STABILIZZAZIONE (CFSE) DI UNO IONE dn
Energia reticolare
degli alogenuri MX2
Raggio ionico
SPETTROSCOPIA ELETTRONICA
Regole di selezione per una transizione elettronica :
•Spin: non può cambiare la molteciplità di spin DS = 0
•Laporte: deve cambiare la parità g/u
Le transizioni “proibite” sono, in realtà, poco probabili
Banda
emax
Spin-proibita
<1
Laporte proibita
10
Laporte permessa
100
A trasferimento di carica
103-104
[Cr(ox)3]3- e [CrF6]3n1 corrisponde a Do
MnF2
D DIPENDE DA:
Dq = 1/6{ze2<r4>/a5} a = dist.internucleare, <r> = dist media di e dal nucleo
• La carica sul metallo:
[Ru(H2O)6]2+
19800 cm-1
[Ru(H2O)6]3+
28600 cm-1
•La geometria del complesso:
Do2 Dt
[VCl4]
7900 cm-1
[VCl6]2-
15400 cm-1
•La natura del metallo:
Mn2+ < Ni2+ < Co2+ < Fe2+ <Fe3+ < Cr3+ <Co 3+
< Mo3+ < Rh3+ < Ru3+ < Pd4+ < Ir3+ < Pt4+
[MnF6]2[TcF6]2[ReF6]2-
21800 cm-1
28400 cm-1
32800 cm-1
•Il legante (serie spettrochimica)
[CoF6]313100 cm-1
[Co(H2O)6]3+
20800 cm-1
[Co(NH3)6]3+
22900 cm-1
LA SERIE SPETTROCHIMICA
Leganti diversi spostano il massimo di
assorbimento nello spettro elettronico.
C > N > O > F > Cl > Br > I
Leganti “misti” hanno effetto “medio”
GLI ORBITALI d NEI COMPLESSI
QUADRATO-PLANARI
IL CAMPO DEI LEGANTI (LIGAND FIELD):
IL CASO OTTAEDRICO
Le possibili combinazioni tra gli
orbitali del metallo e quelli dei
leganti:
Gli orbitali di frontiera (HOMO e
LUMO sono centrati sul metallo):
GLI ELETTRONI DI UN COMPLESSO
GLI ELETTRONI DI UN COMPLESSO
L’EFFETTO p
La sovrapposizione p tra un
orbitale d e un orbitale p:
Orbitali p pieni (leganti
elettronegativi, bassa energia):
Orbitali p vuoti (alta energia):
MAGNETISMO
Diamagnetismo: il moto degli elettroni appaiati viene perturbato da un
campo magnetico.
Si genera una magnetizzazione (I) che si oppone al campo esterno
B = H + 4pI
(I è una grandezza negativa)
Effetto comune a tutte le sostanze.
Paramagnetismo: anche il moto degli elettroni spaiati viene perturbato
da un campo magnetico.
Si genera una magnetizzazione (I) che si oppone al campo esterno
I = kH
(I è positivo, e molto maggiore che nel caso precedente)
Effetto proprio dei composti con elettroni spaiati.
IL MOMENTO MAGNETICO
La suscettività magnetica k è la risposta di
un mezzo al campo magnetico esterno:
k= I/H
Bilancia di Gouy: la differenza di peso tra il
campione ed uno standard dà la suscettività
magnetica per unità di volume, k
La forza (differenza di peso) che agisce sul
volume V è: F = ½kH2V = cost k
La susc. paramagnetica molare è cM =
(k/d)MM- cdia
MAGNETISMO
Bilancia di Gouy: la differenza di peso tra il campione
ed uno standard dà la suscettività magnetica per
unità di volume, k
La suscettività molare è data da cM = k P.M./d
Per molti composti cM è funzione della temperatura
secondo la legge di Curie: cM = C/T
Il momento magnetico è indipendente dalla
temperatura meff = cost (cMT)½ = √[n(n+2)]
LEGGE DI CURIE
Il contributo diam. cdia è dato dal contributo di ogni
atomo (costanti di Pascal)
La susc. Param. è funz della temperatura e del
momento magnetico, perché T tende a disorientare i
dipoli magnetici:
cM = cost(m2/T)
m = 2.828 cMT) = n(n+2)
n è il numero di elettroni spaiati per ogni molecola
ISOMERIA NEI COMPLESSI
Tipo di isomeria
Descrizione
Esempio
Configurazionale
Diversi poliedri di
coordinazione
Tetraedro/quadrato
TBP/SP
Di spin
Alto/basso spin
Fe(II)
Di legame
Diversi atomi donatori
sul legante
O-NO/NO2
Geometrico
Diversa distribuzione
dei leganti
Cis/trans
fac/mer
Ottico
Enantiomeria
D/L
5T
1A
ISOMERIA IN NiCl2(PPh3)2
Verde scuro, paramagnetico
Rosso scuro
ISOMERIA NEI COMPLESSI
Isomeria geometrica: cis/trans, fac/mer
Isomeria ottica: D/L
ISOMERIA DI IONIZZAZIONE
ISOMERIA IN [Cr(en)3][Ni(CN)5] · 1.5 H2O
Temp. e press.
Ambiente
7 kbar
Dopo esser tornati a
press. ambiente
Composto disidratato
(SP)
IDRURI
Donatori di un elettrone
Pochi complessi puramente idrurici: [ReH9]2- , [FeH6]4-
Molti con altri leganti: H2Fe(N2)(PR3)3, H3Co(PPh3)3, H5Ir(PR3)2, H2PtCl2(PR3)2
Occupano un sito di coordinazione, ma consentono ad altri leganti di espandersi
Idruri “non classici”: il legame di
una molecola H2 funziona da donatore
[BH4]- come legante chelante: Cu(PPh3)2BH4
RAGGI X E NEUTRONI
Il complesso di Kubas: dove sono i legami
tra W e H?
