IPOFISI
Il Sistema Ipotalamo-Ipofisario costituisce il principale centro di regolazione del sistema
endocrino. L'ipotalamo e la neuroipofisi fanno parte del sistema nervoso e sono distinte
dall'adenoipofisi, o ipofisi anteriore, di diversa origine embriologica.
Ciascuno degli ormoni dell'ipofisi anteriore, tropine ipofisarie, è secreto da una specifica linea
cellulare regolata da fattori stimolanti o inibenti prodotti a livello ipotalamico.
Le tropine ipofisarie si distinguono in ormoni proteici (GH, PRL e ACTH) e ormoni glicoproteici
(FSH, LH e TSH) e vengono rilasciate nel torrente circolatorio per agire sulle diverse ghiandole
periferiche.
Le alterazioni dei vari assi ipotalamo-ipofisi-ghiandole periferiche si manifestano con Sindromi
da Iper- o Ipofunzione delle ghiandole periferiche interessate.
SINDROMI DA IPERFUNZIONE IPOFISARIA
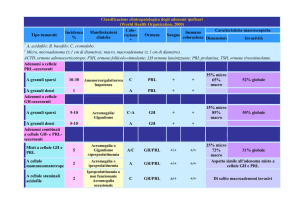
Acromegalia
Quadro clinico da iperproduzione di GH, di solito sostenuto da un adenoma ipofisario, che si
manifesta tra il terzo e il quinto decennio di vita.
La sintomatologia può essere legata a tre diverse condizioni: la situazione locale dell'adenoma
ipofisario, l'ipopituitarismo conseguente alla presenza di un eventuale macroadenoma,
l'ipersecrezione di GH e IGF-I. La presenza della massa tumorale può determinare la comparsa di
cefalea e disturbi visivi, quali alterazioni del campo visivo, visione doppia e paralisi dei nervi
oculari, mentre all'ipersecrezione di GH e IGF-I è dovuto il quadro clinico caratterizzato da
accrescimento delle estremità degli arti, delle ossa del massiccio facciale e delle arcate dentarie
con diastasi. Tipico è il riscontro di macroglossia e cardiomegalia. Si può osservare, inoltre, la
presenza di splancnomegalia, artrosi, ispessimento della cute, iperidrosi, ipertensione arteriosa,
ipertricosi, gozzo multinodulare, alterata tolleranza ai carboidrati, diabete, galattorrea.
La diagnosi si basa sulla valutazione della secrezione di GH e IGF-I nell'arco delle delle 24 ore,
sulla valutazione dinamica della secrezione di GH durante carico orale di glucosio , sulla
eventuale esecuzione di un test al TRH, indicato soprattutto in quei pazienti che non possono
eseguire un carico orale di glucosio e sullo studio morfologico della regione ipofisaria mediante
TC o RMN.
Il trattamento degli adenomi ipofisari GH-secernenti prevede tre diversi approcci: chirurgico,
radiante, medico. La terapia medica prevede l'utilizzo di farmaci ad azione dopamino-agonista
(bromocriptina, cabergolina, quinagolide) e di analoghi della somatostatina (octreotide,
lanreotide, octreotide LAR).
Iperprolattinemia
Quadro clinico caratterizzato da iperproduzione di PRL che può riconoscere cause patologiche,
assunzione di farmaci, ma che può essere legato anche a particolari condizioni fisiologiche.
Le cause patologiche di iperprolattinemia, a loro volta, prevedono: cause ipotalamiche (neoplasie,
granulomi, sezione del peduncolo ipofisario, sindrome della sella vuota), cause ipofisarie
(prolattinomi, adenomi misti), altre (ipotiroidismo, insufficienza renale cronica, sindromi
paraneoplastiche, sindrome dell'ovaio policistico, morbo di Addison, gravidanza isterica,
isterectomia, sindrome di Sheehan, corionepitelioma del testicolo, siringomielia).
I farmaci che possono determinare un aumento della PRL sono: bloccanti dopaminergici, inibitori
della sintesi delle catecolamine, depletori delle catecolamine, inibitori del re-uptake delle
catecolamine, rilascianti le catecolamine, antagonisti del GABA, antistaminici, altre sostanze quali
estrogeni, androgeni, TRH, ossitocina, VIP, oppiacei.
Le condizioni fisiologiche che possono provocare un aumento della secrezione di PRL sono: il
sonno, la pubertà, ma solo nel sesso femminile, la gravidanza, l'allattamento, il periodo neonatale,
il coito e le manipolazioni dei capezzoli.
Le manifestazioni cliniche che caratterizzano l'iperprolattinemia sono, nel sesso maschile,
diminuzione della libido, disfunzione erettile, ginecomastia, galattorrea, oligozoospermia, mentre
nel sesso femminile si riscontra galattorrea, modificazioni dell'areola mammaria, oligomenorrea,
ritenzione idrica, modificazioni dell'umore fino alla depressione.
L'iter diagnostico prevede la valutazione dei livelli ematici di PRL , basale e ritmo, lo studio
dinamico della secrezione mediante test con TRH, agonisti dopaminergici indiretti e antagonisti
dopaminergici, lo studio delle altre tropine ipofisarie e la valutazione morfologica della regione
ipofisaria mediante TC o RMN. Di notevole importanza è un'accurata anamnesi farmacologica.
La terapia si basa fondamentalmente sul trattamento medico che prevede la somministrazione di
farmaci dopamino-agonisti quali bromocriptina e cabergolina, la terapia chirurgica e radiante è
riservata ad un numero assai ristretto di pazienti.
