APPROFONDIMENTI DI
ATTIVITÀ FISICA ADATTATA, INTEGRAZIONE E INCLUSIONE
NELLE ATTIVITÀ MOTORIE
MODULO 01
VARIE TIPOLOGIE DI DISABILITA’
GUIDA ALLE PRINCIPALI PATOLOGIE
Although retarded children may be victims of fate,
they will not be the victims of our neglect.
J. F. Kennedy
CSA-SSIS Veneto A.A. 2006-2007- © Tutti diritti riservati
Donatella Donati –-Pagina 1 di 14
APPROFONDIMENTI DI
ATTIVITÀ FISICA ADATTATA, INTEGRAZIONE E INCLUSIONE
NELLE ATTIVITÀ MOTORIE
INDICE DEI CONTENUTI
INSUFFICIENZA/RITARDO MENTALE
RITARDO MENTALE e MALATTIA MENTALE
EZIOLOGIA -SU BASE ORGANICA- DEL RITARDO MENTALE
Fattori Prenatali
Fattori Perinatali
Fattori Post-Natali
SINDROMI GENETICHE PIÙ NOTE
Sindrome di Down
Sindrome del cromosoma X fragile
LESIONI DEL MIDOLLO SPINALE
Poliomielite Anteriore Acuta - Malattia di Heing-Medin
Traumi Midollari
PARALISI CEREBRALE INFANTILE
DISTROFIE MUSCOLARI
EPILESSIA
AUTISMO
Cosa non è l’autismo
Cos’è l’autismo
PSICOSI
Schizofrenia
NOTIZIE UTILI
Soggetti affetti da DIABETE
Bibliografia essenziale
Note al testo
CSA-SSIS Veneto A.A. 2006-2007- © Tutti diritti riservati
Donatella Donati –-Pagina 2 di 14
APPROFONDIMENTI DI
ATTIVITÀ FISICA ADATTATA, INTEGRAZIONE E INCLUSIONE
NELLE ATTIVITÀ MOTORIE
INSUFFICIENZA/RITARDO MENTALE
Insufficienza mentale è un termine, consigliato nel 1954 dall’O. M. S. è, ancora oggi, spesso usato
genericamente per definire
un deficit delle capacità intellettive. Poiché questo tipo di
classificazione viene usata frequentemente, si ritiene di fondamentale importanza sottolineare che,
soprattutto in ambito pedagogico, questo deve essere utilizzato come uno fra gli indicatori, di un
quadro composito e complesso, di cui fare una lettura ed una interpretazione molto attenta.
Molte sono le classificazioni delle disabilità che esistono in letteratura e altrettanti, quindi, i criteri su
cui si basano.
Ogni quadro teorico, infatti, individuando il proprio livello di analisi delle problematiche che intende
indagare, fornisce un suo modello fondamentale che permette di comprendere il significato dei
fenomeni che vuole spiegare.
Se ne forniscono alcuni esempi
a. anatomopatologica o patogenetica;
b. patologica-sociale: subordinata al criterio di adattabilità all’ambiente sociale;
c. psicometria: si riferisce al Q.I. - Quoziente Intellettivo -;
d. neurologica: basata sulla sede anatomica della lesione;
e. classificazione naturalistico-descrittiva: proposta in termini di sindromi;
in ogni caso, quale sia il modello (medico o psicometrico) ogni sistema di classificazione mescola
quasi sempre in modo insoddisfacente parametri valutativi diversi. In alcuni casi, ad esempio, viene
evidenziato l’aspetto eziologico(classificazione proposta da Grossman), in altri quello relativo alla
gravità (come quello psicometrico), in altri ancora quello fenomenico o sintomatico, in cui ogni
termine viene usato per far riferimento a raggruppamenti di sintomi (Meazzini, 1997, p. 28)
E’ importante comprendere che sono tutte schedature di comodo, sicuramente non inutili dal punto
di vista della ricerca ma poco significative sul piano rieducativo ed educativo se il concetto guida è
la globalità dell’individuo.
Possono essere causa di una vera discriminazione tra i soggetti diversamente abili e determinare
disparità di interventi. Soprattutto in ambito pedagogico-educativo, è necessario andare oltre
l’etichetta diagnostica e conoscere la specificità di ogni individuo se non vogliamo che categorie
linguistiche riduttive rendano falsamente omogenei problemi che sono, invece, diversi.
Si deve rilevare, però che, negli ultimi anni, nelle classificazioni sta prendendo piede il criterio della
“funzionalità” o “adattamento comportamentale1”, che possiamo definire “trasversale” a tutti gli altri.
Poiché la classificazione psicometrica è quella che più da vicino tocca la scuola, se per definire
l’insufficienza mentale si parte dal presupposto delle minori capacità intellettive, è opportuno fare
alcune riflessioni di carattere più generale.
Il concetto di intelligenza è altamente complesso e probabilmente indefinibile in modo univoco. Se
vogliamo considerare intelligente una condotta quando è in sintonia con il contesto situazionale in
cui il soggetto agisce, possiamo affermare che ogni attività è una manifestazione di intelligenza
quando è frutto di un adattamento all’ambiente.
Le capacità cognitive ci consentono di costruire strutture cognitive che permettono: il formarsi di
concetti astratti, il ragionare logicamente o, utilizzando l’esperienza e l’apprendimento, di adattare
quelli già esistenti per affrontare novità.
