FARMACOLOGIA SPECIALE
Fonte prevalente: Katzung, Basic and Clinical Pharmacology, XII edizione.
Altre fonti: slide e sbobine lezioni farmacologia speciale aa2015-16; Harrison.
Buona lettura,
Eddy Andreis, settembre 2016
NOTA BENE: nella presente versione di questo compendio sono presenti alcuni errori che ho notato solo una volta stampato lo stesso, ma che non
inficiano la sua comprensione né la preparazione complessiva.
INDICE
– FANS p. 2;
– ANESTETICI LOCALI p.5;
– FARMACI GASTROINTESTINALI p. 9;
– OPPIOIDI p. 7;
– FARMACI RENALI-CARDIOVASCOLARI p. 12;
– IPERTENSIONE p.12
– DIURETICI p.16
– SIMPATOPLEGICI: p.18
– INIBITORI SRAA p.20
– VASODILATATORI / ANGINA p.23
– INSUFFICIENZA CARDIACA p.25
– Ca2+-ANTAGONISTI o BLOCCANTI CANALI Ca2+
– DIGITALICI;
– ANTIARITMICI p.30
– PATOLOGIE EMOSTASI p.34
– DISLIPIDEMIA p.37
– DIABETE MELLITO p.40
– FARMACI SNC p.43
– SEDATIVO-IPNOTICI p.44
– SCHIZOFRENIA p.45
– DEPRESSIONE p.47
– PARKINSON p.50
– EPILESSIA p.52
– SOSTANZE CHE DANNO DIPENDENZA p.54
Æ
1 / 55
FANS
PREMESSE
– Gli eicosanoidi sono autacoidi lipidici derivanti dall'acido arachidonico (AA).
– L'AA deriva da stress cellulare/ischemia, rilascio da LDL ma soprattutto dall'enzima PLA2, presente
in due forme:
– secreta, attivata da citochine (bloccata da annessine←cortisone);
– citoplasmatica, attivata da aumento [Ca2+]i.
L'AA può esistere in forma libera o esterificata:
– libero può essere elaborato da 3 enzimi: PGH-sintasi, LIPO-ossigenasi, EP-ossigenasi.
– esterificato invece subisce l'effetto diretto dei ROS (senza utilizzo di enzimi) formando
isoprostani, tra cui epi-PGF2alfa, potente vasocostrittore (riperfusione>↑ROS>↑epi-PGF2alfa
>spasmo coronarico).
1) PGH-sintasi (COX):
– è espressa sul reticolo endoplasmatico in maniera costitutiva (COX-1) o inducibile (COX-2);
– produce PGH2, da cui (anche se proviene da altra cellula) derivano PG(D/E/F/I)2 e TXA2.
– è un enzima suicida: dopo la sua stessa attività o il blocco irreversibile è necessaria la risintesi.
– Ha 2 attività: ciclo-ossigenazione e perossidazione. I FANS bloccano la ciclo-ossigenazione con 3
possibili modalità: []-dipendente, tempo-dipendente reversibile, t-dipendente irreversibile. L'azione
perossidasica però rimane e produce endoperossidi ad azione anti-infiammatoria.
2) LIPO-ossigenasi (LO): ne esistono diverse forme, bloccate da inibitori di FLAP (un co-enzima):
– 5-LO: se FANS bloccano COX, LO diventa via preferenziale di processamento di AA. Produce
LTA4, da cui LTB4 (chemotassi neutrofili) e LTC4 (bronco-vaso costrittore). No FANS in asmatici.
– 12-15-LO: modificate da aspirina, formano epi-lipossine, anti-infiammatorie.
RECETTORI PROSTANOIDI (_P) NATURALI (eicosanoidi derivanti da PGH2)
– D → vasodilata, broncocostringe;
– E → contrazione (PGE1), rilassamento (PGE2,4), secrezione gastrica(PGE3);
– F → contrazione utero, vasocostrizione, riduce pressione intraoculare (PGF2alfa);
– I → vasodilata, antiaggrega (PGI2);
– T → vaso/broncocostringe (TXA2).
GENERALITÀ
1. Acidi deboli con buon assorbimento (poco influenzato dal cibo), elevato metabolismo epatico
(CYP3a/2c) e prevalente escrezione renale.
2. Meccanismi azione: ↓ sintesi PG per blocco COX (PGH-sintasi), ↑ epi-lipossine e ↓ NfkB.
3. Proprietà: anti-infiammatori, analgesici, anti-piretici, anti-aggreganti.
CLASSIFICAZIONE
Selettività per COX:
– > COX1: ketoralac, flurbiprofene, ketoprofene, aspirina;
– COX1=2: naprossene, ibuprofene, paracetamolo;
– > COX2: -coxib (solo colecoxib sul mercato), diclofenac, meloxicam.
Emivita:
– breve: diclofenac, ketoprofene, paracetamolo, aspirina, ibuprofene;
– media: ketoralac, flurbiprofene, celecoxib;
– lunga: piroxicam, nabumetone, meloxicam, naprossene.
Æ
2 / 55
PROPRIETÀ
1. ANTI-INFIAMMATORIA:
– bloccano COX quindi ↓ sintesi PG, causa soprattutto di fase infiammatoria acuta. Poco attivi in
infiammazione cronica (dove prevalgono altre citochine IL-1/6, IFN, TNFalfa).
In patologie reumatiche (infiammatorie croniche) sono utilizzati DMARDs (disease modifying
anti-reumatic drugs) ad es. azatioprina, clorochina, ciclofosfamide, ciclosporina, metotrexato,
micofenolato. Tra essi ci sono farmaci “biologici” ad es. rituximab (--| B), abatecept (--| T),
infliximab (--| TNFalfa), tocilizumab (--| IL6).
– meccanismo alternativo è ↑ epi-lipossine (acetilazione COX-2 e LO-12/15) e ↓ NfkB (salicilati);
– paracetamolo e analoghi non efficaci perché inattivati da perossidi in aree infiammate, ma rimane
effetto analgesico e antipiretico per azione ipotalamica.
2. ANALGESICA:
– periferica e in fase acuta, per blocco della sensitivizzazione delle fibre nervose da PG;
– no fase cronica dove domina alterazione centrale (es. gabapentina, oppioidi).
3. ANTI-PIRETICA:
– febbre deriva da IL-1/6/TNF che resettano ipotalamo su temperatura maggiore, raggiunta con
produzione di calore (brividi) e conservazione (vasocostrizione, blocco sudorazione);
– PGE2 è mediatore principale che se bloccato ripristina il set-point consentendo perdita di calore;
– per ridurre eccessiva ipertermia: svestire pz, immergerlo in acqua fredda.
4. ANTI-AGGREGANTE:
– ↓ TXA2 piastrinica, indotta da legame a endotelio, che attiva piastrine;
– ↓ PGI2 endoteliale. Quindi attenzione a effetto trombotico paradosso;
– equilibrio anti-aggregante è possibile perché in piastrine non c'è resintesi COX come in endotelio.
EFFETTI AVVERSI
1. GASTROLESIVITÀ
– ↓PGE2 per blocco COX1 gastrica causa: ↓muco, ↓perfusione, ↑secrezione acida;
– è dose-dipendente, non modificata da assunzione con/senza cibo;
– terapia ≥ 1 settimana necessita gastroprotettore:
– misoprostolo (≈PGE2);
– antiacidi, anti-H2, PPI;
– gastralgia, pirosi, nausea, vomito, diarrea;
– RR ulcera: aumenta per età, precedenti ulcere, ↑dosi, altri FANS, anticoagulanti, corticosteroidi;
– causa 40% emorragie GI superiore. Dipende anche da tipo di FANS:
– ↑ ketoralac, ketoprofene, naprossene, aspirina;
– ↓ ibuprofene, nimesulide, diclofenac;
– selettività COX2 riduce rischio, pur aumentando quello vascolare.
2. EFFETTI SULLA GESTAZIONE
– chiusura prematura dotto arterioso → ipertensione polmonare;
– insufficienza renale → oligoidramnios;
– prolungamento travaglio per blocco PG contrazione utero.
3. TOSSICITÀ RENALE
– riduzione flusso glomerulare per blocco vasodilatazione arteriola afferente.
Æ
3 / 55
4. BRONCOCOSTRIZIONE
– da eccesso di leucotrieni per blocco COX e sovra-utilizzo LIPOSSIGENASI.
5. ALTRE:
– cutanee, idiosincrasiche, sindrome di Reye.
ALCUNI FANS
1. SALICILATI:
– 500mg/4h per effetto FANS, 80mg/d per effetto antiaggregante:
– effetto analgesico mesodermico, poco viscerale;
– effetto antipiretico per azione centrale e disaccoppiamento fosforilazione ossidativa;
– uricosurici solo a dosi >5g/d;
– effetti avversi a dosi elevate (anche dosi 5 g/d per AR): disidratazione e squilibri elettrolitici
(acidosi, poi alcalosi) da stimolo respiratorio indiretto (↑consumo O2) e diretto (centri superiori).
2. PARACETAMOLO
– non è anti-infiammatorio perché inattivato in zone con perossidi;
– attivo in SNC con effetto anti-piretico e analgesico;
– dà tossicità epatica grave se dose singola>7,5g o dosi croniche >2g/die per deplezione del glutatione
ridotto, più facile in pz che abusano d'alcol o assumono altri farmaci con metabolismo epatico. Si
tratta con infusione di acetilcisteina per donazione gruppi sulfidrilici.
3. DERIVATI ACIDO ACETICO:
– diclofenac (Voltaren): COX2>>1, dolore mesodermico patologie infiammatorie;
– ketoralac (Toradol): COX1>>2, potente analgesico postchirurgico.
4. DERIVATI ACIDO PROPIONICO:
– ibuprofene (Moment, Brufen, Nurofen): COX1=2, meno effetti GI di aspirina; a dosi <2400mg/d è
soprattutto analgesico.
– ketoprofene (Oki): >>COX1.
5. ALTRI:
– INDOMETACINA: 40x più potente di aspirina, uso in patologie infiammatorie (spondilite,
osteoatrosi) e analgesia palliativa. Elevati effetti collaterali.
– ENOLOACIDI: piroxicam: lunga emivita.
– FENAMATI: pochi vantaggi terapeutici su altri, tossicità.
– NABUMETONE: unico non acido (poi convertito a derivato ac.acetico), poco efficace, costa molto.
– NIMESULIDE: (Aulin): >COX2 ma sotto osservazione per tossicità epatica.
SALI D'ORO: miscela utilizzata per AR con meccanismo non chiaro, rallenta malattia ma elevata
tossicità, soprattutto cutaneo-mucosa e renale.
Æ
4 / 55
ANESTETICI LOCALI
GENERALITÀ
– causano perdita di sensibilità in una regione limitata del corpo;
– inibiscono i segnali afferenti agendo sulla generazione dell'impulso o sulla sua propagazione:
– nocicettori > fibre C > sostanza di Rolando nel corno posteriore > fascio spino-talamico >
talamo ventrolaterale > corteccia;
– sono somministrati direttamente nei pressi dell'organo bersaglio, spesso con vasocostrittori per
salvaguardarli quanto possibile dalla circolazione sistemica.
CHIMICA
– basi deboli (pKa≈9) sotto forma di sali per ↑solubilità;
– poco liposolubili al pH basso di tessuti infiammati, a volte si aggiunge NaHCO3;
– composti da un gruppo lipofilo (es. anello benzenico) e un gruppo ionizzabile (es. ammina terziaria)
legati da un legame esterico o amidico;
– amino-esteri (procaina, tetracaina): idrolizzati rapidamente da esterasi plasmatiche;
– amino-amidi (lidocaina): idrolizzati più lentamente da enzimi epatici.
FARMACOCINETICA
– la velocità di assorbimento sistemico:
– è un fattore negativo: causa ↑picco di []plasmatica (→effetti avversi) e ↓durata d'azione (per
degradazione);
– dipende da:
– perfusione locale: ↓ adrenalina;
– dose e sede: ↑intercostale, ↓sciatico;
– legame tissutale del farmaco: ↑tetracaina (lunga durata), ↓lidocaina (breve);
– la distribuzione del farmaco:
– dipende dal compartimento dove è somministrato, ad es. dopo diluizione in spazio
subaracnoideo diverse soluzioni subiscono diversi effetti gravità e per questo vengono dette:
iperbariche (↓), isobariche (↔), ipobariche (↑).
– metabolismo ed escrezione: (attenzione a pz. con deficit esterasi/epatici)
– dopo idrolisi plasmatica o epatica avviene escrezione urinaria;
– acidificazione di urine facilita ionizzazione quindi velocizza eliminazione.
FARMACODINAMICA
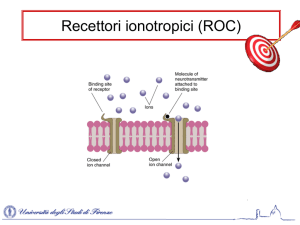
– target: canali volt-dipendenti del Na+:
– ≠diuretici che agiscono su canali Na+ ligando-dipendenti;
– ≠farmaci cardiovascolari --| prevalentemente canali Ca2+, K+;
– dinamica:
1. farmaco entra nella fibra nervosa in forma liposolubile, poi convertito in forma cationica;
2. solo quando il canale si attiva (per depolarizzazione) lo blocca dall'interno;
3. alcuni farmaci possono raggiungere il canale anche per via transmembrana.
– fattori che facilitano l'interazione farmaco-canale dipendono:
– dal farmaco: ↑concentrazione, ↑liposolubilità (tetracaina), ↓ingombro sterico;
– dalla fibra nervosa: ↓diametro, ↑mielinizzazione (a parità di diametro, ed è necessario blocco
3 nodi consecutivi). Più bloccate (in ordine) sono fibre:
– B pregangliari: poco mieliniche;
– C: amieliniche piccole (<1μm), nocicezione;
– A∂: mieliniche piccole (<5μm), termo-nocicezione.
Æ
5 / 55
CLINICA
– modalità possibili di utilizzo:
– applicazione topica (mucosa nasale, margini di incisioni);
– iniezione:
– perineurale presso terminazioni periferiche o grossi tronchi nervosi (“blocco”);
– epidurale (tra legamento e dura) o subaracnoidea (intratecale, “spinale”);
– intravenosa per paralisi regionale di un arto, con blocco del deflusso di sangue;
– durata anestesia dipende da:
– farmaco: tetracaina > lidocaina > procaina;
– sito: plesso brachiale > epidurale > subaracnoidea;
– co-utilizzo adrenalina:
– aumenta durata anestesia per ↓perfusione;
– effetto analgesico diretto (solo in anestesia spinale) per stimolo alfa2 post-sinaptico (uso
clonidina, agonista alfa2, come adiuvante).
EFFETTI AVVERSI E TOSSICITÀ
Da ricordare che i grossi nervi sono composti, dall'esterno all'interno, da:
– fibre motorie > fibre sensitive prossimali > fibre sensitive distali;
– questo può dare problemi in anestesia epidurale e spinale: fase espulsiva travaglio, perdita
sensibilità, ridotto controllo sfinteri, cadute.
Tossicità: da ↑↑[]plasmatica per ↑velocità di assorbimento o erronea iniezione intravascolare;
– CNS (tutti): sedazione, disturbi visivi-uditivi, agitazione. A dosi elevate si possono avere
convulsioni tonico-cloniche per inibizione dei segnali inibitori corticali;
– cardiaca (bupivacaina, ropivacaina): extrasistoli ventricolari per alterazione dei PdA dei
miocardiociti;
– fetale: durante l'epidurale, il passaggio al feto può provocare bradicardia, apnea, convulsioni. Il
rischio aumenta in condizioni di sofferenza fetale per prolungamento del travaglio dovuto alla stessa
anestesia.
Æ
6 / 55
OPPIOIDI
GENERALITÀ
– oppio: da Papaverum somniferum e P. album, contiene il 10% di morfina e altri alcaloidi;
– recettori di oppioidi sono mu (μ), delta (δ) e kappa (κ);
– peptidi oppioidi endogeni: endorfine (>μ), enkefaline (>δ), dinorfine (>κ), derivano da POMC.
– prodotti in risposta a vari comportamenti favorenti la specie come riproduzione,
alimentazione, parto, fratture, attività fisica protratta.
RECETTORI
– μ: analgesia, sedazione, ↓respirazione, ↓GI, modula rilascio ormoni. Attivato ++da endorfine;
– più importante in azione farmacologica: morfina è agonista totale, codeina parziale,
naloxone antagonista.