H-H : 0.75(16) Å (raggi X)
0.74 in H2 libero
0.84 Å (neutroni)
J(H-D) = 33.5 Hz (43.2 Hz in HD molecolare,
2-3 Hz is cis diidruri)
n(HD) = 2360 cm-1
Kubas, Acc. Chem. Res., 1988, 21, 120:
“in an inversion of the normal order,
spectroscopic rather than crystallographic,
data provided the convincing evidence”
H COME LEGANTE
Paramagnetic shielding (correnti elettroniche)
Dihydrogen bond H----H+
IL TEMPO DI RILASSAMENTO T1
L’impulso a 180° inverte la popolazione dei
due livelli di spin. Si diagramma in funzione
del ritardo di acquisizione (delay)
Il tempo di rilassamento dipende anche dalla
distanza di altri nuclei con spin
1H
E 31P NMR
trans-PtHCl(PPh3)2 1J(1H-195Pt) = 1196 Hz; 2J(1H- 31P) = 13 Hz
LEGANTI ALL’AZOTO
•
Ammoniaca e ammine: nessun effetto p,leganti tipici per cationi +2 e +3
•
Deprotonazione di ammoniaca: ammidi (NH2-), nitreni (NH2-) e nitruri (N3-):
p-donatori
•
Ammine aromatiche: piridina, imidazolo, pirazolo e pirazolato. Forti pdonatori, stabilizzano alti stati di ossidazione: [Ag(Py)2]2+
•
Diazoto: N2 es: [Fe(EDTA)N2]-, [Ru(NH3)5N2]2+
•
Basi di Schiff: macrocicli tetra- (o penta-)dentati, con almeno 2 N
•
Pseudoalogenuri: N3-, NCS-, CN-, NO2- (isomeria di legame)
•
Nitrili R-CN (isonitrili R-NC)
•
Nitrosile NO (lineare, piegato, a ponte)
IL LEGANTE N2
I diversi modi di coordinazione possibili.
Tra le fonti di N2 si usa N2O; N2H4, N3-, N2
CO, N2 e HCCH sono (elettronicamente)
simili
LEGANTI AL FOSFORO
•
Fosfine alifatiche: PR3: s-donatori, poveri p-accettori
•
Fosfine aromatiche: PPh3 moderato s-donatore, moderato p-accettore, stabile
all’ossidazione, solida, non volatile, diminuisce la solubilità del composto, ne facilita
la cristallizzazione
•
Fosfine chelanti dppe, dppm: PPh2-(CH2)n-PPh2
•
Fosfine chirali
•
Fosfiti: P(OR)3 scarso s-donatore, forte p-accettore
•
PF3: simile al CO per effetto elettronico e per ingombro sterico
•
Angolo di Tollman:
PMe3 a ponte:
I LEGANTI AL FOSFORO
Il potere p-accettore delle fosfine
Effetti sterici ed elettronici
delle fosfine
INGOMBRO STERICO E INSATURAZIONE
Pt[PPh(t-Bu)2]2
Pt(PPh3)3
Pt(dppp)2
dppp =
Ph2P(CH2)3PPh2
P5 ANALOGO DEL CICLOPENTADIENE
LEGANTI ALL’OSSIGENO
• Acqua, idrosso
• Alcoli, alcolati, eteri, thf, eteri corona
• Legante oxo (O2-) p-donatore; tre formule di risonanza M-O-, M=O, MO+
gruppi M=O sono stabilizzati da stati di ossidazione > +4 e dn < 4
• Diossigeno, superosso O2-, perossido O22• Chetoni, urea, SO2
• Dichetonati [R-C(O)-CH-C(O)-R]-, chelanti, una volta coordinati a M
formali anelli con sistema p delocalizzato
• Ossoanioni (NO3-, SO42-, PO43-,CO32-, carbossilati RCOO- (mono e
bidentati)
• Ossidi delle fosfine (O=PR3)
• Dimetilsolfossido (Me2S=O)
LEGANTI ALL’OSSIGENO: ALCUNI ESEMPI (1)
VO(acac)2
[Cr(O2)4]3-
CuSO45H2O
LEGANTI ALL’OSSIGENO: ALTRI ESEMPI (2)
{Cu(OOCCH3)2 H2O}2
O-Ir-O : 37°
IL DIOSSIGENO COME LEGANTE
O2, O2-, O22-
Lo stato di ossidazione del legante e del metallo si
deduce da dati strutturali (distanza O-O) e spettroscopici(n(O-O))
LEGANTI ALLO ZOLFO
• Solfuri, polisolfuri, tiolati, tioeteri (metalloenzimi)
• Ditioleni S-CR=CR-S. Impossibilità di determinare gli stati di
ossidazione
• SO2 (isomeria di legame)
• Tiocianato (SCN piegato)
• Ditiocarbammato [R2N-CS2]monodentato, a ponte]
[chelante (anello a quattro termini),
SOLFURI, TIOLATI, POLISOLFURI
[Pt(S5)3]2-, un composto
chirale senza carbonio
[Au25(SR)18]+, un colloide
d’oro stabilizzato da tiolati
[Fe4S4(SR)4]2+, un
analogo sintetico delle
ferredossine. Esistono
anche gli stati ossidati
(+3) e ridotti (+1)
[Ni2(dmit)3]2-: un complesso
con un ditiolene
LEGANTI NON INNOCENTI: [ReL3]z
5 stati di ossidazione; z da +1 a -3
Spettro ESR: in ReL32-, l’elettrone spaiato è
centrato sul metallo; 185Re I = 5/2
3 stati di spin
Prisma trigonale
DMSO COME LEGANTE
SO2 COME LEGANTE
ALOGENURI
•
Fluoro: piccolo ingombro, nessun potere riducente, nessun effetto p: alti
numeri di coordinazione ([ZrF8]4-), bassa posizione nella serie
spettrochimica
•
