Appunti di Catalisi Asimmetrica
per il Corso di
Metodi Innovativi in Catalisi Omogenea
Questi appunti sono stati redatti per aiutare lo studente nello studio del Corso di
Metodi Innovativi in Catalisi Omogenea, ma non possono essere considerati sostitutivi
delle lezioni frontali, che rappresentano l’unico modo per acquisirne scopo e contenuto.
il controllo sterico
le funzioni coordinanti
l'etichetta di fase
M
ruff @unina.it
il building block
di origine naturale
Docente: Francesco Ruffo
Introduzione
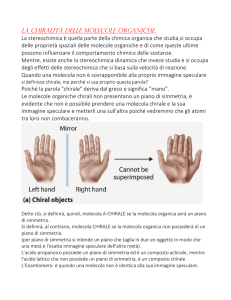
La chiralità, vale a dire la proprietà di un oggetto di non essere sovrapponibile alla sua
immagine speculare, è ubiquitaria nel nostro Universo, ed è centrale nei sistemi viventi.
L’affascinante chimica della vita è infatti basata su interazioni asimmetriche, che coinvolgono
spesso entità altamente organizzate, come gli enzimi. Queste molecole sono in grado di selezionare
substrati attraverso un riconoscimento chirale, e, quindi, un ampio numero di molecole
biologicamente attive, come aminoacidi, carboidrati e vitamine, esistono come singoli enantiomeri.
Lo stesso concetto vale per i principali prodotti artificiali, quali i farmaci, i composti
agrochimici, gli aromi e le fragranze, preparati in larga scala dalla moderna industria chimica.
In questi casi, solo un enantiomero ha le proprietà biologiche desiderate, mentre la sua
immagine speculare non ha alcun effetto, o, peggio, attività indesiderata. Per esempio, l’(S)asparagina è amara, mentre la (R)-asparagina è dolce, l’(R)-limonene sa di arancia, mentre il suo
enantiomero ha sapore di limone (Figura I.1).
O
H2N
(S)
O
O
OH
NH2
(S)-asparagina
H2N
(R)
O
(R)
(S)
OH
NH2
(R)-asparagina
H2C
CH3
(R)-limonene
H2C
CH3
(S)-limonene
Figura I.1
Ovviamente questi comportamenti non sono affatto problematici, e possono essere considerati
come semplice curiosità, a dispetto di quanto riscontrato negli anni Sessanta a carico del farmaco
chirale commercializzato col nome di Talidomide.
In quel periodo, i farmaci chirali erano di solito venduti come miscele raceme (vale a dire
contenenti ambedue gli enantiomeri in ugual quantità), anche se era già noto che solo uno di essi era
dotato di attività terapeutica.
La Talidomide era usata per risolvere problemi di nausea e vomito durante la gravidanza.
Purtroppo, diverse donne, in seguito alla sua assunzione, hanno dato alla luce bambini seriamente
malati. Si è subito capito che questo grave problema era causato dalla tremenda differenza di attività
tra i due enantiomeri (Figura I.2): mentre uno era effettivamente lenitivo del vomito e della nausea,
la sua immagine speculare risultava altamente teratogena.
2
O
O
N
O
N
O
NH
O
NH
O
O
O
S-talidomide
R-talidomide
Figura I.2
La scoperta ebbe un impatto devastante non solo sulla comunità scientifica, ma anche
sull'opinione pubblica, e determinò una svolta importante nella manifattura dei composti chirali.
Furono introdotte nuove e severe norme, tuttora valide, che richiedono la valutazione separata dei
due enantiomeri di un composto biologicamente attivo prima della sua approvazione come farmaco.
Questa disposizione ha, di fatto, gravemente sfavorito la commercializzazione di farmaci in
forma racema, dando grande impulso allo sviluppo di sintesi enantioselettive per la produzione di
composti enantiopuri. Oggi, sono quattro gli approcci generalmente perseguiti per il loro
ottenimento, e tutti si basano sulla considerazione che un composto chirale può essere prodotto
artificialmente solo in presenza di un altro agente chirale (Figura I.3).
catalisi
asimmetrica
separazione di
diastereomeri
sintesi
asimmetrica
uso di
"building block" chirali
Figura I.3
La prima strategia è la separazione di enantiomeri attraverso la risoluzione di diastereoisomeri,
vale a dire isomeri che non sono immagini speculari, e quindi hanno differenti proprietà fisiche.
Questo metodo (Figura I.4) prevede di realizzare una reazione che conduce a una miscela
equimolare di enantiomeri (i fusilli). A questa è aggiunto un agente risolvente chirale (il cucchiaio
elicoidale), che determina la cristallizzazione selettiva di un sale diastereoisomerico (nell’esempio è
l’l-fusillo,l-cucchiaio), dal quale è successivamente possibile ricavare l’enantiomero desiderato.
+
reattore
l-fusillo
+
d-fusillo
l-fusillo,
l-cucchiaio
d-fusillo
Figura I.4
Con questa tecnica sono tuttora prodotti numerosi farmaci chirali, con una percentuale stimata
3
intorno al 50% del totale. Per esempio, nella sintesi della sertralina, noto antidepressivo, gli
enantiomeri sono risolti aggiungendo acido mandelico alla fine del processo (Figura I.5).
NHMe
NHMe
HO
Cl
H
CO2H
Cl
Cl
Cl
acido mandelico
sertralina
Figura I.5
Questa metodologia si avvale di tecniche automatizzate di HPLC chirale, che possono essere
utilmente applicate alla produzione su larga scala.
I suoi principali svantaggi sono il notevole uso di solventi e la presenza di una quantità
equimolare di un enantiomero con la configurazione indesiderata, che deve essere riciclato o
eliminato.
La seconda strategia è quella che sfrutta la chiralità intrinseca di molecole naturali per produrre
i prodotti desiderati (Figura I.6).
reagente
“naturalmente”
chirale
prodotto chirale
Figura I.6
I “building block” di origine naturale possono essere scelti all'interno di una vasta gamma di
categorie, che comprendono carboidrati, aminoacidi, terpeni, idrossiacidi e alcaloidi.
Un esempio è la sintesi di un erbicida a partire da un ben noto idrossiacido di origine naturale,
l’acido lattico (Figura I.7). Dopo la conversione a mesilato, una singola inversione fornisce il
prodotto desiderato con configurazione (R).
HO
(S)
i-PrO2C
CH3
H
Cl
CH3SO3
(S)
i-PrO2C
CH3
H
F
O
N
(R)
i-PrO2C
H
CH3
Figura I.7
Questo approccio è spesso limitato dalla scarsa disponibilità del materiale di partenza, e quindi
di solito è utile per produzioni su piccola scala.
La terza strategia è la sintesi asimmetrica (Figura I.8). In questo caso, un adeguato ausiliare
4
chirale è legato covalentemente a un substrato per guidare la formazione del centro stereogenico
desiderato in un passaggio successivo. L’ausiliare non resta incorporato nella molecola finale, ma è
rimosso dopo la formazione del centro chirale.
+
ausiliare chirale
substrato
+
ausiliare chirale
prodotto chirale
Figura I.8
Ad esempio, un metodo generale per la sintesi di β-amminoacidi (Figura I.9) comporta
addizione di metilbenzilammide chirale a un enone con formazione dello stereocentro desiderato.
Dopo il “quenching” del risultante enolato, l'ausiliare chirale è rimosso per riduzione. Naturalmente,
questo approccio presenta lo svantaggio che sono necessari passaggi addizionali per l’attacco e la
successiva rimozione dell'ausiliare chirale.
Ph
N
Ph
O
Ph
+
Ph
OBu
Ph
Ph
ausiliare chirale
N
NH2 O
O
OBu
Ph
OBu
β-amminoacido
Figura I.9
Infine, il quarto approccio è quello della catalisi asimmetrica, per molti aspetti, la più elegante.
In questo caso, molecole di partenza, prochirali o in miscela racema, vengono convertiti in prodotti
enantiopuri con l'aiuto di una piccola quantità di un adatto catalizzatore chirale (Figura I.10).
catalizzatore chirale
Figura I.10
5
Il grande beneficio economico è che il controllo della stereochimica richiede una piccola
quantità di ausiliare chirale, perché le prestazioni dei catalizzatori spesso consentono di usare
rapporti catalizzatore/substrato molto piccoli.
I catalizzatori
possono
essere sintetici
(chemocatalizzatori) o di
origine naturale
(biocatalizzatori).
In questo ambito, sarà prestata particolare attenzione ai primi (d’ora in poi indicati col nome
generico di catalizzatori), anche se sono note molte reazioni enzimatiche di utilità sintetica che
procedono con grande efficienza.
Un catalizzatore può essere un complesso metallico omogeneo con leganti chirali, una specie
metallica eterogenea modificata con ausiliari chirali, o una specie acido/base organica chirale.
La prima pietra miliare nella storia delle catalisi asimmetrica risale al 1912, quando fu descritta
l’addizione enantioselettiva di HCN alla benzaldeide in presenza di chinina come catalizzatore
chirale. Anche se l'eccesso enantiomerico risultò essere molto basso, questo esperimento dimostrò
per la prima volta la fattibilità di tale strategia (Figura I.11).
OH
H
NC
CHO
CH2 CH
OH
+ H CN
CH3O
N
chinina
Figura I.11
Nel 1940 fu riportato il primo esempio di modifica di un catalizzatore eterogeneo con un
ausiliare chirale, e un doppio legame C=N fu idrogenato in modo asimmetrico usando nero di
platino trattato con acidi chirali (Figura I.12).
H3 C
NOH
+ H2
Ph
NHOH
H3 C
Ph
Figura I.12
Questo lavoro pionieristico motivò la ricerca nel settore, e ben presto altri utili catalizzatori
eterogenei furono preparati e usati con successo nell’idrogenazione di molecole insature.
I primi esempi di reazioni asimmetriche catalizzate da complessi metallici omogenei risalgono
invece al 1966 e al 1968, e riguardano, rispettivamente, la ciclopropanazione di alcheni con
complessi di rame contenenti basi di Schiff , e l'idrogenazione di enammidi con specie fosfiniche di
rodio (Figura I.13).
6
Ph
Ph
+ N2CHCO2Et
N O
Cu
O N
-N2
Ph
CO2Et
Ph
catalizzatori
CO2H
Ph
CO2H
+ H2
NHCOCH3
Ph Ar
P
L
Rh
L
P
Ph Ar
Ph
NHCOCH3
Figura I.13
Le prime rese ottiche furono insoddisfacenti, ma migliorarono molto rapidamente, e già nei
primi anni ‘70 fu raggiunto un eccesso enantiomerico del 95% nell’idrogenazione di enammidi,
usando un complesso di rodio coordinato a una difosfina chirale, oggi universalmente chiamata
DIPAMP (Figura I.14).
CO2H
CO2H
NHCOCH3 + H
2
NHCOCH3
P
CH3O
OCH3
OCH3
H3CO
CH3O
P
OCH3
DIPAMP
Figura I.14
Questa reazione fu rapidamente portata su scala industriale dalla Monsanto, per la produzione di
L-dopa, il farmaco usato contro il morbo di Parkinson.
I risultati impressionarono e stimolarono la comunità scientifica, e nuovi catalizzatori omogenei
furono descritti e applicati con successo in tutti le più importanti reazioni organiche.
Fino al 1985 solo pochi catalizzatori erano in grado di offrire enantioselettività superiori
all’85%. Ora, un gran numero di essi sono in grado di produrre ee vicino al 100%, grazie anche al
contributo di illustri scienziati, quali Jacobsen, Katsuki, Trost, Knowles, Noyori e Sharpless. Nel
2001 Noyori, Sharpless e Knowles sono stati insigniti del Premio Nobel, a riconoscimento
dell’importante lavoro svolto.
Di conseguenza, l'interesse industriale verso la catalisi omogenea enantioselettiva è cresciuta
negli anni, e diverse reazioni sono ora applicate per l’ottenimento di prodotti di chimica fine. Alcuni
esempi sono riportati in Tabella:
7
prodotto
catalizzatore
scala
compagnia
mentolo
Rh-binap
>1000 t/y
Takasako
aspartame
Rh-eniphos
>1000 t/y
Enichem
vitamina E
Ru-binap
300 t/y
Takasago
glicidolo
Os-cincona
> 1 t/y
PPG-Sipsy
carbapenem
Ru-tolbinap
100 t/y
Takasago
epossidi
Mn-salen
piccola
Merck/Chirex
solfossidi
Ti-tartrate
piccola
Lonza
Tuttavia, la sensazione è che ancora oggi la catalisi asimmetrica contribuisca alla produzione
complessiva di sostanze chimiche chirali in misura inferiore alle sue potenzialità, e che la sua
produzione industriale possa essere notevolmente implementata.
Naturalmente, molti sono i fattori critici che determinano la fattibilità industriale di un processo
enantioselettivo. Tra questi, gli aspetti di natura chimica sono:
- l’enantioselettività;
- la chemoselettività e/o la tolleranza ai gruppi funzionali, molto importanti in presenza di
substrati con più funzioni in grado di reagire;
- la produttività, espressa in TON, vale a dire il numero di molecole di substrato convertite per
sito catalitico;
- l'attività, espressa in TOF, cioè il numero di molecole di substrato convertite per sito catalitico
nell’unità di tempo.
Per esempio, per essere adatto alla produzione industriale di un farmaco chirale, l’ee deve essere
superiore al 90%, mentre un ee dell’80% può già essere sufficiente per la produzione di composti
agrochimici. Invece, la produttività e l'attività devono essere maggiori di 1000 e 500 h-1, e maggiori
di 50000 e 10000 h-1, rispettivamente per le produzioni su piccola e larga scala.
Siffatte elevate prestazioni possono essere ottenute solo combinando adatta progettazione dei
catalizzatori con adeguate condizioni di reazione.
Nel corso dei prossimi capitoli, dopo uno sguardo generale alle definizioni importanti nel
dizionario della catalisi asimmetrica (Capitolo 1), verranno presi in considerazione proprio gli
aspetti principali relativi alla realizzazione di un efficiente processo di catalisi asimmetrica.
Nell’ordine, saranno quindi descritti i più semplici modi per ottenere enantioselezione (Capitolo 2),
le classiche modalità di attivazione di substrati nell’ambito di processi catalitici (Capitolo 3), e il
funzionamento stereochimico di importanti classi di catalizzatori asimmetrici (Capitolo 4).
8
1. La chiralità: definizioni
1.1 Unità stereogeniche.
Gli isomeri costituzionali sono quelli che differiscono nella connettività degli atomi (senza
alcuna distinzione che deriva dalla loro disposizione spaziale). Esempi sono l’acetone,
CH3C(O)CH3 e l’alcol etilico CH3CH2OH.
Gli stereoisomeri sono isomeri che possiedono identica costituzione, ma differiscono per
l’arrangiamento dei loro atomi nello spazio. Tra questi si distinguono gli enantiomeri e i
diastereoisomeri.
Gli enantiomeri sono una coppia di isomeri, ciascuna immagine speculare dell’altra, non
sovrapponibili. In questo caso, la molecola gode della proprietà geometrica definita chiralità. Dal
punto di vista della simmetria, le strutture chirali sono caratterizzate dall’assenza di assi di rotazione
impropria (Sn, di cui il piano di riflessione, S1, e il centro di inversione, S2, sono casi speciali).
Per esempio, molecole appartenenti a gruppi di simmetria Cn o Dn sono appunto chirali.
I diastereoisomeri sono invece stereoisomeri non sovrapponibili e non relazionati dalla
simmetria speculare.
Tutte le molecole chirali contengono ben definite unità stereogeniche, che rappresentano la più
piccola porzione di una struttura capace di renderla non sovrapponibile alla sua immagine
speculare. Va comunque rilevato che la presenza di una unità stereogenica è di per sé una
condizione insufficiente per il manifestarsi della chiralità.
L’unità che si incontra più frequentemente (Figura 1.1) è un centro sp3 con quattro diversi
sostituenti. I composti contenenti tale requisito sono spesso denominati centrochirali e il centro è
indicato come unità centrochirale o centro stereogenico.
asse
NMe2
PPh2
O
O
H2N
(R)
(S)
OH
HO
Me
Me
(R)-alanina
NH2
(S)-alanina
Unità Centrochirale
PPh2
Fe
PPh2
(R)-BINAP
(M)-esaelicene
Unità Assiale Chirale
Unità Elicoidale
(R)-ferrocenil fosfina
Unità Planare Chirale
Figura 1.1
Un altro tipo di unità stereogenica è l’unità assiale chirale, associata a un asse stereogenico,
come nel caso della molecola BINAP di Figura 1.
9
E’ poi possibile la presenza di una unità elicoidale chirale, che definisce un’elica chirale, di cui
ne è esempio l’elicene di Figura 1.1.
Ancora, si può manifestare la presenza di unità planare chirale, associata quest’ultima a un
piano stereogenico, come quello riscontrabile nella ferrocenil fosfina di Figura 1.1.
Va notato che se le immagini speculari non sovrapponibili di un composto si interconvertono a
temperatura ambiente attraverso un cambio conformazionale, allora il composto esiste come
miscela racema, e non mostra attività ottica, a dispetto della presenza di una conformazione chirale.
Esempi (Figura 1.2) sono la cis-decalina, che interconverte per inversione di anello, e l’1,1–
binaftile, che può agevolmente ruotare lungo il legame assiale.
cis-decalina
1,1-binaftile
Figura 1.2
Centrochiralità. Il più comune centro stereogenico è un atomo di carbonio con ibridazione sp3
legato a quattro sostituenti diversi. Questi ultimi possono disporsi in due modi possibili facendo sì
che le due strutture risultanti siano immagini speculari non sovrapponibili (enantiomeri). Le
configurazioni dei centri stereogenici sono assegnate seguendo le regole di Cahn-Ingold e Prelog.