– δ: analgesia, rilascio ormoni/neurotrasmettitori. Attivato ++da enkefaline;
– κ: analgesia, effetti psicotropi, ↓GI. Attivato ++da dinorfine.
FARMACOCINETICA
– eccetto codeina e ossicodone hanno elevato primo passaggio epatico quindi dosaggio orale difficile;
– per evitare passaggio epatico si usano vie sottocutanea, intramuscolare, iv, transdermica;
– escrezione renale.
FARMACODINAMICA
– molteplici siti di azione per presenza recettori μ:
– nocicettore: ↓ stimolo doloroso, soprattutto in stati infiammatori;
– pre-sinaptico: ↓ ingresso Ca2+ causa ↓ rilascio neurotrasmettitore;
– post-sinaptico: ↑ uscita K+ causa iperpolarizzazione;
– inibizione interneurone GABAergico causa aumento segnale inibitorio discendente.
– tolleranza:
– è diminuzione di un effetto della sostanza, a parità di dose;
– inizia biologicamente subito dopo la prima assunzione ma si manifesta clinicamente entro 3
settimane, favorita da dosi↑ e frequenti;
– rende necessarie dosi maggiori (curva dose-risposta si sposta a dx: ↓potenza, =efficacia);
– non riguarda tutti gli effetti:
– analgesia, sedazione. euforia, effetti piacevoli e ↓respirazione sono rapidamente
ridotti (60mg morfina causano blocco respiratorio in soggetto non-tollerante, in
tollerante necessari oltre 2000mg);
– stipsi, miosi, convulsioni rimangono costanti → anche in pz dipendenti!
– tipologie:
– acuta, mediata da β-arrestina che internalizza momentaneamente il recettore;
– cronica, mediata da endocitosi e distruzione del recettore, oppure da mancata
endocitosi che determina malfunzionamento del recettore stesso.
EFFETTI ACUTI
– Sensazione di piacere (rush), sedazione/ipnosi, nausea, prurito, bradicardia, dispnea;
EFFETTI CRONICI
– dipendenza fisica e psicologica, determinata da:
Æ
7 / 55
– rinforzo positivo (volontà di riprovare sensazione di piacere contrapposta a “normalità”);
– rinforzo negativo per evitare sindrome di astinenza:
– agitazione, tremori, nausea, iperventilazione, ansia, aggressività, ipertermia,
lacrimazione.
– inizia dopo 6h da ultima assunzione, picco a 48h;
– è precipitabile con antagonista: molto severa e di breve durata (naloxone, dura 1h);
– scompare in 5gg, 2 settimane con metadone (più lieve).
– patologie correlate a modalità di assunzione (es. infettive) o a sinergismo con alcol, benzodiazepine,
barbiturici;
– insufficienza renale, calcoli biliari...
FARMACI
– analgesici:
– morfina, idrossi/ossimorfone, fentanil: agonisti completi di recettore μ;
– codeina, idrocodone: agonisti parziali μ;
– tramadol: agonista debole μ, soprattutto inibitore SERT;
– antitussivi:
– codeina, destrometorfano;
– metadone:
– agonista completo μ, elevata potenza orale (↓ primo passaggio), lunga durata;
– utile per disintossicazione perché:
– uso orale;
– controllo esterno (recarsi dal sanitario),
– tolleranza crociata con eroina (ha meno senso cercarla).
– naloxone:
– antagonista μ, δ, e κ.
– usato per overdose oppioidi, precipita crisi astinenza in dipendenti.
Æ
8 / 55
FARMACI GASTROINTESTINALI
1. ACIDITÀ GASTRICA
2. PROCINETICI, ANTI-DIARROICI, ANTI-EMETICI
3. IBS e IBDs
1. ACIDITÀ GASTRICA
→ GERD, ulcere, Zollinger-Ellison.
GENERALITÀ
1) acidità gastrica dipende da [HCl], che deriva da H+ e Cl- provenienti da cellule parietali;
2) H+ deriva da ATPasi H+/K+ presenti su vescicole ed esposte in membrana secondo 2 vie:
1. ↑ Ca2+:
– ↑ da ACh (recettore M3) rilasciata da neurone parasimpatico intramurale;
– ↑ da gastrina (recettore CCK2) rilasciata da cellule G antrali su stimolo diretto
parasimpatico (ACh), meccanico (distensione) e ↑pH.
2. ↑cAMP:
– ↑ da istamina (recettore H2) rilasciata da cellule EC su stimolo ACh e gastrina;
– ↓ da PGE2 (recettore EP3);
3) Cl- deriva da scambiatore basale Cl-/HCO3- e passa nel lume da canale Cl-K+ (equimolare);
4) K+ luminale consente scambio con H+; insieme, Cl- e H+ formano HCl;
5) feedback negativo grazie a somatostatina prodotta da cellule D antrali per ↓pH e CCK duodenale,
che inibisce rilascio di gastrina da cellule G antrali.
MECCANISMI PROTETTIVI FISIOLOGICI
contro acidità gastrica
– epiteliali: tight junctions, muco e HCO3- (↑ da PGE2 – misoprostolo – e PGI2, su EP3);
– feedback negativo: somatostatina da cellule D (↓pH, CCK duodenale) inibisce cellule G.
POSSIBILI OBIETTIVI INTERVENTO FARMACOLOGICO
1) ↑ di protezione fisica da ↓pH gastrico: antiacidi, citoprotettori, procinetici;
2) ↓ della produzione di HCl tramite azione su:
– recettori: antagonisti di recettori di ACh, H, gastrina; agonisti di recettore PGE2/I2 ;
– ATPasi H+/K+: inibitori di pompa protonica.
1) PROTEZIONE FISICA DA ↓pH GASTRICO
ANTI-ACIDI:
– basi deboli da usare dopo il pasto che tamponano HCl post-prandiale (secreto≈45mEq/h);
– esempi:
– NaHCO3 produce CO2 (→distensione gastrica), NaCl (→ritenzione idrica) e permane una
quota non reattiva che può dare alcalosi;
– combinazioni di AlOH e MgOH sono migliori perché non generano gas, hanno alta efficienza
neutralizzante, e minimizzano i rispettivi effetti avversi (costipazione vs diarrea);
– possibile interazione diretta (legame) o indiretta (per variazione pH) con altri farmaci.
AGENTI CITOPROTETTORI:
– sucralfato è sale di sucrosio combinato con AlOH solfato;
– attivato da pH basso (stomaco vuoto);
– forma polimero vischioso carico negativamente che si lega a zone di mucosa erosa per 6 ore;
Æ
9 / 55
– dà costipazione (Al) e interferisce con altri farmaci.
PROCINETICI:
– domperidone è antagonista recettori D2 (dopamina ha azione opposta a ACh) quindi ACh prevale;
– effetti GI: ↑peristalsi esofagea, ↑pressione LES, ↑svuotamento gastrico. Non ha effetti su intestino;
– effetti SNC: blocco recettori D2 in CTZ ha potente effetto anti-nausea e anti-emetico.
2) RIDUZIONE PRODUZIONE DI HCl
1. RECETTORI
ANTAGONISTI RECETTORE H2
– cimetidina, oggi ranitidina: più comuni farmaci prescritti tra '70 e '90, ridotti dopo H.Pylori e PPI;
– antagonisti competitivi H su recettore H2 (non su H1 e H3) di parietali;
– riducono in parte anche effetto stimolatorio di gastrina e ACh su parietali;
– effetto è lineare dose-dipendente ma soprattutto su secrezione basale (notturna), meno su indotta;
– indicati solo in pirosi occasionale (<3/wk);
– effetti avversi (<3% pz): diarrea, cefalea, mialgie;
– effetti avversi endocrini (solo a lungo termine con alte dosi): ginecomastia e impotenza (M),
galattorrea (F) da blocco azione DHT, ↓ metabolismo estradiolo, ↑ prolattinemia.
ANTAGONISTI RECETTORI ACh e CCK2:
– pirenzepina blocca recettore M1 su neurone pregangliare, riduce secrezione basale 40%;
– antagonista CCK2 in studio.
AGONISTI RECETTORE EP3: AGENTI CITOTROFICI
– misoprostolo è analogo PGE2: usato per ↑ produzione muco e HCO3- e per ↓attività H+/K+;
– effetto soprattutto su secrezione basale, poco su indotta;
– causa diarrea e contrazioni uterine(!!!).
2. INIBITORI POMPA PROTONICA
– sono la categoria oggi più importante: ome/panto/lanso-prazolo;
– estremamente efficaci nel ridurre secrezione acida basale e indotta;
– modalità azione:
– sono pro-farmaci gastroprotetti (se via orale), assorbiti in intestino, raggiungono il circolo;
– concentrati e attivati da pH<1 dei canalicoli di cellule parietali;
– formano legami disolfuro covalenti con ATPasi H+/K+attive, inattivandole irreversibilmente.
– somministrazione:
– 1h prima del pasto per far coincidere picco sierico con massima attività ATPasi;
– efficacia dopo 3-4 giorni per blocco progressivo ATPasi;
– ripristino livelli acidità pre-farmaco dopo sospensione 3-4 giorni (resintesi pompe).
– effetti avversi (<5%):
– diarrea, cefalea, addominalgia;
– ↓ assorbimento B12, Fe2+, Ca2+;
– ↑ infezioni GI;
– ↓ metabolismo (CYP2C19/3A4) warfarin, diazepam, fenitoina, clopidogrel;
– ↑ secrezione gastrina: iperplasia cellule parietali/G: carcinoide, iperacidità di rimbalzo.
Æ
10 / 55
2. PROCINETICI
GENERALITÀ
– motilità GI è regolata estrinsecamente (ACh vs dopamina) e intrinsecamente (istamina);
– il controllo intrinseco si basa sul rilascio di H per distensione del lume, attivazione interneuroni e
rilascio di ACh e CGRP, che promuovono il riflesso di peristalsi.
FARMACI
1) COLINOMIMETICI
– neostigmina iv inibisce Ach-esterasi e migliora svuotamento gastro-intestinale;
– uso in pz ospedalizzati con pseudo-ostruzione acuta colon;
– effetti avversi: salivazione, nausea, vomito, diarrea, bradicardia.
2) DOPAMINA-ANTAGONISTI
– domperidone antagonizza recettore D2 facendo prevalere ruolo ACh;
– effetti GI: ↑peristalsi esofagea, ↑pressione LES, ↑svuotamento gastrico. Non effetti su intestino;
– effetti SNC: blocco D2 in CTZ (--| nausea, emesi).
3) LASSATIVI
– non servono a maggioranza. Importanti fibre, fluidi, esercizio fisico e rispondere a stimolo. Sono:
– colloidi idrofilici (es. psillium),
– ammorbidenti fecali (es. glicerina),
– osmotici (MgOH, sorbitolo, lattulosio, soluzioni bilanciate di polietilene-glicole),
– catartici (aloe, senna, bisacodil),
– attivatori dei canali del Cl- (lubiprostone),
– antagonisti del recettore (mu) degli oppioidi (metilnaltrexone, alvimopan),
– agonisti del recettore 5-HT4 (tegaserod): stimolo interneuroni a rilascio CGRP.
ANTI-DIARROICI
– agonisti oppioidi: loperamide (non attraversa EEB);
– resine leganti sali biliari: colestiramide;
– analoghi somatostatina: octeotride (ad alte dosi) inibisce GI, pancreas, colecisti;
ANTI-EMETICI:
per chemioterapia o eccesso stimolazione vagale (es. post-operatoria).
– antagonisti recettori 5-HT3;
– antagonisti recettori neurokinine;
3. IBS
– antagonisti recettori 5-HT3 (alosetron): ↓ sensibilità da neuroni afferenti, ↓ motilità.
– alcuni lassativi.
IBDs
– aminosalicilati (ASA) es. sulfasalazina;
– glucocorticoidi;
– analoghi purinici (azatioprina) e altri antimetaboliti (metotrexato);
– anti-TNF: infliximab, adalimumab, certolizumab.
Æ
11 / 55
FARMACI CARDIOVASCOLARI e RENALI
PREMESSE
– la relazione fondamentale è Pressione Arteriosa Media = GC * Resistenze Periferiche Totali;
– [Gittata Cardiaca = Gittata Sistolica * Frequenza Cardiaca];
– si può quindi agire in diversi modi per alterare i valori in gioco:
– GC: inotropi, β-bloccanti, bloccanti canali Ca2+,
– RPT: ACEi, diuretici, ARB, vasodilatatori diretti, inibitori renina.
IPERTENSIONE
PREMESSE
– pressione normale è <120/80; 120-135/80-89 è pre-ipertensione; ≥140/90 è ipertensione (HT);
– ogni ↑20mmHg da 115/75mmHg raddoppia rischi danni cardiovascolari, renali, cervello;
– l'eziologia è nel 90% dei casi difficile da stabilire con certezza; quasi sempre però sono presenti
elevate RPT e una normale GG;
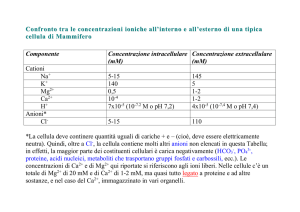
– studi epidemiologici imputano a: genetica, stress psicologico, ↑↑Na+, ↓↓K+, ↓↓Ca2+. Popolazioni
con ↓↓introito Na+ non sviluppano ipertensione con l'età;
GENERALITÀ
– PAM dipende da GC e RPT:
– GC dipende da GS e FC, regolate da pre-carico e stimolo orto/para-simpatico;
– RPT dipendono da arteriole, vasi di capacitanza, cuore e rene (regola volume);
– controllo della PAM è basato:
– a breve termine su barocettori,
– a lungo termine sul controllo del volume, che dipende da:
– osmo-cettori ipotalamici (↑300 mOsm/L) → ADH, senso della sete;
– distensione atriale → PNA (--| renina, aldosterone, ADH), ↑ortosimpatico;
– controllo renale:
– aumento PAM → ↑VFG → natri-diuresi pressoria;
– [Na+] (↓140 mEq/L) → sistema RAA: ↑RPT, ↓VFG, ↑ riass. H2O e Na+.
MANAGEMENT IPERTENSIONE (HT)
– valori >120/80 in almeno 3 diverse visite;
– escludere cause HT secondaria e valutare funzione cardiaca e renale;
– terapia:
– educare pz su patologia e consigliare intervento non farmacologico (↓Na+, ↓peso, ↓alcol);
– iniziare con minima dose ACEi/ARBs/CCBs/BBs/tiazidici, a seconda di comorbidità;
– terapia combinata riduce rischi derivanti da aumento dose singolo farmaco;
– se due farmaci, uno diuretico; se tre, aggiungere vasodilatatore diretto (idralazina, CCBs);
– end-point ottimale è 138/83mmHg, in diabetici anche meno è positivo;
MANAGEMENT CRISI IPERTENSIVA
– spesso accade in pz con HT severa per stop improvviso terapia;
– pz viene ricoverato in terapia intensiva e trattato con sodio nitroprussiato (o altri per via parenterale)
e furosemide per contrastare espansione di volume da vasodilatatori;
Æ
12 / 55
– riduzione graduale (ore) del 25% per evitare ipoperfusione cerebrale, mantenendo diastolica
>100mmHg.
CATEGORIE FARMACI
1. INIBITORI SISTEMA RAA
2. DIURETICI;
3. SIMPATOPLEGICI;
4. VASODILATATORI DIRETTI;
1. INIBITORI SISTEMA RAA
GENERALITÀ
– 20% pz HT essenziale hanno renina ↑↑, sono i pz che rispondono meglio a questi farmaci;
– renina:
– prodotta da JG per: stimolo orto (β1), ↓pressione afferente renale, ↓Na+ tubulo distale;
– taglia angiotensinogeno circolante rilasciando ANG-I;
– ACE endoteliale converte ANG-I in ANG-II → vasocostrittore, rilascio aldosterone;
– ACE cardiaco converte ANG-I in ANG-II → ipertrofia;
– surrene converte ANG-II in ANG-III → rilascio aldosterone;
FARMACI
– possono agire a livello di:
a) renina; b) ACE; c) recettore ANG-II; d) azione aldosterone;
a) inibitori della renina:
– aliskiren: blocca attività enzimatica;
– tossicità: iperK+, teratogeno;
b) ACE-inibitori:
– captopril et al: usati per HT, HF, diabete; ↓ effetti ANG-II, ↑bradichinina;
– tossicità: tosse, angioedema, iperK+, insuff. renale, teratogeni;
c) antagonisti recettore AT1:
– losartan et al.: uso in HT e HF. Non ↑bradichinina;
– tossicità: meno tosse;
d) inibitori aldosterone:
– diuretici risparmiatori di potassio: spironolactone, eplerenone;
2. DIURETICI
– aumentano escrezione di Na+ che causa indurimento e aumentata reattività contrattile vasale;
– inizialmente: ↓volume > ↓ GC > ↓ PAM; (nonostante un transitorio ↑RPT riflesso);
– dopo 8 settimane: ↓ RPT, GC torna nella norma, si mantiene ↓ PAM.
