METABOLISMO
delle
PROTEINE
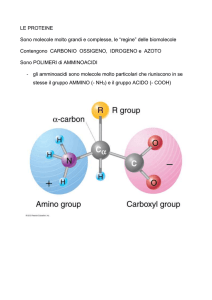
AZOTO essenziale per la vita
- Amminoacidi
- Nucleotidi
In natura
-N2 atmosferico (N.B. N≡N triplo legame, molta energia per
scinderlo)
- ione nitrato NO3– presente nel suolo
Nei sistemi biologici sono presenti le forme ridotte
- ione ammonio NH4+ libero
- gruppo amminico (-NH3+) e gruppo ammidico (-NH-C=O )
presenti in composti organici
GLI ANIMALI DIPENDONO DA BATTERI E PIANTE
PER L’AZOTO (ciclo dell’azoto)
I. Soltanto alcuni batteri anaerobi, simbionti nelle radici delle
leguminose, sono in grado di fissare (ridurre) l’N2
atmosferico con produzione di ammoniaca
II. altri batteri ossidano NH3 a nitrito (NO2– ) e quindi a nitrato
II. Le piante sono in grado di utilizzare NO3– con
produzione di NH4+, che viene quindi incorporato nei
composti organici azotati (punto d’ingresso Glu e Gln)
III. Gli animali assumono composti organici azotati
(amminoacidi)
Fonte primaria di azoto:
amminoacidi forniti dalle proteine
alimentari
Funzioni degli L-α
α-amminoacidi
Substrati per la sintesi proteica
20 a.a - con codone
21 a.a. selenocisteina
riconoscimento via tRNA
seril-tRNA + seleniofosfato Se-cisteinil tRNA
subiscono modificazione post-sintetica
esempi: amminoacidi fosforilati; acido γcarbossiglutammico
Componenti di peptidi
glutatione (GSH) γGlu-Cys-Gly
Intermedi metabolici
ornitina
Fonte energetica
Trasporto di azoto
a.a. glucogenici, a.a. chetogenici
glutammina, alanina
Precursori per la biosintesi degli altri composti
contenti azoto
composti derivati
amminoacidi precursori
––––––––––––––––––––––––––––––––––––––––––––––––––––––––––
Eme
glicina (+ succinil CoA)
Nucleotidi
glutammina, glicina, acido aspartico
Carnitina
lisina, metionina
Creatina
arginina, glicina, metionina
Ammine biogene
,
istidina (
istamina)
triptofano (
serotonina)
Tiroxina, adrenalina
tirosina
Taurina (nei sali biliari)
cisteina
Niacina
triptofano
CLASSIFICAZIONE NUTRIZIONALE
AMMINOACIDI ESSENZIALI :
devono necessariamente essere introdotti preformati con la dieta
valina
leucina
isoleucina
metionina
fenilalanina
triptofano
istidina
lisina
treonina
AMMINOACIDI NON ESSENZIALI
i. semi-indispensabili risparmiano i precursori essenziali
tirosina (sintetizzata da fenilalanina)
cisteina (sintetizzata da metionina)
condizionatamente non essenziali
glicina, serina, prolina, glutammina, arginina
possono non essere sufficienti in alcuni stati particolari quali infezioni,
traumi, bambini prematuri,
non essenziali
alanina, aspartato, asparagina, glutammato
Le reazioni di transaminazione, reversibili, permettono
di ridistribuire il gruppo NH3 fra gli amminoacidi
Vanno comunque integrati con la dieta e l’apporto deve essere
bilanciato in quanto:
- Il pool di amminoacidi non è totalmente riutilizzabile
- NH3 principalmente prodotto di rifiuto, anche se vi è un
riutilizzo limitato a riformare amminoacidi
digestione
Enzimi digestivi secreti come zimogeni inattivi attivati tramite proteolisi nel lume intestinale
I.