Un altro esempio è fornito dalla prolina (Figura 1.3), un amminoacido naturale usato con
successo in sintesi asimmetrica.
(R)
OMe
H
N
O
(R)
O
OH
HO
H
N
N
(S)
(S)
t-Bu
(R)-prolina
(S)-prolina
O
O
(R)
(S)
(S)
Ph
N
(S)
t-Bu
(S,S)-t-Bu-box
Ph
N
(S)
(R)
(R)
OH
N
(R,R)-Ph-bod
(-)-chinina
Figura 1.3
Alcuni composti possono presentare più di un centro stereogenico, come nel caso dei due
leganti (S,S)-t-Bu-box e (R,R)-Ph-bod, illustrati in Figura 1.3. Tali leganti, come molti altri della
stessa tipologia, sono molto apprezzati in catalisi asimmetrica a causa della loro simmetria C2, che,
come risulterà più chiaro in seguito, riduce il numero di modi di coordinazione e di stati di
transizione, rendendo più semplice la progettazione dei catalizzatori e l’interpretazione dei risultati.
Un esempio di molecola dotata di un numero ancora maggiore di stereo centri, e ancora
10
largamente usata in catalisi asimmetrica, è quello della chinina (Figura 1.3), dotata di ben 5 unità
centrochirali.
Unità centrochirali possono essere generate anche da atomi diversi dal carbonio, come silicio,
azoto, fosforo e zolfo.
In pratica, però, solo quelle stabili configurazionalmente sono definibili come unità
centrochirali. Pertanto, molecole chirali contenenti azoto come centro stereogenico sono in numero
estremamente ridotto, dato che atomi di azoto ibridizzati sp3 e con quattro diversi sostituenti (la
coppia solitaria può essere considerata alla stregua di un sostituente) danno tipicamente luogo a
rapida interconversione tra gli enantiomeri attraverso inversione dell’ombrello. Esempi di ammine
stabili configurazionalmente sono le ossaziridine (Figura 1.4), rese tali dalla presenza di un piccolo
ciclo. Inoltre, l’azoto può diventare configurazionalmente stabile qualora la coppia solitaria sia
legata a un centro elettrofilo, come nel caso di sali di ammonio quaternari, di N-ossidi o di centri
metallici (Figura 1.4).
R2
R3
O
ossaziridine
R1
N
ON+
R1
R3
R2
N-ossidi
R4
N+
R1
R3
R2
sali di ammonio
M
N
R1
R3
R2
complessi
Figura 1.4
Nel caso del silicio esiste un certo numero di silani chirali, come quelli illustrati in Figura 1.5.
R4
Si
R1
R3
R2
silano
Si
Ph
Me
silano chirale
PhCH2
H Si Ph
Me
idrosilano chirale
Figura 1.5
Centri stereogenici al fosforo e allo zolfo presentano invece barriera energetica all’inversione
molto più alta di quella dell’azoto. Di conseguenza, questi atomi costituiscono unità centrochirali
qualora sostituiti con quattro diversi sostituenti (anche in questo caso la coppia solitaria vale come
sostituente).
Le fosfine chirali con centro stereogenico al fosforo, come CAMP e DIPAMP (Figura 1.6), sono
di importanza strategica quali leganti in catalisi asimmetrica.
11
OMe
MeO
R1 P R
3
R2
fosfine
P
Ph
P
(R)
P Ph
(R)
MeO
Me
(+)-CAMP
(R,R)-DIPAMP
Figura 1.6
Composti con centro stereogenico allo zolfo includono, tra gli altri, i solfossidi (Figura 1.7), che
sono considerati molto importanti dalla industria farmaceutica e presentano importanti applicazioni
in catalisi asimmetrica. A questo proposito, basti citare l’Omeprazolo e l’Esomeprazolo (Figura
1.7), principi attivi rispettivamente dei farmaci anti-ulcera Prilosec e Nexium, prodotti dalla AstraZeneca. Nel solo 2000 la commercializzazione del primo ha procurato un fatturato di 6.2 miliardi di
dollari.
MeO
S
R1
R2
O
O Me
S
N
H
omeprazolo
solfossidi
t-Bu
N
O
S
N
N
Me
Siam
O
N S t-Bu
OMe
Me
MeO
NO
Me
S
N
H
esomeprazolo
OMe
Me
Me
Ph
Me
Ph
N S
S N
O
O
BISOX
Figura 1.7
Per quel che riguarda invece l’uso in catalisi asimmetrica, esempi di solfossidi chirali usati
come leganti sono Siam e BISOX di Figura 1.7.
Anche centri metallici possono diventare unità centrochirali, e non solo qualora essi presentino
quattro sostituenti. Infatti, è possibile l’ottenimento di enantiomeri per molte comune geometrie di
coordinazione, quali il tetraedro, la bipiramide trigonale, la piramide a base quadrata e l’ottaedro,
eccezion fatta per la quadrato-planare. Addirittura, per alti numeri di coordinazione il numero dei
possibili stereoisomeri può essere molto elevato, e arrivare a 20, 30 e 30, rispettivamente per la
bipiramide trigonale, la piramide a base quadrata e l’ottaedro, qualora tutti i sostituenti siano diversi
(Figura 1.8).
12
identiche
1
2
M
4
3
enantiomeri
enantiomeri
1
2
M
4
3
quadrato-planare
1
4
4
M 3
3 M
5 5
2
2
1
1
5 M 2
4
3
bipiramide trigonale
Fe
Ph3P
1
2 M 5
4
3
enantiomeri
1
3
5
M
4
6
2
1
3
5
M
4
6
2
ottaedro
piramide a base
quadrata
+
OH2
H3N
CN
Co
H2O
CN
NH3
Me
CO O
Figura 1.8
Come nel caso delle unità centrochirali tetraedriche, non è necessario che uno o più leganti
siano chirali al fine di ottenere centrochiralità al centro metallico. Un esempio è relativo al
complesso acilico di ferro di Figura 1.8, che può essere considerato a geometria pseudo-tetraedrica
a configurazione (S).
Anche nel caso di complessi a geometria bipiramidale trigonale è possibile l’ottenimento di
enantiomeri, sebbene la loro applicabilità sia in genere limitata dalla instabilità configurazionale
dovuta alle dinamiche intermolecolari (per esempio la pseudorotazione di Berry).
Al contrario, i complessi ottaedrici sono di gran lunga più stabili configurazionalmente, il che ne
ha consentito l’isolamento di numerose specie sotto forma di enantiomeri puri. Un esempio è il
complesso di Co illustrato in Figura 1.8. Da osservare che in questo caso il metallo è centrochirale
anche nel caso alcuni leganti siano identici.
In presenza di leganti bidentati è possibile avere chiralità anche se tutti i leganti sono identici, a
causa dell’arrangiamento elicoidale che questi ultimi possono assumere nel complesso. La Figura
1.9 mostra schematicamente i due enantiomeri che si hanno nel caso di un complesso tris(chelato).
Le etichette configurazionali ∆ e Λ sono assegnate a seconda che i leganti diano vita a un’elica
destrogira o levogira, qualora il complesso sia osservato lungo (l’unico) asse C3 in esso presente.
N
M
M
∆
Λ
N
∆
Λ
N
2+
N
N
N
Fe
Fe
N
N
∆
2+
N
N
N
N
Λ
Figura 1.9
13
Un esempio reale è fornito dal catione [Fe(2,2’-bipiridile)]2+, i cui enantiomeri sono ancora
illustrati in Figura 1.9.
Uno stereoisomerismo addizionale si può avere con l’etilendiammina (H2NCH2CH2NH2) o altri
leganti bidentati NON planari, come mostrato dalla Figura 1.10.
H2 H2N
3+
N
Co
N
H2
H2N
NH2
H2N
NH2
H2N
∆
NH2 H
2
3+
N
Co
N
H
NH2 2
H2
N
H2
N
N
H2
N
H2
M
M
δ
Λ
λ
Figura 1.10
Infatti, oltre alla chiralità generale del complesso di tipo ∆ e Λ, ciascun legante bidentato può
assumere conformazione δ (twist destrogiro) o λ (twist levogiro). L’insieme di tutte le possibilità,
tra loro combinate, dà luogo a quattro coppie di enantiomeri, così distinguibili:
∆ (δ,δ,δ)
Λ (λ,λ,λ)
∆ (δ,δ,λ)
Λ (λ,λ,δ)
∆ (δ,λ,λ)
Λ (λ,δ,δ)
∆ (λ,λ,λ)
Λ (δ,δ,δ)
In genere, se il legante bidentato è chirale, si osserva una netta preferenza per una
conformazione δ o λ, altrimenti è possibile la formazione di tutte le specie appena descritte.
In linea di principio, complessi con metalli centrochirali dovrebbero essere particolarmente utili
in catalisi asimmetrica, dal momento che il centro metallico gioca un ruolo dominante sul decorso
della reazione. Tuttavia, spesso questa situazione non sempre è la più vantaggiosa perché è
accompagnata da una serie di controindicazioni, quali (i) la difficoltà di ottenere un metallo
centrochirale, (ii) la sua instabilità configurazionale, e (iii) la possibile difficoltà di trasmissione
dell’informazione chirale ai substrati reagenti.
Ad esempio, i complessi metallici, come già fatto notare precedentemente nel caso di quelli a
geometria bipiramidale trigonale, possono essere coinvolti in numerosi riarrangiamenti
intramolecolari che determinano la racemizzazione del metallo centrochirale. Lo stesso effetto
indesiderato può verificarsi attraverso scambio di posizione di leganti mediante meccanismi
dissociativi/associativi.
Inoltre, le unità metalliche centrochirali sono spesso distanti dai centri interessati dalla reazione.
Per esempio, mentre l’ossigeno di un gruppo carbonilico potrebbe ben risentire della chiralità del
14
metallo perchè a esso direttamente legato, l’atomo di carbonio dello stesso gruppo carbonilico
risentirebbe invece maggiormente di un eventuale intorno asimmetrico creato da un legante chirale.
Pertanto, in catalisi asimmetrica spesso è più semplice ed efficace introdurre l’informazione
chirale mediante un legante chirale, che, in seguito, per esempio, alla coordinazione di un substrato
prochirale, porta alla formazione di complessi metallici diastereoisomerici dei quali uno risulterà
più stabile e/o più reattivo.
Chiralità assiale. Come già anticipato, i composti a chiralità assiale sono dotati dell’unità
assiale chirale associata a un asse stereogenico, vale a dire un asse su cui quattro sostituenti si
svolgono in una disposizione che non è sovrapponibile alla propria immagine speculare. Tipici
esempi sono i diarili e gli alleni (Figura 1.11).
OH
1
OH
R1
R1
asse
chirale
R2
R2
bifenili
asse
R1
chirale
C
R2
C
2
(S)-BINOL
(P)
R2
R1
alleni
H
C
Cl
3
OH
4
C
HO
2
H
H
Cl
3
Cl
4
H
(R)-1,3-dicloro1,3-propadiene
(M)
Cl
1
Figura 1.11
Sebbene questi composti appaiano come tetraedri “espansi”, la perdita dell’asse di simmetria C3
comporta il fatto che non sia necessario avere i quattro ipotetici sostituenti diversi. E’ invece
sufficiente che lo sia ciascuna coppia in ognuna delle estremità (R1≠R2).
L’assegnazione della configurazione nei composti a chiralità assiale è fatta attraverso una
convenzione derivata da quella di Cahn, Ingold e Prelog, secondo la quale, osservando la molecola
lungo l’asse chirale, i gruppi più vicini precedono quelli più lontano. Se i tre gruppi a proprietà
maggiore sono disposti in senso orario, allora la configurazione è (aR). Altrimenti, la
configurazione è (aS). Il prefisso a (comunque facoltativo) specifica che si tratta di chiralità assiale.
Le proiezioni dei composti lungo l’asse chirale danno luogo alla stessa configurazione,
indipendentemente dal lato di osservazione.
Alternativamente, le molecole con assi chirali possono essere anche viste come eliche, e allora
15
le loro configurazioni possono essere definite rispettivamente come (P) o (M). Il primo simbolo sta
per plus, e indica il movimento destrogiro che bisogna compiere qualora per andare dal sostituente
vicino a maggior priorità (1) a quello lontano a maggior priorità (3) bisogna compiere una rotazione
oraria, come nell’esempio dell’(S)-BINOL (Figura 1.11). Vice versa, il simbolo (M) denota che lo
stesso movimento deve essere compiuto in rotazione levogira, come per l’(R)-1,3-dicloro-1,2propadiene di Figura 11.
I diarili rotazionalmente ingombrati sono tra i composti chirali più usati, sia come reagenti, sia
come leganti per catalizzatori. In linea di principio, i due enantiomeri possono interconvertirsi per
semplice rotazione lungo l’asse C-C, e, pertanto sono isomeri conformazionali (Figura 1.12).
R2
R1
R2
R1
R2
R1
R2
R2
R2
R1
R1
R1
enantiomero (M)
enantiomero (P)
Figura 1.12
In questo caso i due enantiomeri sono indicati come atropoisomeri. Se la rotazione è
sufficientemente impedita, allora l’unità assiale chirale può essere stabile anche a temperatura
ambiente. Tipicamente, perchè ciò avvenga, è sufficiente che i due anelli siano sostituiti nelle
quattro posizioni orto.
Invece, non sono isomeri conformazionali gli alleni (Figura 11), i quali sono dieni cumulati, e,
come tutti i dieni cumulati a numero di atomi di carbonio dispari, sono potenzialmente dotati di
chiralità assiale.
Chiralità elicoidale. I composti che presentano questo tipo di chiralità non sono dotati di una
specifica unità stereogenica, come un centro, un asse o un piano. Piuttosto, la chiralità proviene
dalla struttura tridimensionale in sé. I composti a chiralità elicoidale sono rari. Esempi ne sono
l’esaelicene e il legante chirale Phelix di Figura 1.13.
Ph2P
(M)-esaelicene
PPh2
Phelix
Figura 1.13
Le configurazioni sono assegnate con i descrittori (P) e (M), che nuovamente denotano un
16
avvitamento rispettivamente destro- o levo-giro.
Chiralità planare. Per presentare chiralità planare un composto deve possedere un piano
definito da atomi appartenenti alla molecola (piano chirale) e almeno un altro sostituente al di fuori
di questo. Tipici composti con chiralità planare presentano due anelli non coplanari sostituiti in
modo asimmetrico con rotazione impedita lungo l’asse che li collega (Figura 1.14).
OC
Me
OC
Cr
CO
PPh2
Cl
Ti
Me
Cl
PPh2
Me
complesso di Cr η6
(ebthi)2TiCl2
(R)-[2.2]PHANEPHOS
Figura 1.14
Anche per questi composti sono definiti descrittori, (pR) e (pS). Tuttavia, la più complessa
procedura di assegnazione non viene qui esposta in dettaglio.
1.2 Enantiomeri e diastereoisomeri
Composti enantiopuri e racemati. Come ben noto, i composti chirali esistono sotto forma di due
enantiomeri, vale a dire due immagini speculari non sovrapponibili.
Se un composto esiste come unico enantiomero, esso allora è definito come enantiomericamente
puro o omochirale. Se esiste come miscela 1:1 dei due enantiomeri, allora è un racemo.
La conversione di un singolo enantiomero nel suo racemo è definita racemizzazione, mentre la
conversione di un enantiomero nell’altro è detta enantiomerizzazione.
Miscele di enantiomeri in rapporto diverso da 1:1 sono indicate come enantiomericamente
arricchite. L’abbondanza relativa dei due enantiomeri viene spesso espressa attraverso l’eccesso
enantiomerico (equazione 1), o il rapporto enantiomerico (equazione 2).
(ent 1) - (ent 2) .
(attività ottica della miscela) .
eccesso
purezza
=
100 = ottica =
100
enantiomerico(%)
(ent 1) + (ent 2)
(attività ottica del singolo)
equazione 1
K(ent 1)
-∆G/RT
rapporto = (ent 1)
=e
=
enantiomerico (ent 2)
K(ent 2)
∆G= GTS1 - GTS2
equazione 2
17
I significati di K, ∆G e GTS saranno chiariti in seguito. Si anticipa solo il fatto che essi sono
legati ai valori delle energie di attivazione di processi che prevedono la formazione dei due
enantiomeri a partire da reagenti allo stesso livello energetico.
Diastereoisomeri.
Isomeri
che
non
sono
enantiomeri
(immagini
speculari)
sono
diastereoisomeri. Esempi sono il cis-2-butene e il trans-2-butene (Figura 1.15), ma anche composti
che presentano due tra le unità stereogeniche descritte nelle sezioni precedenti.
Me
H
diastereoisomeri
OC
OC
Me Cr
PCy2
PPh2
CO
Me
Fe
OAc
HO
cis-2-butene
JOSIPHOS
unità centrochirale +
unità chirale planare
trans-2-butene
complesso di Cr η6
unità assiale chirale +
unità chirale planare
enantiomeri a simmetria C2
HO
H
CO2H
(R)
H
HO2C
(S)
(R)
HO
H
CO2H
HO2C
OH
(S)
OH
H
diastereoisomeri
diastereoisomeri
H
OH
(S)
HO
CO2H
HO
HO2C
(S)
(R)
H
H
(R)
CO2H
HO2C
H
OH
identici (composto meso)
acido tartarico
Figura 1.15
Il numero di isomeri possibile per un dato isomero costituzionale è minore o uguale a 2n, dove n
rappresenta il numero di unità stereogeniche. Per esempio, l’acido tartarico, che contiene due unità
centrochirali, per motivi di simmetria dà luogo a solo tre isomeri. Due di essi, (R,R) e (S,S) sono
enantiomeri, mentre l’(R,S) non è l’immagine speculare di nessuno dei primi due, e quindi è un loro
diastereoisomero.
