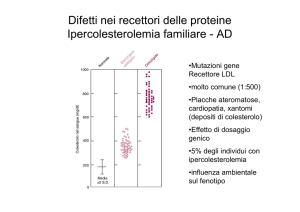
Le malattie recessive
Frequente la perdita di funzione con aplosufficienza
I
Nascono figli affetti in genere
da genitori sani spesso consanguinei
II
che sono eterozigoti
III
ed hanno un rischio di avere
un figlio affetto su quattro
IV
Le malattie recessive
Alcune malattie sono limitate a famiglie di consanguinei
alleli rari o privati
Alcune malattie sono limitate ad alcune popolazioni ristrette
effetto del fondatore - inincrocio o matrimonio endogamico
se è alta la frequenza di portatori
in genere non c’è consanguineità
spesso gli affetti sono eterozigoti composti
è possibile un vantaggio dell’eterozigote e/o un alto tasso di mutazione
Complicazioni del modello autosomico recessivo
La Fibrosi cistica
1:2000-2500
La più frequente patologia autosomica recessiva
in passato era letale nella prima infanzia
sopravvivenza in aumento (30-40 anni)
per le migliorate condizioni igienico-sanitarie
La Fibrosi cistica
Fibrosi polmonare
Muco denso – Ridotta motilità ciliare
Ostruzione delle vie aeree - Danno tissutale
Sostituzione delle cellule morte con tessuto fibroso più rigido
Difficoltà respiratorie e ricorrenza di infezioni polmonari
Effetto pleiotropico di un singolo gene
Deficit delle ghiandole del sistema esocrino
Ostruzione dei dotti pancreatici
Insufficienza pancreatica - Diabete
Ostruzione dei dotti biliari
Difficoltà digestive – Cirrosi epatica
Ileo da meconio – Ostruzione intestinale fetale
Ghiandole sudoripare
Sudore molto salato con grave perdita di elettroliti
Effetto pleiotropico di un singolo gene
Effetti sulla riproduzione
Ridotta fertilità femminile
Ridotta fertilità maschile
oligospermia o azoospermia ostruttiva
CBAVD assenza congenita bilaterale dei vasi deferenti
CBAVD si presenta spesso associata all’allele 5T introne 8
Espressività variabile
I pazienti FC possono presentare tutti i sintomi o solo una parte
ma l’insufficienza pancreatica e la sterilità maschile
possono anche presentarsi
non associate a fibrosi cistica del polmone
Eterogeneità allelica
mutazioni nello stesso gene possono dare fenotipi diversi
da non confondere con
Eterogeneità genetica
mutazioni in geni diversi possono dare lo stesso fenotipo
Diffusione dell’allele
F508
Allele ancestrale originato in Europa dopo l’espansione della popolazione
per la diffusione dell’agricoltura dal Medio Oriente
circa 20-50.000 anni fa (passaggio Paleolitico-Neolitico)
Gradiente Sud-Nord
Vantaggio dell’eterozigote?
Diffusione dell’allele
Non è presente nelle popolazioni di origine NON Europea
Si diffuse nei Paesi extraeuropei
insieme a gruppi di emigrati
di origine prevalentemente Nord-Europea
che si rifugiavano in America per sfuggire alle
persecuzioni religiose durante la Riforma
F508
Diffusione dell’allele
F508
In alcune popolazioni Amish la frequenza di FC è di 1/700 nati o anche più alta
Alto grado di consanguineità e prole numerosa
Effetto del fondatore - Deriva genetica
Inincrocio - Piccole popolazioni fortemente endogamiche
Il gene CFTR
Cromosoma 7
27 esoni - 250 Kb
1480 aa
Un gene duplicato???
La proteina CFTR
Regolatore della conduttanza transmembrana della FC
Appartiene alla famiglia delle ABC (ATP-binding cassette)
trasportatori di membrana
Due domini transmembrana
formano un poro o canale del cloro
Esterno
Interno
Due domini ATP-binding
idrolizzano ATP per l’energia di trasporto
e di apertura/chiusura
Un dominio Regolatorio
fosforilato dalle protein-chinasi cAMP dipendenti
apre o chiude il canale
La proteina CFTR
Espressa nella membrana apicale
delle cellule epiteliali
• polmone
• pancreas
• fegato
• colon
• ghiandole salivari
• ghiandole sudoripare
Un’alterazione dell’espressione
nei diversi tessuti può spiegare
l’espressività variabile della FC??