Esse, quindi, non si estrinsecano solo nella soluzione di problemi percettivi, motori o logici ma
anche nell’affrontare, con competenza, le situazioni sociali, così come dal punto di vista affettivo, si
estrinsecano nell’imparare a controllare gli impulsi e i desideri e a tollerare le frustrazioni.
Se viene considerata l’importanza dei fattori affettivi sullo sviluppo intellettivo, possiamo prendere
atto di un ritardo evolutivo, individuando una inibizione intellettiva senza identificarla
necessariamente come una INSUFFICIENZA, che invece esprime una realtà determinata e
definita.
1
definito come <la capacità mostrata dalla persona di raggiungere i livelli richiesti di autonomia personale e
responsabilità sociale da parte del gruppo sociale di appartenenza, in funzione del diverso livello di età>.
CSA-SSIS Veneto A.A. 2006-2007- © Tutti diritti riservati
Donatella Donati –-Pagina 3 di 14
APPROFONDIMENTI DI
ATTIVITÀ FISICA ADATTATA, INTEGRAZIONE E INCLUSIONE
NELLE ATTIVITÀ MOTORIE
Ciò non significa negare l’esistenza di soggetti con seri ritardi nello sviluppo intellettivo ma,
piuttosto, affermare che le capacità cognitive di quelli che vengono definiti INSUFFICIENTI non
corrispondono alle prestazioni richieste.
Se il criterio diagnostico dell’I.M. si uniforma solamente alle metodologie di misurazione del Q.I.,
considerando possibile che gli individui vengano confrontati secondo una scala obiettiva, rivela
l’idea di un’intelligenza quantitativa, statica e che procede per acquisizioni successive ritenendo,
quindi, che le capacità intellettive aumentino con l’età con un processo di sviluppo che procede
secondo una progressione lineare, passibile di misurazione.
Il più recente concetto di RITARDO MENTALE, considerando il problema in modo più globale, si
discosta dalla valutazione del solo deficit intellettivo e lo rappresenta come una realtà composita in
cui sono implicati fattori fisiologici, psicologici, sociali, culturali ed educativi, definendolo più come
un modo di essere determinato dal rapporto dialettico dell’io del soggetto con i suoi vari deficit
strumentali.
Per esempio, secondo Grossman (Grossman, cit. Meazzini, 1997: 28), durante l’infanzia e la
prima fase adolescenziale, è atteso un adeguato sviluppo nelle abilità:
1. sensomotorie;
2. di comunicazione;
3. autonomia,
4. sociali: capacità di interagire con gli altri;
mentre durante le altre fasi di sviluppo adolescenziale è attesa un’evoluzione per quanto riguarda
le abilità:
1. scolastiche e loro utilizzazione nelle attività quotidiane;
2. cognitve (ragionamento ecc.) nella gestione dei problemi;
3. sociali: partecipazione alle attività di gruppo;
nell’età adulta è attesa la capacità di presa di responsabilità lavorative e civiche.
In questo contesto il grado di ritardo mentale definito con il Q.I.i viene ad essere un elemento di
conoscenza che ci aiuta ad affrontare la complessa analisi dei vari processi evolutivi del soggetto
con cui ci troviamo ad interagire.
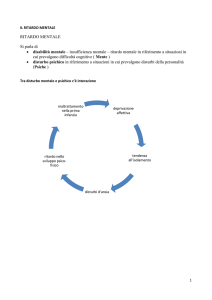
RITARDO MENTALE E MALATTIA MENTALE
L’approssimazione terminologica, purtroppo ancora oggi esistente, ha determinato una
commistione di immagini concettuali, di servizi d’assistenza e di sostegno sociale per i soggetti
portatori di handicap mentale (insufficienti o disabili mentali) e per soggetti malati (disturbati) di
mente.
Anche se, talvolta, l’insufficienza mentale e la malattia mentale si presentano associate nello
stesso soggetto è, tecnicamente e socialmente, opportuno separare le due distinte realtà.
L’INSUFFICIENZA MENTALE: il soggetto insufficiente mentale è “abilitabile” in rapporto
alle sue residue attitudini e capacità (potremmo definire improprio parlare di riabilitazione e
recupero).
LA MALATTIA MENTALE, generalmente, presentando vari tipi e gradi di sviluppo del
comportamento, si tratta più di “devianza dalla norma” che di incapacità di adattamento sociale. Il
malato di mente è funzionalmente “disadatto” ma è
teoricamente recuperabile e riabilitabile, mentre l’insufficiente mentale è “ inadattato” adattabile.
EZIOLOGIA -SU BASE ORGANICA- DEL RITARDO MENTALE
Solo il 20% dei casi di ritardo mentale è dovuto a fattori organici ad eziologia dimostrata, che
possono essere raggruppati, secondo la cronologia, in tre categorie:
CSA-SSIS Veneto A.A. 2006-2007- © Tutti diritti riservati
Donatella Donati –-Pagina 4 di 14
APPROFONDIMENTI DI
ATTIVITÀ FISICA ADATTATA, INTEGRAZIONE E INCLUSIONE
NELLE ATTIVITÀ MOTORIE
FATTORI PRENATALI
SINDROMI MALFORMATIVE CRANIO-ENCEFALICHE
Rientrano in questa categoria diagnostica tutte quelle patologie legate a malformazioni encefaliche,
craniche e cranio- facciali.