Non tutti i diastereoisomeri sono necessariamente chirali. Un esempio è proprio fornito
18
dall’acido (R,S)-tartarico, che ha un piano di simmetria, e, pertanto, è definito come composto meso.
Altri esempi di diastereoisomeri sono quelli del legante JOSIPHOS, che presenta la
combinazione di una unità centrochirale e una planare chirale, o il complesso arenico di cromo, in
cui si distinguono l’unità assiale chirale e l’unità planare chirale (Figura 1.15).
Come nel caso dei diastereoisomeri, è possibile definire l’abbondanza dei diastereoisomeri
presenti in una miscela attraverso sia l’eccesso diastereoisomerico (Equazione 3) sia il rapporto
diastereoisomerico (Equazione 4).
(dias 1) - (dias 2) .
eccesso
diastereoisomerico(%) = (dias 1) + (dias 2) 100
Equazione 3
(dias 1)
K(dias 1)
-∆G/RT
rapporto
=
=e
=
diastereoisomerico (dias 2)
K(dias 2)
∆G= GTS1 - GTS2
Equazione 4
A differenza dell’eccesso enantiomerico, non vi sono misure fisiche correlate all’eccesso
diastereoisomerico. Inoltre, tale espressione non è generalmente utile se vi sono più di due unità
stereogeniche presenti in una molecola.
1.3 Composti prochirali
Un composto è prochirale se la sostituzione di un suo gruppo o l’addizione a una sua faccia dà
luogo a un composto chirale. Per capire se un composto è prochirale o no, si deve verificare la
presenza di facce o gruppi omotopici, enantiotopici o diastereotopici.
Facce e gruppi omotopici. Due gruppi sono omotopici (vale a dire topologicamente equivalenti)
se possono essere scambiati attraverso un qualsiasi asse di rotazione Cn dando luogo a una struttura
equivalente. Per esempio, i due gruppi metili della trans-2,5-dimetilpiperidina (Figura 1.16) o i due
atomi di idrogeno HA e HB della trans-cicloesandiammina sono omotopici.
Z
NH
Me
NH
Me
C2
Me
NH
Me
Z
Me
NH
Me
Me
NH
Z
H1
H2N
C2
inversione
trans-2,5-dimetilpiperidina
H1
H2N
(S)
(S)
NH2
Z
180°
NH2
H2
Z
H2N
NH2
H2
trans-1,2-cicloesandiammina
19
Figura 1.16
Un semplice modo per determinare l’omotopia di due gruppi consiste nel sostituire a uno un
generico gruppo Z. Se il composto che si ottiene è identico a quello che si otterrebbe sostituendo
l’altro gruppo con lo stesso Z, allora i due gruppi sono omotopici.
Le facce omotopiche sono caratterizzate da un piano che contiene un asse di simmetria
coplanare. Due facce di una molecola, tipicamente definite da un doppio legame, sono equivalenti o
omotopiche se l’addizione di un reagente all’una o all’altra dà luogo allo stesso composto. Per
esempio la riduzione del cicloesanone fornisce lo stesso alcol indipendentemente da quella faccia
del doppio legame riceve la molecola di idrogeno (Figura 1.17).
O
H
OH HO
H
NaBH4
cicloesanone
Figura 1.17
Facce e gruppi enantiotopici. Se gruppi o facce non sono omotopici, allora sono eterotopici,
questi ultimi distinguibili in enantiotopici o diastereotopici.
La sostituzione di un gruppo enantiotopico con un generico sostituente Z dà luogo
all’enantiomero del composto che si otterrebbe sostituendo l’altro gruppo con lo stesso Z. Per
esempio, gli atomi di idrogeno HA e HB del t-butil etil chetone di Figura 1.18 sono enantiotopici.
Infatti, la sostituzione di uno di essi fornisce una unità centrochirale, la cui configurazione dipende
da quale dei due è sostituito.
Gruppi enantiotopici possono anche essere presenti in altri tipi di chiralità, come quella assiale.
Così, i due metili MeA e MeB del derivato bifenilico di Figura 1.18 sono enantiotopici, e la loro
selettiva funzionalizzazione dà come prodotti due enantiomeri.
Anche le coppie solitarie possono essere enantiotopiche, come nel caso di MeSEt (Figura 1.18).
I gruppi enantiotopici possono essere etichettati come pro-(R) e pro-(S), a seconda se un
ipotetico prodotto, ottenuto per sostituzione con un gruppo di massima priorità, abbia
configurazione (R) o (S). Ovviamente, non esiste una correlazione diretta tra queste etichette e
quelle dei prodotti che si possono ottenere a partire da un substrato prochirale, come illustrato in
Figura 1.18 per il dimetilchetale dell’acetofenone. I due prodotti, ottenuti nel primo caso per
sostituzione con –Cl e nel secondo per sostituzione con –H, hanno configurazione rispettivamente
(R) e (S), sulla base della convenzione di Cahn, Ingold e Prelog.
20
O
t-butil etil chetone
t-Bu
Br
MeA
Me
(S)
Me
Br
Z HB
O
t-Bu
MeA
O
HA HB
HA
enantiotopici
Me
Me
t-Bu
pro-(S)
pro-(R)
enantiomeri
Me
(R)
enantiomeri
MeB
Br
Br
Z
(R)
Me
Me
(S)
O
S
pro-(S)
Et
pro-(R) pro-(S)
Me
O
Br
Et
S
Me
MeB
(R)
MeO OMe
MeO H
Ph
Ph
Ph
Et
S
pro-(R)
MeO Cl
(R)
Me
Me
(S)
Me
Figura 1.18
La disposizione di gruppi enantiotopici può essere più complessa. Per esempio nel composto NBoc pirrolidina di Figura 1.19 sono quattro gli idrogeni da prendere in considerazione.
HA
Boc
N HC
HB
HD
Boc
N Z
Z
Boc
N HC
immagini HA
speculari
HB
HD
HB
identici
identici
HA
Boc
N HC
immagini
speculari HA
Boc
N HC
Z
Figura 1.19
HD
Z
HB
HD
HA, HD omotopici
HA, HC enantiotopici
HA, HB enantiotopici
HB, HC omotopici
HB, HD enantiotopici
HC, HD enantiotopici
Tra questi esistono due relazioni omotopiche e quattro relazioni enantiotopiche.
Composti meso (vale a dire composti che contengono unità stereogeniche e tuttavia sono
achirali per la presenza di un piano di simmetria) contengono gruppi enantiotopici, come quelli
presenti nella cis-2,5-dimetilpiperidina e nell’ossido di cicloesene (Figura 1.20).
enantiotopici
Me
NH
Me
Z
NH
Me
enantiomeri
Me
NH
Z
(R)
(R)
HO
N3
enantiomeri
(S)
(R)
O
ossido
di cicloesene
cis-2,5-dimetilpiperidina
(S)
(S)
N3
OH
Figura 1.20
21
In modo simile, l’addizione a facce enantiotopiche fornisce enantiomeri. Per esempio,
l’addizione all’acetofenone o al trans-2-butene fornisce molecole chirali che contengono una e due
unità centrochirali, rispettivamente (Figura 1.21).
enantiomeri
enantiomeri
Re
Re
2
3
Me
H
1
O
NaBH4
Ph
Me
2
3
H OH
HO H
Ph
Me
Ph
addizione alla faccia Re
1'
OsO4
Me
1
Me
addizione alla faccia Si
Si
H
Me
3'
2'
H
H
OH
Me
OH
Me
H
addizione dall'alto
alla faccia Re
Re
OH
H
Me
OH
addizione dal basso
alla faccia Si
Figura 1.21
La configurazione del prodotto dipende da quale delle due facce prochirali subisce l’attacco. La
direzione di attacco può essere descritta assegnando la priorità ai sostituenti legati al centro
trigonale coinvolto, mediante le regole di Cahn, Ingold e Prelog. La faccia che vede i tre gruppi
apparire in senso orario è definita Re, l’altra Si (Figura 1.21). Anche in questo non esiste
correlazione tra Re/Si e le configurazioni (R)/(S) del prodotto di attacco.
Se l’attacco genera due nuovi centri stereogenici, come appunto nel caso del trans-2-butene, si
devono assegnare le etichette di attacco ad ambedue i centri trigonali coinvolti. Nel caso illustrato in
Figura 1.21, essi sono entrambi Re qualora l’attacco avvenga dall’alto, e entrambi Si nel caso in cui
si verifichi da parte opposta (da dietro).
La discriminazione tra facce prochirali può dar luogo a enantiomeri con altri tipi di unità
stereogeniche, come nell’esempio di Figura 1.22 in cui il substrato aromatico che possiede facce
enantiotopiche dà luogo a un prodotto dotato di chiralità planare.
enantiomeri
Cr(CO)3
Me
Me
Cr(CO)6
Me
Me
Me
Me
Me
(CO)3Cr
Me
Me
Figura 1.22
Gruppi enantiotopici e facce enantiotopiche non sono interscambiabili da assi di simmetria Cn,
ma devono poterlo essere mediante l’applicazione o di piani σ, o del centro di inversione i o di assi
impropri Sn. Chiaramente, dato che le molecole chirali sono prive di questi elementi di simmetria,
esse non possono contenere gruppi enantiotopici o facce enantiotopiche.
Composti achirali con gruppi enantiotopici o facce enantiotopiche sono detti prochirali, perché
sono immediati precursori di composti chirali.
Facce e gruppi diastereotopici. Due gruppi sono diastereotopici se la sostituzione di uno con un
generico sostituente Z dà luogo a un diastereoisomero del composto che si otterrebbe
22
sostituendo l’altro gruppo con lo stesso Z (Figura 1.23). Per esempio, i protoni metilenici della
serina sono diastereotopici, a causa della presenza contemporanea di una unità stereogenica nella
stessa molecola (Figura 1.23).
Z
Hb
H
(S)
CO2H
H2N
HO
diastereotopici
HA
Hb
H
(S)
CO2H
H2N
HO
diastereoisomeri
HA
HO
Z
H
(S)
CO
H2N
2H
(S)-serina
Figura 1.23
Gruppi diastereotopici sono chimicamente diversi, e non possono essere scambiati da alcun
elemento di simmetria.
Due facce di un composto sono invece diastereotopiche qualora l’addizione di un generico
reagente achirale Z dia luogo a due diastereoisomeri a seconda della faccia su cui avviene
l’addizione (Figura 1.24). Poiché gli stati di transizione degli attacchi alle due facce non sono uguali
(a causa della presenza di un altro centro chirale), allora i due diastereoisomeri possono formarsi in
ammontare diversi. Per esempio, nel caso del substrato chirale (R)-3-idrossibutene di Figura 24,
l’epossidazione fornisce due nuove molecole chirali, tra loro diastereoisomere, ciascuna con due
unità centrochirali, in rapporto 19:1.
(R)
ox
O
O
(R)
(R)
OH
(R)
(S)
OH
19
OH
:
1
Figura 1.24
Qualora si fosse invece partiti dal substrato racemo, ciascun enantiometro di partenza avrebbe
reagito in modo analogo, vale a dire ancora privilegiando l’addizione sin al gruppo OH. I prodotti
sarebbero stati 4, due coppie di enantiomeri in rapporto 19:1 (Figura 1.25).
O
O
(R)
(R)
(R)
(S)
OH
ox
OH
19
O
OH
:
O
(S)
(S)
OH
19
1
(S)
(S)
OH
:
1
Figura 1.25
23
L’epossidazione dell’(S)-2-metil-3-cis-pentenolo fornisce due molecole chirali, tra loro
diastereoisomere, ciascuna con tre unità centrochirali, addirittura in rapporto 400:1 (Figura 1.26).
HO
(S)
ox
Me
(S)
(R)
HO
HO
(R)
Me
Me
(S)-2-metil-3-cis-pentenolo
(R)
(R)
O
(S)
Me
Me
1
O
:
Me
400
Figura 1.26
1.4 Classi di reazioni asimmetriche
Reazioni stereoselettive e reazioni stereospecifiche. Una reazione è stereoselettiva se favorisce
la formazione di uno stereoisomero rispetto a un altro.
OTMS
O
O
+
H
t-BuS
OBn
OH
O
OBn
(S)
t-BuS
99
OTMS
t-BuS
(R)
OBn
(S)
t-BuS
O
OBn
98
O
t-BuS
O
+
H
t-BuS
O
OH
(S)
(R)
t-BuS
:
O
OBn
5
OH
(R)
O
OBn
:
O
t-BuS
(S)
O
O
1
O
OBn
OH
(R)
t-BuS
:
OBn
1.5
OH
(R)
t-BuS
(R)
t-BuS
:
(R)
OBn
OH
(S)
OBn
96
OBn
(S)
5
OH
(S)
O
OH
(R)
t-BuS
45
EtS
OBn
2
OBn
45
(R)
+
H
(R)
OBn
t-BuS
OTMS
(R)
:
(S)
OH
t-BuS
OH
(S)
OTMS
1
OH
(R)
OBn
(R)
t-BuS
:
O
O
+
H
OH
(S)
OBn
1.5
Figura 1.27
Le reazioni possono essere enantioselettive, vale a dire è favorito un enantiomero rispetto a un
altro (primo esempio di Figura 1.27), diastereoselettive, cioè si verifica la prevalenza di un
diastereoisomero (secondo esempio) o di una coppia di enantiomeri (terzo esempio), o possedere
entrambe le qualità (quarto esempio, dove predomina un singolo enantiomero delle due coppie di
enantiomeri).
Una reazione è invece stereospecifica se reagenti con diversa configurazione danno luogo a
prodotti con stereochimica distinta. Di conseguenza, un processo stereospecifico a partire da un
24
singolo stereoisomero è inevitabilmente stereoselettivo, mentre non è necessariamente vero il
contrario.
Un esempio di quanto detto è raffigurato in Figura 1.28. L’idrogenazione (promossa da un
complesso chirale di rodio) delle due enammidi E,Z conduce allo stesso enantiomero. Questa è una
reazione stereoselettiva (enantioselettiva), ma non stereospecifica.
La seconda reazione invece è chiaramente sia stereoselettiva sia stereospecifica, perché
l’isomero E e l’isomero Z di partenza non conducono allo stesso stereoisomero, bensì a due prodotti
distinti, tra loro diastereoisomeri.
Me
H
(Z)
AcHN
H2
Me
(R)
CO2Me
AcHN
CO2Me
> 99 ee
H
Me
(E)
AcHN
H2
stereoselettive
Me
(R)
CO2Me
AcHN
CO2Me
> 99.6%
AcHN
Me
(Z)
BzNH
H2
Me
NHAc
(S)
(S)
CO2Et
BzNH
CO2Et
96% ee
Me
NHAc
(E)
BzNH
CO2Et
H2
Me
(R)
BzNH
stereospecifiche e
stereoselettive
NHAc
(S)
CO2Et
> 98% ee
Figura 1.28
Transfer di chiralità. Il transfer di chiralità si riferisce alla formazione stereoselettiva di un
nuovo elemento stereochimico a scapito di un altro. Anche queste reazioni sono stereospecifiche.
Un classico esempio è la reazione di tipo SN2 in cui si osserva inversione di chiralità alle unità
centrochirali di ciascuna molecola (Figura 1.29). Se il reagente è enantioricco, lo sarà anche il
prodotto.
In contrasto, la reazione con meccanismo SN1 non conserva né inverte la stereochimica
dell’unità centrochirale originaria: il prodotto a partire da un composto enantiomericamente puro
sarà ancora dotata di una unità centrochirale, ma racema (Figura 1.29).
Infine, una eliminazione E2 determinerà completa perdita di centrochiralità e formazione di un
25
composto achirale. Tuttavia, va notato che un nuovo elemento stereochimico, correlato con
l’isomeria E, sarà generato. In questo caso, la reazione è stereoselettiva, ma non stereospecifica,
poiché l’altro enantiomero darà luogo allo stesso alchene (Figura 1.29).
Cl H
Me
Cl
H I
NaI
Me
SN2
H
Me
H OEt
EtOH
EtO H
Me
Me
SN1
Cl H
+
50
:
50
Et3N
Me
E2
Figura 1.29
Il transfer di chiralità può anche consentire la trasformazione di un tipo di unità chirale in
un’altra. Nella reazione del mesilato propargilico con PhZnCl catalizzata da Pd(II) un’unità
centrochirale è scambiata con una assiale (Figura 1.30).
H
OMs
PhZnCl
Ph
F3C
F3C
n-C6H13
n-C6H13
Figura 1.30
Altre selettività: regioselettività e chemoselettività. In una reazione catalitica asimmetrica, si
possono formare anche prodotti che non sono tra loro stereoisomeri. In questo caso, sorge la
necessità di introdurre altri descrittori in aggiunta ai già citati enantioselettività e
diastereoselettività.
La regioselettività si configura come la propensione di un reagente a reagire preferenzialmente
con un sito di una molecola lungo una direzione preferenziale, privilegiando la formazione di un
isomero costituzionale rispetto ad altri. Un esempio è la formazione di una aloidrina a partire dal 2propenilbenzene (Figura 1.31).
OH
Br
Br
NBS
OH
+
80
:
20
Figura 1.31
Un altro tipo di selettività è la chemoselettività, che deve essere specificata quando una reazione
26
può dar luogo a più prodotti. Un esempio è quello di Figura 1.32, in cui l’α-chetoestere potrebbe
reagire con dietilzinco in presenza di un complesso di Ti come catalizzatore, portando alla
formazione o del prodotto di addizione o di quello di riduzione.
O
OMe Et2Zn
O
OH
HO Et
OMe +
O
OMe
O
Figura 1.32
27
2. I Processi Enantioselettivi
A fronte di un grandissimo numero di processi enantioselettivi messi a punto, il numero dei
relativi approcci strategici è decisamente più limitato. I principali includono:
- differenziazione di gruppi enantiotopici o facce enantiotopiche di substrati prochirali;
- conversione di miscele raceme in singoli enantiomeri.
