INTERAZIONI FARMACOCINETICHE CLINICAMENTE RILEVANTI CON
ANTIDEPRESSIVI DI SECONDA GENERAZIONE: UN AGGIORNAMENTO
-
Introduzione
Tra i disturbi psichiatrici, la depressione è classificata come patologia dell’umore caratterizzata da
una serie di segni e sintomi comportamentali che tendono a ridurre gradatamente
il tono
dell’umore, arrivando a compromettere il corretto funzionamento fisico, psichico e sociale.
Oggi sappiamo che la depressione è dovuta ad una combinazione di fattori genetici
(predisposizione familiare e fattori biologici tra cui i livelli dei neurotrasmettitori serotonina,
noradrenalina e dopamina, ipotesi monoaminica o delle amine biogene per i disturbi dell’umore),
fattori ambientali e psicologici (stress, traumi); inoltre, ha un ruolo fondamentale l'asse ipotalamoipofisi-surrene che regola la risposta a lungo termine allo stress, inducendo il surrene al rilascio di
cortisolo. Elevati livelli di cortisolo provocano insonnia, diminuzione dell'appetito, diabete mellito,
osteoporosi, diminuzione dell'interesse sessuale, aumento dell'espressione comportamentale
dell'ansia, immunosoppressione, danni a vasi cerebrali e cardiaci (ipotesi della diatesi da stress).
La terapia farmacologica per il trattamento della depressione si avvale di psicofarmaci con il potere
di normalizzare l'equilibrio alterato dei neurotrasmettitori.
A partire dagli anni Cinquanta, si sono usati gli antidepressivi triciclici o TCA (Tricyclic
antidepressant) che, bloccando la ricaptazione di serotonina e noradrenalina, hanno mostrato
notevole efficacia. Tuttavia questi antidepressivi hanno effetti collaterali, dovuti all'azione
anticolinergica, che non sono tollerati: tachicardia, aritmie, secchezza delle fauci, stipsi, ritenzione
urinaria, offuscamento della vista.
Gli inibitori delle monoamino ossidasi IMAO (monoamine oxidase inhibitors), agiscono come
inibitori della monoaminossidasi, enzima che metabolizza serotonina e catecolamine (adrenalina,
noradrenalina e dopamina) portando ad un aumento della concentrazione di questi
neurotrasmettitori nel sistema nervoso centrale. Essi presentano un'efficacia paragonabile a quella
degli antidepressivi triciclici ma inducono eccitamento, insonnia, tremori, allucinazioni,
ipotensione, sudorazione ridotta; inoltre, producono effetti tossici per interazione con sostanze
contenti elevate dosi di tiramina (formaggi, alcuni vini e birre, fegato, banane, fave, fichi). Gli antidepressivi di seconda generazione, a struttura non triciclica, sono efficaci e meglio tollerati.
Si distinguono cinque gruppi:
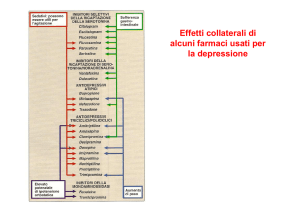
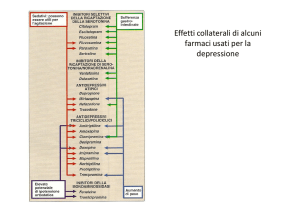
1. Inibitori selettivi della ricaptazione della serotonina (SSRI)
-
Citalopram (Elopram, Seropram in Italia)
-
Escitalopram (Cipralex, Entact in Italia)
-
Fluoxetina (Fluoxerene, Fluoxetina-generica in Italia; Prozac in Italia)
-
Fluvoxamina (Dumirox in Italia)
-
Paroxetina (Daparox, Eutimil, Sereupin in Italia)
-
Sertralina (Zoloft in Italia)
Si possono osservare effetti collaterali come sonnolenza, xerostomia, irritabilità, ansia, diminuzione
dell'appetito e diminuzione della pulsione e capacità sessuale.