DIGESTIONE PROTEINE - STOMACO
pH acido: denatura le proteine alimentari
pH acido: autoattivazione del
PEPSINOGENO PEPSINA + peptidi
Il processo prosegue in modo autocatalitico
Pepsina: endopeptidasi poco specifica ma preferisce
rompere il legame che coinvolge il gruppo
carbossilico di Tyr, Phe, Trp
proteine alimentari + pepsina grandi peptidi
LUME
SANGUE
Cl–
HCO3–
Cl–
Cl–
HCO3–
H+
H+
CO2 + H2O
pompa
H+/K+
metabolismo
ATPasi
K+
membrana
baso-laterale
Cl–
K+
membrana
apicale
II LUME INTESTINALE tramite ENZIMI PANCREATICI
Zimogeni secreti dal pancreas esocrino
Enterochinasi: legata alla membrana apicale degli enterociti
TRIPSINOGENO + enterochinasi TRIPSINA + esapeptidi
CHIMOTRIPSINOGENO + tripsina CHIMOTRIPSINA +2 dipeptidi
PROELASTASI + tripsina ELASTASI
PROCARBOSSIPEPTIDASI A e B + tripsina CARBOSSIPEPTIDASI
endopeptidasi
TRIPSINA - scinde legame COO- di a.a. basici (Arg, Lys)
CHIMOTRIPSINA - scinde legame COO- di a.a. idrofobici (Phe, Tyr, Trp)
ELASTASI - scinde legame COO- di piccoli aa neutri (Gly, Ala, Val)
Esopeptidasi (rilasciano a.a. liberi e oligopeptidi di 2-8 residui)
CARBOSSIPEPTIDASI A - a.a. aromatici
CARBOSSIPEPTIDASI B - a.a basici (Lys, Arg)
III. MUCOSA INTESTINALE
enzimi ancorati alla membrana dell’enterocita
- AMINOPEPTIDASI
- DIPEPTIDASI
PRODOTTI DELLA DIGESTIONE
AMMINOACIDI LIBERI, DI- e TRiPEPTIDI
IV . ENTEROCITA
I peptidi possono entrare nell’enterocita dove sono
scissi da amminopeptidasi citosoliche.
ASSORBIMENTO:
tramite numerosi trasportatori specifici per classi di
a.a. (neutri, dibasici,..), in genere cotrasportatori con
Na+ o H+
ENTEROCITI metabolizzano glutammina (loro principale
fonte energetica), Glu, Asp, Arg per risparmiare glucosio ed acidi
grassi per gli altri tessuti
Circa 1% delle proteine sono parzialmente idrolizzate e
frammenti peptidici possono essere assorbiti come tali tramite
Trasportatore (es H+/PepT1 che importa anche antibiotici
beta lattamici)
Captazione transcellulare per endocitosi - e quindi esporto
tramite esocitosi
Captazione paracellulare tra le cellule, con una permeabilità
non specifica (in particolare in presenza di una mucosa
danneggiata)
Destino degli aminoacidi alimentari dopo un pasto
MUSCOLO
glucosio
-NH2
AA ramificati
alanina
proteine
pool AA
proteine
AA ramificati
α-chetoacidi
urea
FEGATO
alanina
glutammina
CO2
pool AA
SANGUE
serina
glutammina
Interconversione di
AA
NH4+
proteine
α-chetoglutarato
AA alimentari
INTESTINO
RENE
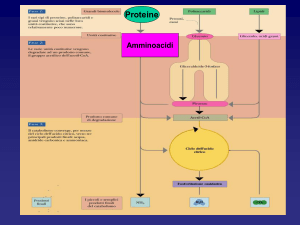
METABOLISMO DELLE PROTEINE
Aminoacidi e proteine sono in rapporto dinamico
Proteine
della dieta
digestione
Quota dei derivati non proteici
minoritaria e non si calcola nel
bilancio azotato;
ma quota significativa in
condizioni di privazione di
proteine
degradazione
sintesi
proteine corporee
Amminoacidi
N
C
Derivati non
proteici
glucosio,
glicogeno
NH3
urea
intermedi del
Ciclo di Krebs
CO2 + energia
acidi grassi
trigliceridi
bilancio di azoto o bilancio proteico: dipende dalla somma
delle velocità di entrata ed uscita dal pool di amminoacidi liberi
a
PROTEINE ALIMENTARI
POOL AA
b
d
PROTEINE CORPOREE
c
POOL DI DERIVATI
flusso in entrata = dieta + degradazione proteica (a + b)
rimozione a.a. = sintesi proteica + ossidazione (c + d)
a+b=c+d
a+d>b+c
costante
mantenimento nell’adulto
bilancio positivo
accrescimento; masse muscolari; gestazione
b+c >a+d
bilancio negativo
insufficiente apporto energia e/o proteine; malattia
UOMO ADULTO: proteine corporee circa 12 Kg
40% nel muscolo di cui 65% miosina ed actina
per locomozione e lavoro muscolare, ma anche come fonte di amminoacidi
in condizioni di stress.