Segue una breve presentazione di questi approcci al fine di generare un singolo enantiomero
dotato di una unità stereogenica.
2.1 Differenziazione di gruppi enantiotopici o facce enantiotopiche di substrati prochirali
Per iniziare, è possibile prendere in considerazione la differenziazione di gruppi enantiotopici.
Come già avvisato, questi spesso sono due sostituenti presenti su un centro di una molecola
achirale. Per esempio, nel precursore prochirale dell’esomeprazolo, gruppi enantiotopici sono le due
coppie solitarie presenti sull’atomo di zolfo (Figura 2.1).
MeO
Me
N
S
N
H
OMe
ox
N O
MeO
Me
OMe
S
N
H
esomeprazolo
Me
Me
Figura 2.1
L’ossidazione selettiva di uno solo di essi, mediante un complesso chirale Ti-tartrato, fornisce
l’esomeprazolo con l’unità centrochirale allo zolfo in elevata purezza enantiomerica.
Anche la desimetrizzazione di composti achirali meso tipicamente procede mediante distinzione
tra gruppi enantiotopici. Un esempio è l’ossidazione enantioselettiva, da parte di un complesso
chirale di rutenio, del diolo meso ciclico di Figura 2.2. In questo caso, il catalizzatore è in grado di
ossidare solo uno dei due gruppi –OH enantiotopici, fornendo un prodotto con un ee del 90%.
OH
OH
OH
O
enantiotopici
Figura 2.2
In modo analogo, è possibile ottenere prodotti enantiomericamente arricchiti attraverso la
differenziazione di facce enantiotopiche. I precursori che più tipicamente sono dotati di tali requisiti
geometrici sono carbonili, immine, e alcheni. Esempi di reazioni coinvolgenti questa classe di
28
composti sono illustrati in Figura 2.3.
O
Ph
OH
Et2Zn
H
N
Ph
Me
AcHN
Ph
CH2Ph
HCN
HN
H
H
H
Ph
H
Et
CH2Ph
CN
Me
H2
CO2Me
AcHN
CO2Me
Figura 2.3
In tutti i casi, il catalizzatore attiva l’addizione del reagente (rispettivamente EtZn-Et, H-CN o
H-H) su una faccia preferenziale, con formazione di un composto arricchito enantiomericamente. Si
fa notare come nel caso della reazione riportata a carico delle immine, i prodotti sono αamminonitrili facilmente convertibili nei corrispondenti α-amminoacidi, precursori di grande
interesse per l’industria di chimica fine e farmaceutica.
Dettagli cinetici. Per poter comprendere più a fondo caratteristiche e problematiche di questo
tipo di strategia, consideriamola in maggior dettaglio. Nel caso ideale, il reagente non è in grado di
reagire con il substrato in assenza di un catalizzatore, vale a dire, l’energia di attivazione del
processo non catalizzato è molto alta (Figura 2.4)
E
reazione non
catalizzata
∆G
S
reazione catalizzata
verso (R)
reazione catalizzata
verso (S)
P
Figura 2.4
L’aggiunta di una quantità substechiometrica di un legante chirale determina la formazione di
una specie catalitica che è in grado di accelerare la reazione. Nella reazione illustrata in Figura 2.5,
il substrato prochirale è l’aldeide e il reagente stechiometrico è il dietilzinco.
29
Me
Me
Me
Me
Et2Zn
Me
NMe2
-etano
OH
Me
Me
Me
N Zn Et
O
Me
N
Et2Zn
Ph acqua
Et H
Et
Zn
O
Et
Zn
PhCHO
Me
Zn
Et
Et
N
Me
Et
Zn
Ph
Et H
stadi di transizione
Ph diastereoisomerici
H
O
O
Me
HO
Me
O
O
n
Me
(S)
Zn
Et
Et
Ph
H
Figura 2.5
Queste due specie, da sole, non sono in grado di reagire a temperatura ambiente. In presenza di
un piccolo ammontare di un β-amminoalcol chirale si ha l’immediata formazione di un addotto tra
quest’ultimo e il dietilzinco, che, a seguito dell’evoluzione di etano, dà luogo a un alcossido di
zinco chirale. Questi non è in grado di trasferire il proprio etile all’aldeide, ma può attivare un’altra
molecola di dietilzinco verso questo processo (in pratica, funge da catalizzatore).
L’intorno chirale generato dal legante riesce, a questo punto, a favorire l’approccio del
dietilzinco a una sola delle due facce prochirali dell’aldeide, perché gli stati di transizione relativi
alle due possibilità (∆GTS1 e ∆GTS2) sono diastereoisomerici, e quindi presentano diverse energie di
attivazione. In particolare, quello relativo all’ottenimento del prodotto a configurazione (S) è
minore, e dunque è privilegiato l’ottenimento di questo enantiomero.
Siccome, nelle condizioni date, la reazione non catalizzata non produce prodotto in quantità
apprezzabili, l’eccesso enantiomerico di quest’ultimo dipenderà esclusivamente dalla differenza in
energia tra i due cammini catalizzati, vale a dire dal valore di ∆G= ∆GTS1 - ∆GTS2. In questo caso,
esso è pari a 2.7 kcal/mol, il che corrisponde a rapporto enantiomerico K pari a circa 99:1, ottenuto
quest’ultimo attraverso la relazione ∆G= RTlnK.
Va sottolineato come questo tipo di valutazione sia valido solo in presenza di due cammini
diastereoisomerici, mentre in casi più complessi la trattazione è più delicata e deve tener conto di un
maggior numero di fattori. Per esempio, qualora la stessa reazione di Figura 2.5 coinvolga il
butanale al posto della benzaldeide, sono ben otto i possibili cammini diastereoisomerici, derivanti
da altrettante possibili direzioni di approccio dell’aldeide al centro attivo. In questo caso, l’eccesso
enantiomerico dipenderà dai valori relativi delle numerose energie in gioco, i cui valori possono
spesso essere predetti mediante calcolo.
30
Si può notare anche come in questa reazione la specie catalitica sia l’addotto alcossido di zinco,
ottenuto per modificazione di una quantità substechiometrica proprio del reagente in seguito
all’aggiunta del legante chirale. In altri casi, tra l’altro molto più comuni, il catalizzatore metallico è
invece un’entità chimica aggiunta ad hoc.
Effetto della temperatura, del solvente, della concentrazione e della pressione. Qualora la
differenza energetica tra i due cammini diastereoisomerici ∆G= ∆GTS1 – ∆GTS2 sia maggiore di 2
kcal/mol, a temperatura ambiente si osservano elevati eccessi enantiomerici. Se tale valore si
riduce, per esempio a 1 kcal/mol, allora lo stesso beneficio può essere ottenuto solo abbassando la
temperatura. E’ facile calcolare che un ee del 95% ottenuto a 25°C con un ∆G di 2.2 kcal/mol,
possa essere ottenuto solo a -78°C qualora tale differenza scenda a 1.4 kcal/mol.
Anche la dipendenza dalla temperatura può essere ben più complicata, e non solo nel caso in cui
siano coinvolti più cammini diastereoisomerici, ma anche qualora la reazione proceda attraverso
diversi meccanismi che diventano a turno prevalenti. Oppure, al crescere della stessa temperatura, la
reazione catalizzata può essere affiancata da quella non catalizzata, che, procedendo ovviamente in
modo non selettivo, può provocare una graduale netta diminuzione dell’eccesso enantiomerico del
prodotto rispetto a quella attesa.
Altri parametri, come la presenza di additivi, il tipo di solvente, la concentrazione e la pressione
possono avere effetti importanti sull’enantioselettività. In particolare, la scelta del solvente può
essere davvero cruciale nel determinare la selettività di un processo, sia a causa delle sue proprietà
di insieme (bulk), come polarità, costante dielettrica, sia per la capacità di partecipare alla reazione
fungendo da legante, da acido o base, e da transfer per reagenti e/o prodotti.
Per quel che riguarda le proprietà bulk, non è facile prevedere l’effetto del solvente, e spesso
questo viene determinato sperimentalmente. E’ comunque possibile fornire alcune indicazioni di
carattere assolutamente generale. In primo luogo, solventi polari non coordinanti privilegiano
tipicamente alti ee rispetto ad altri dotati di minor polarità. Per esempio, la reazione di Figura 6.1
procede con prestazioni migliori usando il toluene in luogo dell’esano. In contrasto, solventi
coordinanti, come il THF, ne determinano una riduzione sensibile, a causa della loro capacità di
inibire il catalizzatore.
Un esempio clamoroso di dipendenza dell’eccesso enantiomerico dal solvente riguarda la
reazione di Diels-Alder, promossa da un complesso chirale di titanio, illustrata in Figura 2.6.
31
O
O
N
H
TiLn
O +
O
N
O
O
Figura 2.6
L’eccesso enantiomerico del prodotto passa da zero in diclorometano all’88% in nitrometano,
solvente dotato di elevata polarità. Studi dettagliati hanno dimostrato come nei due solventi siano
diversi i modi con cui il dienofilo coordina il substrato, e solo nel secondo questo sia vantaggioso
per l’enantioselezione.
In altri casi, tuttavia, sono proprio i solventi dotati di proprietà basiche ad agire come transfer di
reagenti, e ad avere quindi prestazioni più benefiche.
Per quel che riguarda l’effetto della concentrazione, va anticipato che quest’ultima tipicamente
non ha un grosso impatto sull’ee. Tuttavia, si riscontra generalmente un miglioramento al crescere
delle concentrazioni, e questo ha un vantaggio anche perché si riduce l’uso di solventi e di sostanze
ausiliarie.
Qualora sia coinvolto un gas nella reazione, va considerato anche l’effetto della pressione, il
quale comunque non ha andamenti facilmente prevedibili.
Altri importanti effetti. Un altro effetto molto importante, già anticipato precedentemente, è
quello relativo all’eventuale competizione della reazione non catalizzata. Un esempio
paradigmatico è quello relativo all’addizione di reattivi di Grignard a chetoni con ottenimento di
alcoli chirali (Figura 2.7).
E
reazione catalizzata
verso (S)
reazione catalizzata
verso (R)
OH
O
H + RMgX
S
R
OH
+
R
reazione non
catalizzata
P
Figura 2.7
In questo caso, come illustrato dal profilo energetico, la reazione non catalizzata procede con
32
energia di attivazione nettamente inferiore rispetto a quella dei due cammini diastereoisomerici. E
sebbene, tra questi due, sia nettamente favorito il percorso che conduce all’enantiomero (R), proprio
il verificarsi dell’evento non catalitico riduce l’ee, rendendo proibitivo lo sviluppo di questo
processo enantioselettivo.
Un altro tipo di problematica (definito catalisi decelerata dal legante) ha luogo quando, sebbene
la reazione non catalizzata sia silente, il centro metallico sia più attivo in assenza del legante chirale,
secondo un profilo energetico come quello illustrato in Figura 2.8.
reazione non
catalizzata
E
reazione catalizzata
con il legante chirale verso (S)
O
Me
H
+
reazione catalizzata
con il legante chirale verso (R)
S
reazione catalizzata
senza legante chirale
(S)
Me O
P
H
H
(R)
+
O
Me
Figura 2.8
Nel caso in questione, il catalizzatore chirale è un complesso di rame(II) contenente un legante
chirale piridinossazolinico, in grado di promuovere la reazione di Diels-Alder con buona
enantioselettività. Tuttavia, il corrispondente composto di rame(II) Cu(OTf)2, privo del legante
chirale e semplicemente solvatato, risulta più attivo, e questo determina che una parte della reazione
possa decorrere senza alcuna selettività. Per deprimere questa reazione, si utilizza generalmente un
eccesso di legante chirale rispetto al centro metallico, in modo da spostare nettamente verso destra
l’equilibrio di complessazione, e annullare l’effetto della specie “nuda” Cu(OTf)2:
Cu(OTf)2 + legante → [Cu(legante)](OTf)2
2.2 Conversione di miscele raceme in singoli enantiomeri
Invece di partire da un composto achirale e generare un singolo enantiomero con una nuova
unità stereogenica, un approccio alternativo consiste nell’usare miscele raceme di molecole chirali,
già provviste dunque di una unità stereogenica.
Risoluzione cinetica (KR). Miscele raceme possono essere risolte mediante un catalizzatore
chirale, attraverso un processo definito risoluzione cinetica (KR). Nel caso ideale, in questo tipo di
reazione il catalizzatore è in grado di convertire uno dei due enantiomeri del reagente molto più
33
rapidamente dell’altro, dando così luogo a un prodotto enantiomericamente puro, accompagnato da
un substrato altrettanto puro.
Il tipo di diagramma energetico che ne deriva è riportato in Figura 2.9, dove SR e SS sono i due
enantiomeri del substrato, PR e PS i due enantiomeri del prodotto, ∆G‡S e ∆G‡R le energie di
attivazione relative alle conversioni dei due enantiomeri.
E
∆∆G = ∆GR - ∆GS
∆GR
∆GS
SR
SS
PR
PS
Figura 2.9
L’efficienza di una risoluzione cinetica è data dalla velocità relativa con i substrati enantiomeri
SR e SS reagiscono in presenza del catalizzatore chirale per dar luogo ai prodotti PR e PS. La velocità
relativa krel= kfast/kslow o il fattore di selettività, s, sono i parametri usati in genere per qualificare la
bontà del processo, secondo le equazioni cinetiche seguenti:
SS
SR
kS (= kfast)
kR (= kslow)
s = krel =
kfast
kslow
PS
PR
= e∆∆G /RT
La situazione ideale si ha quando krel è molto grande, e la reazione può essere arrestata al 50%
di conversione, consentendo così l’ottenimento di reagente e prodotto enantiomericamente molto
arricchiti.
Il fattore di selettività è strettamente connesso con ∆∆G‡, vale a dire la differenza nelle energie
tra gli stati di transizione diastereoisomerici che determinano la selettività.
Lo svantaggio di questo approccio è che, anche in caso di reazione perfetta, la resa del prodotto
enantiomericamente puro non supera il 50%, e inoltre, esso va separato dal materiale di partenza
34
non convertito. Inoltre, al procedere della reazione, l’enantiomero che reagisce più rapidamente si
consuma, e il substrato si arricchisce via dell’altro enantiomero meno reattivo. Ne consegue che la
velocità con cui essi reagiscono può a un certo punto diventare simile, perché essa dipende sia dalle
costanti cinetiche sia dalle concentrazioni relative dei due enantiomeri. Il risultato è un processo che
diventa via via meno selettivo, a meno di non avere valori molto alti di krel. Pertanto, esso è
tipicamente praticato quando si accompagna ad alte rese, brevi tempi di reazione, catalizzatori
economici, compatibilità con molti gruppi funzionali.
Un classico esempio di KR (Figura 2.10) è la risoluzione cinetica di epossidi racemi mediante
loro idrolisi catalizzata da complessi di cobalto(III) con salen chirali (l’acronimo salen indica
leganti che hanno la struttura di chelanti tetradentati N,N’,O,O’ doppiamente deprotonati
all’ossigeno, del tipo saliciletilendiammina).
N
O
O
+ H2O
Me
OH
+
Me
N
Co
O
OH
Me
t-Bu
O
OAc
t-Bu
catalizzatore
Figura 2.10
Attraverso questa reazione è possibile ottenere epossidi chirali con una resa prossima al valore
massimo teorico del 50% con eccessi enantiomerici superiori al 99%.
La forma attiva del catalizzatore è un complesso di stechiometria [Co(OH)(salen)], in grado di
coordinare ambedue gli epossidi con costanti di legame assolutamente paragonabili. Tuttavia, i
risultanti complessi diastereoisomerici presentano reattività nettamente diversa nei confronti
dell’idrolisi, il che provoca la risoluzione desiderata. Il meccanismo di azione è particolarmente
accattivante, e prevede un’azione sinergica tra due complessi metallici, uno in grado di attivare
l’azione nucleofila dell’idrossido, l’altro, l’epossido nei confronti del primo (Figura 2.11).
OH
Co
O
R
O
O
R
R
OH
Co
O
OH
Co
R
diolo
H2O
OH
Co
OH2
Figura 2.11
Si accenna all’esistenza di una strategia correlata alla KR, ma molto più vantaggiosa, definita
risoluzione cinetica parallela (PKR). In questo caso, si tratta di convertire i due enantiomeri del
35
substrato di partenza in due prodotti diversi, con una velocità paragonabile (kR ≈ kS):
kS
SS
P1
kR
SR
P2
La situazione si traduce nel fatto che, a differenza della KR, nel corso della reazione l’ee del
reagente rimane sempre prossimo a zero, e che quindi, non si verifica come nel caso di
quest’ultima, una progressiva diminuzione dell’efficienza all’aumentare della conversione. A
seconda della natura dei due prodotti la PKR può essere chemodivergente (prodotti non isomeri),
regiodivergente (prodotti regioisomeri, vale a dire il gruppo funzionale è in diverse posizioni),
stereodivergente (prodotti diastereoisomeri).
Risoluzione cinetica dinamica (DKR). Una strategia correlata alla KR, ma ancora più efficiente è
quella definita risoluzione cinetica dinamica (DKR).
La DKR accoppia una KR con una rapida racemizzazione del substrato chirale attraverso un
intermedio o stato di transizione achirale I: S
S
kS
PS
I
SR
kR
PR
Il tipo di diagramma energetico che deriva da questo è del tipo riportato in Figura 2.12 (a
sinistra I è uno stato di transizione, a destra I è un intermedio).