– scelta farmaco:
1. HT moderata senza patologie renali/cardiache > tiazidici;
2. HT severa o patologie renali/cardiache > diuretici dell'ansa (furosemide);
3. risparmiatori di K+;
– tossicità:
Æ
13 / 55
– più comune effetto avverso è deplezione K+, specialmente se non c'è restrizione di Na+.
FARMACI PRINCIPALI
Si distinguono a seconda della zona bersaglio del nefrone:
– tubulo prossimale: osmotici, inibitori anidrasi carbonica, antagonisti adenosina;
– ansa di Henle: osmotici, diuretici dell'ansa (furosemide, torsemide);
– tubulo distale: tiazidici (HCT);
– dotto collettore: antagonisti aldosterone e ADH.
3. SIMPATOPLEGICI:
– possono agire a livello centrale, ganglionare, recettoriale;
– elicitano sempre risposte compensatorie quindi importante usare combinazioni di farmaci;
FARMACI PRINCIPALI
ALFA1-BLOCCANTI
es. prazosina
– blocca recettore alfa1 su vasi di resistenza (arteriole) e di capacitanza (venule) → dilatazione;
– selettività consente feed-back negativo di noradrenalina su recettori alfa2;
– provocano ritenzione di Na+ e H2O, quindi vanno combinati con diuretico +/- β-bloccante.
β-BLOCCANTI
– β1 (↓GC, renina); β2 (↓bronco, ↓vaso); alfa1 (↑vasi).
– propranololo: non selettivo b1=b2; causa ↓GC e ↓produzione renina (β1 renali);
– atenololo: selettività 100:1 per β1 (utile in asmatici);
– pindololo: agonista parziale β2 (↓RPT) > b1 (minor riduzione GC utile in bradiaritmie);
– labetalolo: antagonista non selettivo β e alfa (↓RPT);
– nebivololo: selettivo β1 con proprietà vaso-dilatatoria per ↑NO endoteliale.
4. VASODILATATORI DIRETTI
– riducono RPT per dilatazione arteriole (nitrati/nitroprussiato anche venule);
– la riduzione di post-carico (e pre-carico) è utile in insufficienza cardiaca;
– provocano forti reazioni compensatorie quindi da combinare con diuretico e β-bloccante.
– categorie d'uso:
– terapia HT domiciliare a lungo termine: minoxidil, idralazina, bloccanti canali Ca2+;
– emergenze HT: nitroprussiato, bloccanti canali Ca2+;
– angina: nitrati.
FARMACI PRINCIPALI
– idralazina e minoxidil:
– farmaci orali, usati in pz con HT e insufficienza cardiaca (HF);
– sodio nitroprussiato:
– parenterale per emergenze HT e per HF severa;
– da usare al massimo per un'ora per tossicità da CN-;
– dilata arteriole e venule riducendo RPT (post-carico) e ritorno venoso (pre-carico);
Æ
14 / 55
– GC non cambia in assenza di HF, aumenta in HF per ridotto post-carico;
– bloccanti canali Ca2+:
– usati in aritmia, angina, HT;
– in HT (e in parte angina) funzionano inibendo influsso Ca2+ in muscolo liscio arteriolare;
– verapamil, diltiazem e DHP (amlodipina, nifedipina et al.) hanno stessa efficacia in ↓BP;
– nifedipina e altre DHP sono i CCB più selettivi come vasodilatatori;
– nitrati: vedi avanti tra farmaci per angina.
4.
Æ
15 / 55
FARMACI SISTEMA RENINA-ANGIOTENSINA-ALDOSTERONE
PREMESSA
– agiscono su SRAA, fondamentale per controllo a lungo termine sistema cardiovascolare;
– via fondamentale: renina (cellule JG renali) + angiotensinogeno = ANG-I + ACE1 = ANG-II;
– ANG-II ha effetti multiorgano, soprattutto mediati da recettore AT1:
1) stimolazione simpatica cuore (↑GC) e vasi (↓flusso renale), rimodellamento/ipertrofia;
2) stimola corticale del surrene a rilasciare aldosterone;
3) stimola neuroni ipotalamo a rilasciare ADH e ad ↑ senso della sete.
– complessivamente gli EFFETTI DELL'ATTIVAZIONE DEL SRAA sono:
1) risposta pressoria a breve termine: da ↑RPT per vasocostrizione;
2) risposta pressoria a lungo termine: da controllo di volume (aldosterone);
3) rimodellamento di cuore e vasi: ipertrofia, ↑contrattilità vasale;
COMPONENTI FONDAMENTALI DEL SRAA
1) ANGIOTENSINOGENO;
– proteina plasmatica prodotta costitutivamente da fegato, adipe, rene, SNC;
– ↑in epatatopatie, diabete, estrogeni, ormoni tiroidei, glucocorticoidi, ANG-II;
2) RENINA:
– è una proteasi, la cui produzione (in forma di pro-enzima, prodotto da JG):
– aumenta in 3 situazioni:
– ↓ [Na+] in tubulo distale > ↑segnale macula densa anche tramite PGI2;
– ↓ perfusione renale > attivazione barocettori;
– stimolo diretto ortosimpatico su recettori β1 di JG;
– è ridotta da feedback negativo quando PAM↑, per:
– ↑RPT (ANG-II) o
– ↑volume (aldosterone);
– è alterata per effetto di farmaci:
– ↑ da diuretici (↓ [Na+] in tubulo distale), vasodilatatori (↓ perfusione), ACEi e ARBs
(bloccano effetti ANG-II quindi feedback negativo);
– ↓ da FANS (↓PGI2), agonisti alfa-2 (↓noradrenalina), β-bloccanti (↓β1)
– è attivata per 2 vie:
– catalitica, da proteasi plasmatiche che aumentano in corso di flogosi;
– enzimatica, da recettore della pro-renina (PRR) che attiva anche vie di signalling
intracellulari indipendenti dall'ANG-II (→ fibrosi cardio-vascolare, nefropatia, retinopatia).
– è presente sempre in [] sufficienti a clivare tutto l'angiotensinogeno disponibile, quindi la sua azione
è direttamente proporzionale alla [] di angiotensinogeno presente.
3) ACE: enzima carbossipeptidasico convertitore dell'AT, presente sull'endotelio vasale in tutti gli
organi, in due forme:
– ACE1:
– agisce su ANG-I (→ ANG-II) e su bradichinina (inattivandola, ma sarebbe vasodilatoria!);
– ANG-II ha effetti multi-organo soprattutto tramite recettore AT1 (target di ARBs);
– è target di ACEi che quindi deviano ANG-I in ACE2;
– ACE2:
– agisce su ANG-I e su ANG-II →→ AT (1-7) che ha effetti opposti a ANG-II;
– non agisce su bradichinina (quindi rimane effetto vasodilatatore);
– AT (1-7) agisce tramite fattori Mas e ERK → effetti anti-proliferativo, vasodilatatorio;
– non è inibito da ACEi.
Æ
16 / 55
FARMACI
[
– per mantenere la PAM costante, il bilancio tra input e output di liquidi e Na+ deve essere nullo;
– questo avviene grazie a diuresi pressoria e ↕rilascio renina;
– se ↑introito di Na+ > ↑[Na+] > ↑ sete e ADH > ↑volume > ↑ PAM > diuresi pressoria e ↓renina;
– la curva di eliminazione renale (pressione vs eliminazione Na+) è spostata a destra dall'ANG-II;
– a lungo termine, l'ANG-II sposta la curva di eliminazione ad un set-point maggiore;
]
È possibile inibire il SRAA in 4 punti, agendo su:
1. RENINA ← aliskiren:
– blocca attività plasmatica di renina (PRA) quindi ↓[ANG-I], ↓[ANG-II];
– ↑[] totale di renina con possibili effetti surrenalici;
– farmaco recente ancora poco usato;
– tossicità: iperK+, teratogeno;
2. ACE1 ← ACE1-inibitori:
– selettivi per ACE1, enzima che entra anche in altre cascate proteolitiche (es. bradichinina);
– suddivisi in sulfidrilici (captopril), dicarbossilici (enalapril), fosforici (fosinopril);
– utilizzo:
1) HT essenziale:
– effetto a lungo termine da ↓RPT per ↓ANG-II (vasocostrittore) e ↑bradichinina
(vasodilatatore) portando a downset dei recettori;
– non evocano attivazioni ortosimpatiche riflesse;
– spostano a sx curva pressione/natriuresi, quindi pareggio di bilancio si ha a pressioni
inferiori;
– se si riduce l'intake di Na+ (dieta, diuretici) effetto è maggiore;
– captopril 10mg è sufficiente per HT lieve;
– effetti ipotensivi in 2-3 settimane, più rara risposta rapida (++ se renina↑);
2) insufficienza cardiaca (HF):
– efficienza migliora per ↓post-carico da ↓RPT (↑distensibilità arterie) e ↓ipertrofia cardiaca.
3) diabete e altri fattori di rischio cardiovascolari:
– ↑elasticità arterie riduce nefropatia e retinopatia;
– vantaggi su altri farmaci:
– su β-bloccanti: non hanno effetto simpatico-litico;
– su diuretici: non danno ↓K+, ↑uricemia, ↑glicemia, dislipidemia.
– tossicità:
– tosse, angioedema: forse per accumulo bradichinina;
– ipotensione con pre-sincope: da iper-responsivi, sempre approccio graduale;
– iperK+: rara, presente se associati diuretici risparmiatori di K+;
– aggravamento insuff. renale: NO IN PZ con Cl-Cr < 30ml/min o stenosi arteria renale;
– teratogeni.
3. RECETTORE DI ANG-II ← antagonisti recettore AT1 (ARBs)
– losartan et al.:
– legano irreversibilmente AT1 bloccando tutti gli effetti di ANG-II in maniera prolungata;
– vantaggio su ACEi:
– non ↑bradichinina (teoricamente danno meno tosse);
– evitano accumulo ANG-I quindi metaboliti alternativi;
– bloccando recettore AT1, annullano effetti di vie alternative di sintesi di ANG-II;
– non bloccano recettore AT2 quindi effetti (poco noti) sono mantenuti;
– utilizzo in HT e HF per stesse ragioni di ACEi;
Æ
17 / 55
– tossicità: teratogeni.
4. ALDOSTERONE ← inibitori azione aldosterone: sono diuretici risparmiatori di K+;
– agiscono su DOTTO COLLETTORE che subisce influenza di:
– aldosterone: ↑ATPasi Na/K basali, ↑canali apicali Na (ENaC) e K→riass. Na+, perdita K+;
– ADH: ↑acquaporina-2 → ↑riass. osmotico H2O;
– PNA: ↑GFR; --|secrezione renina, aldosterone, ADH; → perdita Na+ e H2O;
– farmaci:
1) amiloride: blocca canale Na+ (EnaC) così come antagonisti adenosina (in studio);
2) spironolattone ed eplerenone: (↑affinità e specificità per recettore aldosterone, ↑rapidità)
bloccano recettore aldosterone;
3) conivaptan (--| V1, V2) e tolvaptan (--|V2) antagonisti recettori ADH (V2 è renale);
4) peptidi natriuretici ricombinanti: ularitide (urodilatina ricombinante) ed altri, farmaci iv.
– uso:
– iperaldosteronismo primario o da HF e sindrome nefrosica (per ↓volume efficace), ipoK+.
Non come unico diuretico ma associazione con tiazidici per compensare effetti avversi
reciproci;
– tossicità: iperK+, a rischio con associazione farmaci che ↓renina (BBs, FANS, aliskiren) o ANG-II
(ACEi, ARBs).
Æ
18 / 55
DIURETICI
PREMESSA
– filtrato è 125 mL/min, urina 1 mL/min per riassorbimento di ioni;
– >70% Na+, NaHCO3, K+, riassorbiti al tubulo prossimale;
– <30% Na+/K+/Cl-, riassorbiti attivamente in ansa ascendente, tubulo distale e collettore corticale;
– H2O segue Na+ eccetto in ansa ascendente e tubulo distale (impermeabili, diluizione pre-urina);
GENERALITÀ
– aumentano escrezione di Na+ che causa indurimento e aumentata reattività contrattile vasale;
– inizialmente: ↓volume > ↓ GC > ↓ PAM; (nonostante un transitorio ↑RPT riflesso);
– dopo 8 settimane: ↓ RPT, GC torna nella norma, si mantiene ↓ PAM.
– scelta farmaco:
– HT moderata senza patologie renali/cardiache > tiazidici;
– HT severa o patologie renali/cardiache, edemi > diuretici dell'ansa (furo/tor-semide);
– risparmiatori di K+ > amiloride, spironolattone/eplerenone;
– tossicità:
– più comune effetto avverso è deplezione K+, specialmente se non c'è restrizione di Na+;
FARMACI PRINCIPALI
Si distinguono a seconda della zona bersaglio del nefrone:
1. TUBULO PROSSIMALE:
– fisiologia:
– riassorbe 65% Na+/K+/Ca2+/Mg2+; 85% HCO3-; 100% glucosio e AA;
– alta permeabilità ad H2O;
– farmaci:
1) antagonisti adenosina (ancora in studio): rilasciata in ipossia e ↓ATP, adenosina stimola
NHE3 (contro- trasportatore Na/H). Antagonisti quindi ridurrebbero riassorbimento Na+;
2) inibitori anidrasi carbonica (acetazolamide): bloccano riassorbimento di HCO3- perché è
impedita la dissociazione tubulare di H2CO3 in CO2 e H2O.
3) osmotici (mannitolo, isosorbide): farmaci igroscopici non riassorbibili, usati per via ev.
Aumentano osmolarità e quindi volume plasmatico, e sono usati per:
– ↑diuresi pressoria con estrazione di Na+, urea e washout della midollare;
– in emergenza: ↓ edemi (es. cerebrale, glaucoma) e come plasma expanders.
2. ANSA DI HENLE:
– discendente:
– riassorbimento passivo H2O: osmotici impediscono riassorbimento H2O;
– ascendente:
– riassorbimento attivo 25% Na+/K+/2Cl- (NKCC2) e passivo Ca2+ e Mg2+
– impermeabile all'H2O (diluizione pre-urina);
– farmaci: diuretici ad alto tetto (furo/tor-semide) bloccano NKCC2:
– ↑ escrezione Na+, Cl-, K+, ma anche Ca2+ e Mg2+ (per ↓potenziale positivo con lume);
– ↓ ipertonicità midollare rende impossibile concentrare urina (riassorbire H2O);
– uso: anti-edemigeni (edema polmonare acuto, HF cronica, cirrosi), HT severa, ingestione anioni
tossici, iperCa2+, iperK+; torsemide ha effetto più rapido (1h) e duraturo (5h);
– tossicità: ototossicità (+aminoglicosidi), ipovolemia, ipoK+(+digitale), ipoMg2+, iperuricemia,
dislipidemia; effetti ↑da litio e ↓ da probenecid (riduce secrezione in lume).
Æ
19 / 55
3. TUBULO DISTALE:
– fisiologia:
– riassorbimento attivo <8% di Na+/Cl- filtrati tramite cotrasportatore;
– riassorbimento Ca2+ su controllo PTH;
– farmaci: tiazidici (HCT) bloccano cotrasportatore Na/Cl:
– uso: effetto non è molto potente ma importante in:
– ipertensione moderata: ↓riassorbimento Na+ ma anche ↓RPT (senza tolleranza!);
– HF congestizia cronica moderata (altrimenti diuretici del'ansa);
– nefrolitiasi da Ca2+ perché riduce calciuria;
– tossicità: ↓K+, ↑uricemia (tiazidici sono secreti nel TCP dallo stesso trasportatore), dislipidemia.
4. DOTTO COLLETTORE (diuretici risparmiatori di K+):
– influenza di:
– aldosterone: ↑ATPasi Na/K+ basali e ↑canali apicali Na+ e K+ → riass.Na+, perdita K+;
– ADH: ↑acquaporina-2 → ↑riass. osmotico H2O;
– PNA: ↑GFR; --|secrezione renina, aldosterone, ADH; → perdita Na+ e H2O;
– farmaci:
1) amiloride blocca canale Na+ (EnaC) così come antagonisti adenosina (in studio);
2) spironolattone ed eplerenone (↑affinità e specificità per recettore aldosterone, ↑rapidità)
bloccano recettore aldosterone;
3) conivaptan (--| V1, V2) e tolvaptan (--|V2) antagonisti recettori ADH (V2 è renale);
4) peptidi natriuretici ricombinanti: ularitide (urodilatina ricombinante) ed altri, farmaci ev.