L’allele
Una delezione di tre bp nella porzione codificante
porta alla eliminazione dell’aa508 (Fenilalanina)
in un dominio ATP-binding
Esterno
Interno
La proteina viene prodotta ed è parzialmente attiva
a bassa temperatura
ma non viene trasportata sulla membrana
Un difetto di folding o di stabilità della proteina??
F508
Allelia multipla
Più di 1000 mutazioni diverse
• Missense
• Nonsense
• Frameshift
• Siti di splicing
• Delezioni
F508
L’allelia multipla può spiegare l’espressività variabile della FC??
Relazione genotipo-fenotipo
Fibrosi polmonare
Gravità dei sintomi
Normale
I
mancata
sintesi
II
III
Difetto regolazione
del canale
difetto
maturazione
IV
V
ridotta
sintesi
Conduttanza del canale
ridotta
Relazione genotipo-fenotipo
1.7%
Normale
moderata
grave
Sufficienza pancreatica
I
0.3%
II
3.5%
97%
100%
III
IV
V
dominante o recessivo??
un allele di classe IV o V è dominante su di un allele completamente difettoso
Patogenesi
Ridotta esportazione del Cloro e dell’acqua - Aumentato
assorbimento del Sodio nel lume delle vie aeree
Secrezioni deidratate e viscose
Inattivazione delle difensine sensibili alla concentrazione salina
Suscettibilità elevata alle infezioni
Geni modificatori
L’alterazione di altri geni può spiegare la variabilità fenotipica??
Molte proteine intervengono nella maturazione e
nel trasporto della CFTR sulla membrana
L’apertura/chiusura del canale CFTR è regolata dal dominio R
che è (de)fosforilato da protein-chinasi
Il canale CFTR interferisce anche con la regolazione
di un canale del sodio e di un altro canale del cloro
Allelia multipla
Più di 1000 mutazioni diverse
1:20-25 portatori
con frequenze diverse da Paese a Paese
F508 rappresenta circa il 70% di tutti gli alleli
15-20 altri alleli insieme arrivano al 15%
gli altri sono alleli rari o privati
90% porta l’allele
F508
50% in omozigosi
molti affetti sono
50% in combinazione con un altro allele
eterozigoti composti
10% non porta l’allele F508
I saggi diagnostici devono essere progettati Paese per Paese
Interpretazione degli alberi
Dominante o recessivo ?
Interpretazione degli alberi
Dominante o recessivo ?
Eredità dominante X linked
Un affetto ha almeno
può essere scambiato per autosomico dominante
un genitore affetto
Colpite tutte le generazioni
Affetti entrambi i sessi
ma piu’ spesso le femmine
I figli di una donna affetta hanno il 50% di probabilità di essere affetti
indipendentemente dal sesso
Un maschio affetto avra’ tutte le figlie affette e tutti i figli maschi sani
Eredità dominante X linked
Patologie rare - la più frequente è il Rachitismo resistente alla Vit D
Le femmine hanno comunque una sintomatologia più lieve
Lyonizzazione
Mosaicismo funzionale dei geni del cromosoma X
100% affetto se femmina
100% sano se maschio
Eredità dominante X linked
Se il carattere è letale in emizigosi
i maschi non arrivano alla nascita e si trovano solo femmine affette
Eredità recessiva X-linked
Affetti quasi esclusivamente i maschi
emizigoti
Le femmine possono essere affette
se sono omozigoti
il padre è affetto e la madre portatrice
se sono eterozigoti sono affette in forma più lieve
L’inattivazione del cromosoma X può non essere casuale
Eredità recessiva X-linked
I genitori di solito sono sani
La madre di un affetto è certamente
portatrice
puo’ essere asintomatica
Lo stato di portatrice di una donna
e’ certo se ha il padre affetto o un figlio affetto
ha il 50% di probabilità se ha un fratello affetto
50% affetto se maschio
50% portatrice se femmina
Eredità recessiva X-linked
Consanguineita’
Eredità recessiva X-linked
Emofilia
La più antica malattia genetica conosciuta
Difetto della coagulazione del sangue
attivazione della conversione della protrombina in trombina
Due forme principali
• A (deficienza del fattore VIII)
Incidenza 1:5.000 maschi
• B (deficienza del fattore IX)
Incidenza 1:40.000 maschi
Distrofie muscolari
Eterogeneità genetica
Un gruppo di patologie eterogenee
eredità autosomica dominante e recessiva
eredità X-linked recessiva
Eredità recessiva X-linked
Distrofia muscolare di Duchenne DMD