E
E
∆∆G = ∆GR - ∆GS
∆GR
∆∆G = ∆GR - ∆GS
∆GS
I
∆GS
SR
PR
∆GS
I
SS
SR
PR
PS
SS
PS
Figura 2.12
In altre parole, è necessario che i due enantiomeri del reagente possano convertirsi l’uno
nell’altro. Se uno di essi partecipa molto più velocemente a una trasformazione
36
enantioselettiva, allora tutto il materiale di partenza può essere convertito in un singolo
enantiomero.
In genere, una condizione che garantisce elevate prestazioni al processo è il fatto che la
racemizzazione avvenga più velocemente della conversione più rapida (krac > kfast).
E’ importante sottolineare che in una DKR la racemizzazione NON coinvolge il catalizzatore
chirale, bensì avviene attraverso un meccanismo indipendente, che può per esempio prevedere
l’intervento di una base, di un acido, processi termici, sequenze di addizione/eliminazione e così
via.
Un esempio è quello riportato in Figura 2.13, dove un reattivo di Grignard chirale non è
configurazionalmente stabile, e quindi i due enantiomeri sono in rapido equilibrio (mediante un
intermedio achirale che media la racemizzazione). Uno di essi si addiziona molto più rapidamente
al substrato carbonilico in presenza di un catalizzato chirale di palladio, e il risultato è l’ottenimento
di un prodotto con il 95% ee.
Me3Si H
Ph
MgBr Br
H SiMe3
Ph
Ph
Ph
ee 95%
H SiMe3
Ph
MgBr
Figura 2.13
Un altro esempio è la risoluzione cinetica dinamica di composti 1,3-dicarbonilici α-disostituiti
mediante idrogenazione, come quella riportata in Figura 2.14.
O
O
R
OMe
OH O
R
OH O
H2
OMe + R
R
Me
Me
OMe
O
Me
H2
OMe
OH O
OMe + R
R
Me
OMe
Me
OH O
O
R
OH O
Me
OMe
Me
Figura 2.14
I due enantiomeri del substrato racemo possono questa volta interconvertirsi mediante il
passaggio (catalizzato da una base) attraverso l’intermedio achirale planare enolico.
In presenza di un adatto catalizzatore chirale a base di Ru, l’idrogenazione selettiva del
carbonile può condurre alla formazione di uno solo dei quattro possibili prodotti di idrogenazione βidrossicarbonili α-sostituti (reazione diastereo- e enantio-selettiva).
37
Trasformazione asimmetrica cinetica dinamica (DyKat). Sebbene questa sia, per tanti aspetti,
correlata alla DKR, va trattata a parte perché connotata da alcune sostanziali differenze. La più
rilevante riguarda il meccanismo di inversione dello stereocentro pre-esistente nel substrato. Infatti
nella DKR, il fenomeno non interessa il catalizzatore chirale, bensì avviene attraverso un processo
coinvolgente un intermedio o uno stato di transizione achirale, mediato spesso da un altro
catalizzatore.
Invece nella trasformazione asimmetrica cinetica dinamica (DyKat) l’interconversione tra gli
enantiomeri del substrato avviene sul catalizzatore chirale. Per questo motivo, l’inversione di
configurazione sul catalizzatore dà luogo piuttosto a un diastereoisomero, e più che una
racemizzazione è una epimerizzazione. Esistono due sottoclassi di DyKat, definite di tipo A e B:
SS
kScat*
Cat*SS
k"Scat*
PS
SS
k'Scat*
Cat*S
SR
kRcat*
k'Rcat*
k"Rcat*
Cat*SR
kRcat*
kScat*
k'Scat*
PS
Cat*S
k'Rcat*
SR
PR
PR
Tipo A
Tipo B
Nel primo, i due substrati SR e SS si legano al catalizzatore metallico chirale dando vita a due
diastereoisomeri (Cat*SS e Cat*SR). L’epimerizzazione avviene attraverso uno stato di transizione o
un intermedio achirale. In Figura 2.15 è riportato a sinistra il diagramma energetico relativo al caso
in cui Cat*S sia uno stato di transizione, a partire dai due intermedi Cat*SS e Cat*SR, per brevità
indicati con I1 e I2, rispettivamente.
E
E
I1= Cat*SR
I2= Cat*SS
I1
I2
SR
I
SS
PR
SS
SR
PS
PS
PR
Tipo B
Tipo A
Figura 2.15
38
Un esempio di DyKat di tipo A è riportato in Figura 2.16 e si riferisce a una reazione di
alchilazione allilica catalizzata da Pd.
O
R
L*Pd
O
(S)
O
O
O
O
L*Pd
O
O
O
O
PS
O PdL*
ArO-
L*Pd
O
ArO
O
O
I1
SR
ArO
O
O
I2
SS
R
ArO-
L*Pd
O
O
PR
Figura 2.16
In questo caso, i due carbossilati enantiomeri di partenza SS e SR danno luogo a due intermedi πallilici diastereoisomerici I1 e I2, che possono equilibrarsi attraverso un intermedio σ-alcossido.
Nel caso di una DyKat di classe B, i due substrati enantiomeri formano un singolo addotto col
metallo, da cui attraverso cammini diastereoisomerici si perviene all’ottenimento dei prodotti.
Pertanto, la selettività dipende solo dalle velocità relative k’Cat*S/k’Cat*R. Un caso di reazione che
procede in questo modo è l’alchilazione allilica dell’acetato di allile raffigurata in Figura 2.17.
OAc
Ph
(S)
Nu
Ph
Ph
PS
Ph
PdL*
OAc
(R)
veloce
PdL*
PdL*
Ph
Ph
(S)
SS
Nu-= -CH(CO2Me)2
Ph
Nu-
lenta
Nu
Ph
(R)
Ph
SR
Ph
PR
Figura 2.17
Entrambi i substrati reagiscono con il catalizzatore a base di palladio producendo lo stesso
complesso π-allilico, in cui il legante idrocarbilico è chiaramente achirale. La presenza del legante
chirale L* rende gli atomi di carbonio terminali del sistema allilico diastereoisotopici, e quindi
suscettibili di attacco selettivo da parte del nucleofilo con formazione preferenziale del prodotto a
configurazione (S).
39
2.3 Principio di Curtin-Hammett
Un tipo di processo come quello descritto in Figura 2.15 consente di introdurre un postulato
molto importante, che regola i processi multistadio di catalisi enantioselettiva in cui vi siano
intermedi diastereoisomerici (come appunto I1 e I2) che possano essere in rapido equilibrio tra loro.
L’equilibrio può stabilirsi in vari modi, come per esempio attraverso il ritorno ai reagenti, o in
modo diretto, qualora i due intermedi siano isomeri conformazionali e può bastare una semplice
rotazione per passare dall’uno all’altro.
Il principio di Curtin-Hammett afferma che l’enantioselezione dell’intero processo è legata al
valore di ∆G (vedi Figura 2.18), e quindi dipende dalle energie relative degli intermedi I1 e I2 e
dalle energie di attivazione con cui questi si convertono nei prodotti. La matematica da cui si deriva
questa affermazione è semplice ed è illustrata nella Figura (k1 e k2 sono le costanti cinetiche di
ottenimento dei prodotti P1 e P2 a partire da I1 e I2, mentre K è il loro rapporto di equilibrio).
E
d[P2]
= k2[I2]
dt
∆G
d[P1]
= k1[I1] = k1K[I2]
dt
∆G(I1)
d[P1]
dt
d[P2]
dt
∆G(I2)
I2
I1
∆G°
k1K[I2]
k2[I2]
P1
= e-∆G/RT
P2
P2
P1
=
=
k1K
k2
e-∆G(I1)/RTe∆G°/RT
=
-∆G/RT
=e
e-∆G(I2)/RT
∆G= ∆G(I1) - [∆G(I2) + ∆G°]
Figura 2.18
Un esempio di validità del principio può essere riconosciuto nella idrogenazione asimmetrica di
enammidi di Figura 2.19, dove PP è la fosfina chirale nota con l'acronimo di (S,S-diop).
H
N
H
CO2Et
Ph
NHCOMe
Me
CO2Et
+ P
Ph Rh
P
O
I1 (>95%)
+
P + solv
Rh
P solv
H
N
CO2Et
P
+
H
Ph Rh P
O H
H2
lenta
Me
H2
veloce
Me
Me
EtO2C
Ph
(S)
HN
O
P1 (<2.5%)
S= solvente
stadio limitante
H
N
EtO2C
P +
Rh Ph
P
O
I2 (<5%)
CO2Et
P
H + S
Ph Rh P
O H
HN
EtO2C H
N
P
H +
Rh Ph
P
H O
Me
Figura 2.19
EtO2C
NH
P
S +H
Rh Ph
P
H O
CO2Et
Ph
(R)
NH
Me
O
P2 (>97.5%)
40
La reazione produce, con uno stadio limitante che è l’addizione irreversibile di H2, il prodotto
P2 con un eccesso enantiomerico superiore al 95%, sebbene studi spettroscopici abbiano rivelato
che l’equilibrio coinvolgente i due intermedi di reazione, I1 e I2, favorisca nettamente il primo.
In questo caso, l’elevata selettività deriva dalla combinazione di due fattori: le stabilità
termodinamiche relative degli intermedi e la loro reattività. Dal diagramma dell’energia si evince
come in questo caso i due fattori vadano in senso opposto (non è comunque sempre questo il caso),
a dimostrazione di quanto sia fuorviante argomentare che la stereochimica del prodotto principale
derivi necessariamente da quella dell’intermedio più stabile.
Va ricordato che questa è una reazione molto importante perché consente la sintesi di molecole
attive contro il morbo di Parkinson.
2.4 Creazione di chiralità
Creazione di centrochiralità. Tutti i metodi appena descritti sono utili per creare unità
centrochirali. La maggior parte trovano applicazione nell’ottenimento di atomi di carbonio
stereogenici, ma sono validi anche per creare unità centrochirali al silicio, azoto, fosforo e zolfo.
Creazione di chiralità assiale. Composti atropoisomerici enantiomericamente puri possono
essere ottenuti attraverso:
- il metodo introdotto nel punto 2.1 (differenziazione enantiotopica di composti con una unità
assiale prochirale);
- il metodo introdotto nel punto 2.2 (risoluzione cinetica e risoluzione cinetica dinamica);
- creazione della chiralità assiale contemporanea a quella dell’asse chirale;
- il transfer di chiralità.
Un esempio del primo è riportato in Figura 2.20, dove un reattivo di Grignard discrimina tra due
gruppi triflato in relazione enantiotopica in presenza di un catalizzatore chirale di palladio.
TfO
pro-(R)
OTf
pro-(S)
RMgBr
OTf
R
(S)
95
+
:
R
TfO
(R)
5
Figura 2.20
Invece, un esempio di creazione di chiralità assiale contemporanea a quella dell’asse chirale, si
riferisce a un coupling di Suzuki (Figura 2.21), in cui un catalizzatore, sempre chirale e a base di
41
palladio, favorisce l’accoppiamento enantioselettivo del bromoderivato con l’acido boronico.
Et
B(OH)2
Et
Et
+
(R)
P(O)OEt
(S)
+
P(O)OEt
Br
P(O)OEt
96
:
4
Figura 2.21
Infine, un esempio di transfer di chiralità è quello già citato di Figura 1.30.
Creazione di chiralità planare. Mentre la creazione di chiralità planare in composti dotati di
un’altra unità stereogenica, tipicamente centrochirale, è abbastanza semplice (come i leganti di
Figura 2.22), molto più complessa è la creazione di composti enantiomericamente puri dotati della
sola chiralità planare. Per maggiori dettagli, che esulano dagli scopi del corso, si consiglia la visione
di letteratura specializzata.
Me
H (R) PCy2
Fe (S)
PPh2
Figura 2.22
Composti con più unità stereogeniche. Tutte le strategie finora discusse, che consentono
l’ottenimento di un singolo enantiomero di composti chirali dotati di una sola unità stereogenica
sono comunque applicabili alla sintesi di composti enantiomericamente puri contenenti più di una
unità stereogenica. Come esempio rilevante, si può citare l’idrogenazione asimmetrica di enammidi
catalizzata da complessi di rodio (Figura 2.23).
Ph(O)CHN
EtO2C
NHC(O)Me
Me
H2
Ph(O)CHN (S) NHC(O)Me Ph(O)CHN (R) NHC(O)Me
H
+
H (R)
H
H
(S)
EtO
C
Me
EtO2C
Me
2
98
:
2
Figura 2.23
Si può osservare come, attraverso la differenziazione delle enantiofacce, l’addizione di H2
conduca al prodotto chirale con due nuove unità stereogeniche in altissimo eccesso enantiomerico.
42
3. Modi di attivazione del substrato
Ci sono molti modi con cui un catalizzatore metallico può attivare un substrato. I più tipici
prevedono il comportamento del catalizzatore come acido di Lewis, base di Lewis, acido di
Brønsted, base di Brønsted o l’intervento in qualità di specie ioniche, di transfer di gruppi, di agenti
di cross-coupling, di attivatori π. In questo capitolo sarà presentata una breve rassegna di queste
tipologie, accompagnata da una descrizione delle caratteristiche che deve avere un substrato per
sottostare a una efficace attivazione.
3.1 Catalizzatori acidi di Lewis
Di gran lunga, il modo più comune di attivazione di un gruppo funzionale di un substrato ne
prevede l’interazione con un acido di Lewis chirale. In questo caso, un centro metallico in un
precursore LnM (l’acido di Lewis) è tipicamente fatto reagire con un legante chirale L* per dar vita
a un acido di Lewis chirale LnM- L* (Figura 3.1).
LnM + L*
LnM-L*
Me Al
O
O
esempio
Figura 3.1
Se la coordinazione della base chirale restituisce un catalizzatore più attivo dell’acido di Lewis
precursore, allora si è in presenza di un caso propizio di catalisi accelerata dal legante. Questa è
una situazione ottimale, perché non risulta necessaria la presenza di un eccesso di base per inibire
gli effetti negativi che avrebbe sulla catalisi l’azione dell’acido di Lewis privo del legante chirale.
Invece, laddove la coordinazione del legante chirale L* ha l’effetto di rallentare l’azione del
corrispondente acido di Lewis chirale, si parla di catalisi decelerata dal legante. In queste
condizioni è spesso necessario introdurre un eccesso di legante per favorire la formazione
dell’addotto con il metallo.
L’interazione tra l’acido di Lewis chirale e un substrato prevede tipicamente la coordinazione di
un gruppo funzionale di quest’ultimo al centro metallico. In genere, i substrati presentano un’ampia
gamma di funzioni adatte allo scopo, quali quelle eteree, epossidiche, esteree, chetoniche,
aldeidiche, imminiche, nitro, alcoliche e così via (Figura 3.2).
43
MLn(L*)
MLn(L*)
MLn(L*)
O
O
R'
MLn(L*)
N
O
R
H
R
H
Figura 3.2
La coordinazione al centro metallico ha generalmente l’effetto di perturbare i livelli energetici
e/o la distribuzione elettronica nel substrato, con l’effetto di esaltare la elettrofilicità di
quest’ultimo, e renderlo quindi suscettibile di attacco esterno. Generalmente, ciò si verifica perché
si ha un avvicinamento tra l’energia del LUMO del substrato e quella dell’HOMO di un nucleofilo.
Per esempio, come illustrato in Figura 3.3, il LUMO della formaldeide subisce una
destabilizzazione energetica in seguito alla coordinazione del carbonile all’acido di Lewis.
H
MLn(L*)
H
O
O
O
O
H
H
LUMO
LUMO
Figura 3.3
Il modo con cui un substrato può interagire con un acido di Lewis chirale sono numerosi, e una
semplice classificazione può semplicemente essere fatta sulla base del tipo di contatto e dell’azione
che da questi deriva.
Pertanto, distinguiamo tra (a) attivazione diretta via contatto singolo, (b) attivazione diretta via
contatto doppio, (c) attivazione via sistema coniugato con contatto singolo, (d) attivazione via
sistema coniugato con contatto doppio.
Nel primo caso, l’attivazione diretta via contatto singolo, si ha interazione tra l’acido di Lewis e
il centro reattivo dell’elettrofilo, come descritto nell’esempio di Figura 3.4.
R
OR
MLn(L*)
O
O
OR
+
H
R
OR
OR
Figura 3.4
Questo semplicissimo modo di attivazione non è poi così diffuso come si sarebbe portati a
pensare.
Più vantaggiosa risulta l’eventualità in cui il substrato abbia una interazione con l’acido di
Lewis attraverso due punti di contatto, perché spesso ne consegue una ridotta libertà
conformazionale, e quindi un vantaggio per la selettività di reazione. Inoltre, la presenza di un
secondo punto di legame tra substrato e catalizzatore può risultare in una attivazione elettrofila
ancora più efficace. Ancora, un effetto di chelazione stabilizza l’addotto substrato-acido,
aumentando di fatto la concentrazione della specie attiva, e quindi la velocità di reazione.
44
Uno svantaggio può essere quello che il necessario pre-requisito di avere nel substrato gruppi
funzionali vicini può limitare la generalità di un processo.
Molecole che tipicamente partecipano a processi catalitici di questo tipo sono α-chetoeteri
RC(O)CH2OR’ e α-chetoesteri RC(O)CH2CO2R’, come illustrato nella reazione aldolica di Figura
3.5 in cui il substrato HC(O)CH2OBn è attivato attraverso la chelazione al centro metallico che
impegna il gruppo aldeidico (che subisce attacco) e l’ossigeno etereo.
OTMS
+
R
O
O
OBn
H
O
OH
OBn
R
O
R'
R'
N
N
N 2+ Ph
Cu O
Ph O
Bn
H
attacco
Figura 3.5
In contrasto con questi primi due modi, un catalizzatore può attivare un substrato anche in modo
indiretto, per esempio attraverso un sistema coniugato. Anche in questo caso distinguiamo, per
iniziare, l’attivazione via sistema coniugato con contatto singolo. In questo caso, tipicamente
nessuno degli atomi dei legami coinvolti nella reazione è interessato alla coordinazione, perché
l’attivazione procede attraverso un sistema π come illustrato in Figura 3.6.