– uso:
– iperaldosteronismo primario o da HF e sindrome nefrosica (↓volume efficace), ipoK+. Non
come unico diuretico ma associazione con tiazidici per compensare effetti avversi reciproci;
– tossicità: iperK+, a rischio con associazione farmaci che ↓renina (BBs, FANS, aliskiren) o
ANG-II (ACEi, ARBs).
Quindi:
1. antagonisti adenosina --| NHE3;
2. inibitori anidrasi carbonica --| HCO3-;
3. osmotici → H2O;
4. diuretici dell'ansa --| NKCC2;
5. tiazidici --| NaCl;
6. amiloride --| ENaC;
7. spironolattone, eplerenone --| aldosterone;
8. ularitide = PNA;
9. conivaptan, tolvaptan --| ADH
Æ
20 / 55
FARMACI SIMPATOPLEGICI
– sono antagonisti dei recettori adrenergici;
– possono agire a livello centrale, ganglionare, periferico;
– elicitano quasi sempre risposte compensatorie → usare combinazioni di farmaci di diverse classi;
– fisiologia adrenergica di base:
– recettori:
– alfa1: vasocostrizione;
– alfa2: recettore inibitorio;
– β1: ↑GC,↑rilascio renina;
– β2: broncodilatazione e vasodilatazione periferica;
– β3: attivazione lipasi;
– effettori:
– adrenalina agisce su tutti: ↑FC, ↓RPT, PAM costante (↓diastolica,↑sistolica);
– noradrenalina non agisce su β2: ↑RPT, ↑PAM, ↓FC riflessa;
– isoprotenerolo è β-selettivo: ↓↓RPT, ↓PAM, ↑↑FC riflessa.
ALFA-BLOCCANTI
1. NON selettivi: fentolamina (reversibile) e fenossibenzamina (irreversibile):
– usati in feocromocitoma ed eccesso catecolamine, ↓BP ma causano ↑FC riflessa e
ipotensione ortostatica;
2. selettivi ALFA1 (recettore che causa costrizione muscolo liscio): prazosina:
– vasodilata vasi di resistenza (arteriole) e di capacitanza (venule);
– non dà ↑FC riflessa, perché mantiene feed-back negativo noradrenalina su alfa2;
– usato in HT e ipertrofia prostatica benigna (tamsulosina è più selettivo) perché rilassa
muscolo liscio prostata facilitando deflusso urina.
– provocano ritenzione di Na+ e H2O (per ↓volume efficace), quindi vanno combinati con
diuretico +/- β-bloccante per azione anti-HT.
3. selettivi ALFA2 (recettore inibitorio presinaptico): yohimbina:
– ↑BP e FC;
– usato in disfunzione erettile e ipotensione.
β-BLOCCANTI
GENERALITÀ
– utilizzati (vedi dopo) quando sono necessari effetti cardiaci ino+crono+lusi+dromo/tropo negativi:
– HT, angina, aritmie, HF, ipertiroidismo, crisi d'astinenza, attacchi di panico...
– blocco β1 importante anche per ↓ rilascio renina (blocco SRAA);
– ricordare di minimizzare risposte compensatorie tramite:
– bloccanti selettivi β1;
– terapia polifarmacologica a basse dosi e graduale;
PROPRIETÀ DESCRITTIVE
1. Affinità relativa β1 >> β2 (atenololo): importante perché con non-selettivi (propranololo) blocco β2
impedisce broncodilatazione;
2. attività agonista parziale β2>β1 (pindololo): invece di bloccare recettore lo fa lavorare a bassi
livelli, utile per vasodilatare (β2) e mantenere GC (β1) in bradiaritmie.
3. blocco β e alfa1 (labetalolo): riduce risposta riflessa di vasocostrizione;
4. attività di agonista inverso: induce effetti superiori al blocco stesso;
5. stabilizzazione di membrana (propranololo, carvedilolo): effetto antiaritmico per interferenza con
Æ
21 / 55
canali Na+ e ↓eccitabilità;
6. proprietà vaso-dilatatoria: per agonismo β2 (pindololo), ↑NO endoteliale (nebivololo), attività antiossidante (carvedilolo);
7. farmacocinetica (molto confusa): emivita 3-10 ore ma effetti possono perdurare; genotipo di CYP
influenza molto il metabolismo; farmaci liposolubili (propranololo) hanno elevato effetto di primo
passaggio e passano la barriera EE (rischio effetti avversi); gli idrosolubili (atenololo e maggior parte)
vanno assunti a stomaco vuoto perché trasportatori AA sono liberi.
CLASSIFICAZIONE FARMACI
1a generazione (non selettivi):
– propranololo: non selettivo b1=b2; causa ↓GC e ↓produzione renina (β1 renali);
– pindololo: agonista parziale β2 (↓RPT) > b1 (minor riduzione GC utile in bradiaritmie);
2a generazione (selettivi)
– atenololo: selettività 100:1 per β1 (utile in asmatici e BPCO);
– esmololo: emivita brevissima;
3a generazione (azioni aggiuntive):
– carvedilolo: non selettivo, stabilizza membrana ed è antiossidante;
– labetalolo: non selettivo, blocca anche alfa1 riducendo risposta riflessa (↓RPT);
– nebivololo: selettivo β1 con proprietà vaso-dilatatoria per ↑NO endoteliale.
UTILIZZI
1) IMA STEMI/NSTEMI, prevenzione post-infarto, cuore ischemico stabile (angina), insufficienza
cardiaca → riduzione lavoro e vasodilatazione, ↓apoptosi cardiomiociti, ↓formazione placche;
2) HT: per ↓GC, FC, renina. Ricorda però in acuto stimolo alfa1;
3) ARITMIE (flutter, fibrillazione atriale): per effetto stabilizzazione membrana, dromotropo e
lusitropo negativi → evita stasi di sangue in atrio (rischio trombosi) e possibile tachicardia
ventricolare;
4) Glaucoma: ↓ pressione intraoculare per ridotta produzione umore acqueo;
EFFETTI AVVERSI
– cardiaci: eccesso bradicardia (ridotta tolleranza allo sforzo), blocco AV;
– respiratori: mancata broncodilatazione (da non selettivi);
– metabolici: blocco rilascio glucagone e inibizione sintomi ipoglicemia, quindi rischio crisi
ipoglicemica in diabetico;
– SNC (farmaci liposolubili): affaticamento, depressione (femminile), impotenza (maschile).
Æ
22 / 55
VASODILATATORI
PREMESSE
– riducono RPT per dilatazione arteriole (nitroderivati anche venule);
– la riduzione di post-carico (e pre-carico) è utile anche in insufficienza cardiaca;
– provocano forti reazioni compensatorie quindi sono da combinare con diuretici e β-bloccanti;
– utilizzo:
– angina: NITRATI; BLOCCANTI CANALI Ca2+ (CCBs);
– HT: NITROPRUSSIATO (emergenze), minoxidil, idralazina, CCBs;
– tachiaritmie sopraventricolari: CCBs.
ANGINA PECTORIS
– dolore intenso acuto, sintomo di insufficiente irrorazione del miocardio quindi ischemia;
– sito retro-sternale con irradiazione verso braccio, addome o colonna a seconda di zona interessata;
– all'ECG si evidenzia rovesciamento onda T e slivellamento tratto ST;
– può evolvere verso l'infarto;
– strategie di intervento:
– aumentare la perfusione cardiaca tramite:
– vasodilatazione coronarica chirurgica (angioplastica) o farmacologica;
– ↑ tempo di diastole (↓FC):
– ridurre il consumo di ossigeno cardiaco, tramite ↓del lavoro cardiaco grazie a:
– ↓ post-carico tramite ↓RPT (vaso-dilatazione a valle);
– ↓FC (↓eccitabilità) e contrattilità (↓pre-carico, ↓influsso Ca2+).
OSSIDO NITRICO (NO)
– prodotto da NOS:
– I e III: costitutive, Ca2+-dipendenti;
– II: inducibile, Ca2+-indipendente (quando indotta è attiva).
– ha grande importanza perché:
– ↑[cGMP] causa rilassamento del muscolo liscio (defosforila MLCK) → vasodilatazione!
– nitrosila canali K+ rallentando ripolarizzazione → ↓FC!
– nitrosila canali Ca2+ volt-dipendenti riducendo ingresso Ca2+ → ↓contrattilità!
– potenziamento sistema dell'NO può avvenire con:
1. ↑ produzione NO: precursori di sintesi (arginina) o potenziatori di rilascio;
2. apporto diretto di NO: NITRODERIVATI:
– sodio-nitroprussiato: in H2O è instabile e libera direttamente NO;
– nitrati organici: degradati da enzimi su endotelio che ne determinano il sito
particolare di azione. Nitroglicerina è prototipo di coronaro-dilatatori.
3. ↓ degradazione NO: inibitori fosfodiesterasi (viagra), attivatori guanilato-ciclasi;
NITRODERIVATI
– azione: apporto diretto di NO con dilatazione venule di capacitanza e arteriole > ↓RPT;
– effetto: ↓precarico (++venule) e ↓post-carico (++arteriole);
– usati con β-bloccanti o Ca2+-antagonisti per ridurre risposte riflesse es. barorecettoriali;
– farmaci:
1) sodio-nitroprussiato:
– in H2O è instabile e libera direttamente NO;
– somministrazione iv in emergenze ipertensive, in grave insufficienza cardiaca acuta e in
Æ
23 / 55
anestesia per indurre ipotensione;
– elevata tossicità per accumulo tiocianato (non utilizzare oltre 2 giorni).
2) nitrati organici:
– degradati da enzimi su endoteliociti che ne determinano il sito specifico d'azione;
– somministrazione sublinguale/transcutanea per evitare primo passaggio epatico;
– durata d'azione:
– breve: nitroglicerina sublinguale (0,5h);
– lunga: ng a lento rilascio (6-8h), isosorbide mononitrato orale (10h);
– effetti:
– inizialmente detti coronaro-dilatatori, in realtà dilatazione probabilmente è riflessa e
dovuta a riduzione pressione parietale;
– ↓precarico riduce GC (lavoro) e pressione diastolica (↑perfusione);
– ↓post-carico riduce lavoro;
– a lungo termine effetti svaniscono (tolleranza);
– tossicità:
– tachicardia riflessa;
– ipotensione posturale per blocco venocostrizione;
– non efficaci in infarto per “furto”: zona infartuata è già massimamente vasodilatata da
mediatori ischemici, ma non ha pressione di perfusione quindi vasodilatatori hanno
effetto solo su collaterali paradossalmente riducendo la perfusione dell'infarto.
Ca2+-ANTAGONISTI O BLOCCANTI CANALI Ca2+ (CCBs)
[vedi dopo tra farmaci per insufficienza cardiaca]
Æ
24 / 55
INSUFFICIENZA CARDIACA
PREMESSA
– nello scompenso, in risposta ad un calo di GC, si attivano il sistema ortosimpatico e il SRAA, che
tentano di ripristinare la GC agendo su diverse variabili cardiovascolari, fondamentalmente:
– ↑ FC, pre-carico (volume), contrattilità;
– questo funziona a breve termine (compenso);
– a lungo termine la situazione evolve in scompenso cardiaco (↓↓ frazione d'eiezione), per:
– ipertrofia cardiaca, ↑RPT (post-carico).
FARMACI
– l'obbiettivo è aumentare efficacia (frazione d'eiezione) ed efficienza cardiaca;
– questo può avvenire con farmaci che agiscono su diversi aspetti del sistema cardiocircolatorio;
– i farmaci utilizzati possono dividersi (suddivisione del tutto personale, NdR) in:
1. INOTROPI NEGATIVI (↓forza di contrazione):
– ↓ pre-carico: vasodilatazione venosa ← vasodilatatori;
– ↓ post-carico: ↓RPT ← ACEi, ARBs, diuretici, vasodilatatori;
– ↓ contrattilità e ↓FC: ↓ influsso di Ca2+ e ↓stimolazione adrenergica ← CCBs, βBs;
2. INOTROPI POSITIVI (↑ forza di contrazione):
– ↓ FC e nel contempo ↑ perfusione e forza (blocco pompa Na/K) ← digitalici;
– ↑ Ca2+-trigger e SERCA (blocco fosfolambano) ← β-agonisti;
– ↑ [Ca2+]i rimanente per blocco NCX (esporta Ca con gradiente Na) ← derivati amiloride;
– attivatori di miosina a parità di [Ca2+]i ← levosimendan, omecantiv-mecarbil;
– la forza di contrazione cardiaca dipende direttamente dalla [Ca2+]i nella fase di attivazione (che poi
torna a valori bassi e consente la diastole). La [Ca2+] è alterabile in 5 modi;
1) quantità di Ca2+ (trigger) che entra dall'esterno durante plateaux:
– ↑ β1-agonisti, ↓ CCBs;
2) quantità di Ca2+ (activator) che esce da reticolo tramite RYR2:
– ↑ da Ca2+-trigger;
3) recupero Ca2+ in reticolo (SERCA2):
– ↑ β1-agonisti per blocco fosfolambano (inibitore SERCA);
– ↑ farmaci che ↑[Ca2+]i a fine contrazione (blocco NCX e pompa Na/K);
4) esporto Ca2+ da controtrasporatore NCX secondo gradiente Na+:
– ↓ da derivati amiloride per blocco NCX da;
5) esporto Na+ da pompa Na/K per mantenere gradiente Na+:
– digitalici bloccano pompa neutralizzando il gradiente necessario a NCX.
1. INOTROPI NEGATIVI
Già visti in precedenza:
– vasodilatatori;
– ACEi, ARBs, diuretici;
– βBs;
Da vedere:
– Ca2+-ANTAGONISTI o BLOCCANTI CANALI Ca2+ (CCBs)
Æ
25 / 55
Ca2+-ANTAGONISTI o BLOCCANTI CANALI Ca2+ (CCBs)
PREMESSE
– canali del Ca2+:
– tipi importanti sono L (miocardio, muscolo liscio, cervello, vasi) e T (Purkinje);
– CCBs ↓forza e frequenza cardiaca (fase plateaux) e vasodilatano coronarie;
– canali del Na+:
– alterano la depolarizzazione quindi l'inizio del PdA;
– canali del K+:
– alterano la ripolarizzazione quindi la fine del PdA;
USO E MECCANISMO
– usati ++ per angina, ipertensione, tachiaritmie sopraventricolari, insufficienza cardiaca;
– interferiscono con i canali volt-dipendenti del Ca2+ agendo dall'interno della cellula;
– riducono frequenza di apertura in risposta a depolarizzazione quindi ↓ingresso Ca2+;
– hanno specificità tissutale:
– di-idro-piridine: subunità 1alfaCb e canali in stato inattivo → muscolo liscio;
– verapamil, diltiazem: canali molto attivi → canali Ca2+ di miocardio;
EFFETTI
– in muscolo liscio (++ DHP) → vasodilatazione arteriosa (coronarica) per ↑ rilassamento;
– in miocardio (++ verapamil, diltiazem) → riduzione forza e frequenza per:
– ↓ “Ca2+ trigger” da esterno in miocardio di lavoro durante fase di plateaux;
– ↓ frequenza di scarica del nodo SA e ↓ velocità conduzione del nodo AV;
FARMACI
– suddivisione in generazioni successive:
– 1a generazione: breve emivita (verapamil, diltiazem, nicardipina);
– 2a generazione: lento rilascio (nifedipina, nicardipina);
– 3a generazione:
– azione prolungata per lunga emivita o persistenza tissutale (amlodipina);
– effetti aggiuntivi anti-ossidanti e anti-proliferativi;
– effetto protettivo vasodilatatorio su arteriole glomerulari (benidipina, efonidipina);
– farmaci principali:
– DHP:
– amlodipina, benidipina, efonidipina et al;
– effetto prevalente: vaso-dilatatori, da associare a β-bloccanti;
– uso: in angina, HT, HF;
– verapamil, diltiazem: non più usati.
– effetto prevalente: ↓forza e frequenza cardiaca; NON associare a β-bloccanti;
– uso: aritmia, angina, HT, HF.