Alcuni esempi:
• le microcefalie: quando l’indice cranio-cefalico è inferiore a 77 (rapporto tra diametro
trasversale e antero-posteriore X 100). La deformazione cranica è direttamente proporzionale
alla diminuzione del peso del cervello, quando questo é inferiore a 1000 gr.;
• le idrocefalie : l’anomalo accumulo di liquido cefalorachidiano sia nei ventricoli (idrocefalia
interna) che negli spazi sub-aracnoidei (idrocefalia esterna). Importante chiariche che di per sé
non è una malattia ma è l’espressione sintomatologica di differenti condizioni morbose sia
congenite che acquisite, dovute, ad esempio, anche a: infiammazioni, infezioni (es.: la
toxoplasmosi connatale=trasmessa al feto dalla madre ), tumori.
INFEZIONI VIRALI:
Infezioni virali quali:
• embriopatie da rosolia ed
• encefalopatia da toxoplasma.
ENZIMOPATIE EREDITARIE
Sono caratterizzate da un difetto enzimatico che comporta lesioni del sistema nervoso del bambino
il cui cervello, al momento della nascita è ancora in evoluzione. Sono spesso accompagnate da
danni epatici, splenici, cardiaci, vascolari, ossei, cutanei e da lesioni oculari.
Tra le alterazioni del metabolismo degli aminoacidi (amino-acidurie dismetaboliche) la più
conosciuta è:
• FENILCHETONURIA o MALATTIA DI FOLLING anche se rara, la sua precocità di
reperimento che consente di evitarne gli esiti negativi a livello mentale, l’ha resa famosa. Il
bambino alla nascita è normale. Il ritardo può essere evitato se la terapia farmacologica e
dietologica interviene entro le prime settimane di vita. E’ dovuta alla non trasformazione della
fenilalanina in tiroxina e all’accumulo dei precursori (iperfenilalaninemia).
ABERRAZIONI CROMOSOMICHE
Le aberrazioni cromosomiche sono dovute ad errori che si manifestano nel corso della prima o
delle prime divisioni dello zigote. Le alterazioni della meiosi possono determinare sia variazioni del
numero dei cromosomi (alterazioni quantitative) che della loro struttura (alterazioni qualitative).
Quelle numeriche sono dovute, generalmente, ad una non-disgiunzione dei cromosomi. Le
aberrazioni di struttura sono dovute a rotture di uno o più cromosomi e assenza del normale
processo riparatore.
Alcuni esempi:
• aberrazioni numeriche autosomiche = sindrome di Down;
• aberrazioni di struttura =
SINDROME DELL’X FRAGILE (l’estremità del braccio lungo del
⇒
cromosoma X appare stirata);
CSA-SSIS Veneto A.A. 2006-2007- © Tutti diritti riservati
Donatella Donati –-Pagina 5 di 14
APPROFONDIMENTI DI
ATTIVITÀ FISICA ADATTATA, INTEGRAZIONE E INCLUSIONE
NELLE ATTIVITÀ MOTORIE
⇒
SINDROME DEL VERSO DEL GATTO o <<cri du chat>> (delezione
di metà circa delle braccia corte del cromosoma 5 determina
una malformazione della laringe che provoca il grido che
ricorda il miagolio del gatto. Caratterizzata da ritardo
mentale grave, microcefalia, malformazioni cardiache.
EMBRIO-FETOPATIE (=affezioni materne trasmesse al feto)
Oltre alle già citate aggressioni infettive vi sono quelle di origine tossica quali:
• tossiemia gravidica (nefrite ipertensiva della madre);
• fetopatie alcoliche, tabagiche, da farmaci (es.talidomide).
FATTORI PERINATALI
• la sofferenza fetale perinatale generalmente insorge per complicazioni da parto, per trauma
fisico, ma più spesso per anossia dovuta non solo a cause meccaniche (es.: cordone
ombelicale legato attorno al collo) ma anche a cause cardiache e polmonari.
• incompatibilità sanguigna feto-materna: fenomeno di iso-immunizzazione in una madre con
(fattore Rhesus) Rh- incinta di un bimbo Rh+.
FATTORI POST-NATALI
• infettivi: es. encefaliti acute convulsivanti, meningoencefaliti batteriche o virali. Un esempio è
costituito dall’erpete (= simile al virus citomegalico per quanto concerne gli effetti sul feto,
acquisito spesso al momento del parto, provoca meningo-encefalite con cianosi, febbre, vomito,
epatomegalia, coma, convulsioni; l’evoluzione è molto grave.).
• post-vaccinali
• metabolici: ittero nucleare o encefalopatia bilirubinica. Molteplici cause possono determinare
l’aumento della bilirubina libera che oltrepassa la barriera ematoencefalica e si localizza nei
gangli della base, nel midollo allungato, nella corteccia cerebrale. L’ittero non trattato provoca in
seguito spasticità extrapiramidale con sintomatologia coreoatetosica, oligofrenia (microcefalia),
ipoacusia.
SINDROMI GENETICHE PIÙ NOTE
Sindrome di DOWN
CENNI STORICI
Prima comparsa in letteratura medica della sindrome di Down nel 1800.
