Gestione del dolore
come gestire gli analgesici oppiacei
Diritto a non soffrire di dolore inutile
Dolore come V° parametro vitale
Monitorizzare appropriatamente il dolore
Legge n. 38 del 15 Marzo 2010
Oppiaceo / Oppioide
Oppiaceo
morfina ed altri alcaloidi naturali derivati dall’oppio
(codeina, tebaina)
Oppioide
molecola di origine endogena o sintetica con stessi effetti degli
oppiacei (beta-endorfina, leu-enkefalina, meta- enkefalina,
dinorfina)
Narcotici
Sostanza che induce il sonno, in senso lato, sostanza da “ addiction”
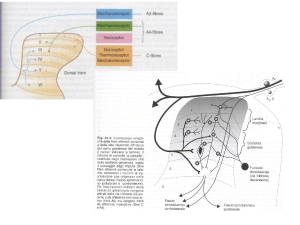
Struttura del recettore accoppiato alla proteina-G
Via ascendente del dolore
Meccanismo di azione degli Oppiacei
OPPIOIDI
+
OPPIOIDI
I peptidi oppioidi endogeni
inibiscono la trasmissione e
l’elaborazione degli impulsi
dolorifici sia a livello del midollo
spinale che delle vie nervose
sopraspinali
Gli analgesici oppioidi sono
agonisti dei recettori per i
peptidi oppioidi endogeni
Tipologia dei Recettori
analgesia sovraspinale, euforia,
depressione respiratoria, miosi, dipendenza
fisica
analgesia spinale, miosi, disforia e sedazione
Probabilmente mediano analgesia spinale ed
attività dei recettori µ
MoA degli oppioidi
La morfina, ed altri farmaci oppioidi, mimano gli oppioidi
endogeni
A livello neuronale presinaptico riducono l’afflusso di ioni calcio
mediante la chiusura dei canali voltaggio dipendenti nelle
afferenze nocicettive primarie con diminuzione del rilascio di
neurotrasmettitori
A livello postsinaptico aumentano la fuoriuscita di ioni potassio
con iperpolarizzazione dei neuroni sensibili agli stimoli nocicettivi,
localizzati nel corno dorsale
MoA degli oppioidi
Effetto analgesico periferico, sulle afferenze nocicettive
primarie
In base all’attività intrinseca si classificano:
AGONISTI
AGONISTI PARZIALI
ANTAGONISTI
Agonismo Recettoriale
puro e parziale
Antagonismo Recettoriale
Effetto di un agonista per i
recettori oppiacei
EFFETTI CENTRALI
Analgesia
Euforia
Sedazione, Depressione respiratoria
Nausea e vomito
Miosi
Effetto antitosse
Diminuzione della pressione arteriosa e frequenza
cardiaca
Effetto di un agonista per i
recettori oppiacei
EFFETTI PERIFERICI
Stipsi (recettori presenti nel tratto GI)
Contrazione e spasmo dello sfintere vescicale
Contrazione delle vie biliari
Analgesia dei tessuti infiammati
Effetto di un agonista per i
recettori oppiacei
EFFETTI COLLATERALI
Gastrointestinali
Psichici
Respiratori
Cardiocircolatori
Genitourinari
Sistemici
Classificazione recettoriale
Principio attivo
Recettore
Attività farmacologica
Morfina
µ [k] (δ)
Agonista
Codeina
µ [k]
Agonista
Petidina
µ [k/δ]
Agonista
µ/k
Agonista
µ
Agonista
Ossicodone
Fentanile
Buprenorfina
µ/k
Agonista parziale µ