2. INOTROPI POSITIVI
– digitalici: ↓ FC e nel contempo ↑ perfusione e forza (blocco pompa Na/K);
– simpatico-mimetici: ↑ Ca2+-trigger e SERCA (blocco fosfolambano);
– derivati amiloride: ↑ [Ca2+]i a fine contrazione per blocco NCX (esporta Ca con gradiente Na);
– levosimendan, omecantiv-mecarbil: attivatori di miosina a parità di [Ca2+]i;
Æ
26 / 55
DIGITALICI
– sono glicosidi cardioattivi in origine derivati da piante del genere Digitalis (digossina);
– la struttura è un nucleo steroideo + lattone + glucide;
– il farmaco agisce su ATPasi Na+/K+ con particolari isoforme di subunità alfa:
– alfa2: forma predominante nel miocardio (il resto è alfa1);
– alfa3: forma neuronale (SNC, plesso GI);
– hanno 2 effetti fondamentali:
1) effetto inotropo positivo:
– elevano [Ca2+] a fine contrazione consentendo un aumentato accumulo nel reticolo da SERCA e
quindi un maggior rilascio in fase di attivazione;
– meccanismo: blocco ATPasi Na+/K+ cardiaca, che di norma esporta Na+ (3Na+/2K+):
– la pompa di norma mantiene un gradiente Na+ (↑esterno) che viene così ridotto;
– riduzione gradiente inibisce attività di esporto Ca2+ da parte del controtrasportatore NCX;
– influenza della [K+] extracellulare:
– farmaco si lega alla pompa in sito extracellulare prossimo al sito del K+, quindi:
– con iper[K+] farmaco si lega meno;
– con ipo[K+] farmaco si lega di più ed ha effetto maggiore a parità di dosaggio. Questa è la
situazione clinica più frequente (sudorazione, vomito, diuretici).
2) effetto cronotropo e dromotropo negativo:
– aumentano l'influsso parasimpatico sul cuore con ↓FC e ↑perfusione;
– meccanismo: blocco ATPasi neuronale:
– in SNC aumenta impulsi vagali;
– ↑ sensibilità dei barocettori e periferica a stimolo parasimpatico.
– effetti tossici dei digitalici
1) cardiaci (sono farmaci aritmogenici):
– ↓ durata PdA: l'aumento di [Ca2+]i determina l'apertura di canali del K+ Ca2+-dipendenti che
facilitano la ripolarizzazione;
– ↓ potenziale di riposo:
– la riduzione di [K+]i causa riduzione del p.d.R., che diventa più vicino alla soglia;
– all'aumentare della dose, a seguito di normali depolarizzazioni compaiono
depolarizzazioni premature, dovute a potenziali oscillatori (DADs) “delayed afterdepolarizations” che riescono a raggiungere soglia;
– se accadono nel fascio di Purkinje si avrà un battito ventricolare prematuro (ritmo
bigemino);
– se i DAD soprasoglia si auto-generano si ha tachicardia→ fibrillazione.
2) extracardiaci (neuronali, centrali e da eccesso parasimpatico):
– malessere, nausea, vomito, anoressia, debolezza;
– alterazione visiva con mancata distinzione contrasti tra oggetti (sfumati);
– delirio digitalico.
INIBITORI FOSFODIESTERASI
– agiscono su PDE cardiovascolari e polmonari;
– PDE3:
– milrinone usato in emergenza per scompenso acuto con ipertensione;
– detto ino-dilatatore: vasodilata per ↑cAMP, inotropo per ↑Ca2+;
Æ
27 / 55
– PDE5:
– sildenafil usato in disfunzione erettile, ipertensione polmonare;
– vasodilata per ↑cGMP.
SIMPATICOMIMETICI
– noradrenalina:
– usata in shock da vasodilatazione per capacità vasocostrittiva
– aumenta RPT ma non FC;
– non usare in shock da perdita di liquidi (c'è già vasocostrizione);
– dobutamina:
– beta1 agonista selettivo usata in scompenso acuto con alta pressione di post-carico;
– forte effetto inotropo con mantenimento/riduzione RPT;
– dopamina:
– usata a dosaggi crescenti per vasodilatazione splancnica/renale, aumento contrattilità
cardiaca, vasocostrizione periferica.
ATTIVATORI MIOSINA CARDIACA
– Levosimendan:
– ↑sensibilità troponina a Ca2+ → ↑forza, = diastole;
– dilata coronarie e altri vasi,
– ↓velocità conduzione nodo AV;
– ↓apoptosi miocardiociti;
– Omecantiv-mecarbil:
– accelera rilascio Pi da miosina rendendo più rapido il powerstroke;
– ↑forza ma ↓diastole.
ALTRI
– istaroxamina: inibisce Na+/K+ cardiaca e stimola SERCA2;
– stabilizzatori RYR2 per evitare perdita di Ca2+ da reticolo in fase di rilassamento.
Æ
28 / 55
ANTIARITMICI
PREMESSE
– aritmie sono alterazioni di origine, frequenza, regolarità o conduzione dello stimolo elettrico;
– stimolo elettrico:
– origina in nodo SA;
– è trasmesso attraverso dischi intercalari (siti intercellulari con molteplici gap-junctions):
– invade rapidamente gli atri;
– rallenta in nodo AV (0,15s) unica conduzione tra atri e ventricoli, permettendo flusso;
– si propaga dalla punta alla base dei ventricoli in meno di 0,1s;
– le fasi del PdA cardiaco:
0: depolarizzazione da soglia fino a picco massimo per corrente (Na+ IN);
– soglia aumentata da blocco canali Na+;
1: ripolarizzazione parziale da corrente K+ (OUT);
– poco influente;
2: plateaux da contrasto tra Ca2+ (IN) e K+ (OUT);
– blocco canali K+ causa ↑ durata di PdA;
3: ripolarizzazione fino a PdR da K+ (chiusura canali Ca2+ e Na+);
– pdR raggiunto è fattore chiave perché canali Na+ si chiudono e tornano disponibili
progressivamente tra -55 e -75 mV, quindi:
– a pdR più negativo ci sono più canali disponibili all'apertura;
→picco PdA più alto, ↑eccitabilità (↓periodo refrattario), conduzione rapida;
– a PdR meno negativo (ischemia, iperK+, blocco di pompa Na+/K+):
→minor picco, ridotta eccitabilità, conduzione lenta.
4: depolarizzazione fino a soglia da corrente funny (Na+ IN);
– corrente funny ↓ da parasimpatico, beta-bloccanti, iperK+;
– ECG (somma PdA di tutte cellule miocardiche);
– onda P (depolarizzazione atri);
– complesso QRS (depolarizzazione ventricoli);
– onda T (ripolarizzazione ventricoli);
CAUSE DI ARITMIE
– ischemia, ipossia, eccessivo stiramento atriale, tessuto cicatriziale, disturbi elettrolitici, stimolazione
adrenergica, tossicità farmacologica;
– in generale, tutti i fattori che causano disturbi di:
1) formazione impulso e/o
2) conduzione impulso;
1) DISTURBI DI FORMAZIONE IMPULSO:
– cellula pacemaker scarica a ritmo regolare, influenzato soprattutto dall'inclinazione della fase 4:
– ↓ da parasimpatico, Beta-bloccanti, iperK+;
– ↑ da stimolazione beta, farmaci cronotropi positivi, ipoK+, acidosi, stiramento;
– tutte le cellule cardiache possono funzionare da pacemaker se depolarizzate in determinate
condizioni (es. ipoK+);
– in alcune condizioni possono avvenire delle depolarizzazioni transitorie che interrompono il PdA:
– in fase 3: dette “early afterdepolarizations” (EADs): più frequenti a basse FC;
– in fase 4: dette “delayed afterdepolarizations” (DADs): più frequenti ad alte FC;
2) DISTURBI DI CONDUZIONE IMPULSO:
– blocchi semplici:
Æ
29 / 55
– interruzione “semplice” della conduzione (es. blocco AV, blocco di branca);
– blocchi parziali a volte risolti da atropina (Ach-antagonista?) se dati da ↑↑parasimpatico.
– rientro:
– situazione patologica in cui un impulso eccita più di una volta la stessa zona di tessuto;
– può avvenire se sussistono 3 situazioni:
– ostacolo a conduzione omogenea dell'impulso;
– conduzione decrementale causa blocco unidirezionale (se bidirezionale non c'è
rientro!);
– il tempo di conduzione del circuito è maggiore del periodo refrattario;
– visivamente: l'impulso biforca; da un lato è bloccato ma non dall'altro; l'impulso
sopravvissuto rientra (da dietro) verso la zona danneggiata, che non lo blocca (perché blocco è
unidirezionale) e quindi eccita nuovamente il tessuto a monte (se non è più refrattario).
– esempi notevoli di aritmie (probabilmente) da rientro:
– fibrillazione atriale: numerosi foci ectopici scaricano in maniera asincrona dando una
contrazione disorganizzata (“a sacco di vermi”) dell'atrio. Il pericolo è la stasi di sangue quindi
formazione di coagulo. Pz vanno in TAO;
– flutter atriale: atrio si contrae con frequenza >200bpm, con il rischio di instaurare una
tachicardia ventricolare. È necessario rallentare la conduzione AV.
FARMACI
GENERALITÀ
– hanno obbiettivi opposti ai fattori aritmici:
1) ridurre attività pacemaker ectopici;
2) bloccare il rientro modificando conduzione o refrattarietà.
– meccanismo funzionamento antiaritmici:
– agiscono preferenzialmente sulle cellule che scaricano più rapidamente, legandosi a canali in
stato attivo o refrattario (non a riposo); tuttavia: all'aumentare delle dosi, in tachicardia,
acidosi, iperK+, ischemia, anche le cellule normali vengono coinvolte;
– riducono la pendenza della fase 4 bloccando i canali di Na+ o Ca2+;
– alzano la soglia di attivazione (→meno negativa) quindi più difficile da raggiungere;
– prolungano la durata del PdA aumentando il periodo refrattario;
– classi di effetti (un farmaco può avere più di una classe di effetti, e sono più efficaci quelli che
agiscono su molteplici canali rispetto a un solo tipo):
1) --| canali Na+ → ↓eccitabilità e conduzione in miocardio comune;
2) --| recettori beta1 → ↓pendenza fase 4 (corrente funny);
3) --| canali K+ → ↑durata PdA quindi periodo refrattario;
4) --| canali Ca2+ → ↓conduzione in nodi SA e AV dove upstroke è Ca2+-dipendente;
FARMACI SPECIFICI
CLASSE 1: --| canali Na+ → ↓eccitabilità e conduzione in miocardio comune;
– 1A (anche effetto classe 3, recupero intermedio);
– procainamide:
– 2a o 3a scelta dopo lidocaina/amiodarone in aritmie ventricolari post-IMA;
--| Na+ → ↓eccitabilità e conduzione in miocardio comune più che in miocardio di
conduzione; effetto di blocco vagale che contrasta in parte rallentamento frequenza;
--| K+ → ↑durata PdA quindi periodo refrattario;
--| gangliare → ipotensione se somministrazione Iv;
Æ
30 / 55
– tossicità: aritmogeno (eccesso rallentamento conduzione), aumento tossicità digitale,
diarrea, sintomi simil-LES reversibili.
– chinidina: usata raramente per eccesso tossicità cardiaca/extracardiaca;
– 1B (rapido recupero):
– lidocaina:
– farmaco iv molto usato per bassa tossicità ed alta efficacia: 1a scelta in
tachicardia/fibrillazione ventricolare dopo cardioversione in IMA (non in profilassi);
– ↓ fase 4 e ↑ soglia riducendo eccitabilità;
– effetto maggiore su cellule Purkinje e ventricolari;
– tossicità neurologica centrale: nausea, cefalea, parestesie.
– mexiletina: come lidocaina ma orale.
– 1C (lento recupero):
– propafenone e flecainide:
– aritmie sopraventricolari (FA);
– propafenone ha debole attività beta-bloccante.
CLASSE 2 (beta-bloccanti, vedi capitolo dedicato):
– riducono corrente funny, [Ca2+], correnti K+;
– usati per effetto combinato su aritmie e RPT;
– effetto minore di anti-aritmici di classe 1 in depolarizzazioni ventricolari ectopiche;
– esempi di anti-aritmici:
– atenololo: beta1-selettivo, non↑durata PdA;
– esmololo: emivita brevissima, usato in fase intraoperatoria;
– sotalolo: non selettivo, ↑durata PdA;
CLASSE 3: --| canali K+ → ↑durata PdA (prolungamento QT in ECG) quindi periodo refrattario;
– problemi potenziali:
– PdR più vicino a soglia → ↑automatismo in cellule di conduzione e danneggiate;
– affinità inversa rispetto a FC→ ↑attività su cellule più lente (rischio torsioni di punta);
– amiodarone:
– farmaco più usato per grande efficacia su molte aritmie;
– effetti positivi oltre a --| canali K+:
– non ha affinità inversa a FC, prolunga uniformemente PdA;
– blocca canali Na+ e in parte canali Ca2+ e recettori beta;
– non causa torsioni di punta nonostante prolungamento QT;
– tossicità:
– cardiaca: bradicardia eccessiva, blocco AV;
– da accumulo tissutale: fibrosi polmonare, epatite, fotodermatite e colorazione blugrigia in zona malare, depositi corneali;
– tiroidea (iper/ipotiroidismo): da blocco conversione periferica T3-T4 e accumulo
iodio: in approvazione dronedarone (analogo senza atomo di Iodio);
– dofetilide (orale) ibutilide (iv):
– blocco puro canali K+;
– usati in ospedale per ripristino e mantenimento ritmo sinusale in pz con FA.
– vernakalant:
– blocca molteplici canali K+;
– ha effetto ↑durata PdA soprattutto su atrio.
CLASSE 4: --| canali Ca2+ → ↓conduzione in nodi SA e AV dove upstroke è Ca2+-dipendente;
– verapamil e diltiazem fanno parte di CCBs, già trattati in precedenza;
– DHP non hanno effetto e possono causare aritmie.
Æ
31 / 55
NON CLASSIFICATI:
– adenosina:
– per emergenza di aritmia sopraventricolare da rientro →ripristina ritmo sinusale;
– somministrazione in bolo iv, con breve durata d'azione;
– blocca canali Ca2+ e K+ inibendo nodo AV e SA.
Æ
32 / 55
PATOLOGIE EMOSTASI
PREMESSE
– danno vascolare instaura fenotipo endoteliale pro-coagulante;
– esposizione collagene e vWF subendoteliale causa adesione piastrine che producono mediatori;
– principali sono TXA2, ADP, 5-HT(serotonina) → attivazione, aggregazione, vasocostrizione;
– integrina αIIb-βIII lega fibrinogeno → TAPPO PIASTRINICO;
– deficit → emorragie superficiali (naso, gengiva, mestruazioni abbondanti);
– cascata coagulazione:
– via intrinseca: cariche negative da danno vasale o microrganismi→12>11>9>8>10;
– via estrinseca: TF endoteliale o plasmatico → 7>10;
– via comune: 10>5>2(trombina)>1(fibrina);
– cascata coagulativa genera trombina attivata (fattore 2a) che:
– lisa fibrinogeno→ RETE FIBRINA;
– deficit → emorragie profonde (articolazioni, retroperitoneo);
– attiva fattore 13 → cross-link fibrina;
– attiva pC → effetto anti-coagulante;
– sistema fibrinolitico (plasmina) rimodella trombo;
– deficit: tromboembolia.
FARMACI
– la patologia più frequente è l'eccesso di emostasi, che porta a trombosi, la quale può essere:
– venosa: trombi rossi costituiti da RBC e fibrina → embolia polmonare;
– arteriosa: trombi bianchi di piastrine → ischemia a valle;
– sul rischio di trombosi si può agire con 3 possibili azioni:
1) antiaggregante;
2) anticoagulante;
3) fibrinolitica.
1) ANTI-AGGREGANTI
– agiscono su funzione piastrinica (2a fase emostasi dopo vasocostrizione) modificabile con:
1. aspirina:
– blocca sintesi PG (TXA2);
– usata in prevenzione secondaria pz con storia vascolare;
2. clopidogrel, prasugrel:
– blocca aggregazione indotta da ADP;
– uso in prevenzione di pz con: futuro impianto stent; ictus; IMA STEMI/NSTEMI;
3. abciximab:
– Ig monoclonale contro integrina αIIb-βIII recettore piastrinico di fibrinogeno e vWF.