Esquirol nel 1838 descrive per la prima volta “idioti” di bassa statura con plica epicantale, testa
piccola, e radice del naso depressa. Nel 1846 Seguin aggiunge alle caratteristiche suddette la
presenza in questi soggetti di lingua grossa e fissurata, una spiccata sensibilità alle infezioni
cutanee polmonari e naso tronco. Nel 1866 nella descrizione del LONDON HOSPITAL REPORTS,
nella classificazione dei deboli mentali che Langdon-Down fa sulla base della morfologia della
faccia, prendendo spunto dalle caratteristiche etniche presentate, collega la sindrome con la razza
MONGOLICA e la denomina “IDIOZIA MONGOLOIDE” o mongolismo . La cultura di tipo
evoluzionistico che a quel tempo poneva la razza bianca come massima espressione
dell’evoluzione porta Down alla spiegazione su base razziale della malattia: era definito segno di
malattia il ritorno ad una razza definita “ meno evoluta”. Nel 1895 Shuttelworth definisce il
bambino affetto da questa sindrome come il bambino non completo “unfinished child”, poiché
questi neonati presentano alcuni segni fisici che ricordano il feto nel 2° trimestre di gravidanza,
CSA-SSIS Veneto A.A. 2006-2007- © Tutti diritti riservati
Donatella Donati –-Pagina 6 di 14
APPROFONDIMENTI DI
ATTIVITÀ FISICA ADATTATA, INTEGRAZIONE E INCLUSIONE
NELLE ATTIVITÀ MOTORIE
taluni dei quali sono conservati anche con la crescita. Nel 1959 J. Lejeune osserva che nel
cariotipo di questi soggetti è presente. nella coppia 21, un cromosoma in eccesso (Lenzini Baccicchetti, op. cit.)
CAUSA DIRETTA
Circa il 95% delle persone affette dalla sindrome di Down ha TRISOMIA 21 LIBERA COMPLETA
ED OMOGENEA, causata da una non-disgiunzione (nondisjunction) cioè un “difetto” di
funzionamento durante la meiosi -divisione cellulare- per cui due cromosomi non si separano e
vengono trasmessi insieme.
Circa il 4% ha la TRISOMIA 21 DA TRASLOCAZIONE (ROBERTSONIANA) cioè il cromosoma
21 invece di essere indipendente viene traslocato- trasferito su di un altro cromosoma
(generalmente sul 14- 15- o 22 ).
Importante sottolineare che circa il 50% di questo tipo di trisomia è di origine familiare, cioè uno dei
due genitori è portatore (carrier) di questo rimaneggiamento.
Circa l’ 1% è affetto da TRISOMIA 21 LIBERA COMPLETA A MOSAICO dovuta in una mancata
disgiunzione di uno dei cromosomi 21 durante i primi stadi di sviluppo dopo il concepimento che
comporta, in uno stesso individuo, la presenza di cellule con completa trisomia del cromosoma 21
e cellule normali (senza trisomia). Si presentano quindi come tessere di un mosaico, distribuite in
percentuali estremamente variabili, cellule con 47 e cellule con 46 cromosomi.
EZIOLOGIA
Le cause indirette responsabili della trisomia 21 o di altri fattori che la condizionano non
sono ancora ben chiariti.
Rimaneggiamenti parentali
I portatori di traslocazioni coinvolgenti il cromosoma 21 sono una categoria ad alto rischio anche
se è dimostrato che il rischio di sbilanciamento cromosomico dipende dal sesso del portatore e dal
tipo di traslocazione. dal 5% al 12% nei maschi - dal 12% al 165 nelle femmine.
Mosaico parentale
Comporta un rischio per la prole il fatto che uno dei due genitori sia portatore di trisomia 21 in
mosaico a bassa frequenza (cioè possiede due linee cellulari: una normale e una con trisomia
21).Mosaici parentali sono stati riscontrati sia nelle madri che nei padri; non è attualmente possibile
stabilire la frequenza nella popolazione di soggetti portatori di mosaicismi, in quanto questo reperto
non può essere associato ad alcun effetto visibile e rimane pertanto sconosciuta fino al momento in
cui si verifica la nascita di un bimbo affetto.
Circa l ‘8% dei casi di ricorrenza familiare di trisomia 21 è dovuta a mosaico parentale.
Effetto intercromosomico
Allo stato attuale non vi sono prove che consentano di accertare un rapporto di causa-effetto tra
un’anomalia cromosomica in un genitore e una completamente diversa nel cariotipo della prole.
Consanguineità dei genitori
I dati sulla’ influenza della consanguineità sulla non-disgiunzione del cromosoma 21 sono ancora
del tutto insufficienti e quindi controversi. Uno studio condotto nel 1980 nella popolazione del
KUWAIT- che ha più del 50% di matrimoni contratti tra consanguinei - ha riscontrato un aumento
del 4% della presenza sella sindrome rispetto ad un campione di controllo.
Radiazioni ionizzanti
Nonostante alcuni studi indichino una probabile correlazione tra radiazioni a basso dosaggio e il
manifestarsi della trisomia 21, al momento attuale non si può dare una risposta definitiva a questo
quesito essendo ancora controversi i dati disponibili.
Contraccettivi (ormoni femminili)
Non si ritiene, a tutt’oggi, che i contraccettivi orali abbiano degli effetti nella non - disgiunzione
cromosomica.
Ruolo dell’ età materna
CSA-SSIS Veneto A.A. 2006-2007- © Tutti diritti riservati
Donatella Donati –-Pagina 7 di 14
APPROFONDIMENTI DI
ATTIVITÀ FISICA ADATTATA, INTEGRAZIONE E INCLUSIONE
NELLE ATTIVITÀ MOTORIE
L’incidenza dell’evento è stimata intorno a 1:100 nelle donne con età superiore ai 38 anni e 1:2000
in quelle intorno ai 20 anni. Non è ancora completamente chiarito il significato del suddetto
rapporto, poiché non si conosce il motivo dell’effetto dell’età materna, sono state formulate diverse
ipotesi:
-la lunga permanenza della cellula uovo allo stato di prima fase meiotica sia responsabile
dell’istaurarsi di eventuali danni dovuti all’accumularsi di eventuali fattori -FISICI-CHIMICI o VIRALI
nocivi, che posterebbero di conseguenza a una segregazione anomala (Ferguson - Smith, 1984).