Pentazocina
µ/k
Agonista κ, antagonista µ
Tramadolo
[µ]
Analgesico oppioide atipico
Naloxone
[µ]
Antagonista
Per evitare rischi, ricordare
– Gli oppiacei sono FARMACI analgesici ad azione centrale
che agiscono su specifici recettori endogeni
– L’interazione farmaco recettore produrrà una serie di
effetti clinici dipendenti dalla affinità farmaco-recettore
Potenza farmacologica
E’ funzione della dose di farmaco che mira ad un determinato
effetto clinico (ad es. l’analgesia)
La potenza dipende:
Superamento della barriera emato-encefalica,
Affinità tra oppioide e recettore
Attività intrinseca (capacità di esprimere l’effetto clinico desiderato)
Classificazione per affinità / potenza
Oppiacei
potenza
durata
Morfina
1
4
Codeina
0.1
4
6
8
Ossicodone
0.8
6
Fentanyl
30
0.3
Buprenorfina
10
8
Tramadolo
0.1
6
Metadone
NB: dal punto di vista farmacologico la definizione di oppiaceo “debole” e “forte” è arbitraria ed è in
funzione della dose dell’oppiaceo. Ad es. 1mg di morfina per os può essere meno efficace di 60 mg di
codeina somministrati per la stessa via
Biodisponibilità (per os)
Morfina: 30-40%
Ossicodone: 60-80%
Fentanyl: 10-25%
Tramadolo: 70-100%
Buprenorfina: 15-20%
Idromorfone < 30%
Metadone: 90%
Distribuzione del farmaco
E’ quel processo dinamico che permette al farmaco di diffondere nei
vari tessuti e di raggiungere il sito specifico di azione
Riguarda e determina quindi sia l’effetto terapeutico che la velocità di
eliminazione
VOLUME DI DISTRIBUZIONE =V = dose
assunta /concentrazione plasmatica
Il legame del farmaco con le proteine plasmatiche
modifica significativamente il processo di distribuzione dei
farmaci
Legame del
farmaco alle
proteine
plasmatiche
Alterazione del
legame del
farmaco alle
proteine
plasmatiche
Metabolismo degli oppiacei
Metabolismo epatico
Escrezione renale
Le malattie epatiche non influiscono in modo significativo
sulla farmacocinetica degli oppioidi, se non in caso di
insufficienza epatica importante
Le malattie renali richiedono la necessità di valutare
attentamente l’utilizzo degli oppioidi
Metabolismo degli oppiacei
Il fegato interviene nella metabolizzazione:
alchilazione,
Coniugazione
idrolisi
Sono coinvolti i sistemi citocromiali (CYP 450) o il
sistema UGM
Occorre ricordare che
– La terapia necessita un monitoraggio per verificare che
non intervengano variazioni cinetiche anche dopo l’avvio
della terapia
– Il farmaco (l’agonista) stimola il recettore endogeno e
quest’ultimo può reagire in maniera diversa per una
predisposzione genetica (up regulation, down regulation)
VIE DI SOMMINISTRAZIONE
Orale
Trans-mucosale
orale / nasale / rettale
Trans-dermica
Intradermica - Sottocutanea
Endovenosa
Intrarachidea
…….