Ma proteine muscolari non sono forma di riserva come glicogeno e lipidi ed
una loro perdita porta a perdita di proteine funzionali.
10% tessuti viscerali (fegato, intestino)
non mobilizzate rapidamente in condizioni di stress per le loro funzioni vitali
30% nelle pelle e nel sangue
lesioni delle pelle ed anemia sono presenti in deficit di proteine alimentari
4 proteine:
miosina, actina, collagene (strutturali) ed emoglobina (trasporto O2)
costituiscono circa la metà di tutte le proteine
CONTINUO RICAMBIO PROTEICO
Serve energia sia per la sintesi che per la degradazione:
15-20 % del bilancio energetico
La continua demolizione e sintesi è fondamentale per
degradare e rimpiazzare proteine danneggiate
modificare la quantità relativa di differenti proteine in base alle
necessità nutrizionali e fisiologiche
rapido adattamento metabolico
La regolazione del turnover proteico è influenzata da:
stato nutrizionale (energetico e proteico)
ormoni (insulina, glucocorticoidi, ormoni tiroidei, ormone della
crescita, citochine)
ORGANISMO
Ricambio giornaliero
Amminoacidi
70-80% riutilizzati
20-30% metabolizzati
Proteine dalla dieta
Proteine metabolizzate
% ricambio
muscolo 30-50%
fegato 25%
leucociti
emoglobina
1-2% proteine totali
70-80 grammi/giorno
250 grammi/giorno
diversa emivita
pochi minuti: proteine regolatorie
300 giorni: collageno
SISTEMI DI PROTEOLISI
ATP-indipendente LISOSOMIALE
contribuisce per il 15%
Enzimi attivi a pH 5
-proteine extracellulari (via endocitosi)
-proteine di membrana
-organelli danneggiati (es mitocondri)
ATP-dipendente CITOSOLICO
sistema ubiquitina-proteasoma
selettivo
- proteine citosoliche
- proteine regolatorie
- proteine difettose (neo -sintetizzate per errori nella sintesi o per
ripiegamento sbagliato; invecchiate)
Premio Nobel 2004 Aaron Ciechanover, Avram Hershko and Irwin Rose
L’ubiquitina come suggerisce il nome è una
proteina presente in tutti gli eucarioti
L’ubiquitina si lega alla proteina da degradare in una via
ATP dipendente che utilizza 3 enzimi
E1
+ ATP E1-Ubiquitina
E2
proteina di trasporto dell’ubiquitina
E3
lega l’ubiquitina attivata alla proteina da
degradare
Come si riconosce la proteina da eliminare?
Varie ipotesi
-amminoacido N-terminale destabilizzante
Arg ~2 min
Tyr, Glu, ~ 10 min
Ile Gln ~ 30 min
oppure stabilizzante
Met. Gly, Ala, Ser, Thr > 20 ore
-particolari sequenze di distruzione
ATP
La proteina marcata va al proteasoma
Proteine regolatorie per il
riconoscimento e selezione di
protine ubiquitilinate
subunità
7α
Proteine degradate dalle subunità
catalitiche β
7β
7β
Attività tipo chimotripsina - a.a. idrofobici
Attività tipo tripsina - a.a. basici
Attività per a.a. acidi
7α
oligopeptidi di 3-25 a.a.
scissi da proteasi citosoliche
L’attività del proteasoma è sotto controllo ormonale
INSULINA inibisce il proteasoma
GLUCOCORTICOIDI attivano il proteasoma
azione coordinata per la mobilizzazione di amminoacidi
muscolari e per la gluconeogenesi epatica
ORMONI TIROIDEI attivano il proteasoma
CITOCHINE attivano il proteasoma
sepsi, febbre, ustioni, cancro,…
Aumento delle proteine della fase acuta ed aumento del
catabolismo proteico delle miofibrille mediato da un
aumento delle citochine TNF-α, IL-1, IL-6
@ Le proteine della fase acuta vengono
sintetizzate dal fegato sotto l’induzione da
parte di citochine e di chemochine.
Le condizioni che portano a un aumento
delle loro concentrazioni plasmatiche sono:
• le infezioni
• i traumi
• gli intervento chirurgici
• le ustioni
• gli infarti di tessuto
• le infiammazioni immumologiche
• le infiammazioni immunologiche (cristalli gottosi)
• il cancro in stadi avanzati
• Gli aa. Non sono conservati nell’organismo,
quelli che eccedono le necessità biosintetiche
sono subito degradati.