Sono però riportati dati che confermerebbero l’aumento delle nascite di soggetti con trisomia 21
anche in madri molto giovani Penrose, 1963; Erickson, 1978). Si ipotizza che vi sia un ruolo dello
squilibrio ormonale che si verifica sia verso la menopausa che nell’età molto giovane.
A tutt’oggi, comunque, nessuna delle ipotesi è in grado di chiarire questa problematica.
Ruolo dell’ età paterna
Non si ritiene che l’età paterna giochi un ruolo rilevante nella non-disgiunzione.
PRINCIPALI SEGNI CLINICI DELLA SINDROME DI DOWN - Jackson et al. 1976
da “la Sindrome di Down” a cura di Lenzini & Baccichetti
1. brachicefalia
2. rime palpebrali orientaleggianti
3. epicanto
4. blefarite o congiuntivite
5. macchie di Brusfield
6. nistagmo
7. bocca semiaperta
8. anomalie dei denti
9. lingua sporta
10. lingua con profonde incisure (scrotale)
11. palato arcuato
12. orecchie poco disegnate
13. collo corto
14. cute sovrabbondante al collo
15. mani corte e tozze
16. 5 dito corto
17.clinodattilia del 5 dito
18. solco palmare trasverso unico
19. aumentata distanza tra il primo ed il secondo dito dei piedi
20. cardiopatia
21. soffio cardiaco
22. ipertensione articolare
23. ipotonia muscolare
Oltre alle caratteristiche somatiche note, di particolare rilevanza per la pratica dell’attività
motoria è la possibile instabilità assiale dell’atlante (non corretto allineamento tra atlante ed
epistrofeo - prime due vertebre cervicali), accertata in una percentuale che va dal 12 al 22%
degli individui, molto più prevalente nelle femmine.
CSA-SSIS Veneto A.A. 2006-2007- © Tutti diritti riservati
Donatella Donati –-Pagina 8 di 14
APPROFONDIMENTI DI
ATTIVITÀ FISICA ADATTATA, INTEGRAZIONE E INCLUSIONE
NELLE ATTIVITÀ MOTORIE
SINDROME DEL CROMOSOMA X
FRAGILE
Il recente sviluppo tecnologico ha portato alla scoperta di una nuova alterazione cromosomica
denominata sindrome del cromosoma X fragile o di Martin-Bell che, secondo Spitz (1988) si
contende con la sindrome di Down il primato di maggior fattore genetico quale causa di ritardo
mentale. La prima osservazione risale al 1969. Sino all’età prepuberale nei soggetti affetti da
questa sindrome non risultano particolarmente evidenti anomalie fisiche. Ciò ne ha reso difficile la
diagnosi e il conseguente ritardo mentale è stato attribuito a cause culturali e/o familiari. Le madri,
portatrici di un’alterazione strutturale di una piccola parte del braccio lungo del cromosoma
sessuale X, denominata “sito fragile”, possono trasmetterla sia ai figli maschi, che risultano affetti
dalla sindrome, sia alle femmine, che potranno essere o portatrici dell’anomalia o affette (solo il
30%) da lieve ritardo mentale. Tale condizione non può essere trasmessa dal padre ai figli maschi.
La frequenza si calcola possa essere di circa 1 caso su 1000 nati maschi, mentre sarebbe di circa
1 su 600 la prevalenza delle femmine “portatrici” (S. Del Gracco n1/98 Handicap Risposte). Pur
non evidenziandosi segni neurologici anormali, il ritardo mentale conseguente a questa sindrome è
mediamente classificato tra il lieve o moderato.
Segni clinici di riconoscimento, che peraltro si evidenziano in età prepuberale sono generalmente:
• mani grandi;
• viso allungato con fronte ampia;
• fronte e mento sporgenti;
• padiglioni auricolari grandi e labbra grosse;
• macrorchidismo
Vi è un ritardo nello sviluppo, ma i piccoli camminano, parlano e generalmente riescono a ben
inserirsi nella società. Tendono a uno scarso contatto oculare e il loro linguaggio si caratterizza per
la ripetizione di suoni o frasi e per la pronuncia veloce e con ritmo sbagliato delle parole. Sviluppo
psicomotorio e attenzione risultano deficitari anche se può esservi iperattività. Talvolta, associati al
ritardo mentale, si possono riscontrare comportamenti di tipo autistico (Blackman,1990), con
stereotipie gestuali e grosse difficoltà di relazione.
Un intervento precoce ed individualizzato di rieducazione logopedica e neuropsicomotoria possono
notevolmente favorire l’inserimento sociale di questi bambini.
LESIONI DEL MIDOLLO SPINALE
POLIOMIELITE ANTERIORE ACUTA- Malattia di Heing-Medin
E’ causata dai poliovirus che producono lesioni infiammatorie distruttive la cui sede elettiva è
rappresentata dalle corna anteriori del midollo spinale. Le lesioni possono estendersi al midollo
allungato, alle formazioni reticolari del ponte, ai nuclei vestibolari, ai centri del cervello ed anche
alla corteccia cerebrale. E’ una malattia infettiva tendenzialmente epidemica, ma oggi rarissima e
sporadica, che provoca paralisi muscolare atrofica. La paralisi flaccida, che interessa i più disparati
gruppi muscolari, ha tuttavia una netta predilezione per la muscolatura degli arti: prevalentemente
quelli inferiori. E’ capricciosa nella sua distribuzione topografica: a volte alcuni gruppi muscolari o
alcuni muscoli di un arto (in prevalenza quelli del tratto prossimale, es.: i glutei, gli adduttori, il
quadricipite femorale) e l’altro arto controlaterale in maniera completa.
