2
basica
O
R"
O
ecc.
neutralizzazione
(acido-base)
C
O
sostituzione
nucleofila
al C saturo
OCH2CH3
C
C
R"
(Na+)
C
HC
C
O
C
sostituzione nucleofila
al C saturo
O
c'è ancora un H acido
(meno di prima )
O
-O
C
O
R X
(CH3)3C O - K+
OCH2CH3
-:CH
Δ
decarbossilazione
O
C CH R
R1
O
R" C CH2 R
31
Scegliendo opportunamente il reagente R-X, si possono ottenere chetoni
con un altro gruppo funzionale
R-X = α-alogenoesteri
OCH2CH3
C
:CH
+
C
H3C
O
O
C
O
CH CH C O CH2 CH3
R CH C O CH2 CH3
Br
C
H3C
OH
Cβ β
O
O
α CH CH C OH
α
γ
2. H3O+
C R
H3C
O
1. NaOH, H2O
OCH2CH3
O
O
R
O
O
CH2 CH C OH
R
C
H
C
CO2
3
O
Δ
CH3 C CH2
O
O
CH C OH
R
γ-chetoacido
R-X = α-alogenochetoni
OCH2CH3
C
:CH
C
O
H3C
OCH2CH3
O
1. NaOH, H2O
CH CH2 C R
2. H3O+
C
H3C
O
O
O
O
C
O
Br CH2 C R
OH
C
O
α CH CH2 C R
βC
H3C
O
Δ
CO2
O
CH3 C CH2 CH2 C R
O
γ-dichetone
R-X = alogenuri acilici
Non si può usare come base CH3CH2ONa in CH3CH2OH, perché l'alogenuro acilico
è molto reattivo (e reagisce con l’etossido ed anche con l'etanolo)
32
OCH2CH3
OCH2CH3
O
C
C
NaH
CH2
:CH
+
C
C
H
O
H3C
2 H3C
O
O
Δ
1. NaOH, H2O
CH3 C CH2
2. H3O+
CO2
OCH2CH3
O
O
C
O
CH C R
R C
Cl
C
H3C
O
C R
O
O
β-dichetone
Con una base molto forte (ammide di sodio o di potassio) si possono
strappare due protoni.
Il dianione può essere fatto reagire con un equivalente di alogenuro alchilico
OCH2CH3
C
- : CH
2 K+
C R
O
CH
.. 2
O
OCH2CH3
C
KNH2
CH2
C
NH3
O
CH3
O
X
OCH2CH3
O
OCH2CH3
O
C
C
NH4+Cl
- : CH
CH2
C
C
O
O
CH2 R
CH2 R
per la neutralizzazione conviene usare l'acido coniugato del solvente
il β-chetoestere con catena allungata può essere utilizzato in tutte le reazioni viste in precedenza
SINTESI "MALONICA"
Sfrutta l'acidità del metilene tra due funzioni esteree e la facile decarbossilazione
degli acidi β-dicarbossilici
-O
O
O
C
C
C
OCH2CH3
+
OCH2CH3
C
:CH
C
O
OCH2CH3
O
CH3CH2O-
OCH2CH3
O
O
propandioato dietilico
(malonato dietilico)
-O
C
C
C
OCH2CH3
C
C
C
OCH2CH3
C
R X
:CH
C
O
X
OCH2CH3
OCH2CH3
C
CH R
C
O
OCH2CH3
O
1. H2O, -OH
2. H O+
3
OH
C
CH R
C
O
OH
O
OCH2CH3
OCH2CH3
"estere malonico"
O
OCH2CH3
R
Δ
CO2
CH2
C
OH
O
acido acetico
α-sostituito
33
OCH2CH3
OCH2CH3
O
C
(CH3)3CO-K+
C
CH R
R C:
R' X
C
C
O
O
OCH2CH3
OCH2CH3
O
OCH2CH3
C
1. H2O, -OH
R C R'
2. H3O+
C
O
OCH2CH3 3. Δ , - CO
O
2
R'
O
C
R CH
OH
acido acetico α,α-disostituito
La reazione si può effettuare con tutti gli alogeno derivati utilizzati con il 3ossobutanoato di etile (estere acetoacetico)
alogenuri alchilici, alogenuri acilici,
α-alogenochetoni, α-alogenoesteri
altri esempi (che si possono usare anche nella sintesi acetoacetica:
R-X = α,ω-dialogenoalcani
O
C
C
C
O
OCH2CH3
+
CH3CH2
O
O-
Na+
Na+
OCH2CH3
-C
: CH
C
O
OCH2CH3
OCH2CH3
OCH2CH3
O
C
(CH3)3CO-K+
C
CH R
R C:
R' X
C
C
O
O
OCH2CH3
OCH2CH3
O
OCH2CH3
OCH2CH3
C
1. H2O, -OH
R C R'
2. H3O+
C
O
OCH2CH3 3. Δ , - CO
O
2
R'
O
R CH C
OH
acido acetico α,α-disostituito
La reazione si può effettuare con tutti gli alogeno derivati utilizzati con il 3ossobutanoato di etile (estere acetoacetico)
alogenuri alchilici, alogenuri acilici,
α-alogenochetoni, α-alogenoesteri
altri esempi (che si possono usare anche nella sintesi acetoacetica:
R-X = α,ω-dialogenoalcani
O
O
C
C
C
OCH2CH3
+
CH3CH2O- Na+
O
Na+
OCH2CH3
-C
: CH
C
OCH2CH3
O
OCH2CH3
34
a) in eccesso di estere
OCH2CH3
OCH2CH3
O
C C
:CH
:CH
Br ( CH2)n Br
C
C
O
O
OCH2CH3
OCH2CH3
O
1) H2O, OH2) H+
OH
O
OH
C
C
CH ( CH2 )n CH
C
C
O
O
OH
OH
O
Δ
OCH2CH3
O
OCH2CH3
C
C
CH ( CH2 )n CH
C
C
O
O
OCH2CH3
OCH2CH3
O
O
HO
CH2 ( CH2)n CH2 C
C
OH
O
2 CO2
acido bicarbossilico
b) in quantità equimolari
O
OCH2CH3
C :CH Br ( CH )n Br
2
C
O
O
O
OCH2CH3
C
C
C
OCH2CH3
CH3CH2O-- O C OCH2CH3
C
CH ( CH2)n Br
:C ( C )n Br
C
C
O
O
OCH2CH3
OCH2CH3
O
OCH2CH3
(CH2)n
1) H2O, OH-
O
2) H+
OCH2CH3
O
C
C
C
OH
(CH2)n
Δ
OH
(CH2)n C
CO2
OH
C
n = 2,3,4,5
O
acidi cicloalcancarbossilici
35