2. Inibitori selettivi della ricaptazione della serotonina e della noradrenalina (NSRI o SNRI)
-
Desvenlafaxina (Pristiq)
-
Duloxetina (Cymbalta, Xeristar in Italia)
-
Venlafaxina (Efexor, Zarelis)
-
Nefazodone
3. Antidepressivi serotoninergici specifici e noradrenergici (NaSSA)
-
Mianserina (Lantanon in Italia)
-
Mirtazapina (Remeron, Avanza, Zispin)
Questi bloccano i recettori adrenergici presinaptici alfa-2 e allo stesso tempo alcuni recettori della
serotonina. Possono presentarsi effetti collaterali come sonnolenza, aumento dell'appetito e di peso.
4. Inibitori selettivi della ricaptazione della noradrenalina (NaRI)
-
Atomoxetina (Strattera)
-
Mazindolo (Mazanor, Sanorex)
-
Reboxetina (Davedax, Edronax)
5. NDRI (inibitori della ricaptazione della noradrenalina e della dopamina).
-
Bupropione (Wellbutrin, Zyban)
6. Agonista della melatonina e antagonista della serotonina, disinibisce la trasmissione
noradrenergica e dopaminergica:
-
Agomelatina.
CLASSE
PRINCIPI ATTIVI
Imipramina, amitriptilina, clomipramina, doxepina,
dosulepina, trimipramina, nortriptilina
TCA
Tranilcipromina, fenelzina,isocarboxazide
IMAO
Citalopram, Escitalopram, Paroxetina, fluoxetina,
fluvoxamina, sertralina
SSRI
Reboxetina
NaRI
Venlafaxina, duloxetina
SNRI
Bupropione
NDRI
Mirtazapina, agomelatina, trazodone, nefazodone.
ALTRI
I gravi effetti collaterali degli antidepressivi di prima generazione, ed il ben documentato rischio di
interazioni farmacologiche con altri farmaci co-somministrati, hanno contribuito al loro graduale
declino nella pratica clinica per lasciare il posto agli antidepressivi di seconda generazione il cui
buon profilo di tollerabilità ed il meccanismo d’azione maggiormente selettivo rappresentano punti
di forza.
-
Interazioni farmacologiche
Poiché gli antidepressivi di seconda generazione sono comunemente prescritti in associazione con
altri farmaci usati per il trattamento di malattie psichiatriche o somatiche concomitanti
(antimicrobici, farmaci cardiovascolari, ansiolitici, antipsicotici) potrebbero verificarsi interazioni
farmacologiche; sebbene presentino un basso rischio di interazioni farmacodinamiche per merito
della loro selettività d’azione, mostrano capacità inibitoria sugli isoenzimi CYP del citocromo P450,
pertanto sono associati ad interazioni di tipo farmacocinetico clinicamente rilevanti soprattutto a
livello della biotrasformazione metabolica.
(Vedi tabella “Principali interazioni farmacocinetiche degli antidepressivi di II generazione” nella
home page di www.fvcalabria.unicz.it).
Il processo di metabolizzazione di una sostanza introdotta nell’organismo avviene a carico di
enzimi localizzati a livello di fegato, intestino, reni, polmoni, cute, plasma e sistema nervoso
centrale. Le reazioni di biotrasformazione sono di due tipi:
Reazioni di fase I: reazioni di ossidoriduzione e di idrolisi ad opera dei citocromi;
Reazione di fase II : reazioni di coniugazione, mediate da diversi enzimi e cofattori, che
aggiungono diversi gruppi funzionali per rendere più facilmente eliminabile il farmaco; con
acido glicuronico (o glicuronoconiugazione), acetilazione, con amminoacidi (soprattutto
glicina, taurina e glutammina), con solfato (o solfoconiugazione) e metilazione.