Di particolare importanza è l’inalterata sensibilità dei muscoli colpiti, non essendo le corna
posteriori del midollo spinale sede elettiva dei poliovirus;
dal punto di vista
dell’apprendimento motorio è sicuramente un vantaggio non trascurabile avere coscienza
della posizione di un segmento corporeo pur non potendolo muovere.
CSA-SSIS Veneto A.A. 2006-2007- © Tutti diritti riservati
Donatella Donati –-Pagina 9 di 14
APPROFONDIMENTI DI
ATTIVITÀ FISICA ADATTATA, INTEGRAZIONE E INCLUSIONE
NELLE ATTIVITÀ MOTORIE
TRAUMI MIDOLLARI
Lesioni midollari provocate da cause accidentali, quali ad esempio lussazioni e/o fratture vertebrali
da incidenti d’auto o tuffi errati, provocano l’interruzione della conduzione dell’impulso nervoso
necessario al movimento volontario, con la conseguente paralisi della muscolatura.
Il livello della lesione e la sua completezza - totalità o parzialità - incompletezza, determinano la
parte di corpo non collegata e quindi paralizzata.
Le minuscole porzioni di midollo spinale comprese tra due corpi vertebrali, denominate metameri,
raramente, infatti, vengono lesionate da un trauma in modo completo, cioè con sezione trasversa
perfetta; quasi sempre la lesione avviene in modo “imperfetto”, consentendo un ponte di
collegamento al di sopra o al di sotto di essa. Questo ponte porta ad un alterato tono
muscolare(ipertono); la lesione totale ne provoca, invece, la mancanza assoluta cioè, la paralisi
flaccida.
Generalmente, se la lesione è compresa tra la IV vertebra cervicale e la I toracica vi è una paralisi
di tutti e quattro gli arti, cioè, tetraplegia; se è compresa tra la II toracica e la II sacrale vi è una
paralisi degli arti inferiori, cioè, paraplegia.
PARALISI CEREBRALE INFANTILE
E’ descritta come un disturbo neurologico cronico, non progressivo derivante da una lesione
cerebrale insorta durante uno stadio di sviluppo precoce (prenatale, perinatale, neonatale). Può
essere associata a ritardo mentale; HEALY(1990) ritiene che il 60% - 70% delle persone con CP
abbia qualche grado di ritardo mentale.
Classificazioni della paralisi cerebrale
Circa il 97% dei casi ricade sotto le tre maggiori categorie :
• Paralisi cerebrale di tipo spastico - 60% - 65% Dovuta, principalmente, ad “alterazione” delle vie piramidali. E’ caratterizzata da difficoltà di
generare movimenti volontari rapidi e precisi. Vi è infatti l’incapacità di rilassare, nei distretti colpiti, i
muscoli ad esso antagonisti. Il disturbo tonico-motorio si caratterizza, prevalentemente, con
retrazioni tendinee che provocano: ipertonia estensoria agli arti inferiori, ipertonia flessoria e
adduttoria agli atri superiori. Con la crescita, le ipertonie che interessano alcuni distretti muscolari,
provocano posture sbagliate e deformazioni articolari.
• Paralisi cerebrale di tipo atetosico - 25% Dal greco thetos (=posizione fissa) a privativa; dovuta principalmente ad “alterazioni “ delle vie
extrapiramidali . HEALLY ( 1990) più accuratamente descrive i seguenti tipi di CP: Discinesia è il
termine che descrive la paralisi cerebrale caratterizzata da movimenti spontanei, involontari; questi
includono i movimenti lenti che ricordano lo scrivere o lo strisciare dei vermi, in particolare del
polso e delle dita (ATETOSICI), che possono essere accompagnati da altri più a scatto e repentini
(COREOATETOSICI). Questi, alternandosi al movimento volontario, lo rendono funzionalmente
incoordinato e inefficiente. Un’altra forma di discinesia (DISTONIA) comprende movimenti, lenti e
ritmici che interessano il tronco o una estremità. BLECK (1982) che usa il termine più antico di
atetosi identifica come maggiore sua caratteristica i “movimenti caldi: “I movimenti sono, a volte,
rotatori (torsione e rotazione degli arti), distonici (posizione distorta degli arti, collo, o del tronco,
che è tenuta per alcuni secondi, e poi rilassata). Se sollecitato da ragioni emotive il movimento di
un atetosico può assomigliare a quello di un burattino. Generalmente chi è atetosico ha uno scarso
controllo delle labbra e dei muscoli della lingua; ciò rende estremamente difficoltoso il parlare ed è
spesso causa di balbuzie. Questa inabilità a parlare, dovuta a questo accentuatissimo
balbettamento, porta a pensare che questi soggetti abbiano un forte ritardo mentale; molti bambini
con atetosi hanno invece capacità intellettuali medie o superiori alla media.
• Paralisi cerebrale di tipo atassico - 7% -
CSA-SSIS Veneto A.A. 2006-2007- © Tutti diritti riservati
Donatella Donati –-Pagina 10 di 14
APPROFONDIMENTI DI
ATTIVITÀ FISICA ADATTATA, INTEGRAZIONE E INCLUSIONE
NELLE ATTIVITÀ MOTORIE
Dal greco taxis (ordine - equilibrio ) a privativa. Dipende generalmente da alterazioni del
cervelletto e delle sue vie. Caratterizzata da disturbi della coordinazione dei movimenti volontari
e dell’equilibrio, barcollamento nel cammino, non è generalmente associata a ritardo mentale.