Altri alogeni: stabilizzano complessi tetraedrici, possono fungere da ponti
(donatori di tre e cinque elettroni)
Cu4I4py4
REAZIONI DI COMPOSTI
ORGANICI ED INORGANICI (1)
REAZIONI DI COMPOSTI
ORGANICI ED INORGANICI (2)
VELOCITÀ DI SCAMBIO: COMPLESSI
LABILI ED INERTI
MECCANISMI DI SOSTITUZIONE
E
U
E
U
E
U
E
U
Associativo
Interscambio
a carattere
associativo
Interscambio
a carattere
dissociativo
Dissociativo
A
Ia
Id
D
ENTALPIA ED ENTROPIA DI
ATTIVAZIONE: DH‡ E DS‡
Equazione di Arrhenius: k = A exp(-Ea/RT)
Equazione di Eyring: k =(kBT/h)exp(-DG‡/RT) =
(kBT/h)exp(-DH‡/RT + DS‡/R )
ln k/T = - DH‡/RT + ln (kB/h) + DS‡/R
Plot di Arrhenius (●) e di Eyring (■)
per la reazione di scambio di una
molecola di solvente in
[Ga(dmso)6]3+
VOLUME DI ATTIVAZIONE: DV‡
d lnk/d P = -DV‡/RT
DV‡ è positivo per reazioni dissociative, è negativo per reazioni
associative
EQUAZIONI CINETICHE
MECCANISMO ASSOCIATIVO:
U-M + E U-M-E
v = -d[M-U]/dt = k[U-M][E] (dipende da E)
MECCANISMO DISSOCIATIVO:
U-M M + U k1, k-1
M+E M-E
k2
v = -d[M-U]/dt = k1 k2[M-U][E]/(k-1[U]+ k2[E])
Se k2[E] >> k-1[U] v = k1[M-U] primo ordine, non dipende da E
Se k-1[U] >> k2[E] v = k1 k2[M-U][E]/k-1[U] si ha effetto inibitore di U
Se k-1[U] k2[E] si deve usare l’espressione completa
MECCANISMO DI INTERSCAMBIO:
U-M + E
U-M--E K0
U-M--E U--M-E k3
U--M-E M-E + U k2, veloce
v = -d[M-U]/dt = K0k3[M-U][E]/(1+ K0[E])
Se K0[E] >> 1 v = k1[M-U] primo ordine, non dipende da E
Se K0[E] << 1 v = K0k3[M-U][E] secondo ordine, dipende da E
Se k-1[U] k2[E] si ha dipendenza complessa
REAZIONI DI SOSTITUZIONE:
I COMPLESSI QUADRATO - PLANARI
Pt(py)2Cl2 + L Pt(py)2(Cl)L
Due meccanismi paralleli: kobs = k1 + k2[L]
EFFETTO E INFLUENZA TRANS
EFFETTO E INFLUENZA TRANS
L’influenza trans (donazione s) è un
fenomeno termodinamico
(destabilizza il reagente). T e X
competono per uno stesso orbitale
L’effetto trans (retrodonazione p)è un
fenomeno cinetico (stabilizza lo stato
di transizione). Un accettore rimuove
l’eccesso di elettroni
L’EFFETTO TRANS: IMPLICAZIONI
SINTETICHE
CN-, C2H4, CO, NO >PR3H- > tu > CH3- >Ph > SCN- > NO2- > I- >Br- >
Cl- > NH3 > OH- > H2O
Sintesi di cis-Pt(NH3)2Cl2
[PtCl4]2- + NH3 [Pt(NH3)Cl3]- cis-Pt(NH3)2Cl2
Sintesi di trans-Pt(NH3)2Cl2
[Pt(NH3)4]2+ +Cl- [Pt(NH3)3Cl]- trans-Pt(NH3)2Cl2
LA SCALA DI NUCLEOFILICITÀ
log (kL/k0) = shPt
hPt, fattore di nucleofilicità, è determinato per reazioni di sostituzione su
[Pt(py)2Cl2], ma vale per molti complessi di platino:
s è il fattore di discriminazione
CRISTAL FIELD ACTIVATION ENERGY
Velocità (sperimentale)
Velocità (calcolata con MO, AO)
Velocità (calcolata con CF)
CRISTAL FIELD ACTIVATION ENERGY
Si calcola la stabilizzazion del campo cristallino nel reagente e nello
stato di transizione (in unità Dq): CFAE = (CFSE)‡ - CFSE
Le velocità di scambio di solvente negli acquocomplessi:
Ione
Config.
CFAE (Dq)
k (s-1)
DV‡(cm3/mol)
DH‡(KJ/mol)
V2+
t2g3
1.80 (OC)
87
-4.1
62
Cr2+
t2g3 eg1
-3.14 (SP)
> 108
Mn2+
t2g3 eg2
0
2 107
-5.4
33
Fe2+
t2g4 eg2
-2.08(OC)
4.4 106
+3.8
41
Co2+
t2g5 eg2
-2.56(PP)
3.2 106
+6.1
47
Ni2+
t2g6 eg2
1.80(OC)
3.2 104
+7.2
57
Cu2+
t2g6 eg3
-3.14(SP)
>107
Zn2+
d10
0
>107
Si riesce a prevedere quali ioni sono labili e quali inerti. Meno efficace per
determinare l’ordine di reattività
CORRELAZIONE LINEARE
DI ENERGIA LIBERA (L.F.E.R.)
Keq = kd / ki
DG° = DG‡i - DG‡d
Se DG‡i = cost
log Keq + C = log ki
[Co(NH3)5 X]2+ + H2O
[Co(NH3)5 H2O]3+ + X-
SOSTITUZIONE IN COMPLESSI [CrL6]3+
Il complesso [Cr(H2O)6]3+ da’ sostituzioni associative, il complesso
[Cr(NH3)5(H2O)]3+ dissociative; DS‡ sono però confrontabili
MECCANISMO DI INTERSCAMBIO
Raramente si riesce a osservare una variazione
dell’ordine in funzione della concentrazione di
legante entrante
Esempio: [Cr(NH3)5(H2O)]3+ + SCN-
[Cr(NH3)5SCN]2+ + H2O
Per le reazioni di sostituzione [Ni(H2O)6]2+ + L, k3 = kf/K0
SOSTITUZIONI CATALIZZATE
1.
DA BASI
[Co(NH3)5X]2+ + OH- [Co(NH3)4(NH2)X] + + H2O
[Co(NH3)4(NH2)X] + [Co(NH3)4(NH2)] 2+ + X[Co(NH3)4(NH2)] 2+ + H2O [Co(NH3)5OH] 2+
2.