2) ANTI-COAGULANTI
1. eparina e fondaparinux:
– inibitori indiretti della trombina, agiscono attivando antitrombina-III;
– AT è inibitore suicida che agisce su trombina (fattore 2a) e fattore 10a;
– in seguito a distacco da AT i farmaci sono di nuovo disponibili;
– uso: trombosi venosa profonda, embolia polmonare, post-intervento chirurgico;
– eparina è miscela di mucopolisaccaridi solforati (a.glucuronico e glucosamina solfato) di origine
estrattiva animale, presente in 2 forme:
– HMWH: eparina ad alto peso molecolare (5/30.000 Kda) o non-frazionata (UFH):
Æ
33 / 55
– lega AT inattiva (attivandola), trombina e fattore Xa;
– accelera 1000 volte azione di AT;
– LMWH: eparina a basso peso molecolare:
– lega AT (attivandola) e trombina;
– fondaparinux:
– farmaco di sintesi;
– lega solo l'AT attivandola;
– l'attività dei farmaci si misura valutando aPTT (via intrinseca) e attività anti-Xa;
– tossicità:
– emorragia da sovradosaggio (pz con insufficienza renale) controllabile con protamina,
sostanza con cariche positive, che neutralizza eparina;
– reazioni allergiche;
– trombocitopenia da reazione immune (++UFH di origine bovina);
2. lepiruden e altri;
– sono inibitori diretti della trombina;
– derivano da irudina (in saliva sanguisughe per fluidificare sangue).
3. dicumarolo:
– farmaco naturale è dicumarolo, warfarin è analogo sintetico;
– è antagonista della vitamina K. Ne blocca la riattivazione (riduzione epossido a idrochinone);
– deficit vitK impedisce carbossilazione fattori 2, 7, 9, 10 (→inattivabili in coagulazione);
– meccanismo è indiretto quindi effetto avviene dopo 8-12h, in seguito a consumo dei fattori già
attivabili (fattore 7 ha emivita più breve – 6h);
– terapia è complicata:
– assorbimento disomogeneo;
– >95% legata a proteine plasmatiche;
– vitamina K è un prodotto alimentare ed è prodotto anche da batteri intestinali;
– si inizia con 5-10mg/d e si valuta PT (via estrinseca) dopo una settimana;
– obiettivo è PT = 25% del normale (INR = 2-3);
– terapia di mantenimento di solito è 5-7mg/d ma INR va controllato regolarmente;
– in caso di sovradosaggio si usano concentrati di protrombina, 7a ricombinante, plasma;
– ci sono interazioni farmacologiche: barbiturici, antibiotici, salicilati.
4. Nuovi anticoagulanti orali (NOA):
– costo elevato ma finestra terapeutica è molto ampia quindi ridotta necessità controlli;
– dabigatran etexilato:
– primo approvato da FDA nel 2010, in EU per prevenzione tromboembolia post-protesi
anca/ginocchio;
– inibitore della trombina (fattore 2a) ne blocca reversibilmente il sito attivo;
– apixaban, rivaroxaban:
– Ig monoclonali contro fattore 10a;
– uso in prevenzione e trattamento tromboembolia, e prevenzione stroke in pz con FA (se si
forma coagulo in atrio sx poi passa in ventricolo, va in aorta e può arrivare al cervello).
3) FIBRINOLITICI
– catalizzano formazione di plasmina (impossibile uso diretto plasmina per inibitore plasmatico);
– creano stato litico generalizzato per degradazione di fibrina;
– eccesso può causare lisi di trombi emostatici quindi emorragia;
– fondamentali per pz infartuati (entro 6h), soprattutto in strutture senza PCI (Percutaneous Coronary
Intervention) dove non è possibile intervento rapido di cateterizzazione+stent.
– farmaci:
Æ
34 / 55
– streptochinasi (da Streptococchi), urochinasi (sintesi renale):
– catalizza direttamente trasformazione di plasminogeno in plasmina;
– alteplasi, reteplasi, tenecteplasi:
– attivatore tissutale del plasminogeno (tPA) in forma ricombinante.
Æ
35 / 55
DISLIPIDEMIA
PREMESSA
– per dislipidemia si intendono valori (1mmol/L=40mg/dL):
– LDL-C ≥ 200mg/dL e/o;
– HDL-C ≤ 40mg/dL e/o
– trigliceridi ≥150mg/dL .
– conseguenze cliniche dirette di iperlipidemia sono pancreatite acuta e aterosclerosi;
– fattori di rischio per aterosclerosi:
– dislipidemia, fumo, ipertensione, diabete, familiarità, età (M>45aa, F>55aa), obesità,
inattività, dieta aterogenica, livelli di Lp(a), fattori pro-trombotici e pro-infiammatori, glicemia
alterata a digiuno;
– cause secondarie di dislipidemia: diabete, sindrome nefrosica, alcolici, contraccettivi ormonali,
glucocorticoidi, ipotiroidismo;
– lipidi plasmatici sono complessati in lipoproteine:
– aterogeniche sono:
– VLDL, LDL, IDL e Lp[a] (lipoproteina a) che contengono apoB-100;
– remnants di chilomicroni, che contengono apoB-48;
– anti-aterogeniche sono:
– HDL: recuperano lipidi da periferia a fegato;
– placca aterosclerotica:
– causata da accumulo endoteliale (recettori scavenger su macrofagi) di lipoproteine ossidate
da ROS;
– successiva modifica causa accumulo “foam cells”, collagene, fibrina, Ca2+;
– rottura di placca causa attivazione piastrinica e formazione di trombi occlusivi;
– controllo di lipidemia passa per combinazione di tre fattori:
1) dieta povera in colesterolo e grassi saturi/trans, esercizio fisico, perdita di peso;
2) statine: ↓LDL-C (25–50%), ↑HDL-C (5–9%), ↓trigliceridi (5–30%) con effetto potente su
aterosclerosi e malattia ischemica cardiaca, indipendentemente da LDL-C precedente;
3) fibrati o niacina per ↑HDL-C e ↓TG;
– antilipemici permettono:
– ↓LDL e TG, ↑HDL e blocco ossidazione lipidi che ristabilisce funzione endoteliale;
– stabilizzazione/regressione placche.
LINEE GUIDA DISLIPIDEMIA 2013
di American Heart Association (da Harrison):
– includono tra farmaci utili solo le statine (unici con Randomized Controlled Trials) le quali:
– stabilizzano la placca aterosclerotica (non la riducono molto);
– riducono il rischio di eventi cardiovascolari (ma non lo annullano);
– l'aggiunta di Ig monoclonali anti-PSCK9 riduce ulteriormente LDL (in studio);
– target preciso di LDL-C non è più obiettivo di terapia;
– dose dipende da livello di rischio del pz;
– farmaco non sostituisce vita sana (2h30' attività fisica a settimana, dieta vegetale e bassi
grassi saturi);
– individuano 4 gruppi di pz che beneficiano di statine:
– pregressa malattia aterosclerotica cardiovascolare (ASCVD);
– LDL≥190 mg/dL;
– diabetici con LDL 70-189 mg/dL;
– pz 40-75aa con LDL 70-189 mg/dL e rischio aterosclerotico ≥7,5%;
– non consigliano farmaci (ma terapia non-farmacologica) per aumento HDL-C :
– valori HDL-C <1mmol/L (40mg/dL) correlano direttamente con rischio cv;
Æ
36 / 55
– aumentata prevalenza diabete o sindrome metabolica causa aumento pz con ↓HDL;
– perdita peso e attività fisica elevano HDL e riducono rischio cv;
– farmaci che ↑HDL ma che non hanno dimostrato efficacia su eventi cv (a volte anche effetti
negativi):
– acido nicotinico;
– agonisti PPARalfa e gamma (recettore nucleare attivato da proliferatore di
perossisomi);
– inibitori di CETP (transferasi di esteri di colesterolo).
FARMACI
1. farmaci che riducono produzione LDL
1) INIBITORI HMG-CoA reduttasi: STATINE (atorvastatina, simvastatina, rosuvastatina):
– unica tipologia farmaci consigliata in linee guida AHA-2013 (vedi sopra); gli altri farmaci possono
andar bene come associazione a statine;
– meccanismo:
– inibitori competitivi enzima HMG-CoA (idrossi-metilglutaril-CoA) reduttasi;
– quindi inibizione sintesi mevalonato, precursore colesterolo;
– blocco produzione causa ↑ espressione recettori LDL quindi ↓ LDL plasmatico;
– isolate inizialmente da aspergillo, poi di sintesi;
– effetti dose-dipendenti:
– ↓LDL (anche >50%) e indirettamente elevano HDL (5-9%) e ↓TG (5-30%);
– importante soprattutto stabilizzazione placca, blocco proliferazione muscolo liscio vasale,
riduzione ossidazione LDL, riduzione tromboembolia (rosuvastatina);
– farmacocinetica:
– buon assorbimento, elevato primo passaggio, emivita breve (eccetto rosuvastatina, 30h);
– somministrazione serale è migliore perché sintesi maggiore notturna.
– tossicità: epatica (1%), miopatie;
– interazioni farmacologiche su CYP-3A4 e 2C9: in particolare
– gemfibrozil, amiodarone, verapamil aumentano rischio miopatie;
– macrolidi, ketoconazolo, farmaci HIV, pompelmo aumentano livelli plasmatici;
– fenitoina, barbiturici, riducono livelli plasmatici.
2) NIACINA (vit. B3):
– meccanismo:
– ↓ lipolisi da adipe quindi trasporto lipidi al fegato;
– ↓ secrezione VLDL quindi produzione LDL;
– ↑ attività lipoproteina lipasi quindi deposito periferico;
– effetti:
– ↑↑HDL (+40%); ↓VLDL, LDL (-20%) e Lp(a);
– tossicità:
– dosi elevate necessarie (2-3 grammi/d);
– vampate (ridotte da aspirina), dispepsia, epatotossicità;
– possibili ↑acido urico (attenzione in gotta), ↑insulino-resistenza (attenzione in diabete).
3) ATTIVATORI PPARalfa (clofibrato, gemfibrozil):
– utili in pz con elevato VLDL-C: effetto principale è ↓VLDL, inferiore è il calo di LDL;
– attivano recettore nucleare PPARalfa la cui trascrizione determina:
– ↑lipoproteina lipasi e ↓apoC3 (inibitore lipolisi) →deposito extraepatico;
– aumento attività ossidazione lipidi muscolare e epatica;
– lieve ↑apoA1 e A2 quindi ↑HDL;
– tossicità è rara: rash, miopatie, aritmie.
Æ
37 / 55
4) ANTICORPI MONOCLONALI
– anti-PCSK9:
– proteina plasmatica che si lega a LDL-R causandone internalizzazione e degradazione;
– se bloccata permette di mantenere il recettore in membrana;
– i livelli di LDL plasmatici calano.
2. farmaci che diminuiscono assorbimento di colesterolo
1) RESINE CHELANTI ACIDI BILIARI (colestiramina):
– meccanismo:
– chelano acidi biliari intestinali impedendone il riassorbimento;
– riducono ricircolo acidi biliari aumentando necessità di produrre colesterolo;
– effetti: ↓LDL (massimo 25%) ma nel contempo ↑TG;
– tossicità: ↑TG, gonfiore, dispepsia, interferenza con assorbimento intestinale di farmaci e elementi.
2) INIBITORI ASSORBIMENTO COLESTEROLO (ezetimibe)
– meccanismo: blocca proteina NPC1L1, trasportatore colesterolo su enterociti, quindi blocca
assorbimento colesterolo alimentare e della bile;
– effetto: ↓LDL (20%) ma aumento sintesi epatico (associazione con statine).
Æ
38 / 55
DIABETE MELLITO
PREMESSA
– parametri normali:
– glicemia a digiuno < 100 mg/dL (5.6mmol/L), diabete >125 mg/dL;
– glicemia dopo carico di glucosio <140 mg/dL, diabete ≥200 mg/dL;
– HbA1C<5.6%, diabete ≥6.5%;
– glucosio è trasportato:
– da intestino a plasma, controgradiente, tramite SGLT (Na+-dipendente);
– da plasma a cellule tramite trasportatori GLUT (12 domini transmembrana):
– 1: SNC (endotelio barriera EE) e RBC, ↑ capacità e ↑affinità;
– 2: cellule beta e fegato, ↑ capacità ma ↓ affinità;
– relazione lineare tra glicemia e secrezione insulina fino a 180-200mg/mL;
– 3: neuroni, ↑ capacità e ↑affinità;
– 4: muscolo, adipe, miocardio: upregolato da insulina e esercizio fisico.;
– legame con recettore causa esposizione in membrana di GLUT4;
– regolazione glicemia si basa su:
1) insulina (immagazzinata in vescicole in cellule beta pancreas)
– rilascio insulina:
1. ↑[glucosio]p post-prandiale che entra in cellula beta da recettore GLUT2;
2. ↑[ATP] causa blocco canale K+ ATP-sensibile per legame a subunità Kir 6.2;
– canale è etero-ottamero con subunità SUR1 e Kir6.2;
– subunità SUR: bloccata da sulfoniluree e meglitinidi; attivata da diazossido;
3. depolarizzazione causa ↑[Ca2+]i per ingresso da canale volt-dipendente;
4. ↑[Ca2+]i causa esocitosi;
5. altri fattori che determinano secrezione insulina tramite ↑[cAMP] o [Ca2+]:
– effetto incretinico (GIP, GLP-1) determina maggior rilascio di insulina da assunzione orale
rispetto ad infusione iv di pari quantità glucosio;
– GIP: peptide insulinotropico glucosio-dipendente (cellule L duodeno);
– insufficienza renale causa ↑[GIP] per ridotta clearance di esso;
– GLP-1: glucagon-like polipeptide (cellule K tenue-colon);
– rilascio modulato positivamente da vago;
– glucagone (recettore GPCR>Gs);
– AA gluconeogenici;
– colecistochinina;
– elevate [FFA];
– attività beta-adrenergica;
– effetti insulina (emivita 4 minuti, rilascio con picchi a alta-bassa frequenza):
– ↑uptake tissutale glucosio (upregolazione GLUT4) ed effetti anabolici: glicogenogenesi,
lipogenesi, sintesi proteica;
– ↓gluconeogenesi (GNG);
2) ormoni controinsulari (sono 4, quindi evolutivamente è più importante uscire da ipoglicemia!):
– rilascio dipende da ↓[glucosio]p e consente utilizzo glucosio, FFA e AA;
– glucagone: ↑glicogenolisi e GNG;
– adrenalina: ↑lipolisi e resistenza insulinica (effetti rapidi);
– GH: ↑lipolisi e resistenza insulinica (effetti lenti);
– cortisolo: ↑catabolismo muscolare per GNG, ↑lipolisi.
Æ
39 / 55
DIABETE MELLITO
– è stato patologico definibile tramite tre parametri:
– glicemia a digiuno: pre-diabete se tra 100 e 125 mg/dL, diabete oltre 125mg/dL;
– glicemia dopo carico di glucosio: pre-diabete se 140-199 mg/dL, diabete ≥200;
– HbA1C: pre-diabete se ≥5.7%, diabete se ≥6.5%;
– dovuto a insufficiente secrezione assoluta o relativa di insulina;
– categorie:
– tipo1: insulino-dipendente:
– distruzione selettiva di cellule beta causa deficienza insulinica totale o severa;
– causa autoimmune e diagnosi <30aa sono più frequenti;
– incidenza maggiore in nord Europa e Sardegna; familiarità positiva nel 15% del casi;
– terapia insulinica sostitutiva è vitale per evitare livelli fatali di chetoacidosi;
– tipo2: non insulino-dipendente:
– combinazione di resistenza insulinica e deficit relativo (non assoluto) di secrezione;
– può essere più o meno grave e presentarsi con gradi diversi di resistenza e deficit;
– glicemia non è più controllata e aumentano TG (per ↓lipogenesi);
– terapia insulinica non è vitale ma necessaria in 30% pz per controllare glicemia;
– 15% dei pz tipo2 hanno in realtà LADA (tipo1 latente autoimmune);
– possono andare in chetoacidosi ma rischio maggiore è coma iperosmolare non-chetotico
(glicemia raggiunge livelli 5-20 volte la norma);
– tipo3: iperglicemia causata da altre cause specifiche (pancreasectomia, pancreatite, ecc.);
– tipo4: gestazionale:
– resistenza insulinica determinata da ormoni placentari;
– si verifica in 7% gravidanze USA;
– screening a 24 settimane in donne a basso rischio.
TERAPIA
– obiettivi terapia diabete:
– controllo glicemico:
– HbA1C <7% (<5.6% nel sano) ← indicatore fondamentale di efficacia terapia.