• 75% degli aa è usato per biosintesi, il 25% per
altri composti azotati. In una alimentazione
corretta sarebbe sufficiente integrare questo
25% (pari circa a 1g/Kg peso corporeo)
• Al pool aminoacidico concorrono: aa da proteine
della dieta; aa da proteine tessutali; aa
sintetizzati de novo.Il pool è di circa 100g.
cheto-omologo
o transaminasi
Per essere ossidati devono
perdere il gruppo aminico:
per transaminazione o
deaminazione ossidativa.
Tutti gli aa con l’eccezione di lisina e
treonina vanno incontro a
transaminazione, l’accettore è l’αchetoglutarato
L’AST, aspartato aminoTasi. è un
eccezione perché l’accettore non è l’alfachetoglutarato, ma l’ossalacetato che
sarà portatore del 2°gruppo aminico
nella sintesi dell’urea.
L’alanina aminotransferasi (ALT) ex GPT
Lisina e treonina subiscono la deaminazione ossidativa
AST
Ex GOT
Meccanismo d’azione delle aminotransferasi:
Tutte richiedono il piridossalfosfato (vit B6)
legato con legame ε-aminico ad una lys del sito
attivo. La reazione è una reazione bimolecolare a
ping pong. Infatti il primo substrato lascia il
gruppo amminico al PDF ed esce come
chetoacido, mentre il secondo chetoacido entra e
lega il gruppo amminico dalla piridossamina
fosfato ed esce come amminoacido.
La K di equilibrio della reazione è quasi 1, così la
reazione può decorrere in tutti e due i sensi,
rispondendo alle diverse necessità della cellula.
Le reazioni di transaminasi assolvono a due compiti:
•
Promuovono l’interconversione degli aa adeguandone le quantità alle
esigenze metaboliche ed ovviando agli eventuali squilibri dietetici.
•
Indirizzanol’eccesso di aa verso il loro utilizzo salvaguardando la quota
richiesta per la biosintesi proteica.
Questo controllo è assicurato da due meccanismi:
1. Induzione delle transaminasi epatiche e intestinali da eccesso di proteine
dietetiche
2. Scarsa affinità delle transaminasi per gli aa (Km: 1-50mM). Così le
transaminasi si attivano solo oltre una certa soglia di concentrazione
amminoacidica.
Le transaminasi rivestono valore diagnostico in quanto la loro elevata
concentrazione nel plasma è indice di lesione d’organo, in particolare la GOT e
la GPT rispettivamente indicative di danno cardiaco ed epatico.
Valore diagnostico delle
amminotransferasi plasmatiche
O (GPT)
Andamento dell’ALT e della
bilirubina sierica nel monitoraggio
dell’avvelenamento da Amanita
Phalloides
Nel loro insieme le reazioni di transaminasi tendono a convogliare il gruppo
amminico verso l’α-chetoglutarato per formare glutammato, il quale viene restituito
alla sua funzione di collettore di gruppi amminici dalla deaminazione ossidativa, che
ripristina l’α-chetoglutarato liberando l’NH3. Nelle cellule la GOT esiste in due
isoforme: citosolica e mitocondriale. La forma citosolica facilita la formazione del
glutammato, quella mitocondriale dell’ α-chetoglutarato
Deaminazione
• La deaminazione può essere ossidativa e non
ossidativa.
• La D. Ossidativa è catalizzata dalle amminoacido
ossidasi, la più importante è la glutammico
deidrogenasi (oltre alla L e la D-amminoacido
ossidasi).
NAD
• Glutammato
NADH
H2O
Imminoglutammato
NH3
α-chetoglutarato
La L e D amminoacido Ossidasi sono FMN e FAD Dipendenti la D interviene su
D aa prodotti dalla flora e assorbiti occasionalmente, la L ha attività
bassissima e scarso rilievo fisiologico
Nel loro insieme le reazioni di transaminasi tendono a convogliare il gruppo
amminico verso l’α-chetoglutarato per formare glutammato, il quale viene restituito
alla sua funzione di collettore di gruppi amminici dalla deaminazione ossidativa, che
ripristina l’α-chetoglutarato liberando l’NH3. Nelle cellule la GOT esiste in due
isoforme: citosolica e mitocondriale. La forma citosolica facilita la formazione del
glutammato, quella mitocondriale dell’ α-chetoglutarato
la GDeidrogenasi: il gruppo amminico della maggior parte degli aa è indirizzato al glutammato per
mezzo della transaminazione dell’α-chetoglutarato, che poi produrrà l’NH3
La reazione è reversibile, nel verso riduttivo usa NADPH. Il verso della
reazione dipende dal rapporto [NAD]/[NAD(P)H. Poiché le transaminasi
citosoliche sono più attive di quelle mitocondriali e poiché la glutammato
deidrogenasi è solo mitocondriale, α-chetoglutarato colleziona gruppi ammici a
livello citosolico, mentre il glutammato mitocondriale si libererà dell’ NH3 nel
processo di deaminazione ossidativa.