La via orale
Non invasiva
Semplice ed accettata dal malato
Indicata come ideale dalle linee guida internazionali e dall’OMS per
il trattamento antalgico con oppiacei
Biodisponibilità ridotta rispetto alle vie parenterali (first pass
epatico)
Non può essere usata nei pazienti con impossibilità a deglutire
La via nasale
Permette un più basso dosaggio rispetto alla via orale
Veloce via di assorbimento
Evita il first pass epatico
Rapido passaggio della BEE
Non invasiva e di facile accesso anche per il paziente
La via transdermica
Non invasiva
I farmaci utilizzati devono essere molto liposolubili e a
basso peso molecolare
Solo fentanyl e buprenorfina sono ad oggi disponibili
Prima della diffusione sistemica il farmaco si accumula
a livello cutaneo per cui si verifica un ritardo di diverse
ore (anche più di 12 ore) prima di ottenere una
concentrazione plasmatica efficace
Ricordare che esistono sistemi citocromiali nel
sottocutaneo
Vantaggi
Svantaggi
economica, comoda,
non invasiva
Onset
entro 20 – 40 minuti
rapido onset (2-5 minuti )
invasiva, non sempre praticabile
Praticabile dal paziente
Lento onset e cinetica imprevedibile
Transmucosale
orale
(fentanyl)
comoda, non invasiva,
rapido onset analgesico (10 –
15 minuti)
Limitata praticabilità in pazienti con
gravi mucositi o xerostomia
Transmucosale
nasale
(fentanil)
comoda, non invasiva,
rapido onset (5 minuti)
Sgocciolamento delle soluzioni
acquose (maggiore stabilità della
dose erogata con le formulazioni a
base di pectina)
Orale
(ad es morfina,
ossicodone, ecc)
Endovenosa
/sottocutanea
(morfina, ecc)
Transcutanea
(ad es fentanil))
CODEINA
Profarmaco (metil mofina)
Bassa affinità per i recettori μ
Potenza farmacologica 10 volte inferiore alla morfina
Buona disponibilità per os (media 40%); Emivita circa 2,5 ore
(mediana)
Metabolismo epatico: 6 metaboliti attivi (morfina e codeina 6glucuronide);
Nel metabolismo della codeina è coinvolto il CYP2D6
Eliminazione urinaria
“Effetto tetto” per dosi complessive giornaliere di 360 mg
Stessi eventi avversi degli oppiacei
TRAMADOLO
Debole attività agonista oppioide (recettore µ)
Inibizione re-uptake Serotonina e Noradrenalina
Potenza farmacologica: 1/5-1/10 della Morfina
Biodisponibilità orale: 70-100%
Emivita: 5-7 h
11-22 metaboliti attivi (CYP2D6 / CYP2D4) ;
10 % caucasici scarsi metabolizzatori: maggior rischio di effetti
collaterali)
Metabolismo epatico / Eliminazione renale
“Effetto tetto” per dosi complessive giornaliere di 400-600 mg.
OSSICODONE
affinità recettoriale per recettori μ e κ
Struttura simile alla codeina, ma con attività analgesica superiore
Biodisponibilità (60-87%), maggiore nelle donne
Equianalgesia ossicodone morfina orale va da 1-1,5:1 a 2:1
Emivita 3.5 - 4 ore
Assenza di metaboliti con attività farmacologica significativa (CYP3A4/5)
Eliminazione urinaria
Assenza di effetto tetto
sinergia con paracetamolo (325mg per compressa)
MORFINA
Alta affinità per i recettori µ [k] (δ)
Bassa riserva recettoriale
Biodisponibilità per os circa 33% con onset 20-30 minuti
Tempo di emivita: 2-3 ore
Il metabolismo prevalentemente epatico, dove si coniuga
con l’acido glicuronico con 2 metaboliti, M-3-G e M-6-G
Eliminazione per via renale
Assenza di “effetto tetto”
BUPRENORFINA
Agonista parziale con elevata affinità per i recettori μ
Antagonista sui recettori k (25-50 volte più potente
della morfina, ma meno efficace).
sostanza lipofila, a basso peso molecolare
Metabolismo CYP2C8, CYP3A5/7 e UGT1A1
effetti collaterali, parzialmente reversibili con naloxone.