MLn(L*)
O
O
+
H
H
Figura 3.6
Ad esempio, questo fenomeno si riscontra con i carbonili α,β-insaturi, che a seguito della
interazione con l’acido di Lewis, sono attivati verso reazioni di Diels-Alder e, in genere, addizioni
coniugate.
La mobilità conformazionale permessa dalla presenza di un unico punto di contatto può essere
fonte di problemi, come illustrato in Figura 3.7. In questo caso, infatti, non solo la posizione
dell’acido di Lewis è flessibile, ma anche la posizione relativa dell’alchene e del carbonile.
MLn(L*)
(*L)LnM
O
O
X
X
R
H
(E) s-trans
MLn(L*)
H
R
H
(Z) s-trans
O
R
X
(E) s-cis
(*L)LnM
H
O
R
X
(Z) s-cis
Figura 3.7
Tuttavia, esistono esempi in cui questo approccio è risultato fruttuoso, come ad esempio, la
reazione di Diels-Alder a carico di aldeidi α,β-insaturi di Figura 3.8, che, catalizzata da un
45
opportuno complesso di alluminio, porta a un prodotto con ee fino al 98% con alta
diastereoselettività (eso:endo 97:3).
O
Me
H
H
(R)
+
O
Me
Figura 3.8
Anche nel caso dell’attivazione indiretta è possibile che essa proceda via due punti di contatto.
In questa situazione esistono gli stessi vantaggi e svantaggi descritti precedentemente nel caso
dell’attivazione diretta, con l’aggiunta che le restrizioni conformazionali sono ancora più gradite
visto il gran numero di isomeri possibili nei sistemi coniugati.
Un esempio virtuoso di questo tipo di attivazione è riportato in Figura 3.9, e si riferisce a una
Diels-Alder tra diciclopentadiene e un ossazolidinone α,β-insaturo.
O
O
Me
N
H
O +
O
N
Me
O
O
Figura 3.9
Il complesso meccanismo di questo processo, catalizzato da Ti, sarà illustrato nel Capitolo
successivo.
3.2 Catalizzatori basici di Lewis.
Sebbene inizialmente meno apprezzate degli acidi di Lewis, le basi di Lewis stanno destando
interesse crescente nel ruolo di catalizzatori, e scenari sempre più promettenti si stanno rapidamente
profilando in seguito al loro uso.
I catalizzatori basici di Lewis agiscono attraverso interazioni col substrato di tipo sia covalente
sia non covalente, come illustrato schematicamente in Figura 3.10.
B*
B*
S1'-B* + S2
S1 + S2
P
interazione covalente
B*
S1 + S2
B*
S1'...B* + S2
P...B*
P
interazione non covalente
Figura 3.10
Il primo modo di attivazione è di gran lunga il più usato, e prevede la reazione di una base di
Lewis chirale B* con un substrato S1 a formare un addotto covalente S1’-B*. Se la porzione S1’ è
46
achirale, allora si verifica tipicamente trasferimento di chiralità quando l’addotto reagisce con un
secondo substrato S2 a formare un prodotto P enantiomericamente arricchito.
Meno frequente è invece la formazione di un addotto non covalente S1’…B*, che deve risultare
più reattivo del reagente isolato (secondo esempio di Figura 3.10). Il tipo di ciclo è del tutto simile a
quello illustrato nel caso di interazione covalente.
Sebbene, come anticipato, l’uso delle basi di Lewis (anche noto come organocatalisi) sia molto
efficace ed esistano molti esempi di processi catalitici con grande efficienza, ulteriori
approfondimenti esulano dagli scopi didattici di questi appunti, che sono incentrati prevalentemente
sull’uso di catalizzatori metallici (e quindi acidi di Lewis).
3.3 Catalizzatori acidi di Brønsted
Come nel caso delle basi di Lewis, recentemente è sorto un significativo interesse anche nei
confronti della catalisi promossa da acidi di Brønsted. Secondo il meccanismo di azione, questi
catalizzatori possono essere divisi in due grandi categorie: la prima, cui appartengono acidi che
attivano il substrato stabilendo con esso un legame a idrogeno reversibile; la seconda, nella quale
si distinguono i catalizzatori acidi che danno luogo a una protonazione (enantioselettiva) del
substrato (prochirale). Anche in questo caso, si presenterà una rapida panoramica dei due possibili
scenari, rimandando a letture più specializzate ulteriori eventuali approfondimenti.
Attivazione mediante legame a idrogeno. Il comportamento catalitico di un acido protico è a
tutti gli effetti assimilabile a quello di un acido di Lewis, il cui uso è stato descritto all’inizio di
questo Capitolo. La differenza principale consta nel fatto che con gli acidi di Lewis è possibile
modulare l’attività e la stereochimica del centro attivo con una scelta appropriata del metallo e del
suo intorno di coordinazione, adattando così il catalizzatore a ogni particolare substrato (in Figura
3.11 è riportato un esempio di interazione tra un acido di Lewis e un acido protico con lo stesso
substrato).
BRn
MLn
H
O
R'
O
H
R'
H
Figura 3.11
Nel caso in cui l’interazione coinvolga il protone, la minor versatilità pone gli acidi di Brønsted
in una condizione di svantaggio nei confronti degli acidi di Lewis, sebbene la possibilità di usarli in
soluzioni acquose, unita all’attitudine a promuovere reazioni in condizioni blande e senza l’uso di
metalli, ne può far preferire la scelta in più di una occasione.
47
La natura del legame a idrogeno che si istaura tra il catalizzatore acido di Brønsted e il substrato
può essere molto varia. I legami a idrogeno hanno una forza che spazia da 1 a 40 kcal/mol, con
relativi angoli compresi tra 90 e 180°. Nella maggior parte delle applicazioni catalitiche, il legame a
idrogeno ha una forza moderata, compresa tra 4 e 15 kcal/mol, cui corrispondono angoli di legame
tra 140 e 180°.
I catalizzatori più tipicamente usati sono quelli riportati nella Figura 3.12.
OH
O
OH
O
P
R
OH
acido fosforico da BINOL,
pka= 2
BINOL, pKa= 9
N
H
O
O
+
CO2H
H
prolina, pka= 2
N
N
H
N
H
R
urea, pKa= 2
Ar Ar
CO2Me
H
R
O
R
O
OH
OH
Ar Ar
taddolo, pka= 12
prolina metil estere,
pka= 11
Figura 3.12
Un esempio di applicazione in catalisi asimmetrica può essere quello relativo all’uso di un
derivato del BINOL nella reazione di Morita-Baylis-Hillman di Figura 3.13, accoppiamento tra
un’aldeide e una specie elettron-attrattrice α,β-insatura con formazione di un alcol allilico.
In questo specifico caso, il bifenolo chirale (indicato con B-H) stabilisce un legame a idrogeno
con l’enone e catalizza la formazione di una specie zwitterionica derivante dall’attacco della fosfina
al doppio legame. L’intorno stereochimico della specie risultante favorisce l’addizione
stereoselettiva al substrato aldeidico. La successiva eliminazione della fosfina restituisce il protone
all’alcol consentendogli di partecipare al ciclo successivo.
O
-
H-B
O
+
Et3P
O
R
B-H + PEt3
B-H
OH
O
O-
H
O
R
R
+
Et3P
Figura 3.13
48
Attivazione mediante protonazione del substrato. L’esempio riportato nel paragrafo precedente
mostra come un acido protico possa comportarsi alla stregua di un acido di Lewis, attivando il
substrato attraverso una interazione reversibile. Dopo l’evento reattivo enantiodiscriminante, il
legame a idrogeno, che con il prodotto risulta indebolito, è ristabilito con una successiva molecola
di substrato, permettendo così l’iterazione del ciclo.
Invece, qualora l’acido protico abbia il ruolo di introdurre (enantioselettivamente) il protone nel
substrato prochirale, risulta più delicata la sua restituzione alla fine del ciclo catalitico.
Anche per questo motivo, l’approccio risulta oggi ancora confinato a un numero limitato di
applicazioni, che prevedono sempre il compimento di un delicato bilancio tra le proprietà acidobase di tutti gli ingredienti, e spesso sono limitate da prestazioni non esaltanti e dalla scarsa
tolleranza dei gruppi funzionali alle condizioni di reazione.
3.4 Catalizzatori basici di Brønsted
Come nel caso dei catalizzatori acidi corrispondenti, l’uso delle basi di Brønsted in catalisi
asimmetrica è ancora sporadico, soprattutto perché, per definizione, la loro interazione con acidi è
limitata al semplice protone.
Tipicamente, le basi di Brønsted partecipano a cicli catalitici complessi, con più stadi di
attivazione, uno dei quali prevede il loro coinvolgimento. Un esempio è quello della reazione
aldolica catalizzata da un complesso eterobimetallico derivato dal BINOL e contenente Li+ e un
lantanide Ln nello stato di ossidazione III (Figura 3.14).
Li
O
O
Ln
O
O
O
Li
O
R1
Li
O
O
O
Ln
O
O
O
Li
H
O
R1
LiO
O
H
H
O
Li
sito
basico
R2
thf
Li
LiO
O
O
O
Ln
O
H
O
O
R2
Li
Li
O
O
O
Ln
O
O
O
Li
R1
Li thf
thf
OH O
R2
R1
Li
H
O
O
O
O
Ln
O
O
O
R2
Li
Li
catalizzatore
O
R1
Figura 3.14
49
Il catalizzatore combina una funzione basica di Brønsted (l’ossigeno dell’alcolato) con una
funzione acida di Lewis (il metallo centrale). La prima serve per la deprotonazione del substrato
chetonico, con formazione di un enolato. Questi attacca la molecola di aldeide, a sua volta attivata
dalla coordinazione del carbonile al lantanide acido di Lewis. L’addotto organico, riacquistando il
protone, dà luogo al prodotto chirale e restituisce il catalizzatore intatto.
3.5 Catalizzatori a coppia ionica
In una reazione di catalisi asimmetrica sono spesso coinvolti substrati dotati di carica, sia
cationici sia anionici. Pertanto, in ambedue i casi, essi sono accompagnati da controioni di segno
opposto, il cui ruolo è spesso trascurato. Può però accadere che questi ultimi siano specie chirali
capaci di trasmettere la propria informazione stereochimica, consentendo di promuovere la reazione
in modo asimmetrico.
Il punto chiave per realizzare con successo questo tipo di trasformazione risiede nell’efficace
recupero del controione chirale per poter riavviare un nuovo ciclo catalitico (come schematizzato in
Figura 3.15, dove X*+ e Y*- sono rispettivamente un catione e un anione chirale).
catalizzatore cationico chirale
prodotto
neutro
reagente
catalizzatore anionico chirale
X*+Ysubstrato
prodotto
neutro
reagente
substrato modificato- X*+
X+Y*substrato
substrato modificato+ Y*-
Figura 3.15
Un esempio seminale di applicazione di questo tipo di catalisi è quello descritto in Figura 3.16,
e si riferisce alla reazione di enolati in condizioni bifasiche acqua/solvente organico.
Il ciclo comincia con la deprotonazione in soluzione acquosa del chetone con formazione del
sale sodico dell’enolato, solubile solo in acqua. Questo si trasferisce all’interfaccia dove incontra la
specie Y+Cl- (dove Y+ è un catione chirale derivato dalla cincona), rilasciando NaCl in acqua, e
dando luogo a una coppia ionica solubile nel solvente organico. In questo ambiente, l’enolato
incontra il reagente MeCl che dà luogo all’alchilazione, il cui decorso sarà enantioselettivo per via
dell’intorno chirale creato dal catione.
In questo modo, si forma un prodotto neutro, solubile nel solvente organico, e si rigenera il
catalizzatore che può avviare il ciclo successivo.
50
Cl
O
Cl
Cl-
N+
Me
Cl
OH
Ph
MeO
N
CF3
OH Cl
O
-
Ph
Y*+
Cl
O-Na+
Cl
MeO
Ph
MeO
solubile solo in acqua
Cl
MeCl
O-Y*+
NaCl
Cl
Ph
MeO
solubile solo in solvente organico
Figura 3.16
Il tipo di catalizzatore cationico usato più di frequente è basato sulla struttura della cincona,
anche se ovviamente non vi sono preclusioni sulla sua natura.
La strategia illustrata si applica bene anche al caso in cui l’informazione chirale sia portata
dall’anione. Questo approccio si può utilizzare con substrati che normalmente reagiscono in forma
neutra, ma che, per opportuna modifica, possono dar luogo alla stessa reazione come cationi.
Oppure, l’anione chirale può essere il controione di un complesso achirale, il cui centro metallico
catalizza la reazione in modo enantioselettivo per effetto dell’intorno asimmetrico creato
dall’anione. Per esempio, questo è stato realizzato in processi promossi da composti fosfinici
cationici di oro(I), contenenti l’anione chirale fosfato derivato dal BINOL (Figura 3.17).
i-Pr
Me
i-Pr
i-Pr
O
P
O
O
i-Pr
+
Me P Au
Ph
O
i-Pr
i-Pr
Figura 3.17
3.6 Catalizzatori transfer di gruppi
I catalizzatori transfer di gruppi sono quelli che promuovono reazioni che avvengono in due fasi
(Figura 3.18).
51
substrato
catalizzatore
reagente attivatore
catalizzatore
reagente disattivato
gruppo
substrato
gruppo
Figura 3.18
Nella prima, il catalizzatore chirale reagisce con un reagente achirale con formazione di una
specie attivata contenente un gruppo “trasferibile”. Nella seconda, la specie attivata trasferisce in
modo enantioselettivo il gruppo a un substrato prochirale, spesso senza coordinazione di
quest’ultimo, dando luogo al prodotto chirale e rigenerando la specie catalitica.
Molte sono le reazioni che procedono con questo meccanismo: ciclopropanazioni,
epossidazioni, diidrossilazioni, aziridinazioni. A titolo di esempio, si riporta il meccanismo
proposto per l’epossidazione di alcheni prochirali cis catalizzata da complessi di manganese
contenenti leganti chirali tipo salen, reazione che conduce a un prodotto trans e un prodotto cis,
entrambi chirali (Figura 3.19).
enantiomeri
(R)
ox
Me
O
Me
Ph
(R)
Me
(R)
Ph
O
(S)
Ph
(S)
O
Me
(S)
Me
(S)
Ph
O
(R)
Ph
enantiomeri
Figura 3.19
Secondo gli studi più accreditati, il complesso di Mn(III) subisce un’ossidazione da parte
dell’ossidante (per esempio NaClO), con formazione di un osso complesso di Mn(V). Questo stadio
costituisce la prima fase, e il gruppo “osso” è quello “trasferibile” (Figura 3.20).
52
O
N
O
ox
N
O
Mn
Ph
N
O
Mn
N
O
Ph
Ph
Me
Me
O
Me
N
O
O
prodotto cis
Mn
N
O
Figura 3.20
Successivamente, l’alchene prochirale si approssima a questa specie, generando un radicale
benzilico. L’immediata chiusura di quest’ultimo fornisce l’epossido cis, che sarà chirale qualora
l’alchene venga invitato, dall’intorno asimmetrico generato dal legante, ad avvicinarsi con una sola
delle due enantiofacce. In Figura 3.20, per comodità, è rappresentata un’unica possibilità. Nel
prossimo capitolo sarà invece spiegata in maggior dettaglio l’origine dell’enantioselettività.
Se la chiusura dell’anello epossidico è anticipata dalla rotazione lungo il legame singolo C-C,
allora l’epossido avrà configurazione trans (Figura 3.21).
O
N
O
ox
N
O
Mn
Ph
N
O
Mn
N
O
Ph
Ph
Me
O
Me
Me
Me
O
N
O
O
prodotto cis
Ph
prodotto trans
Mn
N
O
Me
O
N
O
Ph N
Mn
O
Figura 3.21
3.7 Catalizzatori di cross-coupling
Un processo affine a quello presentato nella sezione precedente è il cross-coupling, in cui due
gruppi sono accoppiati da un catalizzatore che promuove la formazione di un nuovo legame.
In questo caso (Figura 3.22), il ciclo catalitico prevede uno stadio in cui ambedue i gruppi
53
risultano legati al centro metallico simultaneamente. Perché ciò avvenga è quindi necessaria la loro
introduzione sequenziale sul centro metallico, seguita dall’eliminazione del prodotto attraverso il
loro accoppiamento.
gruppo 1
catalizzatore
sottoprodotto 1
reagente gruppo 2
sottoprodotto 2
reagente gruppo 1
catalizzatore
catalizzatore
gruppo 1 gruppo 2
gruppo 1
gruppo 2
Figura 3.22
Un classico esempio di accoppiamento è quello relativo alla reazione di Suzuki, che prevede la
formazione di un legame C-C a partire da un alogenuro arilico e un acido arilboronico:
Ar-I + Ar’-B(OH)2 → Ar-Ar’ + I-B(OH)2
Il meccanismo semplificato della reazione è proposto in Figura 3.22, e coinvolge l’addizione
ossidativo dell’alogenuro arilico al complesso di palladio(0) con formazione di un intermedio
arilico di palladio(II). A questo punto, si ha lo scambio dell’alogenuro con il secondo frammento
arilico Ar’ ceduto dal derivato boronico, seguito dall’accoppiamento Ar-Ar’, che fornisce il
prodotto restituendo il catalizzatore al ciclo.
Ar2
L Pd Ar1
X
L
HO
B
OH
X
Ar1 X
HO
L
Pd
L
B
OH
L Pd Ar1
Ar2
L
Ar1
Ar2
Figura 3.23
Un esempio di coupling di Suzuki asimmetrico è offerto dalla Figura 2.20, in cui
l’enantioselettività del processo dipende dalla conformazione relativa dei due frammenti arilici
legati al palladio prima della loro eliminazione.