Il sistema CYP negli uomini consiste in più di 50 enzimi localizzati sulla membrana del reticolo
endoplasmatico liscio degli epatociti ed in molti altri tessuti. Questi enzimi sono responsabili delle
reazioni ossidative di fase uno di molti farmaci, nutrienti, tossine e sostanze endogene, sono divisi
in famiglie e sottofamiglie in base alle similitudini delle sequenze aminoacidiche; ogni enzima è
distinto attraverso il CYP seguito da un numero che indica la famiglia, una lettere che indica la
sottofamiglia ed un altro numero che denota la specifica isoforma.
Gli isoenzimi CYP che giocano il ruolo maggiore nella biotrasformazione degli agenti terapeutici
sono CYP1A2, CYP2C9, CYP2C19, CYP2D6 e CYP3A4. Minori ma clinicamente rilevanti
CYP2A6, CYP2B6, CYP2C8 e CYP2E1.
Molti farmaci sono substrato per una sola isoforma di CYP, mentre altri sono metabolizzati da più
isoforme risultando in una serie di metaboliti.
Esiste una grande variabilità di espressione e di attività di questi enzimi che porta a delle differenze
interindividuali manifestate con l’esposizione al farmaco; la variabilità deriva da fattori genetici,
fisiopatologici, ambientali (inclusa la cosomministrazione di altri farmaci). I polimorfismi dei CYP
hanno importanti implicazioni cliniche e riguardano CYP2C9, CYP2C19 e CYP2D6.
Le interazioni farmacologiche che coinvolgono le isoforme di CYP generalmente derivano da una
loro inibizione o induzione.
-
L’inibizione enzimatica consiste nella competizione con un altro farmaco per il legame al
sito bersaglio e la quota metabolizzata di un agente somministrato concomitatamente
diminuisce mentre la concentrazione plasmatica e gli effetti farmacologici aumentano.
Quando si sospende la somministrazione dell’inibitore, le condizioni precedenti si
ristabiliscono in base alla quota di eliminazione del farmaco influenzato e dell’inibitore.
-
L’induzione enzimatica porta alla riduzione della concentrazione plasmatica del composto
attivo e alla perdita di efficacia terapeutica. E’ un processo lento dose- e tempo-dipendente.
Poiché di solito il processo richiede la sintesi di nuovi enzimi, l’induzione si verifica con un
leggero ritardo rispetto alla esposizione dell’agente induttore, in genere da pochi giorni a
una o due settimane. Il tempo che intercorre per la de- induzione è allo stesso modo graduale
e dipendente dalla quota di degradazione dell’enzima e dal tempo richiesto per
l’eliminazione del farmaco induttore. L’attività degli enzimi che metabolizzano i farmaci
potrebbe essere incrementata anche dalla somministrazione cronica di uno xenobiotico,
alcool, componenti assunti con la dieta e fumo.
Le interazioni farmacocinetiche sono studiate inizialmente in vitro per predirne l’importanza in
vivo, ma non tutte le potenziali interazioni in vitro si verificano in vivo. Quando si valuta il rischio
potenziale, l’estensione ed il significato clinico di un’interazione farmacocinetica è necessario
considerare diversi tipi di fattori:
-
Fattori correlati al farmaco: potenza, dose/concentrazione dell’inibitore/induttore, indice
terapeutico del substrato, estensione del metabolismo del substrato, la presenza di metaboliti
attivi o tossici.
-
Fattori correlati al paziente: predisposizione genetica, suscettibilità agli eventi avversi come
nel caso di pazienti anziani.
-
Fattori epidemiologici: probabilità di interazione per due farmaci prescritti insieme.
Generalmente, ci si aspetta un’interazione farmacocinetica clinicamente rilevante quando un
farmaco con basso indice terapeutico è somministrato con un potente inibitore o induttore della
maggiore via di metabolizzazione che il farmaco utilizza. Al contrario, poiché molti farmaci hanno
diverse vie metaboliche, l’inibizione di un enzima che gioca un ruolo marginale in tutta
l’eliminazione del farmaco somministrato può avere effetti limitati sulla sua disponibilità e ci si
aspetta solo un minimo incremento delle concentrazioni plasmatiche: un’altra isoforma può
provvedere all’eliminazione attraverso una via metabolica secondaria ma adeguata.