La glutammato deidrogenasi possiede regolazioni allosteriche multiple: La direzione della reazione
dipende dalle esigenze della cellula. I regolatori allosterici sono: ATP,GTP e ADP,GDP, e dai
coenzimi NAD e NADPH Questo è in relazione con la richiesta di α-chetoglutarato per Krebs
Deaminazione non ossidativa
Enzimi inducibili piridossal fosfato dipendenti
3^ lezione
Destino metabolico dell’ammoniaca
• L’NH3 che si libera dalla deaminazione degli aa e degli
altri composti azotati (es. l’adenilato deaminasi molto
attiva nel muscolo card. e schel., libera NH3 da AMP)
deve essere eliminata perché molto tossica,l’organismo
la incorpora subito in composti atossici che
rappresentano una forma di trasporto e di
preeliminazione. Negli organismi uricotelici l’NH3 si
elimina come Urea .
Tre sono i processi di organicazione dell’NH3:
• Formazione dell’a-chetoglutarato
• Formazione della glutammina
• Sintesi del carbamilfosfato
La reazione è reversibile, nel verso riduttivo usa NADPH. Il verso della
reazione dipende dal rapporto [NAD]/[NAD(P)H. Poiché le transaminasi
citosoliche sono più attive di quelle mitocondriali e poiché la glutammato
deidrogenasi è solo mitocondriale, α-chetoglutarato colleziona gruppi ammici a
livello citosolico, mentre il glutammato mitocondriale si libererà dell’ NH3 nel
processo di deaminazione ossidativa.
La glutammato deidrogenasi possiede regolazioni allosteriche multiple: La direzione della reazione
dipende dalle esigenze della cellula. I regolatori allosterici sono: ATP,GTP e ADP,GDP, e dai
coenzimi NAD e NADPH Questo è in relazione con la richiesta di α-chetoglutarato per Krebs
La Glutammina sintetasi:
più attiva nei muscoli scheletrici e cardiaci, nell’intestino e
nel cervello, che non può sintetizzare carbamilfosfato.
7-8mg/dl è la quantità di glutammina nel sangue, pari a
circa il doppio degli altri aa, in maggior parte derivata dai
muscoli, non è tossica, è priva di carica, quindi passa le
membrane, è utilizzata soprattutto dai reni, ricchi di
glutamminasi (enzima mitocondriale).
L’NH3 che si forma nei tubuli renali passa nel liquido luminale si combina con i protoni
H+ e forma ione ammonio NH4+ eliminato con le urine, risparmiando l’equivalente
quantità di cationi. Questo meccanismo permette al rene di eliminare l’eccesso di H+ per
il mantenimento dell’equilibrio acido-base.
L’importanza di questo meccanismo è confermata dall’aumento della G.sintetasi
muscolare e della glutamminasi renale in condizioni di acidosi.
rene
•
Il 3°processo di organicazione dell’NH 3 è la sintesi del carbamilfosfato. La carbamilP-sintetasi è
attiva solo in presenza di N-acetil-glutammato, dipende da NH3, è intramitocondriale di concerto
con la glutammato deidrogenasi, incorporando l’NH3 man mano che si forma ad opera della
Glutammato Deidrogenasi. Si forma carbamil fosfato anche nel citosol, dove non è usato per la
sintesi dell’urea, ma per i nucleotidi pirimidinici. I due E coinvolti nelle due vie sono
rispettivamente l’aspartato transcarbamilasi citosolica e la ornitina transcarbamilasi
mitocondriale. La carbamilfosfato sintetasi citosolica è più attiva nei tessuti in accrescimento
(tumori, fegato rigenerante). La sintesi dell’urea è invece solo del fegato.