DA RIDUCENTI
[Cr(II)(H2O)6]2+ + X- [Cr(II)(H2O)5X]+
[Cr(II)(H2O)5X]+ + [Cr(III)(H2O)6]3+ [Cr(III)(H2O)5X]2+ + [Cr(II)(H2O)6]2+
Complessi di Co(III) sostituiscono più rapidamente in presenza di riducenti (carbone)
1983, for his work on the mechanisms of electron transfer reactions,
especially in metal complexes
1992, for his contributions to the theory of electron transfer reactions in
chemical systems
REAZIONI REDOX: MECCANISMO A
SFERA ESTERNA
PRINCIPIO DI FRANCK-CONDON: il movimento dei nuclei è molto più
lento del movimento degli elettroni
DG‡ è funzione di DG°
Il DG‡ di attivazione minore si ha
dove le due curve si intersecano;
È funzione della ripidità della
parabola e della ampiezza della
deformazione
Anche transizioni elettroniche possono contribuire in modo significativo alle barriere di
attivazione; la velocità di autoscambio di Co(NH3)62/3+ è molto bassa
Co(II) passa ad uno stato
eccitato “a basso spin”
Il trasferimento elettronico tra
Co(II) in basso spin e Co(III)
è spin-permesso
There is a disagreement between our, 6 10-3 (M s)-1, and the experimental rate, 5 10-5 (M s)-1. Either something is
not correct with the theoretical estimate or the experimental rate constant is too small. In the more likely case that the
theory misses some details……
REAZIONI DI AUTOSCAMBIO (DG° = 0)
Si può calcolare l’energia di attivazione DG‡ di una reazione redox
valutando:
1. Il lavoro necessario per avvicinare i reagenti
2. L’energia per ricostruire la sfera di idratazione
3. L’energia per deformare i parametri strutturali dei reagenti
L’accordo è discreto (entro un fattore 100) considerando il gran
numero di parametri da calcolare
REAZIONI REDOX DI AUTOSCAMBIO
Le reazioni che implicano maggiori differenze
di distanze di legame (Dd, elettroni eg) sono le più lente:
DG‡ (LA VELOCITÀ) DIPENDE DA:
•
La forma della curva (=le costanti
di forza dei legami, l’ordine di legame M-L)
•
La distanza tra i minimi
(= l’entità della deformazione)
•
La differenza tra i minimi
(= il DG° di reazione)
REAZIONI REDOX ASIMMETRICHE (DG0)
le deformazioni subite dai due reagenti sono simili a quelle nell’autoscambio:
DG(AB‡) = ½[DG(AA‡) + DG(BB‡)]
REAZIONI REDOX ASIMMETRICHE (EQ. DI
MARCUS SEMPLIFICATA)
k12 = (k11k22K12f)1/2
DG12‡ = ½ (DG11‡ + DG22‡ + DG12°)
REAZIONI REDOX: MECCANISMO A
SFERA INTERNA
I complessi condividono un legante. L’esperimento di Taube:
[Co(NH3)5Cl]2+ + [Cr(H2O)6]2+ [Cr(H2O)5Cl]2+ + [Co(H2O)6]2+ + 5 NH3
La reazione redox è molto più veloce della sostituzione del Cr(III)
Si forma un “complesso precursore”:
Se, nell’ambiente di reazione, sono presenti ioni Cl- isotopicamente marcati,
questi non vengono incorporati nel complesso di Cr(III)
COMPLESSO PRECURSORE E
SUCCESSORE
MO-L + M’R MO-L-M’R complesso precursore
MO-L-M’R MR-L-M’O complesso successore. Raramente è il
passaggio lento. Può esser generato in situ da MO-L-M’O
MR-L-M’O MR + L-M’O
si formano i prodotti finali
ATTACCO VICINO E ATTACCO LONTANO
Leganti multiatomici possono aver più siti di attacco. Possono
presentarsi diverse possibilità per il complesso pontato.
[(NH3)5CoNCS]2+ + Cr2+ Co2+ + Cr-SCN
Solo S ha un doppietto disponibile, si forma un unico isomero
[(NH3)5CoSCN]2+ + Cr2+ Cr-SCN (meno stabile, 30 %) + Cr-NCS
Si formano entrambi gli isomeri
O
Co
N
+
NH2
Cr2+
Co2+ +
O
N
Qual è il meccanismo di trasferimento dell’elettrone?
NH2
Cr
MECCANISMO CHIMICO O DI
RISONANZA?
MO-L-M’R MO-L--M’O MR-LM’O Meccanismo chimico (a
stadi). Si forma il legante ridotto
MO-L-M’R MR-L-M’O
Trasferimento per risonanza.
(trasferimento di atomi neutri)
•La velocità dipende dalla distanza e dalla coniugazione nel legante
•Se l’elettrone rimane intrappolato dal legante, si vede via ESR
SFERA ESTERNA O SFERA INTERNA?
1. Identificare i prodotti di sostituzione o gli intermedi
2. Confronto tra velocità di sostituzione e velocità della
reazione redox
3. Assenza di leganti con coppie di non legame
(ammoniaca, fenantrolina)
4. Dipendenza della velocità dal pH (differente capacità
pontante di H2O/OH)
5. Scala di reattività con leganti diversi (F/Cl/Br, SCN/N3)
6. Corrispondenza tra k (e DG‡) sperimentali e calcolati
COMPOSTI DI INTERVALENZA
Il complesso di Creutz:
FILI METALLICI
Il sale di Krogmann:
K2[Pt(CN)4]X0.3·(H2O)n
(X = Cl, Br)
REAZIONI DI ISOMERIZZAZIONE:
ISOMERIA DI LEGAME
[Co(NH3)5-17O-N-O]2+ [Co(NH3)5-O-N-17O]2+ [Co(NH3)5-NO17O]2+
Lo scambio O-17O ha velocità confrontabile con lo scambio O-N.
Meccanismo intermolecolare con intermedio p
Isomeria di legame in
[Os(NH3)5NR2Ph]2/3+.
L’isomerizzazione è attivata da
redox elettrochimiche.
Il complesso di Os(II) presenta
entrambi gli isomeri, quello di
Os(III) solo quello legato a N
DISSOCIAZIONE DI UN CHELANTE
a) inversione e isomerizzazione
avvengono con la stessa velocità
b) In [Rh(ox)3]3- racemizzazione e
scambio di 18O interno col solvente
avvengono con la stessa velocità
REAZIONI DI ISOMERIZZAZIONE: I
COMPLESSI OTTAEDRICI
Rotazione di Bailar: devono
deformarsi gli angoli interni di tutti
gli anelli
Rotazione di Ray-Dutt: non
richiede deformazione degli anelli
PROCESSI DINAMICI (FLUSSIONALITÀ)
(a) Stato limite a bassa T (w0)
(b) Regime di scambio lento k = p(wE-w0)
(d) Coalescenza k = (Dn0)p/2
(e) Regime di scambio veloce k = p(Dn0)2/2(wE-w0)
(f) Stato limite ad alta T (w0)
ISOMERIZZAZIONE DI fac- e mer-RuQ3
Q = di-tbut-chinone
+25°C
-85°C
Ray-Dutt twist:
ISOMERIZZAZIONE IN COMPLESSI
QUADRATO-PLANARI
1.
Doppia sostituzione (catalizzata da L)
2.
3.
Pseudorotazione diretta (A → B)
Dissociazione
ISOMERIA IN BIPIRAMIDI TRIGONALI
Alta T: una sola J
media
Bassa T: una J
grande e una
piccola
HRhL4 L=P(OEt)3
1H NMR
J(1H-31P) e J(1H-103Rh)
Co(CF3)(PF3)(CO)3, due isomeri con
CF3 assiale, PF3 ass. ed eq.
NMR di CF3: J(19F-19F) e J(19F-31P)
REAZIONI DI ISOMERIZZAZIONE: I
COMPLESSI TETRAEDRICI
Complessi del tipo Ni(PR3)2X2
isomerizzano da tetraedrico a
quadrato planare.
Ni(dpp)2Cl2 esiste, a temperatura
ambiente in MeCN, nei due
isomeri:
Planare
tetraedrico
k1, k-1, K1
La composizione della miscela di
equilibrio può essere alterata per
irraggiamento.