Nella posizione seduta questi soggetti possono ben allenare la parte superiore del corpo
(annullando quindi, problemi di equilibrio statico e dinamico) tanto da poter essere sottoposti anche
a carichi pesanti. L’ergometro a manovella è, per loro, ottimo strumento per migliorare la capacità
cardiorespiratoria.
• Paralisi cerebrale di tipo misto
Non essendo sempre possibile classificare tutti i casi nelle suddette categorie, poiché presentano
caratteristiche che appartengono a diverse classi, viene utilizzato il termine “tipo misto” ( HALEY).
La localizzazione della lesione definisce la classificazione con termini usati in Italia in modo diverso
dagli altri paesi.(vedi tabella)
DISTROFIE MUSCOLARI
A questa categoria appartiene un grande gruppo di malattie con eziologia e quadri clinici
eterogenei. Sono caratterizzati dalla degenerazione progressiva dei muscoli scheletrici. In alcune
forme, dette miogene, il processo degenerativo è a carico principalmente delle fibre muscolari; in
altre, dette neurogene, esso è a carico delle fibre nervose, conseguentemente a ciò avviene la
progressiva degenerazione muscolare. In genere hanno carattere ereditario.
EPILESSIA
Epilessia è il nome della sindrome caratterizzata da crisi che tendono a ripetersi in modo
cronico a causa di una anomalia durevole del funzionamento cerebrale.
Epilessia lesionale: viene denominata quella causata da alterazioni strutturali del cervello
evidenziabili.
Epilessia non lesionale o criptogenetica: è denominata quella in cui non sono evidenti anomalie
anatomiche del cervello.
La crisi epilettica (= manifestazione clinica parossistica, cioè di massima intensità) può
manifestarsi in vario modo ed è la conseguenza di una anormale attività di una parte della
sostanza grigia cerebrale, espressione biologica dell’ipersincrono funzionamento di una massa di
neuroni corticali. Tale attività viene abitualmente registrata come una alterazione specifica del
tracciato elettroencefalografico.
La malattia epilettica è data dalla ripetizione delle crisi dovuta a circostanze scatenanti somatiche
e psichiche.
L’epilettico è colui la cui organizzazione psichica utilizza il ripetersi delle crisi come via di scarico
di pulsioni, sia in modo massivo che investendo di un valore rappresentativo funzionale le
manifestazioni cliniche, valore di rappresentazione funzionale, affettiva o fantasmatica che le
manifestazioni cliniche in origine non possiedono.
CSA-SSIS Veneto A.A. 2006-2007- © Tutti diritti riservati
Donatella Donati –-Pagina 11 di 14
APPROFONDIMENTI DI
ATTIVITÀ FISICA ADATTATA, INTEGRAZIONE E INCLUSIONE
NELLE ATTIVITÀ MOTORIE
E’ importante sottolineare la rarità di disturbi mentali cronici nell’epilettico. Con le precauzioni
determinate dalla possibilità di una crisi, sotto il controllo del trattamento farmacologico, la
maggioranza di essi può condurre una vita normale.
I casi di ammalati gravi, bisognosi di assistenza per vivere, sono soggetti colpiti sin dall’infanzia ed
è quindi particolarmente difficile capire quanto della loro condizione trova fattore eziologico nella
malattia epilettica - sia come causa organica che dal trattamento - o in carenze educative o di
relazione.
Per quanto riguarda la personalità dell’epilettico, risulta evidente che esiste un <<vissuto>>
dell’epilessia che, in una parte dei soggetti, stabilisce delle relazioni tra la malattia e lo sviluppo
della loro personalità.
Moltissimi sono gli studi - psicometrici, fenomenologici e psicoanalitici - che tentano di determinare
il confine dell’epilessia verso le nevrosi e i disturbi psico-affettivi.
AUTISMO
A questa sindrome sarà dedicato un modulo. In questo si danno solo le informazioni basilari.
Il concetto di autismo infantile nasce nel 1943 con LEO KANNER che descrive e definisce un
gruppo di 11 bambini con questo termine. Per questo autore ..” tutti questi bambini sono
indubbiamente dotati di buone potenzialità cognitive”...
Kanner utilizzò il termine autistico per significare l’incapacità di rapportarsi con gli altri e il
desiderio di essere lasciati soli.
COSA NON E’ L’AUTISMO
Non è la conseguenza di un alterato rapporto tra un bambino nato sano e l’ambiente, soprattutto
familiare, incapace di accettarlo. Non è un disturbo psicologico dovuto alla “patogena” relazione
affettiva madre - figlio, l’effetto di una <<madre frigorifero>>.
CHE COS’E’ L’AUTISMO
• Conoscenze scientifiche ci hanno consentito di capire che l’autismo è un disturbo
generalizzato dello sviluppo che coinvolge diverse funzioni cerebrali e perdura per tutta la
vita.
• per descriverlo viene utilizzato il termine sindrome perché le cause che provocano
caratteristiche cliniche e disturbi dello sviluppo, comuni nelle persone ne soffrono, sono diverse
e sconosciute.
• La comunità scientifica internazionale (classificazione ICD 10 dell’OMS e DSM IV) lo considera
un disturbo pervasivo dello sviluppo che si manifesta entro il terzo anno di età con deficit in tre
aree:
1. comunicazione,
2. interazione sociale,
3. immaginazione.