Un esempio che descrive bene le due attività è fornito dal fegato rigenerante:
Durante la fase rigeneratrice prevale la via n.1, ma appena il fegato è stato riformato
L’attività dell’aspartato transcarbamilasi si riduce mentre aumenta quella dell’Ornitina
Transcarbamilasi.
Ciclo dell’alanina muscolo fegato, forma in cui l’NH3 viene portata in circolo in forma atossica, il
fegato utilizza il piruvato per fare glucosio e incorpora l’NH3 nell’urea. Il muscolo rilascia anche
glutammina, usata dai reni e aa ramificati, usati prevalentemente dal cervello.
Gli esseri umani usano due meccanismi per
trasportare l’NH3 dai tessuti periferici al fegato
dove si produce l’urea: molti tessuti attraverso la
glutammina (glutammina sintetasi) che funge da
tampone per l’NH3, una forma atossica dell’NH3.
Il secondo meccanismo lo utilizza essenzialmente
il t. muscolare che transamina il piruvato (dal glucosio)
ad alanina. l’ala raggiunge il fegato viene transaminata,
si forma piruvato e glutammato. (ciclo glucosio
alanina)
La carbamil-P-sintetasi Idipende da NH3 ed è attiva solo in presenza di N-acetil-Glu, (effettore
allosterico), è intramitocondriale, opero di concerto con la glutammato deidrogenasi. Nel citosol
CPSII, produce CarbamilP a partire da glutammina ed è insensibile all’Nacetil-Glu
CPSI: carbamilfosfato sintetasi NH3 dipendente
CPSII: carbamilfosfato sintetasi glutammina dipendente
•La carbamil-P-sintetasi è attiva solo in presenza di N-acetil-glutammato, dipende da NH3, è
intramitocondriale di concerto con la glutammato deidrogenasi, incorporando l’NH3 man mano che
si forma ad opera della Glutammato Deidrogenasi. Si forma carbamil fosfato anche nel citosol,
dove non è usato per la sintesi dell’urea, ma per i nucleotidi pirimidinici. I due E coinvolti nelle due
vie sono rispettivamente l’aspartato transcarbamilasi citosolica e la ornitina transcarbamilasi
mitocondriale. La carbamilfosfato sintetasi citosolica è più attiva nei tessuti in accrescimento
(tumori, fegato rigenerante). La sintesi dell’urea è invece solo del fegato.
Il ciclo dell'urea richiede un'elevata
quantità di energia (4 ATP per ogni
molecola di urea prodotta).
P. Champe, R. Harvey, D. R. Ferrier, LE BASI DELLA BIOCHIMICA, Zanichelli Editore S.p.A. Copyright © 2006
L’NH3 non entra come tale nel ciclo dell’urea, ma come carbamilfosfato e come aspartato,
quindi le quantità di carbamilfosfato, nel mitocondrio e di aspartato nel citosol costituiscono
Il primo fattore di regolazione, poi ci sono una regolazione:
e una regolazione:
La capacità ureogenetica del fegato garantisce un livello di NH3 nel sangue minore di
5µm (0,1mg/100ml). Contribuisce all’uricemia l’assorbimento di NH3 dell’intestino
crasso di derivazione batterica.
Il livello di NH3 nel sangue può arrivare a 250 µmoli/l, cui segue stato
confusionale,coma e morte, come ad es per grave insufficienza epatica.
I livelli normali si aggirano tra 2,5-6 µm /l valori più elevati sono indice di alterata
filtrazione glomerulare (glomerulonefrite), o di bassa pressione ematica. Bassi valori di
urea si riscontrano in difetti congeniti dell’ureagenesi e in gravi epatopatie si fa diagnosi
per la presenza di ac orotico nelle urine: L’ac. Orotico è un intermedio della biosintesi
delle basi puriniche. (carbamilfosfato citoplasmatico).
CATABOLISMO DELLO SCHELETRO
CARBONIOSO DEGLI AMMINOACIDI
Come riportato in figura gli scheletri carboniosi convergono in sette
composti in grado di entrare direttamente o indirettamente nel ciclo di
Krebs: piruvato, acetilCoA, acetoacetilCoA, α -chetoglutarato, succinilCoA,
fumarato, ossalacetato.
Gli amminoacidi che vengono degradati ad acetilCoA o acetoacetilCoA sono
detti chetogenetici e sono i precursori dei corpi chetonici .