-
Potenziali interazioni farmacocinetiche con antidepressivi di II generazione
Tutti i farmaci antidepressivi di seconda generazione sono sottoposti ad intensa biotrasformazione
epatica e ciascuno presenta un particolare profilo di rischio di interazioni farmacocinetiche. (Vedi
tabella “Isoenzimi coinvolti nel metabolismo degli antidepressivi di seconda generazione ed enzimi
da loro inibiti” nella sezione “Farmaci e ADR”- antidepressivi sul sito www.fvcalabria.unicz.it).
La somministrazione concomitante di farmaci che agiscono da inibitori o induttori degli enzimi
coinvolti nella biotrasformazione degli antidepressivi di seconda generazione possono influenzare la
loro eliminazione, portando a modifiche nelle concentrazioni plasmatiche e conseguenti effetti
clinici. Comunque, la maggior parte degli antidepressivi di seconda generazione ha un largo indice
terapeutico quindi l’inibizione o l’induzione del loro metabolismo solo raramente conduce ad
interazioni clinicamente rilevanti. D’altra parte, possono essere associati ad inibizioni clinicamente
rilevanti degli isoenzimi CYP e richiedere cautela quando aggiunti ad un regime di politerapia. Gli
antidepressivi di seconda generazione differiscono in maniera considerevole nel loro potenziale di
inibizione individuale degli isoenzimi CYP ed influiscono in maniera diversa sulla concentrazione
plasmatica del farmaco cosomministrato.
SSRI
FLUOXETINA
È metabolizzata principalmente attraverso la N-demetilazione a metabolita attivo norfluoxetina. I
risultati di studi sia in vitro sia in vivo indicano che il maggiore responsabile della reazione è
CYP2D6, con contributo aggiuntivo degli altri. Gli studi indicano che sia la fluoxetina che il suo
metabolita sono potenti inibitori del CYP2D6 ed in maniera moderata degli altri. Pertanto, la
fluoxetina presenta un alto rischio potenziale di interazioni farmacocinetiche con altri agenti
cosomministrati e metabolizzati dallo stesso CYP. Inoltre, poiché la fluoxetina ed il suo metabolita
presentano una lunga emivita (7-14 giorni) l’inibizione dell’isoenzima si protrae per settimane oltre
la sospensione del trattamento.
PAROXETINA
Metabolizzata nel fegato tramite ossidazione e metilazione in prodotti inattivi che sono coniugati
con acido glucuronico o sulfurico. Le isoforme coinvolte nella ossidazione sono CYP2D6 ad alta
affintà, la cui saturazione è responsabile della cinetica non lineare dei farmaci, ed il CYP3A4 a
bassa affinità. La paroxetina è un potente inibitore del CYP2D6 in vitro, mentre ha effetti minimi
sulle altre isoforme. Pertanto, la paroxetina può aumentare la tossicità di vari agenti psicotropi che
sono metabolizzati principalmente dal CYP2D6.
FLUVOXAMINA
È sottoposta ad un intenso metabolismo epatico che comprende de metilazione ossidativa e
deaminazione. Il CYP1A2 e il CYP2D6 sembrano essere le isoforme implicate in queste reazioni.
La fluvoxamina è un potente inibitore di CYP1A2 e CYP2C19, in maniera moderata inibisce gli
altri. Conseguenza dell’inibizione non selettiva è la possibilità di interagire con farmaci di diverse
categorie.
SERTRALINA
La maggiore via metabolica per la sertralina è rappresentata dalla N-demetilazione a Ndemetilsertralina, che è meno attiva come bloccante della ricaptazione di serotonina rispetto al
composto originario. I dati degli studi in vitro indicano nella reazione sono coinvolti diversi
isoenzimi. La sertralina possiede capacità inibitoria sul CYP2D6, di tipo dose dipendente.
CITALOPRAM
Si tratta di una miscela racemica degli enantiomero S attivo ed R inattivo. La via metabolica
principale è rappresentata dalla N-demetilazione ad N- desmetilcitalopram, reazione mediata dagli
isoenzimi CYP2C19, CYP2D6 e CYP3A4. Il citalopram ha il potere di inibire solo debolmente
CYP2D6.