Adenomi Ipofisari Gonadotropino-secernenti
Gli adenomi a cellule gonadotrope costituiscono un gruppo di tumori estremamente raro. Sono
colpiti soggetti di età medio-avanzata, quasi esclusivamente di sesso maschile.
Si tratta quasi esclusivamente di macroadenomi con tendenza all'invasività. Clinicamente i
pazienti giungono all'osservazione del medico per cefalea e disturbi visivi. Dal punto di vista
ormonale si osserva un aumento dei valori basali di LH e FSH associato a livelli non ridotti di
steroidi sessuali.
La diagnosi si basa fondamentalmente sui dati biochimici, sulla sintomatologia neurologica e
sulla dimostrazione radiologica della lesione, e comunque deve essere confermata
dall'imunoistochimica.
La terapia è chirurgica, seguita da trattamento radiante. In alcuni casi si è dimostrata efficace la
terapia a base di dopamino-agonisti. Attualmente è in studio l'impiego di agonisti del GnRH.
Adenomi Ipofisari TSH-secernenti
Si tratta anche in questo caso di adenomi assai rari. Essi determinano un eccesso di TSH in circolo
con conseguente iperstimolazione tiroidea e ipertiroidismo.
Il quadro clinico è quello di un ipertiroidismo moderato associato a gozzo resistente alla terapia
tireostatica e mancanza di esoftalmo. In caso di macroadenoma possono associarsi deficit di altre
tropine ipofisarie o segni neurologici da compressione.
La diagnosi è data dal riscontro di elevati livelli di ormoni tiroidei associati a un TSH non
soppresso e dalla mancata risposta alla prova con TRH.
La terapia di elezione è quella chirurgica, seguita da eventuale radioterapia.
Adenomi Ipofisari ACTH-secernenti
Descritto per la prima volta nel 1932 dal neurochirurgo Harvey Cushing, il morbo di Cushing
viene definito come un ipercortisolismo ACTH-dipendente dovuto alla presenza di un adenoma
ipofisario corticotropo. Come tutte le forme di ipercortisolismo cronico, il morbo di Cushing
rappresenta una malattia grave che espone il paziente a varie complicanze cardiovascolari,
metaboliche, psichiche, reumatologiche ed infettive che ne riducono significativamente la qualità
e l'aspettativa di vita. La diagnosi differenziale con le altre forme di ipercortisolismo è
fondamentale per il corretto indirizzo terapeutico del paziente.
Per le informazioni relative agli aspetti clinici, diagnostici e terapeutici del morbo di Cushing si
rimanda al capitolo riguardante il surrene.
SINDROMI DA IPOFUNZIONE IPOFISARIA
L'ipopituitarismo è una condizione clinica conseguente a diverse cause che determinano
un'insufficienza parziale o completa della secrezione ormonale dell'ipofisi anteriore. Viene
distinto un ipopituitarismo primitivo, se è causato da una patologia ipofisaria, ed ipopituitarismo
secondario, se è determinato da disturbi ipotalamici. Se è compromessa la secrezione di tutte le
tropine si parlerà di panipopituitarismo, mentre se sono carenti una o alcune tropine si definirà
ipopituitarismo parziale.
L'esordio si può avere sia in età infantile che adulta ed è in genere permanente richiedendo
quindi una o più terapie sostitutive ormonali.
Le manifestazioni cliniche dell'ipopituitarismo variano in relazione all'età del soggetto, alla
patologia primitiva, alla rapidità di insorgenza e al tipo e al numero degli ormoni deficitari.
CARENZA di ACTH
Acuta - astenia, adinamia, depressione, nausea, ipotensione arteriosa, disidratazione.
Cronica - affaticabilità, sonnolenza, pallore, anoressia, perdita di peso, ipoglicemia.
In età prepuberale - difetto della crescita.
Nell'adulto - astenia, intolleranza al freddo, cute secca e sottile, depressione.
CARENZA di TSH
Acuta - astenia, bradicardia, stipsi.
Cronica - adinamia, sonnolenza, edema, pallore, anoressia, aumento di peso, bradilalia.
Nel neonato - ipoattività, ipotermia, stipsi, ittero, disturbi della suzione.
Nell'età prepuberale - difetto della crescita e cretinismo.
Nell'adulto - menometrorragie, voce rauca, astenia, cute secca, edema, fragilità dei capelli.
CARENZA di LH e FSH
Nell'età prepuberale - pubertà ritardata.
Nella donna - disturbi del ciclo fino ad amenorrea, infertilità, riduzione della libido, dispareunia,
ipoplasia genitale, caduta dei peli pubici e ascellari, instabilità emotiva, aterosclerosi prematura e
osteoporosi, nella carenza cronica.
Nell'uomo - oligospermia, infertilità, riduzione del volume dei testicoli, della prostata e delle
vescicole seminali, riduzione dei caratteri sessuali secondari (barba, peli ascellari e pubici),
riduzione della forza muscolare, riduzione dell'eritropoiesi con anemia, depressione.
CARENZA di GH
Nell'età prepuberale - ritardo della crescita.
Nell'adulto - ridotta massa e forza muscolare, aumentato tessuto adiposo viscerale, disturbi del
tono dell'umore, aterosclerosi prematura ed osteoporosi, nella carenza cronica.
La diagnosi di ipopituitarismo si basa sulla valutazione degli ormoni ipofisari in condizioni basali
e dopo prove dinamiche. Per la diagnosi di causa di ipopituitarismo sono importanti le indagini
strumentali (TC e RMN) della regione ipotalamo-ipofisaria.
Per ripristinare la funzione compromessa dal deficit di una o più tropine ipofisarie
si attua un'adeguata terapia sostitutiva mediante somministrazione dell'ormone prodotto dalla
ghiandola bersaglio.