K1 = 0.75 k1 = 4.5 105 s-1
k-1 = 6 105 s-1
Lo spettro NMR di Ni(PMePh2)2Br2 a Temp. variabile
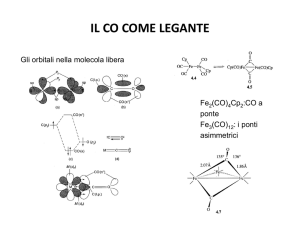
IL CO COME LEGANTE
Gli orbitali nella molecola libera
Fe2(CO)4Cp2:CO a
ponte
Fe3(CO)12: i ponti
asimmetrici
SINTESI DEI METALLO CARBONILI
• Per reazione diretta:
Ni + 4 CO
Ni(CO)4
Fe + 5 CO Fe(CO)5
• Riduzione di un sale in presenza di CO:
CoX2 + H2 +CO Co2(CO)8
CrCl3 + Al + 6 CO Cr(CO)6 + AlCl3
• Riduzione operata dal CO (carbonilazione riduttiva):
Re2O7 + 17 CO Re2(CO)10 + 7 CO2
6 RuCl3 + 33 CO + 27 OH- 2 Ru3(CO)12 + 9 HCO3- + 18 Cl- + 9 H2O
REAZIONI DEI METALLO CARBONILI
•
Cr(CO)6 + L Cr(CO)5L + CO
Sostituzione:
Mo(CO)6 + Me3C6H3 Mo(CO)3(Me3C6H3)
•
Fotosostituzione
Cr(CO)6 + thf (hn) Cr(CO)5thf + CO
•
Sostituzione catalizzata
Cr(CO)6 + L +Me3NO Cr(CO)5L + CO2
•
Attacco elettrofilo all’ossigeno:
M-CO + AlCl3 M-CO-AlCl3
•
Attacco nucleofilo al carbonio:
M-CO + Nu M=C(O-)-Nu (Nu = OH-, NR2-, RO-, R-)
•
Inserzione:
•
Riduzione:
MeMn(CO)5 + L Me-C(O)Mn(CO)4L
Co2(CO)8 + 2 Na 2 Na[Co(CO)4]
Fe(CO)5 + OH- [Fe(CO)4(COOH)]- [HFe(CO)4]-+ CO2
Fe2(CO)9 + 4 OH- [Fe2(CO)8]2- + CO32- + 2 H2O
ATTACCO NUCLEOFILO ED ELETTROFILO AL CO
Re2(CO)10 + MeO-
I CO a ponte sono più basici:
[Fe4(CO)13]2- + MeSO3F
WATER GAS SHIFT REACTION
NO COME LEGANTE
NO piegato: NO-, donatore di un elettrone,
M-N-O, M in alti stati di ossidazione
NO lineare: NO+, donatore di tre elettroni,
M-N-O, M in bassi stati di ossidazione
The Coordination Chemistry of Nitric Oxide and
Its Significance for Metabolism, Signaling, and
Toxicity in Biology
characterizing the exact
electronic structures of
transition metal nitrosyls has
been difficult, which led to the
establishment of the famous
Enemark- Feltham* notation
that allows for a general
classification of transition
metal nitrosyls without the
need to define an exact
electronic structure.
* {MNO}n
ISONITRILI (CN-R) COME LEGANTI
[Co(CNR)4]-, [Co2(CNR)8], [Ta(CNR)7]+
Isonitrili terminali e a ponte, l’angolo R-NC è 130-180°, per effetto della
retrodonazione
(R-N=C=M).
Danno facilmente reazioni di attacco
nucleofilo al C e reazioni di inserzione:
SINTESI DEI METALLO-ALCHILI
•
Attacco nucleofilo al metallo:
WCl6 + 6 LiMe WMe6 + 6 LiCl
(transmetallazione)
•
Attacco elettrofilo al metallo:
[Mn(CO)5]- + CH3I MeMn(CO)5 + I•
Somma ossidativa:
Ir(CO)Cl(PPh3)2 + MeI Me-IrI(CO)Cl(PPh3)2
2 Cr2+ + MeI Me-Cr2+ + CrI2+
•
Inserzione:
PtHCl(PEt3)2 + C2H4 PtEtCl(PEt3)2
L’energia di legame M-C è
160-300 kJ/mol:
LA b ELIMINAZIONE
Un possibile intermedio
per l’eliminazione: il
legame agostico
Per la b-eliminazione occorrono:
•H in b
•Catena alchilica flessibile
•Sito di coordinazione vuoto
•Un orbitale pieno per retrodonazione
SINTESI E REAZIONI DEI METALLO-IDRURI
•
Sostituzione nucleofila con H-:
Cr(CO)6 + BH4- [HCr(CO)5]- [(m-H)Cr2(CO)10]-
•
Attacco elettrofilo al metallo:
[Mn(CO)5]- + H+ HMn(CO)5
•
Somma ossidativa:
Ir(CO)Cl(PPh3)2 + HX H-IrX(CO)Cl(PPh3)2
Ir(CO)Cl(PPh3)2 + H2 (H)2Ir(CO)Cl(PPh3)2
•
H-transfer da un legante preesistente (b-eliminazione)
GLI ALCHENI COME LEGANTI
Il modello Dewar-Chatt- Ducanson (1953) per
il legame tra metallo e doppio legame:
donazione s e retrodonazione p
Le due situazioni limite: legame
M-alchene e metallaciclopropano
Il sale di Zeiss: C-C 1.36 Å
diedro CH2/CH2 146°
A destra: angoli diedri
CH2/CH2: 138°
CF2/CF2: 106°
SINTESI DEI METALLO ALCHENI
• Per sostituzione diretta:
[PtCl4]2- + C2H4 [PtCl3(C2H4)] - + Cl• Riduzione di un sale in presenza di alchene:
Pt(PPh3)2Cl2 + C2H4 + BH4- Pt(PPh3)2(C2H4)
• Da un alchile per b-eliminazione:
MLn-CH2-CH2-R HMLn-1-CH2=CH-R + L
• Protonazione di un allile:
Mn(CO)5-(h1-CH2-CH=CH2) + H+ [Mn(CO)5-(h2-CH2=CH-CH3)]+
REAZIONI DEI METALLO ALCHENI
•
Inserzione in un legame M-H:
M-H + CH2=CH2 M-CH2-CH3
(PPh3)AuMe + CF2=CF2 Au(PPh3)-CF2-CF2-Me
•
Attacco nucleofilo:
(NHMe2)PtCl2(C2H4) + NHMe2 (NHMe2)Pt(-)Cl2-CH2-CH2-N(+)HMe2
[CpFe(CO)2(C2H4)]+ + Nu- CpFe(CO)2-CH2CH2-Nu
GLI ALCHINI COME LEGANTI
Due formule limite: metalla-ciclopropene e di-carbene
Ciclo-esino e benzino stabilizzati da coordinazione
l’alchino può donare 4 elettroni
ALLILI E DIENI CONIUGATI
ISOMERIA DEI METALLO-ALLILI
L’isomerizzazione syn-anti avviene
tramite intermedio h1-allile
Pd2Cl2(C3H5)2 E CATALISI
1983
1985
1987
1989
1991
1993
1995
1997
1999
2001
2003
2005
2007
2009
2011
2013
0
20
40
60
80
100
120
140
160
SINTESI E REAZIONI DEI METALLO-ALLILI
•
Attacco nucleofilo al metallo:
NiBr2 + 2 C3H5MgBr (h3-all)2Ni + 2
MgBr2
•
Attacco elettrofilo al metallo:
[Mn(CO)5]- + C3H5I (h1-all)Mn(CO)5
+ I- (h3-all)Mn(CO)4 + CO
•
Sale metallico, allil cloruro e riducente
•
Olefina e alogenuro metallico, con
eliminazione di HX
Gli allili reagiscono con nucleofili (più
spesso) e con elettrofili.