54
3.8 Catalizzatori che attivano attraverso coordinazione π.
I principali modi di coordinazione π riguardano i sistemi η2-olefinici, η3-allilici, η4-butadienici
e η6-arenici (Figura 3.24).
M
attivazione η2
M
M
M
attivazione η3
attivazione η4
attivazione η6
Figura 3.24
L’attivazione π per eccellenza è ovviamente quella a carico degli alcheni, che, in seguito alla
coordinazione η2, subiscono una profonda modifica della densità elettronica π, attraverso il ben
noto meccanismo di σ-donazione e π-retrodonazione (Figura 3.25).
M
M
π-retrodonazione
σ-donazione
Figura 3.25
Questo consente la loro attivazione nei confronti di un ventaglio di reazioni, che tipicamente li
coinvolgono in processi di inserzione, e come elettrofili.
Catalizzatori che attivano attraverso coordinazione π di tipo η2. Questo tipo di attivazione è di
gran lunga la più classica e applicata nell’ambito delle reazioni catalitiche coinvolgenti alcheni.
Trova infatti riscontro nell’idrogenazione, nell’idroformilazione, nel processo Wacker, nella
reazione di Heck e così via.
Un primo esempio può essere fatto proprio relativamente alla reazione di Heck (catalizzata da
complessi di palladio), che, nella sua classica versione, procede attraverso la sostituzione di un
atomo di idrogeno con un gruppo arilico:
RCH=CHR’ + Ar-X → RCH=C(Ar)R’ + H-X
Nella sua versione asimmetrica, tipicamente si usano come substrati anelli insaturi a 5-7 atomi,
e il meccanismo è quello proposto in Figura 3.26.
55
L2Pd
O
OTf
OTf
L2Pd
base
L2Pd
TfO-
L2Pd+
base.HOTf
O
H
OTf
OTf
L2Pd
O
O
Figura 3.26
Il meccanismo prevede addizione ossidativa del legame C-X (in questo caso del fenil triflato)
con ottenimento di una specie arilica di palladio(II). Segue coordinazione dell’alchene prochirale
che dà luogo a inserzione nel legame Pd-C. A questo punto, il frammento idrocarbilico è coinvolto
in una β-eliminazione, che risulta nella formazione del prodotto chirale. Il catalizzatore di
palladio(0) è ripristinato per reazione con una base B presente nel sistema di reazione.
E’ interessante osservare che la posizione del doppio legame nel prodotto non è quella iniziale,
per motivi legati alla stereochimica dell’anello coinvolto nella reazione di β-eliminazione. Infatti,
per ripristinare la posizione originaria dovrebbe essere coinvolto l’atomo di idrogeno geminale al
fenile entrante, che però si trova in posizione trans rispetto al palladio e non può β-eliminare.
L’eccesso enantiomerico del prodotto è legato alla capacità del legante chirale nel creare un
intorno asimmetrico capace di indurre coordinazione selettiva di una sola delle due facce
dell’alchene prochirale.
Un altro esempio rilevante, già descritto in Figura 2.19, è quello relativo all’idrogenazione di
enammidi catalizzata da complessi di rodio. In questo caso, come spiegato in dettaglio
precedentemente, è interessante osservare come l’eccesso enantiomerico del prodotto non dipenda
dalla capacità del legante a indurre coordinazione selettiva dell’alchene prochirale, ma piuttosto
dalle velocità relative con cui gli addotti olefinici diastereoisomerici danno luogo ad addizione
ossidativa di idrogeno.
56
Quelli citati sono solo due esempi dimostrativi della grande importanza che riveste l’attivazione
di alcheni attraverso coordinazione π. Tra l’altro, essa è ortogonale ad altri modi, e quindi presenta
grande tolleranza nei confronti di altri gruppi funzionali, garantendo in genere elevata selettività.
Catalizzatori che attivano attraverso coordinazione π di tipo η3. La coordinazione η3 è tipica
dei sistemi π-allilici, il cui meccanismo di legame prevede una donazione di due coppie elettroniche
dal legante idrocarbilico anionico al centro metallico (Figura 3.27).
M
M
σ-donazione
π-donazione
Figura 3.27
Questo tipo di interazione attiva il legante allilico (in particolare i due atomo di carbonio
terminali) nei confronti di nucleofili, fenomeno che trova la sua applicazione più suggestiva nella
sostituzione allilica promossa da palladio, il cui ciclo catalitico può essere descritto semplicemente
X
come in Figura 3.28.
Nu
R
R
-
+ Nu
R
+ X-
R
R
L2Pd
X
X
R
R
R
R
L2Pd+
L2Pd
R
R
R
Nu
R
X-
Nu-
L2Pd
Nu
R
Figura 3.28
Tipicamente, il catalizzatore di palladio(0) subisce coordinazione η2 da parte del substrato
allilico, e successiva addizione ossidativa da parte di quest’ultimo con ottenimento di un complesso
π-allilico. Questi subisce attacco da parte del nucleofilo esterno Nu-, con formazione del prodotto, a
sua volta coordinato η2, che è successivamente rilasciato, consentendo al ciclo di ripartire.
Secondo la natura del substrato allilico di partenza (racemo, achirale, meso) esistono varie
57
possibilità di induzione di asimmetria. Nell’esempio riportato in Figura 3.27, ad esempio, si parte da
un substrato chirale racemo, che, in seguito ad addizione ossidativa, dà luogo a un complesso in cui
l’allile è achirale. I due atomi di carbonio terminali del sistema allilico sono però resi enantiotopici
dalla presenza del legante chirale sul palladio, e quindi l’attacco selettivo su uno di essi restituisce
un prodotto chirale. Nel prossimo capitolo questo aspetto verrà spiegato in maggior dettaglio.
58
4. L’induzione di asimmetria
4.1 I leganti privilegiati
Uno degli obiettivi principali della catalisi asimmetrica è la progettazione razionale di nuovi
catalizzatori. Il raggiungimento di questo traguardo è in genere un compito molto arduo, soprattutto
perché il livello di comprensione dei meccanismi della catalisi e delle specie attive coinvolte è
spesso scarso e di difficile previsione. Inoltre, la differenza in energia tra i vari possibili stati di
transizione che conducono a prodotti enantiomerici è spesso così esigua da rendere proibitiva ogni
facile razionalizzazione.
Per questi motivi, l’ottimizzazione di un catalizzatore è spesso frutto di un cocktail di fattori,
che comprende progettazione, intuizione e serendipità.
In ogni caso, il metodo più comune con cui un legante trasferisce la propria informazione
chirale al substrato è attraverso il controllo sterico, e quindi, per capirne le modalità, è necessaria la
conoscenza della struttura tridimensionale delle specie coinvolte, esercizio che diventa
particolarmente complesso qualora il legante presenti molti gradi di flessibilità, e sia
conformazionalmente dinamico.
Questo aspetto, unito alla pluralità di reazioni catalitiche oggetto di studio e applicazione, si
manifesta in un’intensa attività di ricerca, volta a progettare e valutare in catalisi nuovi leganti
chirali, e a definirne le modalità di coordinazione ai centri metallici coinvolti nelle catalisi.
Tra le migliaia di leganti oggi noti, un gruppo selezionato è stato indicato come privilegiato
(Figura 4.1), termine indicato dal chimico americano Jacobsen per definire strutture di vasta e
comprovata abilità, usate di regola per scoprire nuovi processi enantioselettivi e per migliorare
l’efficienza di quelli già conosciuti.
N
N
OH
R
BINAP (X= PPh2)
BINOL (X= OH)
R
N
O
O
X
X
R
N
LEGANTE DI BRINTZINGER CINCHONIDINE
BOX
P
OHHO
R
R
N
O
OH HO
P
R
DUPHOS
N
R
R
SALEN
O
TADDOLO
E DERIVATI
Figura 4.1
59
Nelle prossime pagine verrà presentata una breve descrizione del funzionamento di alcuni di
essi, e di come la loro struttura tridimensionale risulti determinante nell’indirizzare
l’enantioselettività di processi selezionati di rilevanza sintetica. Gli esempi serviranno anche da
spunto per introdurre o ribadire alcuni concetti base.
Bis(ossazoline). Le bis(ossazoline) sono leganti bidentati a simmetria C2 (Figura 4.2).
O
O
N
N
N
N
R
R
O
O
M
R
BOX
L
R
L
Figura 4.2
Trovano applicazione in un gran numero di processi catalitici, quali le ciclopropanazioni, le
aziridinazioni, le reazioni di Diels-Alder, le alchilazioni alliliche. In seguito a coordinazione, danno
luogo a un anello ciclometallato a sei membri, quasi planare. L’aspetto stereochimico caratteristico
è la presenza di due sostituenti R ingombranti sugli atomi stereogenici degli anelli ossazolinici, che
si posizionano in stretta vicinanza del centro metallico e dei siti di coordinazione del substrato,
come
appare
evidente
dalla
struttura
del
complesso
rappresentativo
[{(S,S)-t-Bu-
BOX)}Cu(OH2)2](SbF6)2, riportata in Figura 4.3.
Figura 4.3
E’ palese dalla figura come i due sostituenti t-butili si estendano verso le posizioni occupabili da
un eventuale substrato, ingombrando selettivamente due settori dei quadranti con cui si è soliti
suddividere lo spazio intorno al metallo.
A questo proposito, infatti, vale la pena ricordare che, per una immediata rappresentazione
dell’occupazione sterica intorno al metallo, e quindi per dedurre la stereoselettività di coordinazione
di un substrato, l’intorno di coordinazione è suddiviso in quattro quadranti, la cui linea di
demarcazione orizzontale coincide con il piano o lo pseudopiano del catalizzatore. In particolare, i
60
quadranti sono colorati in grigio qualora siano occupati da sostituenti del legante chirale, mentre
sono incolori nel caso in cui le regioni di spazio siano nettamente più sgombre. La Figura 4.4
mostra, a titolo di esempio, lo schema per un catalizzatore C2 simmetrico contenente l’ossazolina di
Figura 4.3.
R'
R'
M
R'
R'
(b)
(a)
(c)
Figura 4.4
Appare evidente dalla Figura 4.3 come i sostituenti t-butili vadano a occupare i due settori in
alto a sinistra e in basso a destra (dando luogo al diagramma di Figura 4.4a), favorendo così
l’avvicinamento di un substrato prochirale, come per esempio un’olefina trans, attraverso una sola
delle due facce (Figura 4.4b), svantaggiando l’altra (Figura 4.4c).
Un’applicazione notevole delle bis(ossazoline) è nell’alchilazione allilica asimmetrica promossa
da Pd (già schematizzata in Figura 3.27), il cui decorso stereochimico può essere proficuamente
proposto in Figura 4.5, qualora il legante sia (S,S)-Bn-BOX (Bn= benzile).
H OAc
AcO H
Ph
Bn
N
Ph
H
Nu
Ph
+
Pd
Nu-
meno stabile
Ph
[Pd(Bz-BOX)]
[Pd(Bz-BOX)]
Nu H
Bn Ph
+
N Pd
(R)
Ph
Ph
(S)
H
N
Ph Bn
Nu-
H
N
Bn
H Nu
Ph
H +
N Pd N
Bn
Ph
Ph
H
più stabile
H
Ph
Bn
Nu
Ph
Ph
più abbondante
meno abbondante
Figura 4.5
61
Indipendentemente dalla sua configurazione, l’addizione ossidativa del substrato (presente come
racemo) al precursore di Pd(0) contenente il legante bis(ossazolinico) fornisce un intermedio in cui
il legante π-allilico è simmetrico.
A questo punto, il nucleofilo esterno Nu- può attaccare i due atomi di carbonio terminali, tra loro
enantiotopici, dando luogo rispettivamente ai due prodotti enantiomerici, inizialmente ancora
coordinati η2 al centro metallico. Appare evidente come uno dei due complessi intermedi olefinici
(e quindi più precisamente lo stato di transizione che ad esso conduce) sia reso meno stabile dalla
presenza di contatti sterici sfavorevoli tra i sostituenti sull’alchene e il benzile del legante bidentato.
Questa situazione avvantaggia il percorso illustrato a destra, favorendo l’ottenimento
preferenziale di uno dei due enantiomeri.
E’ interessante osservare come l’addizione ossidativa del substrato conduca all’ottenimento di
un unico intermedio π-allilico, indipendentemente dalla orientazione (M o W) del legante
idrocarbilico rispetto al piano di coordinazione (Figura 4.6).
Bn
N
Ph
H
+
Pd
H
Bn Ph
N
N
Ph Bn
H
Pd
+
H
Ph
N
Bn
orientazione W
orientazione M
Figura 4.6
In questo caso, infatti, è facile intuire come le due possibili orientazioni diano vita allo stesso
composto, a causa della simmetria C2 del legante bidentato. Non altrettanto sarebbe accaduto
qualora il legante avesse avuto simmetria C1, come per esempio si osserva nel caso di chelanti con
due atomi donatori diversi, quali, per esempio, N e P (Figura 4.7)
N
Ph
H
+
Pd
H
Ph
N
P
Ph
H
Pd
+
H
Ph
P
orientazione W
orientazione M
Figura 4.7
Ora, i due composti non sono più sovrapponibili, e, pertanto sono tra loro diastereoisomeri. La
loro interconversione può avvenire attraverso il classico meccanismo η3-η1-η3, che prevede il
passaggio del legante idrocarbilico attraverso una forma coordinata attraverso un solo atomo di
carbonio, spesso assistita da un legante esterno (Figura 4.8).
N
Ph
H
+
Pd
H
P
Ph
orientazione M
+L
H
Ph
Ph
N
+
Pd
P
L
-L
Ph
N
H
Pd
+
H
Ph
P
H
H
orientazione W
62
Figura 4.8
L’aspetto principale di questa considerazione stereochimica è che in questo caso entrambi gli
intermedi devono essere tenuti in considerazione nel meccanismo di reazione, e, quindi la sua
interpretazione e la previsione dell’esito stereochimico della reazione si complica notevolmente. Va
anche rilevato come ora gli attacchi ai due atomi di carbonio terminali del substrato allilico siano
inequivalenti, non solo più stericamente, ma anche elettronicamente, in quanto uno avverrebbe in
trans all’atomo di P e l’altro in trans a quello di N.
Questo esempio vale come testimonianza di una valutazione generale riguardo la simmetria dei
leganti chirali, vale a dire che nella loro scelta si è sempre tipicamente preferito mantenere la
simmetria C2 allo scopo di ridurre il numero di possibili intermedi e stati di transizione, anche per
garantire maggiore selettività al processo. Va anche detto però che, recentemente, è stata invece
dimostrata la validità di moltissime tipologie di leganti a simmetria C1, il che parzialmente
smentisce le osservazioni appena menzionate.
BINOL e derivati. Una classe molto importante di leganti privilegiati è quella derivata dall’alcol
atropoisomerico BINOL (1,1’-bi-2-naftolo, Figura 4.9)
1
1'
2
2'
OH
OH
1
1'
3
R
2
2'
OH
OH
3'
R
BINOL e derivati 3,3'-disostituiti
Figura 4.9
Il BINOL è stabile nei confronti della racemizzazione perché la rotazione intorno al legame C1C1’ è impedita da una barriera di 37-38 kcal/mol. Ne consegue che non si osserva racemizzazione
neppure a 100°C in una miscela acqua/diossano in un intervallo di tempo pari a 24h.
Il BINOL e i suoi derivati sono ottimi leganti di centri metallici, e i relativi complessi risultano
spesso eccellenti catalizzatori per processi asimmetrici. Gran parte dell’abilità del legante a indurre
enantioselettività è imputabile alla conformazione “skew” che il legante adotta in seguito alla
coordinazione, come ben si evince dalla rappresentazione schematica di Figura 4.10, in cui è anche
riportata la relativa occupazione nello spazio con il diagramma dei quadranti.
R
O
O
R
L
M L
M
63
Figura 4.10
I due sostituenti R nelle posizioni 3 e 3’ si protendono in avanti e vanno a occupare due
quadranti opposti, lasciando liberi gli altri due. In questa situazione, ad esempio in un catalizzatore
tetraedrico, un substrato planare prochirale L’ può essere facilmente attaccato solo da una delle due
facce, dando così luogo a un prodotto enantiomericamente arricchito (Figura 4.11). Infatti, come si
deduce dalla figura, l’attacco da sopra è ostacolato dal gruppo R (in questo caso un fenile), mentre
non vi è alcuna inibizione per un nucleofilo proveniente dal di sotto del piano del foglio.
attacco libero
L'
O
M
O
L
attacco inibito
Figura 4.11
BINAP. Il BINAP, al pari del BINOL, è un legante assiale chirale, che presenta una
conformazione non co-planare degli anelli binaftilici (Figura 4.12).
1
1'
2
2'
PPh2
PPh2
(S,S)-BINAP
Figura 4.12
Anche in questo caso, la rotazione lungo il legame centrale C-C è impedita, e quindi il legante è
stabile nei confronti della racemizzazione.
Il BINAP è estremamente versatile, e può accomodare metalli di diverso raggio, ottimizzando il
legame per rotazione intorno al legame C-C appena citato, e intorno ai legami tra gli atomi di
fosforo e quelli di carbonio 2 e 2’. Tuttavia, una volta stabilita la coordinazione, la struttura
metallaciclica risultante diventa molto rigida.
Il legante trasmette asimmetria al substrato coordinato al centro metallico tramite la posizione e
l’orientamento dei quattro gruppi fenili diastereotopici. Infatti, due di questi assumono una
conformazione pseudo-assiale, gli altri due pseudo-equatoriale, come appare chiaro dalla struttura
mostrata in Figura 4.13 del complesso con il Ru(acetato)2 (gli ioni acetato non sono mostrati per
maggior chiarezza).
