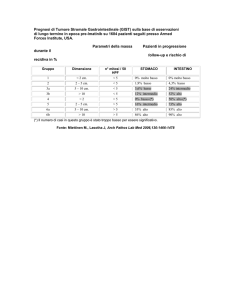
COSA SONO I MICRO-GIST E PERCHE’ STUDIARLI
AIUTERA’ A CAPIRE COME CURARE I GIST
Comprendere il meccanismo di sviluppo dei Tumori Stromali Gastrointestinali è l'obiettivo
dei ricercatori, per scoprire nuove terapie per la cura dei GIST.
I dottori Sebastian Bauer e Jonathan Fletcher, in un articolo pubblicato sul giornale del Life
Raft Group (LRG), il 23 gennaio 2013 (“Understanding how GISTS develop enables discovery of new terapie”) parlano dei GIST di piccolissime dimensioni (microGIST), di come
questo tipo di tumore si sviluppa e che cosa può derivare da queste conoscenze.
Pubblichiamo l’articolo di LRG, tradotto in italiano, nello spirito di condivisione che unisce la comunità internazionale dei pazienti GIST.
Anche se non si tratta di una facile lettura per i non addetti ai lavori, pensiamo che la conoscenza della malattia in ogni suo aspetto aiuti i pazienti ad abbattere la
paura e a combattere meglio la malattia.
A.I.G. Associazione Italiana GIST Onlus
L'articolo originale in lingua inglese si può leggere al link
http://liferaftgroup.org/2013/01/understanding-how-gists-develop/
Traduzione e sintesi italiana a cura di A.I.G.
“Comprendere come i GIST si sviluppano consente la scoperta di nuove terapie”.
Dr. Sebastian Bauer, West German Cancer Center, University of Essen, Garmany
Dr. Jonathan Fletcher, Brigam & Women's Hospital, Harvard University, LRG Research
Team.
La gran parte di noi ha alcuni 'nei' sparsi nella cute
del proprio corpo. La maggior parte di questi “nei“
sono simili a neoplasie benigne, molto comuni
nella popolazione, ma sappiamo tutti che,
raramente, questi si trasformano in tumori maligni
della pelle. Ciò che pochi sanno è che i GIST si
potrebbero descrivere come 'nei ' dello stomaco.
Una persona su tre ha un minuscolo GIST nello
stomaco. Conosciamo questo dato grazie al lavoro
diligente dei dottori Kaori Kawanowa, Shinji Sak e
colleghi, i quali, in diversi centri medici giapponesi,
hanno studiato attentamente 100 stomaci, che erano stati rimossi chirurgicamente da
pazienti con tumori gastrici (non GIST). La lunghezza media di uno stomaco adulto è di 25
cm e questi dottori hanno sezionato gli stomaci a intervalli di 5 mm, il che ha permesso
loro di esaminare al microscopio una quantità pari a centinaia o anche a migliaia di fette di
stomaco da ciascun paziente. Con grande sorpresa di molti, il Dr. Kawanowa ha
identificato 50 piccolissimi GIST in 35 degli stomaci esaminati.
Tuttavia, nessuno di questi minuscoli GIST sarebbe da considerarsi clinicamente rilevante
o pericoloso (1). Ciò che colpisce per i pazienti affetti da GIST è che questi piccoli GIST
innocui presentano le medesime mutazioni dei geni KIT o PDGFRA (cioè le mutazioni che
costituiscono i principali meccanismi oncogeni nei GIST) che si riscontrano nella malattia
quando viene diagnosticata (2).
Fortunatamente la mutazione di un singolo gene nei geni KIT e PDGFRA, da sola, non
trasforma una cellula gastrointestinale normale in un tumore GIST, altrimenti avremmo due
miliardi di pazienti con GIST in tutto il mondo. Infatti, vi è una forte evidenza scientifica, in
base all’epidemiologia e agli studi di laboratorio, che sono necessarie mutazioni multiple,
che coinvolgano geni importanti nello sviluppo cellulare, per produrre la maggior parte dei
tumori.
Questi concetti sono stati diffusi per la prima volta dal Dr. Carl Nording, finlandese, il quale
pur essendo architetto e storico ha speso molto del suo tempo su problemi che erano al di
fuori del suo campo professionale. Nel 1953 egli ha pubblicato una teoria “multi mutazione” con l'obiettivo di spiegare perché i tumori diventano sempre più comuni con
l’avanzare dell'età delle persone (3). Ha ipotizzato che l'aumento del cancro con
l'invecchiamento potrebbe essere spiegato col fatto che la maggior parte dei tumori
richiede sei mutazioni sequenziali e che, quindi, un tempo considerevole è necessario
perché queste mutazioni casuali si sviluppino in una precisa lesione pre-tumorale. La sua
teoria fu poi perfezionata dal Dr. Alfred Knudson, il quale riteneva che la maggior parte dei
tumori richiedessero almeno due mutazioni genetiche cruciali (4).