Incidenza 1:3.000-3500 nei maschi
Atrofia e debolezza muscolare con esordio infantile
morte entro la terza decade di vita
deficit mentale
Il gene della Distrofina (DMD) è lungo 2,5 Mb
uno dei più lunghi geni conosciuti
1% sequenze esoniche con un mRNA di 14 Kb
Distrofia muscolare di Duchenne DMD
La distrofina
Laminina
Complesso dei
sarcoglicani
Sarcolemma
Distrofina
Actina
La proteina di 427 kDa si lega ad un complesso glicoproteico nella membrana muscolare
che collega la laminina extracellulare alla actina intracellulare
Alterazioni di questo complesso proteico sono responsabili di altre distrofie
Distrofia muscolare di Duchenne DMD
Gli affetti non si riproducono
quasi tutti i casi sono dovuti a nuove mutazioni
Il tasso di mutazione è molto alto (circa 1:10.000)
2/3 degli affetti porta una delezione parziale del gene DMD
diverse mutazioni per posizione ed estensione
un “hot spot” tra gli esoni 43 e 55
insorgono quasi esclusivamente durante la gametogenesi materna
probabilmente per crossing-over ineguale
Le mutazioni puntiformi avvengono invece prevalentemente
durante la gametogenesi maschile
Eterogeneità allelica
Distrofia muscolare di Becker BMD
Incidenza 1:20.000 nei maschi
dovuta al difetto dello stesso gene
La sintomatologia è più lieve e la speranza di vita quasi normale
L’ “hot spot” tra gli esoni 43 e 55
43 44 45 46 47 48 49 50 51 52 53 54 55
BMD
Delezioni in frame
Mutazioni missense
Delezioni non in frame
Mutazioni non senso
DMD
Frameshift
Siti di splicing
Mutazioni del
promotore
Delezione senza
slittamento del modulo di lettura
Delezione con
slittamento del modulo di lettura
Distrofia muscolare di Duchenne DMD
Inattivazione non casuale del cromosoma X
Molte femmine affette sono eterozigoti
per una traslocazione bilanciata (X;autosoma) con un punto di rottura in Xp22
X
X
X;21
X
21
21 21
21;X
Distrofia muscolare di Duchenne DMD
Inattivazione non casuale del cromosoma X
Nelle cellule in cui viene inattivato il cromosoma X traslocato
si ha la perdita funzionale di una regione autosomica
Queste cellule sono perciò svantaggiate
Le cellule in cui viene inattivato il cromosoma X normale
sono bilanciate per la regione autosomica e sono perciò avvantaggiate
ma hanno attivo il cromosoma X traslocato in cui il gene DMD è interrotto
Femmine affette
Eredità recessiva X-linked
Sindrome dell’X fragile o sindrome di Martin-Bell
Incidenza 1:1000-1250 nei maschi
1:2500 nelle femmine
Ritardo mentale
associato alla mancata espressione del gene FMR1
Una proteina di trasporto (shuttle) degli RNA tra nucleo e citoplasma
Sindrome dell’X fragile o sindrome di Martin-Bell
FRAXA si evidenzia citologicamente
come una fragilità nel tratto distale del braccio lungo
del cromosoma X (Xq27.3-ter)
coltivando le cellule in carenza di acido folico
Penetranza incompleta e...
80% dei maschi emizigoti sono affetti
...dominanza incompleta
30% delle femmine eterozigoti sono affette
Sindrome dell’X fragile o sindrome di Martin-Bell
Una serie di triplette (CCG)n localizzate a monte del gene FMR1
sono espanse in molti soggetti affetti (>>200)
Instabilità della regione
Affetto >2OO
Portatore >50-60
Normale 5-50
FMR1
CCGn
Sindrome dell’X fragile o sindrome di Martin-Bell
L’espansione avviene preferenzialmente nella linea germinale femminile
Se un maschio eredita l’X-fragile da
una donna affetta sarà affetto
22-29
82
29-80
Madri eterozigoti non affette possono
avere figli maschi e femmine affette
22-90
500
22-83
Padri emizigoti non affetti hanno figlie femmine normali
le quali però avranno figli affetti
>200
>200
Sindrome dell’X fragile o sindrome di Martin-Bell
L’espansione della regione extragenica potrebbe disturbare
l’espressione del gene FMR1
Alterazione della struttura della cromatina
Silenziamento del gene
Metilazione anomala delle sequenze di regolazione
Lo stesso fenotipo è associato a mutazioni puntiformi del gene FMR1
Eredità recessiva Y-linked
Eredità oloandrica
Colpisce solo i maschi
I maschi affetti hanno il padre affetto
Tutti i figli maschi di un uomo affetto sono affetti