Isomerizzazione (catalitica) di olefine:
•
Somma ossidativa
REAZIONI STEREOSELETTIVE
•
•
•
Mo è centro stereogenico; il
neomentile genera due
diastereoisomeri
La sostituzione CO/NO genera un
complesso cationico, soggetto ad
attacco nucleofilo
Si forma un unico enantiomero
•
•
I due enantiomeri vengono
separati mediante sostituzione
Cl/canforsulfonato
(diastereoisomeri)
Nel complesso neutro, l’allile è il
nucleofilo
LEGANTI DIENICI: PtLCl2 E FeL(CO)3
C3-C4 1.42 Å
C4-C5 1.43 Å
I METALLOCENI (MCp2)
I METALLOCICLOPENTADIENI
APPLICAZIONI DEI
METALLOCICLOPENTADIENI
Composti a trasferimento
di carica, con allineamento
degli spin
Sequestro e rilascio di ioni regolato dalla
ossidazione del Fe
Terapia antitumorale
I METALLO-INDENILI E LA
POLIMERIZZAZIONE
PROCESSI DINAMICI
Fe(CO)2(h5-C5H5)(s-C5H5)
Ti(h5-C5H5)2(s-C5H5)2
LEGANTI ANALOGHI AL Cp
Cp* = Me5C5; maggior ingombro
sterico, impossibilità di Interazione
coi legami C-H, campo più forte:
Es.: MnCp2 alto spin,
Mn(Cp*)2 basso spin
I METALLOARENI M(C6H6)2
•
3 CrCl3 + 2 Al + 2 C6H6 3 [Cr(C6H6)2]+
[Cr(C6H6)2]+ + S2O42- [Cr(C6H6)2]
Le distanze C-C sono tutte
uguali. I due anelli sono
“eclipsed”
La reattività è modificata, in
accordo con una minor densità
elettronica
LEGAME QUINTUPLO Ar-Cr-Cr-Ar
Cr-Cr 1.85 Å
POLIENI SUPERIORI (C>6)
C60 COME LEGANTE
Os3(CO)11(h2-C60)
Os3(CO)8(PPh3)(m3-h6-C60)
Os5C(CO)12(PPh3)(m2-h4-C60)
Fe(C5H5)(h5-C60Me5)
CARBENI =CR2 E CARBINI CR
Carbene di Fischer R = sostituenti elettronattrattori, C elettrofilo
Carbene di Schrock R = alchile o idrogeno, C nucleofilo
SINTESI DI CARBENI DI FISCHER
1- Un nucleofilo, seguito da un
elettrofilo; reazione più facile
sugli isonitrili (CN-R)
2- Estrazione di H+ o H- da un
alchile
3 - Attacco del carbene,
(generato da diazometano)
REAZIONI DEI CARBENI DI FISCHER
1 - Sostituzione nucleofila al
carbonio
3 - Alchilazione in b
2 - Ossidazione a lattame
4 – Coupling (dec. Termica)
SINTESI DEI CARBENI DI SCHROCK
Alchilazione e a-eliminazione;
il metallo deve essere molto
ingombrato
I complessi dei metalli di prima transizione non
raggiungono i 18 elettroni.
Possono essere stabilizzati da legami agostici:
Astrazione di Me- e H+
REAZIONI DEI CARBENI DI SCHROCK
Con un alchene:
metallaciclobutano e
somma
Con un chetone: (cf. Wittig)
Con un estere (reag. di Tebbe)
Un carbene “a metà strada”
Applicazioni:
catalizzatori organometallici, anche
con sostituenti chirali, dalle caratteristiche
modificabili giocando sull’ingombro
sterico
Complessi NHC-M studiati per proprietà
di luminescenza, catalisi e costruzione
di strutture supramolecolari
CARBENI E CARBINI A PONTE
CH2I2 + [Fe2(CO)8]2- Fe2(CO)8(m-CH2)+ 2 ICo2(CO)8 + CHCl3 Co3(CO)9(m3-CH)
LA SOMMA OSSIDATIVA: LnM + A-B B-LnM-A
Di solito, lo stato di ossidazione e il numero di coordinazione del metallo
aumentano di due unità
Il “complesso di Vaska”: Ir(CO)Cl(PPh3)2
Si sommano i legami H-H, H-X, X-X, C-X, O-O, S-H, Si-H; Hg-Cl, S-S, Sn-X
Si sommano i legami C-C
tensionati:
Un complesso a 18 elettroni deve dissociare per lasciare posto ai due leganti
entranti: Pt(PPh3)4 + Ph3SnCl [(PPh3)2PtClSnPh3] + 2 PPh3
In un complesso a 17 elettroni, può avvenire una reazione binucleare:
MeI + 2 [Co(CN)5]3- Co(CN)5I]3- + [Co(CN)5Me]3-
IL MECCANISMO: 1 ADDIZIONE
CONCERTATA
l’addizione avviene in cis,
il complesso sigma è
l’intermedio, il CO rimane
nel piano equatoriale
dell’intermedio TBP
Nell’addizione di diossigeno si forma uno ione
superossido. L’addizione è reversibile se X = Cl,
irreversibile se X = I
IL MECCANISMO: 2 SOSTITUZIONE
NUCLEOFILA
[Rh(CO)2I2]- + CH3I [trans MeRh(CO)2I3]-
Il metallo funge da base di Lewis. Se la reazione si ferma al primo
intermedio è più corretto definirla addizione elettrofila.