Somministrato con un agonista puro può antagonizzare gli effetti
dell’agonista
A dosaggi terapeutici non ha effetto tetto: tale effetto compare a
dosaggi > 4 mg/die
IDROMORFONE
Agonista i recettori μ e k
più potente della morfina, ma con effetto analgesico
meno duraturo
Biodisponibilità inferiore al 30%, assorbimento piccolo
intestino
Metabolizzato a livello epatico
esteso primo metabolismo epatico; non produce
metaboliti attivi (metabolita 3-glicuronide è inattivo)
FENTANYL
Agonista puro dei recettori μ
Azione analgesica 75-125 volte maggiore della morfina
Elevata lipofilia con rapido passaggio della barriera ematoecefalica ed elevata rapidità
d’azione (30’’ ev) e limitata durata (30-60’ ev)
Assenza di effetto tetto
Al contrario della morfina, fentanil e principalmente metabolizzato dall’enzima CYP450
3A4 responsabile della metabolizzazione di oltre il 35% dei farmaci attualmente disponibili
Catabolismo epatico senza formazione di metaboliti attivi:
norfentanil
(maggiore) e
despropionilfentanil, idrossifentanil e idrossinorfentanil (minori)
Se somministrato per via trans dermica per raggiungere lo steady state sono necessarie
fino a 24 h
METADONE
Azione agonista sui recettori μ e δ (isomero destrogiro)
Azione antagonista sui recettori NMDA (entrambe le forme isomeriche)
Alta biodisponibilità orale (40%-99%)
Picco plasmatico raggiunto in circa 4 ore
Effetto analgesico precoce grazie alla elevata lipofilia
Assenza di effetto tetto
La C max si mantiene costante per molte ore
Emivita plasmatica fino a 75 ore
Metabolismo epatico per demetilazione (senza metaboliti attivi)
Eliminazione anche attraverso la via renale
Prodotto
Indicazioni
Jurnista
OxyContin
MS Contin
Oramorph
Co-efferalgan
Trattamento del dolore severo
Trattamento del dolore intenso
Remissione dei dolori prolungati, gravi e ribelli
Dolori cronici intensi e/o resistenti agli altri antidolorifici
Trattamento delle esacerbazioni transitorie di dolore nei pazienti oncologici adulti (Breakthrough Pain Dolore Episodico Intenso - DEI)
Trattamento delle esacerbazioni transitorie di dolore nei pazienti oncologici adulti (Breakthrough Pain Dolore Episodico Intenso - DEI)
Trattamento delle esacerbazioni transitorie di dolore nei pazienti oncologici adulti (Breakthrough Pain Dolore Episodico Intenso - DEI)
Trattamento delle esacerbazioni transitorie di dolore nei pazienti oncologici adulti (Breakthrough Pain Dolore Episodico Intenso − DEI)
Trattamento del dolore cronico da cancro e del dolore ribelle che necessita di un'analgesia a base di
sostanze oppiacee.
Trattamento del dolore oncologico di intensità da moderata a severa e del dolore severo che non
risponde agli analgesici non oppioidi.
Dolore severo cronico, che può essere trattato adeguatamente solo con analgesici oppiacei
Dolore severo cronico, che può essere trattato adeguatamente solo con analgesici oppiacei
Trattamento del dolore da moderato a grave in corso di malattie muscolo-osteoarticolari non controllato
da (FANS)/paracetamolo utilizzati da soli
Trattamento sintomatico del dolore da moderato a severo che non risponde al trattamento con
analgesici non oppioidi utilizzati da soli.
Tachidol
Trattamento sintomatico delle affezioni dolorose acute e croniche anche accompagnate da iperpiressia
Contramal
Stati dolorosi acuti e cronici di diverso tipo e causa e di media e grave intensità, come pure in dolori
indotti da interventi diagnostici e chirurgici.