– glicemia pre-prandiale <130mg/dL;
– glicemia post-prandiale <180mg/dL;
– pressione <130/80;
– controllo dislipidemia:
– LDL <100, HDL >40, TG<150 mg/mL;
– scelta farmaci:
– prima linea: metformina in monoterapia;
– seconda linea: aggiunta di secondo agente orale o insulina (sulfoniluree o insulina sono
preferite per minor costo e effetti avversi);
– terza linea: aggiunta altri agenti orali;
– quarta linea: insulina con o senza metformina.
FARMACI
1. ipoglicemizzanti orali
1) BIGUANIDI (metformina);
– farmaco euglicemico di prima scelta;
– agisce indipendentemente da funzionalità di cellule beta;
– effetti:
– principale: ↓ produzione glucosio epatico tramite attivazione AMPK;
Æ
40 / 55
– secondari: ↑trasporto glucosio in muscolo, ↓GNG renale, ↓assorbimento intestinale glucosio,
↓glucagone.
– effetti avversi:
– nausea, vomito, diarrea in 20%: dose-dipendenti, tendono a scomparire (eccetto 3-5%);
– ridotto assorbimento b12 (controllo annuo e integrazione);
– acidosi lattica (non frequente) per inibizione metabolismo epatico. Non usare in malattie
renali, polmonari-cardiovascolari, epatiche, alcolismo (rischio ipossia).
2) SECRETAGOGHI:
– sulfoniluree (es. glicazide) e glinidi (es. nateglinide);
– bloccano canale K-ATP su cellule beta interagendo con subunità SUR-1 (ABC-C8);
– indicati per controllo glicemia post-prandiale (vanno associati a metformina per glicemia di base);
3) TIAZOLIDINEDIONI:
– attivano recettori PPARgamma in adipe, muscolo e fegato riducendo resistenza insulinica;
– efficaci ma eccessivi effetti avversi: ritenzione idrica, cardiovascolari, demineralizzazione;
4) INIBITORI DI alfa-GLUCOSIDASI (acarbosio-Glicobay® e miglitolo):
– bloccano α-glucosidasi su orletto a spazzola di enterociti riducendo digestione a monosaccaridi;
– riducono (non totalmente perché α-amilasi è poco inibita) assorbimento di glucidi;
– efficaci in riduzione glicemia (-25mg preprandiale/-50mg postprandiale) e HbA1C (-1%);
– effetti avversi: flatulenza, diarrea, crampi addominali. A lungo termine aumenta espressione enzimi
in ileo distale;
5) MODULATORI SISTEMA INCRETINICO
– analoghi di GLP-1 (exenatide):
– potenziano secrezione insulinica in base a glicemia (effetto anti-apoptotico cellule β);
– rallentano svuotamento gastrico: ↑senso di sazietà, ↓calo dell'appetito, ↓peso;
– sopprimono rilascio glucagone (quindi glucosio) post-prandiale;
– antagonisti di DPP-4, enzima che degrada incretine (saxagliptina):
– aumentano livelli di GIP e GLP-1 potenziando secrezione insulinica in base a glicemia;
– sopprimono rilascio glucagone (quindi glucosio) post-prandiale;
– effetto anti-apoptotico cellule β;
– effetti avversi: nasofaringiti, cefalea, edema, pancreatite acuta.
2. insuline
– obiettivo è replicare la secrezione endogena anche se è difficile da ottenere;
– regimi intensivi prevedono uso di:
– insulina a lenta azione (livelli basali) +insulina rapida (livelli post-prandiali);
– infusori di insulina ultrarapida;
– tipologie di insulina (rapidità è misurata su somministrazione parenterale intramuscolo):
– ultrarapide: lispro, aspart, glulisin:
– picco a < 30';
– molto usate perché adatte a somministrazione subito prima di pasto e ad infusore;
– rapide: regular (insulina ricombinante):
– primo effetto a 30', picco a 2h, durata 5h;
– relativamente difficile far combinare glicemia con effetto farmaco;
– unica utilizzabile in emergenza per via intravenosa (ketoacidosi diabetica);
– intermedie:
– NPH (neutral protamine Hagedorn), è insulina combinata con protamina per
rallentarne il rilascio;
– primo effetto a 2h, durata 4-12h;
Æ
41 / 55
– oggi poco usata per alta variabilità (50%) di assorbimento e azione;
– lente: glargine, detemir:
– determina formazione deposito cristallino sottocutaneo che rilascia gradualmente;
– ha curva “senza picco”: effetto inizia dopo 2h e ha picco a 5h che dura >12h;
– somministrazione bid consente adeguati livelli di insulinemia basali.
Æ
42 / 55
FARMACI DEL SNC
PREMESSE
– sinapsi è sito di trasformazione di impulso neuroelettrico in messaggio neurochimico e
successivamente da neurochimico a elettrico;
– tale trasformazione è utile per modulare il messaggio attraverso feedback negativi e positivi;
– la frequenza dei PdA in arrivo (eccitatori vs inibitori) determina la quantità di neurotrasmettitore
rilasciato;
– la rapidità di risposta al neurotrasmettitore dipende da:
– tipo di recettore: canale ionico > GPCR > trascrizione genica;
– quantità di neurotrasmettitore e sua permanenza nella fessura sinaptica;
– neuromodulatori;
– sistemi neurotrasmettitoriali cerebrali:
1) eccitatori:
– glutammato: sistema con maggior rilevanza;
– iperattività causa neurodegenerazione;
– ACh: effetto minore, forse legato ad alternanza sonno-veglia e regolazione vigilanza;
2) inibitori:
– GABA: da interneuroni in SNC e midollo spinale;
– iperattività deprime centri respiratori;
– glicina: da interneuroni in tronco encefalico e midollo spinale;
3) monoamine:
1. dopaminergico:
– DA è prodotta in sostanza nera e tegmento ventrale di mesencefalo;
– funzioni:
– regolazione umore e processi comportamentali per bisogni primari;
– coordinazione motoria nei nuclei della base;
2. noradrenalinergico:
– NA è prodotta in locus ceruleus; (adrenalina non entra in cervello)
– funzioni:
– risposta allo stress ambientale (es. lotta o fuga);
– se iperattivo, causa disturbo d'ansia generalizzato (es. ansia per colazione);
3. serotoninergico:
– 5-HT prodotta in rafe dorsale;
– funzioni:
– regolazione risposte neurovegetative;
– reazioni affettive-emotive che danno valenza ad eventi;
4) istamina;
– H prodotta in ipotalamo ventrale posteriore;
– funzioni:
– eccitazione, temperatura corporea, vasodilatazione;
– inibizione recettore H1 causa sonnolenza;
– i farmaci:
– hanno struttura simile al trasmettitore naturale;
– spesso impediscono degradazione o ricaptazione degli stessi trasmettitori;
– agiscono ovunque (non sono secreti in sito specifico);
– dipendono da farmacocinetica (non da attività neuronale);
Æ
43 / 55
SEDATIVO-IPNOTICI
PREMESSA
– sistema noradrenergico:
– attivato da amigdala in risposta a situazioni stressorie/emotigene a breve termine
(soppiantato da asse ipotalamo-ipofisi-surrene a lungo termine);
– iperattivato in stati d'ansia generalizzati;
– sistema GABAergico:
– consente inibizione sistema noradrenergico tramite interneuroni GABA diffusi;
– apre canali ionici che determinano influsso Cl- quindi potenziali inibitori;
– potenziato da sedativo-ipnotici:
– modulatori allosterici positivi del GABA-A;
– benzodiazepine: aumentano frequenza di apertura del canale;
– barbiturici: aumentano durata di apertura del canale;
– utilizzi:
– ansia, insonnia, interventi medico-chirurgici (sedazione, amnesia anterograda, anestesia
generale), convulsioni.
FARMACI:
BENZODIAZEPINE E BARBITURICI
– modulatori allosterici positivi del recettore GABA-A (B è legato da spasmolitico baclofen):
– recettore pentamerico con diverse isoforme;
– legato da BZ tra subunità alfa1 e gamma2, da barbiturici in altro sito;
– legato da etanolo e altri farmaci che causano effetti additivi;
– meccanismo di azione:
– benzodiazepine:
– aumentano la frequenza di apertura del canale in presenza del ligando endogeno;
– barbiturici:
– aumentano la durata di apertura del canale;
– curva dose-risposta è più ripida che benzodiazepine;
– effetti depressivi dose-dipendenti sul SNC (curva dose-risposta è più ripida in barbiturici):
– sedazione, effetto ansiolitico (con depressione funzione psico-motoria);
– amnesia anterograda (durante effetto di farmaco);
– ipnosi (riduzione latenza di addormentamento);
– anestesia;
– coma e depressione centro respiratorio midollare;
– uso in terapia ansiolitica:
– BDZ usate in stati acuti (es. alprazolam, ma tutte simile efficacia) mentre in disturbo d'ansia
generalizzato e a lungo termine sono meglio nuovi antidepressivi inibitori selettivi del reuptake
di serotonina/noradrenalina (SSRIs e SNRIs);
– rischi di dipendenza, depressione funzioni SNC, effetti amnestici;
– ricordare interazione con etanolo.
– utilizzo a lungo termine:
– non è motivato;
– causa dipendenza (stato fisiologico alterato che richiede somministrazione continua per
evitare sindrome da astinenza) maggiore di oppioidi;
– interruzione causa sindrome da astinenza: ansia, insonnia, ipereccitabilità, convulsioni.
Æ
44 / 55
SCHIZOFRENIA
PREMESSA
– diagnosi con presenza, per almeno 1 mese continuo, per almeno 6 mesi totali, di almeno 2 tra:
– illusioni/allucinazioni;
– disorganizzazione del discorso/del comportamento;
– disfunzione occupazionale/sociale;
– psicosi è reazione psico-comportamentale indotta da alterata percezione dell'ambiente:
– sistemi sensitivi sono intatti e funzionali, ma non down-regolati;
– sensory gating (filtro di attenzione) è ridotto → immersione in cacofonia di suoni e immagini
che induce presenza di allucinazioni, visioni, ecc.
– lo psicotico crea fantasticherie su quello che vede nella realtà: un rigonfiamento nello zaino è
interpretato come un coltello, le persone parlano alle spalle, ecc.
– distinta da neurosi, disturbo d'ansia legato a situazioni oggettive correttamente interpretate dal pz.
– fattori di rischio:
– ereditari: crescita assoni, mielinizzazione;
– legati a eventi personali: traumi psico-fisici, contesto sociale, esposizione alla cannabis;
– trattamento integrato:
– terapia farmacologica (vedi dopo):
– farmaci tipici e atipici sono egualmente efficaci su sintomi positivi (deliri,
allucinazioni);
– farmaci atipici funzionano meglio anche su sintomi negativi (anedonia, appiattimento
affettivo, perdita di volontà);
– terapia psico-sociale.
IPOTESI CAUSALI
1) iperattività SISTEMA DOPAMINERGICO:
– inizialmente si notò che la reserpina (inibitore recettore dopamina) era capace di indurre in
soggetti sani una sindrome depressogena che veniva risolta da somministrazione di DOPA; è
un'ipotesi oggi poco in voga perché:
– in psicosi coesistono ↓attività dopaminergica corticale e ↑attività dopaminergica limbica;
– antipsicotici atipici funzionano meglio di neurolettici e non alterano funzioni extrapiramidali,
avendo poco effetto su recettori D2 e molto su recettori 5-HT-2A.
2) iperattività SISTEMA SEROTONINERGICO (5-HT):
– allucinogeni LSD e mescalina sono agonisti di recettori 5-HT-2A/2C;
– antipsicotici atipici: clozapina e derivati (olanzapina, risperidone) sono agonisti inversi del
2A e sono più efficaci di neurolettici;
– il blocco dell'attività costitutiva del recettore modula negativamente il rilascio di DOPA, NA,
Glut, GABA, ACh.
3) disfunzione recettori NMDA di GLUTAMMATO:
– ipofunzionalità di NMDA su interneuroni GABAergici → ridotta inibizione a valle;
– iperattività conseguente causerebbe iperstimolazione corticale;
4) disfunzione recettore alfa7 di ACh:
– sistema ACh (recettore nicotinico alfa7 in giro dentato) è deficitario in patologie cognitive
(memoria, attenzione, capacità verbali, funzioni esecutive);
– disfunzione recettore causerebbe deficit cognitivo-attenzionale alla base di altri sintomi.
Æ
45 / 55
FARMACI
– due categorie fondamentali:
1. neurolettici:
– primi farmaci scoperti (clorpromazina, aloperidolo);
– agiscono bloccando soprattutto recettore dopamina (D2);
– causano:
– importanti effetti extra-piramidali (aloperidolo): distonie, rigidità espressive, discinesia;
– sedazione e ipotensione (clorpromazina).
2. antipsicotici atipici:
– scoperti a fine anni ‘50 (clozapina);
– agiscono bloccando:
– poco il recettore D2;
– soprattutto i recettori 5-HT-2A , istaminico, adrenergico alfa2 (effetti combinati):
– risperidone --| 5-HT-2A;
– quetiapina --| H1 >> alfa2 >> 5-HT-2A;
– olanzapina --| 5-HT-2A >> H1;
– effetto limitato su recettore alfa7 nicotinico dell'ACh (90% schizofrenici sono fumatori).
Æ
46 / 55
DEPRESSIONE
PREMESSA
– sintomi:
– sofferenza psichica, apatia, anedonia, alterata percezione della realtà, irritabilità, pensieri
negativi verso sé stessi e futuro, ruminazione, amnesie, scarsa concentrazione, ideazioni
suicidarie, sensi di colpa;
– sintomi fisici correlati: insonnia, anoressia, affaticabilità;
– terapia:
– psico-comportamentale: rimozione cause ambientali;
– farmacologica: azione centrale su SNC;
IPOTESI CAUSALI
1. ipotesi monoaminergica:
– deficit di monoamine (dopamina, NA, 5-HT) è alla base della patologia;
– ipotesi sostenuta da:
– effetto positivo di farmaci che ↑[monoamine] in fessura sinaptica;
– aumentata suscettibilità a depressione in soggetti con deficit SERT;
– effetto pro-depressivo di reserpina e di dieta priva di triptofano, che causano
deplezione di monoamine;
– oggi è integrata dall'ipotesi neurotrofica;
2. ipotesi neurotrofica:
– è ipotesi attuale, con sostanziali evidenze a favore;
– si pensa che un calo di fattori di crescita neuronali (neurotrofine) tra cui BDNF (brainderived neurotrophic factor) causi ridotta plasticità neuronale, resilienza e neurogenesi,
soprattutto in strutture quali:
– l'ippocampo;
– la corteccia frontale mediale;
– il cingolato anteriore:
– integra la risposta limbica (emotiva) con quella prefrontale (cognitiva);
– pz con iperattività in questo sito rispondono a terapia farmacologica;
– tale riduzione si registra anche in situazioni di stress e dolore;
– l'espressione di BDNF sarebbe riattivabile tramite farmaci che ne inducono la produzione
indirettamente, agendo sui livelli di serotonina, NA, Dopamina, Glutammato.
3. Ipotesi neuroendocrina:
– disregolazione asse ipotalamo-ipofisario: deficit tiroidei, eccesso ACTH e cortisolo;
– deficit di estrogeni: post-parto, post-menopausa;
FARMACI
In ordine preferenziale di utilizzo:
1. SSRIs:
– inibitori selettivi del reuptake della serotonina (fluoxetina, paroxetina);
– bloccano il trasportatore SERT aumentando la concentrazione nella fessura sinaptica;
– tossicità: ben tollerati ma possono causare disfunzione sessuale;
2. SNRIs:
– inibitori selettivi reuptake serotonina e noradrenalina (venlafaxina, duloxetina);
– bloccano SERT e NET (trasportatore norepinefrina);
– tossicità: anticolinergici (accomodamento visivo, secchezza fauci), sedativi.
Æ
47 / 55
3. triciclici:
– oltre a blocco SERT e NET: ↑dopamina, ↓ACh... (imipramina, doxepina, amitriptilina);
– effetti alfa-bloccanti, sedativi, aumento di peso, aritmie, letali in overdose;
4. antagonisti 5-HT-2A:
– blocco recettore serotonina;
– tradozone, nefadozone;
5. tetraciclici (mirtazapina) e uniciclici (bupropione):
– ↑azione NA e dopamina;
– tossicità: ↓soglia convulsioni, aumento di peso;
6. inibitori monoaminoossidasi (MAOIs):
– bloccano degradazione monoamine;
– fenelzina (MAO A-B), selegilina (MAO-B);
– tossicità: ipotensione, insonnia;
7. ketamina:
– usato per indurre stato catalettico (anestesia dissociativa, pz ha occhi aperti e fissi) con
amnesia e analgesia senza perdita di coscienza;
– dà crisi allucinatorie all' “emersione” in 50% adulti, 10% bambini (uso ++ pediatrico);
– agisce stimolando sistema glutammatergico (eccitatorio): è soprattutto antagonista non
competitivo recettori NMDA del glutammato, causando relativo aumento di attività dei
recettori AMPA;
– sembra funzionare (in studio) in infusione lenta in pz depressi non responsivi.