Gli altri sono glucogenetici e possono, una volta convertiti in piruvato ed
ossalacetato, formare glucosio attraverso la gluconeogenesi.
P. Champe, R. Harvey, D. R. Ferrier, LE BASI DELLA BIOCHIMICA, Zanichelli Editore S.p.A. Copyright © 2006
Lo scheletro carbonioso degli amminoacidi viene utilizzato nel ciclo di Krebs
per produrre energia.
piruvato, acetilCoA, acetoacetilCoA, α -chetoglutarato, succinilCoA, fumarato, ossalacetato
P. Champe, R. Harvey, D. R. Ferrier, LE BASI DELLA BIOCHIMICA, Zanichelli Editore S.p.A. Copyright © 2006
Sabato, 12 novembre 2011 ore 12:37
Biochimica degli amminoacidi
Le proteine ingerite con gli alimenti vengono idrolizzate nello stomaco e nell'intestino tenue per produrre
amminoacidi liberi ed oligopeptidi. Questi prodotti vengono assorbiti dalle cellule dell'intestino tenue e riversati
nel circolo sanguigno; la maggior parte degli amminoacidi viene quindi utilizzata dai vari organi e tessuti per i
processi di rinnovamento cellulare (turnover proteico).
DEGRADAZIONE DEGLI AMMINOACIDI
Gli amminoacidi vanno incontro a degradazione:
1) per normale turnover delle proteine
2) quando il loro apporto con la dieta è eccessivo
3) in carenza di carboidrati
La prima tappa del catabolismo degli amminoacidi prevede l'allontanamento del gruppo amminico. Lo
scheletro carbonioso viene così utilizzato nel ciclo di Krebs o nella gluconeogenesi.
Le amminotransferasi o transaminasi rappresentano gli enzimi chiave nella rimozione del gruppo amminico
degli amminoacidi.
Le reazioni di transaminazione consistono nel trasferimento di un gruppo amminico da un amminoacido
donatore all'alfa-chetoglutarato per formare glutammato. Durante questa reazioni il gruppo amminico donatore
è convertito in α- chetoacido. Il glutammato convoglia i gruppi amminici verso il ciclo dell'urea o verso le vie
biosintetiche degli amminoacidi.
Coenzima delle transaminasi è il piridossalfosfato, un enzima prodotto a partire dalla piridossina (Vitamina B6
).
Le transaminazioni sono reversibili e possono funzionare nei due sensi, a seconda delle necessità della
cellula .
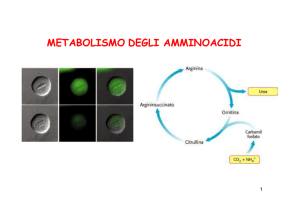
IL CICLO DELL'UREA
Il ciclo dell'urea inizia con la formazione del carbamil fosfato ad opera dell'enzima carbamil-fosfato sintasi.
Durante questa reazione vengono spese due molecole di ATP.
Le successive reazioni del ciclo dell'urea sono rappresentate in figura.
P. Champe, R. Harvey, D. R. Ferrier, LE BASI DELLA BIOCHIMICA, Zanichelli Editore S.p.A. Copyright © 2006
La glutammina entra nel circolo sanguigno e raggiunge il fegato dove, all'interno dei
mitocondri epatici, viene riconvertita a glutammato con liberazione dello ione ammonio
NH4 + .
L'alanina rappresenta il principale trasportatore di gruppi amminici dal muscolo al
fegato. Essa viene formata per trasferimento del gruppo amminico dal glutammato
all'acido piruvico o piruvato. Similmente a quanto avviene per la glutammina, una volta
giunta all'interno dei mitocondri epatici, l'alanina libera il proprio ione ammonio
generando glutammato e piruvato. Il piruvato è necessario al fegato nel processo
chiamato gluconeogenesi.
Lo ione ammonio NH4 + è tossico per le cellule del corpo ed in particolare per il
cervello. Come abbiamo visto, in sede extraepatica lo ione ammonio viene
neutralizzato tramite il legame con il glutammato o con il piruvato. Nel fegato l'NH4 +
viene incorporato nella molecola atossica dell'urea. L'urea prodotta dal fegato viene
trasportata attraverso il sangue ai reni per l'escrezione urinaria.
P. Champe, R. Harvey, D. R. Ferrier, LE BASI DELLA BIOCHIMICA, Zanichelli Editore S.p.A. Copyright © 2006