Trattamento sintomatico del dolore acuto da lieve a moderato
Trattamento sintomatico del dolore acuto da lieve a moderato
Actiq
PecFent
Effentora
Abstral
Durogesic
Transtec
Matrifen
Quatrofen
Depalgos
Patrol
Kolibri
Interazioni farmacologiche
effetti dell’induzione / inibizione enzimatica sulla farmacocinetica
Concentrazione plasmatica
Rischio di tossicità (inibizione enzimatica)
Inefficacia (induzione enzimatica)
Massima dose tollerata
Finestra
terapeutica
Minima dose efficace
tempo
P. Geppetti , S Benebei Clin Drug Invest. 2009; 29 (1): 9-16
Parametri utili per la gestione
farmacologica del dolore
Criteri farmacologici
– Biodisponibilità / metabolizzazione
Variabilità individuale della farmacocinetica
Dosaggio personalizzato
– Affinità recettoriale / potenza analgesica
Risposta individuale no n prevedibile
– Presenza di “effetto tetto”
Prevedere la necessità di ruotare a farmaco più efficace
– Vie di somministrazione / tecniche farmaceutiche
Formulazioni ad immediato rilascio per avviare la terapia
Formulazioni a lento rilascio per il trattamento di mantenimento
Parametri utili per la gestione
dell’oppiaceo
Criteri clinici
– Indicazioni delle schede tecniche
Molti farmaci non sono indicati nell’età pediatrica
Farmaci a lento rilascio non devono essere masticati
– Interazioni farmacologiche
Predisposizione genetica
Minima dose interagente
Minima dose tollerata
– Risposta analgesica individuale
Necessità di un avvio graduale dell’oppiaceo
Per evitare rischi, ricordare
I FANS causano un danno d’organo diretto
Gli oppiacei inducono stipsi, nausea,
sonnolenza, depressione del SNC, ecc
vomito,
tolleranza,
Tali effetti sono correlati al meccanismo di azione generico
dei farmaci oppiacei
Parametri utili per la gestione
dell’oppiaceo
Criteri farmacologici
– Avviare con la terapia con oppiaceo a basse dosi e/o in
combinazione con paracetamolo
– Avviare la terapia con farmaci ad immediato rilascio fino
alla stabilizzazione del dolore prima di passare ad
oppiacei a lento rilascio (mantenimento)
– Ruotare l’oppiaceo piuttosto che aumentare le dosi di
oppiaceo
– Cambiare la via di somministrazione piuttosto che
aumentare la dose sopra quella massima tollerata
STRATEGIA PER IL CONTROLLO
DEGLI EFFETTI COLLATERALI
Riduzione della dose
Idratazione
Sospensione di 1-2 somministrazioni
Eliminazione o riduzione di farmaci concomitanti che
interagiscono con gli oppioidi
Somministrazione di farmaci sintomatici per gli effetti
indesiderati
Cambiamento della via di somministrazione
Rotazione degli oppioidi
Utilizzo farmaci antagonisti
ROTAZIONE DEGLI OPPIOIDI
Rotazione= passaggio da un oppioide ad un altro, quando il grado di
analgesia ottenuto è limitato dalla comparsa di effetti avversi.
Questo approccio si basa sull’osservazione che la risposta del pz varia
da oppioide a oppioide, sia per quanto riguarda l’analgesia che per gli
effetti avversi.
La rotazione degli oppioidi si basa sulla tabelle di equianalgesia, che
forniscono valori, basati sull’evidenza scientifica, delle potenze relative
dei differenti oppioidi.
Solitamente, quando si passa dall’oppioide A al B è
prudente diminuire la dose del secondo oppiaceo
Effetti degli oppioidi
Alcuni effetti sono
legati all’inizio del
trattamento, o in
caso di aumento di
dose
Altri effetti sono
più tipici dell’uso
prolungato
(mantenimento)
Effetti iniziali
nausea, vomito
sedazione
secchezza delle fauci
stipsi
prurito
Effetti nel mantenimento I
stipsi
sedazione (minore)
secchezza delle fauci
allucinazioni
mioclonie
Effetti tardivi
alterazioni cognitive
disforia
depressione respiratoria
ritenzione urinaria
edema polmonare
miosi
Nausea e vomito
Trattamento farmacologico / recettore coinvolto:
metoclopramide (D2-5HT4), cisapride (5HT4)
domperidone (D2 periferici), aloperidolo (D2)
fenotiazine (D2, muscarinici, H1, 5HT2)
setronici (5HT3)
steroidi (?)