• Secondo stime recenti l’autismo colpisce 1 persona su 1000; 2 su 1000 sono quelle che ne
presentano alcuni sintomi e, pertanto, vengono incluse nello “spettro autistico”.
CSA-SSIS Veneto A.A. 2006-2007- © Tutti diritti riservati
Donatella Donati –-Pagina 12 di 14
APPROFONDIMENTI DI
ATTIVITÀ FISICA ADATTATA, INTEGRAZIONE E INCLUSIONE
NELLE ATTIVITÀ MOTORIE
PSICOSI
Potremmo definirla un modo di essere abnorme della psiche; l’IO del soggetto è disturbato ed egli
non ha piena coscienza di ciò che gli accade e lo disturba. E’ ancora oggi ritenuta valida la
distinzione tradizionale tra psicosi organiche ed endogene o funzionali.
PSICOSI ORGANICHE: sindromi psicopatologiche di tipo psicotico conseguenti a disturbi organici
del funzionamento cerebrale e si dividono in croniche e acute. Le prime si identificano con le
demenze essendo caratterizzate da diminuzione di memoria e intelligenza; le seconde sono
caratterizzate da una compromissione più o meno marcata della lucidità della coscienza e sono
conseguenti ad un alterato funzionamento cerebrale per cause tossiche, infettive o traumatiche
(es.: al risveglio da un coma per trauma cranico).
PSICOSI FUNZIONALI: sono un insieme non omogeneo di disturbi di tipo psicotico per i quali è
assente o non accertata una patologia cerebrale, o comunque somatica.
La psicosi funzionale più tipica è la schizofrenia
SCHIZOFRENIA
Secondo il DSM III la malattia schizofrenica consiste in:
<<disturbi mentali con tendenza alla cronicità, con diminuzione delle funzioni e caratterizzati da
sintomi psicotici riguardanti disturbi del pensiero, dell’affettività e del comportamento>>.
Cause: risultano molto importanti fattori ambientali stressanti e carenze affettive ed educative; si
ammette che esista una predisposizione genetica. Molti soggetti rivelano in precedenza tratti
caratterologici particolari quali: asocialità, chiusura affettiva, atteggiamento persegutivo.
Sintomatologia: vi è una progressiva disorganizzazione delle funzioni psichiche che in fase acuta
risultano tutte compromesse: il linguaggio, il pensiero, la percezione, il sentimento di sé e il
rapporto con gli altri, gli affetti. Molto raramente vi è la coscienza del proprio stato.
Forme cliniche: LATENTE include i casi definiti al limite o “borderline”
ATTENUATA
EBEFRENICA in cui l’impoverimento demenziale della personalità è l’elemento
prevalente;
CATATONICA in cui prevalgono i disturbi del movimento
PARANOIDE caratterizzata da comportamenti violenti poiché i soggetti si
sentono perseguitati e minacciati
Può esservi agitazione psicomotoria o, al contrario, blocco totale dei movimenti. Alcuni soggetti
possono manifestare movimenti bizzarri, smorfie, atteggiamenti enfatizzati. La percezione è
alterata da “allucinazioni” , da “ voci” che vengono udite in assenza di ogni stimolazione obiettiva. Il
pensiero è confuso, caotico, pieno di astrazioni, simbolismi, strutture deliranti. Può esservi la
convinzione che il proprio pensiero sia dominato da altri.
CSA-SSIS Veneto A.A. 2006-2007- © Tutti diritti riservati
Donatella Donati –-Pagina 13 di 14
APPROFONDIMENTI DI
ATTIVITÀ FISICA ADATTATA, INTEGRAZIONE E INCLUSIONE
NELLE ATTIVITÀ MOTORIE
Notizie utili
Soggetti affetti da DIABETE
Se il soggetto è correttamente trattato può praticare qualsiasi attività fisica o sportiva.
Si possono tuttavia verificare delle manifestazioni anormali dovute a varie cause tipo insufficienza
o eccesso trattamento insulinico somministrato, o per una non corretta alimentazione. Se il
trattamento insulinico è insufficiente può verificarsi un imperioso bisogno di urinare o una sete
incoercibile. Per eccesso di insulinoterapia si possono verificare dei malesseri ipoglicemici con
manifestazioni variabili da soggetto a soggetto ma costanti nello stesso con sole variazioni
d’intensità.
Queste manifestazioni generalmente sono le seguenti:
• sensazione di fame
• fame imperiosa con sensazione di dolenza allo stomaco “crampi”
• malessere spesso descritto come “impressione di cervello vuoto”, accompagnato da abbondanti
sudorazioni, pallore, cefalea, vertigine e anche piccoli tremori;
• dolenza alle gambe con impossibilità di effettuare il minimo sforzo, deambulazione da ubriaco;
• turbe del comportamento quali: assoluta indifferenza o insolita eccitazione, collera improvvisa,
cattivo umore;
• sonnolenza che può sfociare in una perdita di coscienza più o meno completa, accompagnata
talvolta da movimenti convulsivi.
Primo immediato intervento è quello di somministrare glucosio sotto qualsiasi forma.
i
Quoziente intellettivo: è il quoziente numerico che esprime la misura dell’intelligenza individuale, ottenuto dal
rapporto tra:
Q.I.= età mentale (risultante da test psicometrici)
età cronologica
X 100
CSA-SSIS Veneto A.A. 2006-2007- © Tutti diritti riservati
Donatella Donati –-Pagina 14 di 14