[….] Simili osservazioni hanno fatto Brian Rubin della Cleveland Clinic, membro del team
di ricerca di Life Raft Group e Peter Besmer del Memorial Sloan- Kettering Cancer Center,
scopritore del gene KIT, i quali in base ad esperimenti di laboratorio sui topi (6), hanno
confermato che l'ipotesi del dottor Knudson si applica al GIST.
Sappiamo che i pazienti con GIST diffuso possono rispondere all'imatinib per diversi anni,
ma questo trattamento di solito non guarisce completamente la malattia.
L'importanza dei piccoli GIST, altrimenti detti "microGIST", sta nel fatto che essi ci danno
un punto di partenza molto precoce nello sviluppo del GIST, per individuare le varie
mutazioni geniche, aggiuntive e decisive, necessarie perché insorga un GIST clinicamente
rilevante. Inoltre, poiché sappiamo che in pratica tutti i microGIST perdurano come piccoli
tumori benigni, o vanno incontro ad autodistruzione (invece di progredire e svilupparsi in
tumori maligni), questi studi dovrebbero permettere di identificare gli ostacoli biologici alla
progressione maligna in pazienti con GIST.
La comprensione di tali ostacoli, potrebbe rivelare nuovi bersagli terapeutici nei GIST, tra i
quali proteine anormali che impediscono alle cellule GIST di morire, anche in presenza di
un’efficace inibizione di KIT /PDGFRA.
Che cosa si sa già delle mutazioni genetiche aggiuntive- vale a dire, quelle ben oltre le
mutazioni di KIT o PDGFRA - che sono necessarie per produrre un tumore GIST? Un
modo tradizionale per identificare queste mutazioni è mediante cariotipizzazione[1] fatta
su cellule GIST prelevate da biopsie chirurgiche che sono coltivate in laboratorio e sulle
quali i cromosomi sono identificati mediante colorazioni speciali e osservati al microscopio.
I cromosomi contengono il DNA delle cellule, e la cariotipizzazione è lo stesso approccio
con cui si valutano i cromosomi di cellule fetali quando si fa l’amniocentesi, per vedere se
ci sono anormalità associate a malattie e per determinare il sesso del feto. Una cellula
normale ha 46 cromosomi, ma i cariotipi (cioè il "corredo cromosomico" caratteristico di
una determinata specie.) in molti GIST mostrano perdite di diversi cromosomi e spesso di
particolari parti di un cromosoma. Queste anomalie cromosomiche, molto ricorrenti,
identificano le posizioni dei geni che controllano la crescita e altre importanti proprietà
delle cellule GIST: geni che normalmente mantengono sotto controllo le cellule precursori
dei GIST. Quando i geni che controllano il GIST sono perduti per delezione[2] del
cromosoma o mutazione, questo fa perdere il controllo alle cellule GIST innocue,
permettendo loro di crescere e di comportarsi in modo più aggressivo.
La cariotipizzazione del GIST e altri studi sui cromosomi o sul DNA, fino ad oggi, hanno
localizzato nel genoma umano regioni che contengono più di cinque geni fondamentali che
causano il GIST e molti di questi geni sono stati identificati. L'identificazione di altri geni
coinvolti nei GIST costituisce un’importante priorità di ricerca, poiché queste scoperte,
probabilmente, porteranno a progressi nelle terapie per curare i GIST.
Diversi ricercatori hanno osservato che delezioni di parte del cromosoma 9 sono viste
raramente nei GIST a basso rischio, ma sono comuni nei GIST ad alto rischio di recidiva. Il
gene bersaglio diqueste delezioni cromosomiche è CDKN2A (7, 8), la cui funzione
è quella di inibire il ciclo cellularenelle cellule interstiziali di Cajal (quelle cellule da
cui derivano GIST) e in altre cellule. CDKN2A regola la replicazione cellulare.
I pazienti affetti da GIST, che hanno perso CDKN2A, hanno un rischio di gran
lunga maggiore di sviluppare progressione di malattia rispetto a quelli che non l’hanno
perso.