DS < 0 in un processo associativo
IL MECCANISMO: 2 SOSTITUZIONE
NUCLEOFILA
RX + Ir(CO)Cl{P(p-C6H4Y)3}2
La velocità di reazione dipende dai sostituenti Y
delle fosfine
●MeI
○PhCH2Cl
LA STEROCHIMICA DELLA
SOSTITUZIONE NUCLEOFILA
R e X sommano in trans al metallo. Si ha inversione di configurazione al
carbonio
Si determina la configurazione al carbonio nel prodotto finale. L’inversione
avviene nello stadio di somma ossidativa
IL MECCANISMO: 3 PROCESSO
RADICALICO
• Electron transfer (redox a sfera esterna):
NiL4 NiL3 + L
NiL3 + R-X {NiL3+, RX-}
{Ni(I)L3+, RX-} RNiL2X + L
• A gabbia (terminazione immediata)
PtL3 PtL2 + L
PtL2 + RX X-PtL2▪ + R▪ X-PtL2-R
• A catena:
Ir(I)(CO)ClL2 + RX R-Ir(II)(CO)ClL2
R-Ir(II)(CO)ClL2 + R-X RIr(III)(CO)ClL2X + R▪
SOMMA OSSIDATIVA DI LEGAMI C-H
A solution of (h-C5H5)Ir(CO)2 in
perfluorohexane under methane (ca. 10 atm)
was irradiated for 6 h. From IR and NMR the
only new compound in solution was
(h-C5H5)Ir(CO)(H)CH3 (3a). From NMR
intensities, the yield was ca. 20%
COMPLESSI CICLOMETALLATI PER GLI OLED
La ciclometallazione:
All cyclometalates are anionic, so that these ligands offer very strong M–C covalent interactions as well as highly stabilized
ligand-field. One important consequence regarding the photophysics is that the energy of the higher lying metal-centered d–d
excited states in these complexes, which generally serves as a major nonradiative deactivating channel, is raised substantially
Preparation of highly efficient, true-blue phosphorescent complexes has been long considered as a formidable challenge.
This task is far more difficult than those for preparing longer wavelength emission deriving from green, orange and red
phosphors
ELIMINAZIONE RIDUTTIVA.1
B-LnM-A LnM + A-B
È difficile isolare prodotti stabili:
2 cis-PtH(Me)L2 Pt + PtL4 + 2 CH4 (-25°)
L’eliminazione può essere indotta da dissociazione di un legante (l’intermedio
pentacoordinato è meno rigido):
ELIMINAZIONE RIDUTTIVA.2
Possono eliminare solo due gruppi in cis.
>
>
In trans-PdL2(Me)2 l’isomerizzazione precede l’eliminazione:
ELIMINAZIONE RIDUTTIVA.3
L’eliminazione può essere indotta dal CO, gli acili eliminano più facilmente
degli alchili:
CpCoMe2L + CO CpCo(COMe)MeL
CpCo(COMe)MeL + CO CpCo(CO)L + MeCO-Me
Eliminazione binucleare:
DOs(CD3)(CO)4 {DOs(COCD3)(CO)3}
DOs(COCD3)(CO)3-H-Os(Me)(CO)4 DOs(CO)4Os(Me)(CO)4 + CD3-H
Coupling ossidativo due alcheni ( o alchini) coordinati formano un legame C-C:
INSERZIONE
Inserzione 1,1 o 1,2.
In genere i leganti h1 danno inserzione 1,1 (Es. CO)
I leganti h2 danno inserzione 1,2 (Es. alcheni)
SO2 dà entrambi i tipi di inserzione.
Lo stato di ossidazione del metallo non cambia, il conteggio elettronico
diminuisce di due unità
CINETICA DELL’INSERZIONE
Stato stazionario: L’’intermedio non si accumula d[Int]/dt = 0
k1[Rgt]- k-1[Int]- k2[Int][L] = 0
[Int] = k1[Rgt]/{k-1+ k2[L]}
v = k2[Int][L] = k2 k1[L] [Rgt]/{k-1+ k2[L]}
Due casi limite (cfr. con meccanismo dissociativo):
k-1 >> k2[L] Cinetica del secondo ordine, lo stadio lento è l’attacco di L
k-1 << k2[L] Cinetica del primo ordine v = k1[Rgt]
Se L = 13CO, il nuovo legante entra in cis.
La retro-inserzione permette di determinare qual è il gruppo che migra:
Lo studio originale (1967)
è stato fatto per IR
Gruppi elettron-attrattori rallentano la reazione. Acidi di Lewis (AlCl3, Na+,
etc.) si coordinano al CO e la accelerano. La configurazione al carbonio
non si inverte.
Alchile > Benzile > Vinile, Arile
L’INSERZIONE SEGUITA VIA 13C-NMR
In seguito a migrazione, il *CO marcato va
in posizioni specifiche:
Lo spettro 13C-NMR dell’alchile (a) e dell
acile (b): senza arricchimento preferenziale,
il rapporto tra i segnali cis/trans = 4
[Mn(CO)5]- + Me*COCl
Mn(CO)5(*COMe)
cis-Mn(Me)*CO(CO)4 + CO
Il rapporto
sperimentale è
Cis/trans = 2 ± 0.1,
compatibile con la
migrazione del Me
INSERZIONE STEREOSPECIFICA
CO e R migrano, in funzione del solvente.
La reazione è catalizzata da acidi di Lewis
(BF3, ultime righe)
ALTRE INSERZIONI
Alcheni: H e alchene
devono essere in cis,
quasi coplanari
La rarità di inserzione degli
alcheni nei legami M-C
(oligomerizzazione) è dovuta a
fattori cinetici : nello stato di
transizione si destabilizza un
orbitale d (per perdita di
retrodonazione) . L’effetto è
ininfluente per Metalli d0
ALTRE INSERZIONI
SO2: attacco elettrofilo di un acido di Lewis al carbonio. Non è necessario
un sito libero su M. Si formano due isomeri
Inserzione 1,2 di CO2: [MeW(CO)5]- + CO2 [(CO)5W-O-C(O)-Me]Gli isocianati danno facilmente poli-inserzione:
CATALISI E DFT
ISOMERIZZAZIONE DI ALCHENI
Tutti gli stadi sono reversibili: si
produce una miscela in cui
prevalgono i prodotti
termodinamicamente più stabili
(alcheni interni invece di terminali).
Occorrono un idruro ed un sito
vacante (meccanismo a)
oppure due siti vacanti (mecc. b)
IDROGENAZIONE DI ALCHENI
Il catalizzatore di
Wilkinson: RhCl(PPh3)3.