Æ
48 / 55
MALATTIE NEURODEGENERATIVE
– date da perdita di popolazioni neuronali in aree specifiche del SNC;
– cause:
– intrinseca vulnerabilità cellulare al danno di alcune popolazioni neuronali;
– tossine ambientali, metabolismo ed invecchiamento;
→ danni da eccitotossicità e da stress ossidativo;
– malattie più comuni:
– Alzheimer:
– lesioni in ippocampo e aree corticali specifiche da oligomeri di amiloide beta;
– più frequente causa demenza anziano: colpisce 3%>65aa, 20%>75, 45%>84aa;
– Parkinson:
– degenerazione neuroni dopaminergici sostanza nigra pars compacta per deficit
ubiquitinazione alfa-sinucleina (corpi di Lewy) che si associa a dopamina;
– colpisce 1% >65 anni;
PARKINSON
PREMESSA
– degenerazione neuroni dopaminergici di sostanza nigra pars compacta:
– si manifesta quando persi oltre 70% (perdita inferiore è comune in anziani asintomatici);
– tali neuroni hanno di norma effetto disinibitorio a valle →lesioni riducono eccitabilità;
– cause:
– idiopatico;
– post-ictus;
– iatrogeno: intossicazione da antagonisti dopaminergici o assunzione anti-psicotici tipici;
– patogenesi:
– eccesso produzione/deficit ubiquitinazione alfa-sinucleina (accumuli in “corpi di Lewy”);
– si associa a dopamina (prodotta in sostanza nigra) creando fibrille tossiche;
– neuroni di s.n. pars compacta hanno di norma un effetto disinibitorio a valle tramite:
– stimolo via diretta: eccitano i neuroni spinosi medi (nsm) che da corpo striato
proiettano al segmento interno del globus pallidus;
– inibizione via indiretta: inibiscono i nsm che proiettano al segmento esterno del gp;
– lesioni riducono quindi l'eccitabilità a valle;
– segni e sintomi:
– bradicinesia, rigidità muscolare, tremori, alterazioni dell'equilibrio, perdita mimica facciale,
saccadi ridotte;
– non trattato causa acinesia totale e morte entro 10 anni;
TERAPIA
– obiettivo è ristabilire sistema dopaminergico;
– sintesi dopamina: Phe → Tyr > step limitante > L-idrossi-Phe (L-DOPA) → DOPA;
– recettori dopamina (D1-D5) sono presenti in corpo striato, corteccia ma anche ippocampo e
ipotalamo (implicazione dopamina in psicosi e depressione);
– possibili strade:
– terapia rigenerativa o stimolazione cellule residue;
– aumento livelli dopamina → attivazione diretta recettori dopaminergici;
– riduzione livelli ACh → inibizione diretta sistema colinergico.
Æ
49 / 55
FARMACI
1. Levo-DOPA:
– precursore di DA (via decarbossilazione);
– obbiettivo è ri-caricare i neuroni dopaminergici residui: effetto si riduce nel tempo per
degenerazione progressiva;
– farmaco è somministrato iv e passa barriera EE (al contrario di DA);
– somministrato con inibitore di decarbossilasi plasmatica (carbidopa): blocca conversione plasmatica
in DA (inotropo, vasodilatatore splancnico) e quindi riduce effetti sistemici e degradazione del
farmaco (dose ridotta del 75%);
– effetti terapia:
– a breve termine:
– carbidopa 25mg + levodopa 100mg 3x/die; poi si aggiunge agonista DA;
– 1/3 pz ha miglioramenti netti, 1/3 scarsi, 1/3 non responsivi-effetti avversi;
– infusione intraduodenale sembra essere la miglior metodica;
– a lungo termine:
– diminuiscono dopo 3-4 anni, a prescindere da risposta iniziale, quindi necessario
iniziare terapia quando sintomi sono importanti;
– aumentano reazioni di “wearing-off” (effetto dura poco) e fluttuazioni on-off (periodi
di acinesia alternati a mobilità con forte dis-cinesia) in risposta a terapia;
– tossicità:
– senza carbidopa o con dosi elevate: nausea, effetti GI, ipotensione, aritmie;
– con carbidopa: allucinazioni, confusione, ansia, ipereccitabilità;
– con inibitori aspecifici MAO: ipertensione.
2. Agonisti dopaminergici:
– pramipexolo → D3;
– ropinirolo → D2;
– derivati dell'ergot, non più usati: bromocriptina → D2; pergolide → D1, D2;
– indicati come prima linea (monoterapia) o aggiunti a levodopa se fenomeni wearing-off o on-off;
– agiscono direttamente sui recettori DA senza richiedere conversione da neuroni dopaminergici;
– viene quindi attivato il sistema dopaminergico in maniera progressiva (ma non fisiologica);
– effetti avversi: GI, ipotensione, discinesie, allucinazioni, comportamenti compulsivi per inibizione di
controllo.
3. Inibitori COMT:
– tolcapone, entacapone;
– inibiscono enzima alternativo che degrada L-DOPA a livello plasmatico e cerebrale;
4. Inibitori MAO-B:
– MAO-B specifica per DA, MAO-A tutte monoamine;
– selegilina è inibitore selettivo irreversibile;
– usato in aggiunta a levodopa;
5. Antimuscarinici:
– poco effetto su bradicinesia, possono migliorare tremore e rigidità.
Æ
50 / 55
EPILESSIA
PREMESSA
– patologia neurologica cronica che coinvolge 1% popolazione (la più frequente dopo stroke);
– si presenta con episodi di crisi comiziali ripetute nel tempo, derivanti da scariche anomale di neuroni
cerebrali;
– eziologia: infezione, neoplasia, trauma, ereditarietà;
– tipi di crisi epilettiche:
– PARZIALI:
– si distinguono in semplici (senza perdita di coscienza) e complesse (con perdita di
coscienza, coinvolgono ippocampo);
– hanno origine corticale focale (++ temporale);
– causano manifestazioni localizzate e diverse (es. clonie/parestesie), a seconda
dell'area corticale colpita (es. corteccia motoria/sensitiva);
– possono divenire generalizzate;
– GENERALIZZATE:
– coinvolgono entrambi gli emisferi e quindi possono essere estese;
– “grand mal”: crisi tonico-cloniche generalizzate, con perdita di coscienza, di durata 12 minuti;
– “petit mal”: crisi di assenza, ossia improvvisa alterazione di coscienza, associata con
fissazione oculare e interruzione dell'attività in corso, che dura ≈30s;
– crisi toniche/atoniche/cloniche/miocloniche/spasmi infantili: possono colpire singole
parti del corpo o essere generalizzate;
TERAPIA
– in caso di crisi occasionali senza riscontri EEG/RM né familiarità, si può non trattare;
– si inizia con monoterapia a seconda del tipo di crisi (tonico-clonica e grande male vs assenza);
– durata terapia è attorno ai 2 anni: se crisi scompaiono, si prova a sospendere.
FARMACI
Si dividono in farmaci usati in:
CRISI PARZIALI E DI GRANDE MALE
1. azione prevalente di blocco Na+;
→ impedisce verificarsi di PdA ad alta frequenza;
1) fenitoina:
– effetti principali: blocco corrente Na+ e Ca2+, ↓ rilascio glutammato e ↑ GABA;
– finestra terapeutica ridotta (→ aggiustamento dosi deve essere graduale e lento);
– tossicità: nistagmo, diplopia, atassia, irsutismo; implicato in aritmia (anti-aritmico secondo
Katzung).
– sostituita da lamotrigina (nuovo farmaco): ha stessi effetti.
2) carbamazepina:
– triciclico usato in nevralgia trigeminale;
– riduce corrente Na+ e aumenta corrente K+;
2. azione prevalente di potenziamento GABAergico:
→ aumenta inibizione
Æ
51 / 55
1) fenobarbital:
– più vecchio antiepilettico disponibile, oggi sostituito (eccetto infanti) causa effetto sedativo;
– modulatore allosterico di GABA-A, prolunga tempo di apertura (ingresso Cl-);
2) gabapentin / pregabalin:
– analogo GABA;
– usato in crisi parziali e in dolore cronico neuropatico;
3) tiagabina:
– inibitore di uptake GABA;
4) valproato:
– usato in crisi generalizzate e in assenze;
– inibisce degradazione GABA e ne aumenta sintesi;
– riduce correnti Na+.
5) levetiracetam: (nuovo farmaco)
– lega proteina vescicolare sinaptica modificando il rilascio di glutammato e GABA;
CRISI DI ASSENZA E ALTRE CRISI GENERALIZZATE
1) etosuccimide:
– blocca canali Ca2+ soprattutto ipotalamici: si pensa siano origine di corrente pacemaker che causa
scariche ritmiche verso corteccia durante assenze;
2) valproato:
– usato in crisi generalizzate e in assenze;
– inibisce degradazione GABA e ne aumenta sintesi;
– riduce correnti Na+.
Æ
52 / 55
SOSTANZE CHE CREANO DIPENDENZA
PREMESSA: LA DIPENDENZA
– disturbo del comportamento dovuto all'uso e abuso di sostanze psicoattive più o meno lecite;
– implica l'incapacità di interrompere un uso protratto o ad alta frequenza di una sostanza:
– a causa della sindrome da astinenza che ciò comporta;
– indipendentemente dai suoi effetti negativi;
– deriva dall'interrelazione di 3 fattori concomitanti:
1) individuali: fattori genetici e di personalità;
2) farmacologici: effetti acuti e cronici (es. tolleranza, astinenza) propri della sostanza;
– ad es. il rischio di dipendenza a 10 anni da uso occasionale di cannabis e cocaina è
15%, di eroina 25%, di tabacco 30%;
3) ambientali: contesto familiare, socio-economico:
– sono fondamentali sia a breve termine che a lungo termine: un'esperienza positiva in
un momento di vita “normale” può essere recuperata come valvola di sfogo in una
situazione di difficoltà, innescando la dipendenza.
– il ciclo della sostanza prevede:
– uso > abuso > astinenza > riassunzione;
– il ciclo della dipendenza prevede:
– acquisizione > mantenimento > disassuefazione > ricaduta;
→ domande tipiche sono: quante volte? Hai mai provato a smettere?
– l'escalation della dipendenza prevede diverse fasi:
0) assunzione ripetuta per effetti positivi (rinforzo positivo);
1) tolleranza al farmaco: è necessario aumentare le dosi per lo stesso effetto;
2) sintomi di astinenza: più o meno marcati;
3) consapevolezza della compulsione all'uso;
4) comportamento focalizzato di ricerca della droga;
5) assunzione della sostanza per evitare i sintomi spiacevoli (rinforzo negativo);
6) tentativo di cessazione e ricaduta;
– ricaduta:
– 85% di pz dipendenti provano almeno una volta a smettere;
– cessazione coatta è relativamente semplice (pz chiuso in clinica);
– è molto probabile:
– per sindrome di astinenza (a breve termine);
– per persistenza di fattori individuali e ambientali (a lungo termine) che inducono
craving (bramosia) della sostanza;
– la durata media della dipendenza da sostanze è:
– 10 anni in oppioidi;
– 20 anni da alcol;
– 40 anni da tabacco.
PATHWAY IN SNC
– principale sistema coinvolto in dipendenza è il sistema dopaminergico mesolimbico;
– origina dall'area tegmentale ventrale e proietta a nucleo accumbens, amigdala, ippocampo e
corteccia prefrontale;
– assunzione di droga causa potente rilascio dopamina che determina rinforzo positivo;
– droghe di abuso che non stimolano il sistema dopaminergico (allucinogeni: LSD, ketamina) non
causano dipendenza (cit. Katzung che intende dipendenza come intrinsecamente fisica, legata a
sindrome da astinenza).
PRINCIPALI SOSTANZE CHE CREANO DIPENDENZA
Æ
53 / 55
→ AUMENTANO RILASCIO DOPAMINA da VTA
CLASSE 1: azione su GPRC (recettori metabotropi);
1) oppioidi:
– derivati dell'oppio che agiscono su recettori endogeni, responsabili di effetti farmacologici
(vedi capitolo dedicato) e di dipendenza (sono anche VTA-area tegmentale ventrale);
– causano aumentata produzione di DA per inibizione di interneuroni GABAergici in VTA;
– morfina (più basica, passa meglio EEB), codeina (tosse, analgesica), citometadone (farmaco
orale contro dipendenza), fentanil, idr+ossimorfone, ossicodone;
– rischio di dipendenza 4 su 5;
– naloxone per overdose;
2) THC (tetra-idro-cannabinolo):
– da infiorescenze di cannabis sativa (marijuana) o resina da esse prodotte (hashish);
– causa aumento di produzione dopamina per inibizione presinaptica di neuroni GABA;
– astinenza breve e lieve;
– rischio di dipendenza 2 su 5;
3) GHB (acido gamma-idrossi-butirrico):
– agisce su GABA-B come debole agonista;
4) allucinogeni:
– mescalina, LSD, psilocibina;
– inducono rilascio corticale di glutammato agendo su recettori presinaptici 5-HT;
– alterano percezioni in maniera vivida e imprevedibile;
– possono indurre sintomi psicotici;
CLASSE 2: azione su canali (recettori ionotropi);
1) benzodiazepine (es. tamazepam);
– modulatori positivi GABA-A (vedi capitolo sedativo-ipnotici);
– agiscono su interneuroni GABAergici di VTA (neuroni dopaminergici non hanno subunità
alfa-1);
– elevato rischio dipendenza perché utilizzo domiciliare;
2) etanolo:
– modula positivamente GABA-A e altri recettori determinando aumento dopamina;
– astinenza è grave (convulsioni, allucinazioni, delirium tremens) e richiede trattamento
supportivo con benzodiazepine;
– elevato rischio dipendenza e effetti avversi per sviluppo tolleranza;
3) barbiturici (es. nembutal)
– modulatori positivi di GABA-A, aumentando tempo di apertura (vedi sedativo-ipnotici);
– utilizzo solo intra-ospedaliero limita rischio diffusione;
4) nicotina:
– agonista selettivo di recettore nicotinico di ACh (eccitatorio);
– recettore è presente su neuroni DOPAminergici dell'ATV, che vengono eccitati;
– migliora umore, funzioni cognitive, ansiolitico;
5) ketamina:
– anestetico dissociativo con potenziale allucinogeno (vedi tra antidepressivi)
– rischio 1/5 di dipendenza ma esposizione cronica può causare psicosi;
CLASSE 3: azione su trasportatori;
1) cocaina:
– alcaloide di Erytroxylon coca disponibile in duverse forme:
– cocaina cloridrato (idrosolubile): iniettabile o assorbibile da membrane es. sniffata;
– cocaina crack: deriva da riscaldamento in soluzione alcalina, può essere fumata;
Æ
54 / 55
– inibisce reuptake di DA (→effetto ricompensa), NA (→iperattivazione simpatica) e 5-HT;
– rischio 5/5 di dipendenza (in individui predisposti, anche dopo poche dosi);
– non esistono antagonisti o trattamenti farmacologici per disintossicarsi;
2) anfetamine:
– sostanze simpatico-mimetiche sintetiche che causano aumentato rilascio di DA (→euforia,
possibile psicosi), NA (→eccitabilità) e 5-HT (→ anoressia, ipertermia);
– agiscono bloccando il sistema di caricamento vescicolare intraneuronale; ciò causa ↑[]i e
azione inversa da parte del trasportatore responsabile di reuptake →aumenta secrezione nonvescicolare, diminuisce quella vescicolare;
– via orale e ev;
– es. MDMA (metilen-diossi-met-anfetamina) “ecstasy”:
– affinità particolare per SERT → aumenta livelli 5-HT quindi empatia e socialità;
– è il prototipo di “designer drug” usata in rave party;
– tossicità: deplezione di 5-HT per 24h e possibile anche a lungo termine.
– tossicità:
– neurotossicità mediata da NMDA, soprattutto per neuroni dopa e serotoninergici;
– ipertermia.
Æ
55 / 55