Modificazione via di somministrazione o oppioide
Stipsi da oppioidi
- lattulosio: effetto osmotico, aumenta il contenuto idrico
delle feci (voluminose e morbide)
- senna: stimola il plesso mioenterico, induce la peristalsi
- idratazione
TOLLERANZA
La tolleranza farmacologica è una diminuzione
degli effetti oppioidi causata da una esposizione
ripetuta al farmaco con bisogno di aumentare la
dose di oppioide per mantenere il medesimo
grado di analgesia
DIPENDENZA FISICA
l’interruzione improvvisa dell’esposizione cronica a
questi farmaci può esitare nello sviluppo dei sintomi
di una sindrome d’astinenza, tra i quali è incluso il
dolore.
la sindrome d’astinenza può essere provocata dalla
somministrazione di naloxone o di oppioidi con
azione antagonista/agonista a pazienti con
dipendenza da oppioide con azione agonista pura.
ADDICTION
Addiction = dipendenza fisica e psichica
L’addiction è un ‘alterazione cronica neurobiologica,
influenzata da fattori genetici, psicosociali ed ambientali.
La comparsa dell’ addiction durante la terapia con oppioidi
può essere attribuita ad un errato controllo dell’uso del
farmaco, ad un uso compulsivo, all’uso continuato
nonostante gli effetti nocivi e al craving.
Craving = impegno di tutte le energie fisiche e mentali del
paziente nella ricerca ossessiva del farmaco
Protocollo terapeutico con oppioidi
Terapia Step by Step
Paziente naive agli oppiacei e con dolore
lieve moderato
p
Primo ste
step
Secondo
p
Terzo ste
Razionale della combinazione farmacologica
Combinazione tra due farmaci con effetti
– Additivi
– sinergici (di potenziamento)
Razionale della combinazione farmacologica
Miaskowski C et Al, Pain 1992; 49: 137-144
Paracetamolo
Inibisce una cox centrale
Stimolazione delle vie serotoninergiche inibitorie discendenti
Stimolazione dei recettori CB1
Gatti A. Sabato E, Di Paolo AR, Mammucari M, Sabato AF Clin. Drug Invest 2010 ; 30 (2):3-14
Razionale della combinazione farmacologica
Gatti A. Sabato AF, Carucci A, Bertini L, Mammucari M, Occhioni R Clin. Drug Invest 2009; 29 (1):31-40
Razionale della combinazione farmacologica
Gatti A. Sabato AF, Carucci A, Bertini L, Mammucari M, Occhioni R Clin. Drug Invest 2009; 29 (1):31-40
Razionale della combinazione farmacologica
10
p< 0.0005
9
8
Basale
2 settimane
4 settimane
7,7
6,5
7
5,6
6
5
6,6
6,4
5,1
4,4
4
4,7
4,4
3,8
3,3
2,6
3
2
1
D
ol
or
e
at
tu
a
le
m
ed
io
D
ol
or
e
do
M
in
or
Pe
gg
io
r
do
lo
re
lo
re
0
Gammaitoni (2003)
Razionale della combinazione farmacologica
10
Basale
2 settimane
4 settimane
p< 0.0004
9
8
7
6,2
6
5
5,2
4,6
4,5
4
6,1
6
3,4
3,6
3,3
2,6
3
4,3
3,5
3,6
3,2
2,4
3,6
2,8
2,7
2
1
er
e
o
G
us
to
di
vi
v
So
nn
zio
ne
Re
la
la
vo
ra
tiv
a
ità
M
ov
i
or
e
Um
m
en
to
At
tiv
At
tiv
ità
ge
ne
ra
l
i
0
Gammaitoni (2003)
Tempi di azione della terapia e
relativi usi nella pratica clinica
3.
Somministrazione
Nasale per il doore
grave e
imprevedibile
1. Terapia del dolore
cronico con dose
fissa ad orari fissi
2. Terapia con
oppiaceo
forte per os
Dolore di base
Tempo
Usare i farmaci ad azione centrale secondo le
schede tecniche