ESCITALOPRAM
Enantiometro S attivo del citalopram. Secondo gli studi in vitro, CYP2C19, CYP2D6 e CYP3A4
contribuiscono nella stessa misura alla formazione di S-desmetilcitalopram (escitalopram), mentre
solo CYP2D6 è responsabile della produzione di S-didesmetilcitalopram. Gli effetti di inibizione
sugli isoenzimi sono deboli, il che suggerisce un basso rischio potenziale di interazioni
farmacologiche.
SNRI
VENLAFAXINA
Metabolizzata nel fegato nel suo maggiore metabolita attivo O- desmetilvenlafaxina e metabolita
inattivo N- desmetilvenlafaxina. Le evidenze sia in vitro sia in vivo suggeriscono che il CYP2D6
sia l’enzima responsabile della de metilazione, mentre il CYP3A4 lo è della N-demetilazione.
Secondo gli studi in vitro, la venlafaxina è un debole inibitore del CYP2D6 ed ha un effetto minimo
o nullo sugli altri enzimi. A causa del suo effetto inibitorio sul sistema citocromiale epatico, la
venlafaxina ha una bassa predisposizione per le interazioni farmacocinetiche con gli altri farmaci
somministrati contemporaneamente.
DULOXETINA
Sottoposta a metabolismo epatico mediante il CYP2D6 e il CYP1A2 da cui derivano diversi
metaboliti ossidati e coniugati inattivi ed eliminati principalmente nelle urine. La duloxetina è
subsrtato di CYP2D6 ed è anche suo inibitore seppure moderato, e di CYP1A2.
Altri antidepressivi di seconda generazione
MIRTAZAPINA
Antidepressivo noradrenergico e serotoninergico metabolizzato a livello epatico attraverso la 8idrossilazione ed N-demetilazione che avvengono per CYP2D6 e CYP3A4 rispettivamente.
L’inibizione è debole su entrambi i CYP. Gli studi suggeriscono che la mirtazapina abbia un proflio
favorevole di interazione farmacologica, e che siano poco probabili le influenze sugli altri farmaci
cosomministrati.
BUPROPIONE
Antidepressivo che inibisce il reuptake neuronale di norepinefrina e dopamina e non ha
apprezzabile affinità per i recettori post sinaptici. Il bupropione è metabolizzato nel fegato per
idrossilazione ed il suo principale metabolita idrossibupropione ssembra essere farmacologicamente
attivo. Studi in vitro indicano che il CYP2B6 sia coinvolto nella metabolizzazione del bupropione.
Mentre i dati in vitro suggeriscono che il bupropione ed il suo metabolita hanno bassa attività di
inibizione sul CYP2D6.
NEFAZODONE
Antagonista del recettore 5-HT2 della serotonina che inibisce il reuptake sia della serotonina che
della norepinefrina. È metabolizzato nel fegato nel metabolita idrossinefazodone che ha una attività
farmacologica smile a quella del farmaco originario e in triazoledione e metaclorofenilpiperazina,
entrambi meno attivi del nefazodone. Studi in vitro indicano che il nefazodone è un potente
inibitore di CYP3A4 e debole di CYP2D6. Basandoci su dati biochimici, il nefazodone potrebbe
avere significative probabilità di interagire quando somministrato in combinazione con substrati di
CYP3A4 con basso indice terapeutico.
La conoscenza degli effetti dovuti alla inibizione di una particolare isoforma di CYP, da parte degli
antidepressivi di seconda generazione, può indirizzare il medico proscrittore verso la scelta più
appropriata nel singolo caso del paziente da trattare in base a fattori costituzionali e alla presenza di
altri farmaci assunti contemporaneamente.
Clinically relevant pharmacokinetic drug interactions with second-generation antidepressants: an
update. E. Spina, V. santoro, C. D’Arrigo. Clinical Therapeutics/vol. 30, Num 7, 2008.