64
Figura 4.13
Gli anelli pseudo-equatoriali si estendono al di là del centro metallico, fiancheggiandolo, mentre
quelli pseudo-assiali si orientano da parte opposta. Il motivo chirale che deriva da questo
arrangiamento sterico determina l’enantiocontrollo sul sito di coordinazione del substrato. Una
rappresentazione più immediata è ovviamente offerta dal modello a quadranti, in cui viene
evidenziato il modo con cui i fenili pseudo-equatoriali vadano a intralciare le regioni in alto a destra
e in basso a sinistra (Figura 4.14).
M
P M P
Figura 4.14
Leganti di Brintzinger. Questi leganti danno vita alla categoria di complessi ansa-metalloceni,
tipicamente contenenti metalli di transizione del gruppo 4b (Ti, Zr e Hf), e il suo esemplare più
rappresentativo è il legante C2 simmetrico, etilene-1,2-bis(η5-4,5,6,7-tetraidro-1-indenile),
abbreviato in EBTHI, di cui è riportata in Figura 4.15 la rappresentazione di un complesso da due
diversi punti di vista.
X
M
X
M
X
X
Figura 4.15
Tra le molte reazioni che questa classe di composti è in grado di promuovere, si può citare la
idrosililazione di immine cicliche catalizzata dal complesso Ti(EBTHI)F2 (Figura 4.16).
65
N
H
N
H-[Si]
MeOH
Figura 4.16
Il decorso stereochimico può essere facilmente previsto sulla base dell’intorno sterico creato dal
legante bidentato, reso rigido dalla presenza del ponte etilenico che collega i frammenti
idrocarbilici. Senza entrare nel dettaglio del meccanismo, si può semplicemente partire
dall’osservazione che lo stadio enantiodeterminante consiste nell’inserzione dell’immina prochirale
in un legame Ti-H generato da un adatto silano, come H3SiPh, i cui stadi di transizione
diastereoisomerici sono rappresentati in Figura 4.17.
contatto
sterico
H
N
(S)
H
M
N
H
M
N
H
N
(R)
Figura 4.17
Dalla Figura si evince chiaramente come la coordinazione di una delle due facce prochirali
dell’immina sia destabilizzata dalla presenza di un contatto sterico proibitivo tra il fenile del
substrato e uno degli anelli indenilici. E’ questo che determina un vantaggio energetico per l’altro
cammino, e quindi favorisce l’ottenimento dell’enantiomero (S).
Leganti derivati dal TADDOLO. Una famiglia di leganti ampiamente sfruttata in catalisi
asimmetrica è quella dei leganti derivati dal TADDOLO, la cui struttura prende origine dall’acido
tartarico (Figura 4.18).
Ph Ph
OH
O
OH
Ph Ph
Ph Ph
O
O
M
O
O
Ph Ph
TADDOLO
TADDOLato
O
Figura 4.18
In seguito a deprotonazione degli atomi di ossigeno, i leganti diventano ottimi coordinanti O,O’dianionici per una varietà di metalli di transizione, principalmente appartenenti ai primi gruppi della
serie (Figura 4.18).
La conformazione adottata dai TADDOLati è ben nota e prevede la formazione di anello
66
biciclico a 10 membri, i cui stereocentri sono però a una certa distanza dal sito di coordinazione di
eventuali substrati. Tuttavia, essi impartiscono una precisa conformazione al metallaciclo adiacente,
facendo assumere ai gruppi fenilici diastereotopici un arrangiamento pseudo-equatoriale/pseudoassiale. I due anelli pseudo-assiali si dispongono antiperiplanari rispetto ai contigui legami C-H
diossolanici. In questo modo, la asimmetria dei centri stereogenici è trasferita in avanti, verso il
metallo e verso i siti di coordinazione del substrato.
L’intorno chirale che si viene a creare è descritto in Figura 4.19, dove sono riportate due visioni
parziali di una struttura di un complesso [TiCl2(TADDOLato)]. Appare evidente come i due anelli
pseudo-assiali siano responsabili del trasferimento di chiralità verso il centro metallico, mentre
quelli pseudo-equatoriali siano invece nettamente più arretrati e il loro effetto quasi nullo.
Figura 4.19
Per illustrare un uso virtuoso in catalisi di questa famiglia di leganti si può prendere in
considerazione la reazione di Diels-Alder tra il diciclopentadiene e l’N-acilossazolidinone di Figura
4.20, catalizzata da un complesso di titanio(IV).
O
O
N
O
+
R
H
[TiCl2(TADDOLato)]
(S)
R
O
N
O
O
Figura 4.20
In questo processo si ritiene che il dienofilo sia attivato attraverso la sua coordinazione
bidentata al titanio, con formazione di un addotto ottaedrico, come quello di cui la Figura 4.21
riporta la struttura ottenuta da cristallo singolo.
67
Figura 4.21
In questo caso, gli ossigeni del TADDOLato sono in posizione equatoriale, mentre i due atomi
di cloro occupano le due posizioni trans-assiali.
Originariamente era stato proposto che proprio questa fosse la struttura dell’intermedio attivo in
catalisi, nonostante alcune evidenti contraddizioni. Innanzi tutto, il legame C=C del dienofilo
impegnato nella reazione si trova ben distante dall’intorno chirale promosso dal legante, e questo
non chiarisce come la reazione possa procedere con alta enenatioselettività. Inoltre, entrambi i
carbonili del dienofilo sono in trans ai due atomi di ossigeno di natura alcossidica, e quindi basica,
del TADDOLato, il che non favorisce l’attivazione del substrato. Un ragionamento plausibile,
infatti, suggerirebbe un’attivazione più efficace qualora il carbonile adiacente al doppio legame
reattivo fosse in trans al cloruro.
Per questo motivo, lo studio del meccanismo della reazione è stato approfondito, e studi teorici
hanno rivelato che ben cinque sono i possibili stereoisomeri coinvolti nel processo di attivazione del
dienofilo. Questi intermedi sono rappresentati in Figura 4.22, dove le lineette verticali sul legante
chirale rappresentano le orientazioni dei fenili pseudo-assiali.
R
R
O
O
Cl
O
O
Ti
Cl
A
O
O
O
N
R
N
O
O
O
O
Ti
O
Cl
O
O
N
Ti ClO
O
O
O
O
Ti
O
N
O
Cl
O
R
O
O
N
Ti ClO
Cl
Cl
Cl
Cl
B
C
D
E
R
Figura 4.22
Nell’isomero A tutti gli atomi di ossigeno sono in posizione equatoriale, i diastereoisomeri B e
C mostrano l’atomo di ossigeno del carbonile adiacente al doppio legame in trans rispetto al
cloruro, mentre in D ed E questi è in trans rispetto a un atomo di ossigeno del TADDOLato.
68
Uno studio approfondito ha anche dimostrato che solo tre dei cinque isomeri sono
effettivamente presenti in soluzione, in rapporto 70:24:6, e il più abbondante è proprio l’isomero A
di cui è stata proposta la struttura in Figura 4.21. Questi è anche il più stabile perché
l’arrangiamento relativo dei leganti minimizza i contatti di non legame.
I tentativi di assegnare la geometria agli altri due isomeri presenti in soluzione non hanno
portato a risultati incontrovertibili.
Tuttavia, è apparso chiaro che più importante dell’abbondanza relativa dei vari isomeri, è il
grado con cui ciascuno di essi riesce ad attivare il substrato. Studi teorici supplementari hanno così
rivelato che il massimo grado di attivazione si riscontra nelle geometrie B e C, seguito da quello
offerto dalle geometrie D e E, ambedue più reattive di A, come dimostrato dal fatto che
quest’ultimo può essere sia osservato in soluzione, sia addirittura isolato allo stato solido in cristalli
singoli.
Pertanto, uno scenario plausibile per il meccanismo di reazione coinvolge un rapido equilibrio
di scambio di substrato tra tutti e cinque gli isomeri, prevedibile sulla base della scarsa energia di
legame prevista per la coordinazione del substrato al centro metallico (15 kcal/mol).
Questo scambio è più rapido della reazione di cicloaddizione, che rappresenta lo stadio
enantiodeterminante. Ne consegue che la reazione si trova nelle condizioni di applicabilità del
principio di Curtin-Hammett (paragrafo 2.3), e quindi sono proprio gli intermedi B e C a condurre
alla formazione del prodotto.
In entrambi questi isomeri la faccia Si del substrato è schermata dal gruppo fenile pseudo-assiale
del legante TADDOLato, e quindi l’attacco del diene è favorito sulla faccia Re, come proposto in
Figura 4.20, e in accordo con l’esito stereochimico della reazione.
R
attacco alla faccia Si
attacco alla faccia Re
O
O
O
N
O
O
Ti
Cl
Cl
B
Figura 4.23
Questo esempio, così articolato, è paradigmatico per molti aspetti che riguardano lo studio di
una reazione catalitica: mostra come siano possibili più intermedi attivi nel corso di una reazione
catalitica, ciascuno con una propria reattività, dimostra come il principio di Curtin-Hammett possa
69
essere determinante nel capire il controllo stereochimico della reazione, e suggerisce l’importanza
di studi combinati teorico-sperimentali per arrivare a una proposta verosimile del meccanismo di
reazione.
Leganti tipo salen. Questi leganti sono derivati chirali della saliciletilendiammina, acronimo
salen, di cui la classe ideata da Jacobsen, basata sulla trans-1,2-cicloesandiammina, rappresenta
l’esempio applicato con più efficacia in catalisi asimmetrica (Figura 4.24).
N
N
OH HO
(S)
(S)
N
N
OH HO
t-Bu
salen
t-Bu
legante di Jacobsen
Figura 4.24
Essi, tipicamente, coordinano come leganti tetradentati dianionici N,N’,O,O’, in seguito a
deprotonazione degli ossidrili. La Figura 4.25 mostra un esempio di complesso cationico di Mn(III).
(S)
N
(S)
+
N
Mn
O
t-Bu
O
t-Bu
Figura 4.25
Numerose sono le reazioni asimmetriche catalizzate da complessi metallici contenenti questi
leganti. Si possono citare le addizioni ai carbonili, le aperture di epossidi e l’ossidazione di alcheni
ad epossidi chirali. In questo ambito, è interessante descrivere proprio il decorso stereochimico di
quest’ultima reazione, il cui ciclo catalitico è stato già illustrato in Figura 3.21 nel caso del cis-βmetilstirene.
Come citato precedentemente, la reazione procede con attacco dell’alchene al gruppo osso di un
intermedio di Mn(V), formato in seguito all’ossidazione del precursore catalitico di Mn(III) di
Figura 4.25.
L’accesso all’alchene è però governato dal motivo sterico imposto dal legante, che assume la
forma lenticolare schematizzata in Figura 4.26. Questo motivo sterico inibisce l’accesso
dell’alchene da sinistra, e parimenti ne è ostacolato l’ingresso frontale dai due gruppi t-butili posti
sugli anelli del legante. Risulta quindi nettamente preferito solo l’accesso da destra.
70
accesso impedito
O
N
O
Mn
N
accesso libero
O
accesso impedito
Figura 4.26
A questo punto, l’alchene potrebbe accedere al gruppo osso con ciascuna delle due facce
prochirali, dando luogo rispettivamente ai due epossidi chirali in configurazione cis (Figura 4.27).
Tuttavia, gli stessi gruppi t-butili sul legante giocano ancora un ruolo importante, inibendo, a causa
di un contatto sterico sfavorevole con il fenile dell’alchene, l’ingresso di una faccia, come in Figura
4.27a.
Me
O
N
O
N
Mn
Me
Ph
Ph
O
contatto sterico
O
N
O
cis-(1R,2S)
N
Mn
Ph
Ph
Me
O
Me
cis-(1S,2R)
O
enantiomero favorito
O
enantiomero sfavorito
(a)
(b)
Figura 4.27
Ne consegue l’ottenimento preferenziale con alto ee dell’enantiomero a configurazione cis(1S,2R). Il discorso è stato ovviamente semplificato, perché, come illustrato in Figura 3.22, la
reazione può anche prevedere l’ottenimento della coppia di enantiomeri trans.
Gli esempi riportati nelle pagine precedenti rappresentano piccoli spunti per comprendere
l’importanza della comprensione del riconoscimento esercitato dai leganti chirali per poterne
progettare di nuovi e più efficienti. Sicuramente, questa rappresenta sempre una sfida insidiosa,
perché spesso, come già citato, la differenza in energia tra stati di transizione diastereoisomerici
risulta piccola, e spesso di difficile predizione e calcolo. Pertanto, l’unica strada ancora perseguibile
consiste nel ciclo iterativo di progettazione del catalizzatore, esame delle sue prestazioni, e
conseguente opportuna modifica della sua struttura. Senza dimenticare che, come diceva Louis
Pasteur, chance favors the prepared mind.
71
4.2 L’approccio alla catalisi asimmetrica bifasica nel gruppo di Chimica Metallorganica
dell’Università di Napoli “Federico II”. La strategia del gruppo di ricerca in questo ambito si basa
sull’ipotesi che leganti chirali per catalizzatori omogenei possano essere molto convenientemente
preparati a partire da molecole di origine naturale.
Un primo vantaggio di questa scelta deriva dall’ampia e varia offerta di molecole naturali, che
permette di selezionare “building block” polifunzionali con gli stessi accurati e precisi motivi sterici
dei leganti privilegiati, vale a dire le strutture coordinanti di vasta applicabilità, citate nei paragrafi
precedenti.
Un ulteriore vantaggio è la possibilità di creare librerie di leganti modulari, che presentano precise
differenze nella stereochimica di coordinazione, nella struttura del legante o in ambedue.
In aggiunta, il building block naturale polifunzionale può consentire l’introduzione di specifiche
etichette di fase, per definire le proprietà chimico-fisiche del catalizzatore, e renderlo versatile e
adattabile a più condizioni.
Infine, proprio la natura polifunzionale consente di godere di siti di coordinazione emilabili, che
possono avere il ruolo di riconoscere o avvicinare un substrato, rendendo così il sistema più attivo
e/o selettivo. La versatilità della strategia è illustrata in Figura 4.28.
il building block
i componenti
il catalizzatore
una funzione coordinante
il controllo sterico
le funzioni coordinanti
l'etichetta di fase
un centro metallico
+
un gruppo di
adatto ingombro
un building block
naturale
un catalizzatore
modulare polifunzionale
un'etichetta di fase
Figura 4.28
Il building block chirale di origine naturale è rappresentato da un pallone a tre colli, e ciascun collo
descrive una funzione organica. Gli altri oggetti di comune vetreria (testa di Claisen, imbuto,
condensatore) simboleggiano possibili derivatizzazioni (rispettivamente una funzione coordinante,
un gruppo di adatto ingombro sterico, una etichetta di fase). Un graduale e appropriato
72
assemblaggio di tutti questi componenti può consentire l’ottenimento di ampie librerie di leganti,
adattabili a diverse condizioni e reazioni. Per esempio, attraverso l’opportuna scelta dell’etichetta di
fase, il corrispondente catalizzatore può risultare solubile in solventi non convenzionali, quali
acqua, liquidi ionici, solventi supercritici o fluorurati. Oppure, la stessa etichetta può essere usata
per consentire l’ancoraggio del catalizzatore a una matrice solida.
Concretamente, l’attività di ricerca prevede la progettazione di due classi di leganti pseudoenantiomeriche, denominate Naplephos e elpaNphos (Figura 4.29). Queste librerie presentano il
glucosio quale “building block” di origine naturale, e la loro struttura è stata ottimizzata al fine di
garantirne facilità sintetica e elevate prestazioni in catalisi.
phase tag
phase tag O
O
O
O
R
O
O
O HN
OR'
O
P
naplephos Ph Ph
phase tag
O
O
O
O
NH HN
R
elpanphos
P
Ph Ph
Figura 4.29
In particolare, l’anello a sei membri offerto dal glucosio ricorda il motivo dei leganti privilegiati
derivati dalla 1,2-cicloesandiammina (vedi Figura 4.1). Nelle Naplephos, in posizione 2 è presente
un braccio rigido difenilfosfinoammidico, funzione coordinante di grande potenzialità. La posizione
1 presenta un α-benzile (R’= CH2Ph), di immediata e selettiva introduzione. Le posizioni 4 e 6 sono
quelle deputate all’eventuale inserimento della etichetta di fase, utile per definire le proprietà
chimico-fisiche del legante. La posizione 3 è invece destinata al controllo sterico/elettronico nelle
prossimità del centro metallico.
La libreria elpaNphos rappresenta la controparte chirale di quella Naplephos, ed è stata ottenuta
per semplice introduzione della stessa funzione coordinante difenilfosfinoammidica, nella posizione
1 in luogo della 2.
Va inoltre sottolineato come tutte le funzioni ossigenate, naturalmente presenti nella struttura
nativa, siano potenziali siti di coordinazione emilabili, utili per benefiche interazioni secondarie con
un substrato e/o altri centri metallici.
73
Le strutture Naplephos e elpaNphos sono già state adattate con successo a diversi processi
enantioselettivi, realizzati in condizioni tradizionali, dando così chiara dimostrazione della loro
versatilità. Inoltre, come atteso, le due librerie, essendo pseudo-enantiomeriche, conducono a
prodotti (a elevata purezza ottica) con configurazione opposta.
In alcuni casi è stato possibile estenderne l’applicazione anche in catalisi bifasica, sia dotando il
legante di un’etichetta di fase polare, come in Figura 4.30a, sia ancorando lo stesso a una matrice
polimerica, mediante un robusto legame covalente (Figura 4.30b).
O O
n-Bu4N - P
O O
O
+
6
O
5 O
4
1
3 2
O
O
O
OCH2Ph
O
6
5 O
4
1
3 2
O
HN
OCH2Ph
O
HN
P
Ph Ph
P
Ph Ph
b
a
Figura 4.30
74