L’etilene forma un
complesso stabile e non
viene idrogenato. Alcheni
ingombrati non si
coordinano. Se la fosfina è
poco ingombrante, il
catalizzatore è meno attivo
IDROGENAZIONE DI ALCHENI
In questo schema, le sostanze all’interno della linea tratteggiata sono gli
intermedi proposti del ciclo catalitico. Tutti i composti che sono stati isolati o
individuati in soluzione stanno al di fuori del ciclo principale. Questo non è
generale; in molti altri casi sono stati osservate specie che appartengono
direttamente al ciclo catalitico principale. Comunque, questo studio dimostra
che l’assenza di dati cinetici può portare a interpretazioni scorrette. «Only
when kinetic and thermodynamic measurements define the role of complexes
along the actual reaction path can the mechanism be defined».
PROCESSI DINAMICI: RhCl(PPh3)3
A: il cat di Wilkinson.
B: dopo la somma ossidativa di H2
B’: come B, a bassa T
C: in atmosfera inerte (si torna ad A)
In nessun caso è presente PPh3 libera
LA STEREOCHIMICA DEGLI INTERMEDI
IDROGENAZIONE ASIMMETRICA
Si usano fosfine chelanti chirali. La chiralità è di solito al carbonio:
In seguito a coordinazione
si formano più centri
stereogenici; tra i vari
diasteroisomeri, uno
reagisce più velocemente
BINAP
IDROGENAZIONE ASIMMETRICA
Per idrogenazione del doppio legame si
ottiene L-DOPA
In questo caso il complesso più stabile (più
abbondante) è il meno attivo:
DDG‡ = 3.7 Kcal/mol; e.e = 90 %
The Nobel Prize in Chemistry 2001
S. Knowles and Ryoji Noyori "for their work on chirally catalysed
hydrogenation reactions"
IDROFORMILAZIONE
R-CH=CH2 + CO + H2 R-CH2-CH2-CHO + CH3-CHR-CHO
Attualmente, si usano complessi di rodio e fosfiti P(OR)3, ottenendo aldeidi lineari
IL PARAIDROGENO
p-H2
I segnali NMR dei protoni, subito dopo reazione con p-H2, hanno intensità
aumentata (fino a 30000 volte) e fase invertita. Es: idroformilazione con Ir
CARBONILAZIONE DEL METANOLO
CH3OH + CO CH3-COOH
Il catalizzatore è [Rh(CO)2I2]-, il co-catalizzatore HI
PROCESSO WACKER
CH2=CH2 + ½ O2 CH3-CHO
v = k[C2H4][PdCl42-]/[H+][Cl-]2
La marcatura isotopica ha dimostrato che
la configurazione al C si inverte, e che
non si scambiano H col solvente
Quattro processi redox, O2 ossidante
stechiometrico, Cu2+ catalitico
PROCESSO WACKER: CICLO COMPLETO
L’effetto trans dell’etilene è
maggiore dell’effetto trans
del Cl, si deve formare
trans-Pd(H2O)(C2H4)Cl2
The Nobel Prize in Chemistry 2010
R. F. Heck, E. Negishi, A. Suzuki
Richard F. Heck Ei-ichi Negishi, Purdue University, West Lafayette, Indiana, USA,
and Professor (emeritus) Akira Suzuki, Hokkaido University, Sapporo, Japan. The
Royal Swedish Academy of Sciences is rewarding the three chemists for:
“palladium-catalyzed cross couplings in organic synthesis”. The discoveries by the
three organic chemists have had a great impact on academic research, the
development of new drugs and materials, and are used in many industrial chemical
processes for the synthesis of pharmaceuticals and other biologically active
compounds.
24 riferimenti di letteratura Ni(PPh3)2Cl2 and Heck/Suzuki
LA METATESI DI ALCHENI
Scambio di sostituenti di due alcheni. È catalizzata da complessi dei metalli
dei gruppi 5-6. Es MoCl6, WCl6; co-catalizzatori agenti alchilanti quali AlMe3; o
da carbeni preformati.
APPLICAZIONI DELLA METATESI
• Sintesi di olefine terminali:
R-CH=CH-R’ + CH2=CH2 CH2=CH-R’ + CH2=CH-R
• Sintesi di olefine di interesse commerciale:
IL MECCANISMO DELLA METATESI
Due meccanismi possibili: a) ciclobutano coordinato b) metallaciclobutano (di
Chauvin)
METATHESIS DANCES
http://www.hitteam.net/index.php?lang=en&page=
metathesis
IL MECCANISMO DELLA METATESI
Anche all’inizio si forma il
diene misto (C14):
EVOLUZIONE DEI CATALIZZATORI
Catalizzatori a base di Ru resistenti all’ossigeno e all’umidità, compatibili
con altri gruppi funzionali.
EVOLUZIONE DELLA METATESI
Catalisi enantioselettiva: (ROM, RCM):
Alkyne & Enyne metathesis:
Cross metathesis:
PROCESSI DINAMICI (FLUSSIONALITÀ)
La costante di velocità dello scambio può
essere ricavata dalla larghezza dei picchi
in presenza (wE) ed in assenza di scambio
(w0)
Scambio lento: k = p(wE-w0)
Coalescenza k = Dn0)p/2
Scambio veloce k = 4p(Dn0)2/(wE-w0)
NUCLEI QUADRUPOLARI S > 1/2
I nuclei quadrupolari consentono
un rapido rilassamento. Le bande
si allargano, salvo nel caso di
molecole molto simmetriche.
Banda centrale: molecole con 14N;
Satelliti: molecole con 15N
NMR di un composto paramagnetico: Ni(PPh3)2Cl2
1H
NMR di Ni(PPh3)2Cl2: segnali
larghi e a chemical shift anomali;
il “contact shift” misura la
delocalizzazione di elettroni
spaiati sul legante (effetto dp-pp)
L’aggiunta di PPh3 dimostra lo
scambio veloce di legante:
ELECTRON SPIN RESONANCE
Spettroscopia degli elettroni spaiati: valgono i principi dell’NMR
(gli elettroni hanno spin ±½). Però
DE = ghB
g(rapp. giromagnetico dell’elettrone è -1760 rad/Ts)
g (rapp. giromagnetico di H è 26.75)
La frequenza della radiazione elettromagnetica corrisponde alle microonde
( e non alle onde radio)
N = N0exp(-DE/kT)
La diff. di popolazione tra stato fondamentale (N0) e stato eccitato (N) è
molto grande: maggior sensibilità (10-8 M)
EPR di Cr(acac)3
EPR di 53Cr3+ (10 %, s=3/2)
ESR DI Fe(NO)(S2C-NEt2)2
14N
(s = 1)
A = 36.5 MHz