Nell’ambito del progetto di ricerca del Life Raft Group una serie di GIST sono stati
analizzati non solo guardando i cromosomi, ma anche attraverso il sequenziamento di
ogni gene (genoma intero e sequenziamento dell’ esoma intero)[3]. Un'osservazione
importante che deriva da questi studi è che i GIST più aggressivi presentano mutazioni
genetiche con coinvolgimento di CDKN2A e di altri geni correlati, che portano a una
disregolazione[4] del ciclo cellulare. Questi studi indicano che quasi tutti i GIST richiedono
una o più mutazioni che aumentino l'attività del ciclo cellulare, per passare da un GIST a
basso rischio a uno ad alto rischio di recidiva. Per confermare queste scoperte sono in
corso studi su un gruppo più ampio di campioni di GIST, utilizzando ricerche
all’avanguardia. In particolare, le mutazioni che interessano i geni correlati al ciclo cellulare
non sembrano rendere le cellule GIST meno sensibili agli inibitori di KIT / PDGFRA come
imatinib. Ciononostante, queste scoperte potrebbero essere rilevanti in ambito clinico,
giacché vari inibitori terapeutici del ciclo cellulare, compresi inibitori di CDK4/6, potrebbero
ripristinare il controllo del ciclo cellulare nei GIST con delezioni di CDKN2A, o con altri
difetti del ciclo cellulare.
Alcuni dei difetti del ciclo cellulare dei GIST sono effetto della soppressione di una
proteina, che è considerata il principale regolatore e controllore del ciclo cellulare, la
proteina p53 che molto spesso determina se un danno genetico alla cellula può essere
riparato o se la cellula deve andare incontro ad apoptosi, cioè a morte cellulare
programmata.
Trattando le cellule GIST con imatinib e poi aggiungendo farmaci che "svegliano", per così
dire, il principale regolatore del ciclo cellulare, p53 aumenta notevolmente il numero di
cellule apoptotiche (9). Tuttavia modificare p53 nei pazienti potrebbe essere pericoloso,
perché modificare p53 può anche avere effetti sulle cellule normali. Studi clinici in corso
stanno indagando con molta cautela se questi trattamenti possono essere fatti sui pazienti
con sicurezza.
Ci auguriamo che nei prossimi anni si possa fare più luce sulle differenze genetiche e
biologiche tra i "microGIST” e i GIST e così ci possiamo avvicinare all’obiettivo finale che è
quello di curare con successo questo tumore.
[1] nr, la cariotipizzazione è un test per esaminare i cromosomi in un campione di cellule e può
aiutare ad identificare problemi genetici come causa di malattie; il test consiste nel contare il
numero dei cromosomi e osservare i cambiamenti strutturali nei cromosomi. Può essere fatto su
quasi tutti i tessuti, compreso il liquido amniotico come si fa per l’amniocentesi
[2] nr aberrazione o mutazione cromosomica, che consiste nell’assenza di un tratto di un
cromosoma con conseguente perdita di materiale genetico
[3] nr esoma è la porzione del genoma che contiene le istruzioni per fabbricare una determinata
proteina
[4] deterioramento del meccanismo fisiologico di regolazione
Bibliografia
1.
Kawanowa K, Sakuma Y, Sakurai S, et al.: High incidence of microscopic gastrointestinal stromal
tumors in the stomach. Hum Pathol 37:1527-1535, 2006
2.
Corless CL, McGreevey L, Haley A, et al.: KIT mutations are common in incidental gastrointestinal
stromal tumors one centimeter or less in size. Am J Pathol 160:1567-1572, 2002
3.
NORDLING CO: A new theory on cancer-inducing mechanism. Br J Cancer 7:68-72, 1953 4.
Knudson AG, Jr.: Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A
68:820-823, 1971
5.
Rubin BP, Antonescu CR, Scott-Browne JP, et al.: A knock-in mouse model of gastrointestinal
stromal tumor harboring kit K641E. Cancer Res 65:6631-6639, 2005
6.
Sommer G, Agosti V, Ehlers I, et al.: Gastrointestinal stromal tumors in a mouse model by
targeted mutation of the Kit receptor tyrosine kinase. Proc Natl Acad Sci U S A 100:6706-6711, 2003
7.
Schneider-Stock R, Boltze C, Lasota J, et al.: High prognostic value of p16INK4 alterations in
gastrointestinal stromal tumors. J Clin Oncol 21:1688-1697, 2003
8.
Lagarde P, Perot G, Kauffmann A, et al.: Mitotic checkpoints and chromosome instability are strong
predictors of clinical outcome in gastrointestinal stromal tumors. Clin Cancer Res 8:826-838, 2012
9.
Henze J, Muhlenberg T, Simon S, et al.: p53 modulation as a therapeutic strategy in
gastrointestinal stromal tumors. PLoS One 7:e37776, 2012