c
tri y
a
i
ed olog y
P
g
r
n
s i ente colo
DI
w
R A ACI
Ne stro rma
U
A C CA P
Ga Pha
NI
O
M
Disordine linfoproliferativo post
trapianto nei bambini: diagnosi
precoce, gestione e terapie innovative
SILVIA RIVA, FRANCESCO CIRILLO, MARCO SCIVERES
Epatologia Pediatrica e Trapianto di Fegato, ISMETT - University of Pittsburgh Medical Center di Palermo
INTRODUZIONE
I disordini linfoproliferativi post-trapianto (PTLD) costituiscono un
gruppo clinicamente ed istologicamente eterogeneo di malattie che hanno a fattore comune l’insorgenza dopo un trapianto d’organo solido o ematologico (1).
La cellula d’origine (T o B), l’attitudine proliferativa, il grado di atipia cellulare
e il potenziale aggressivo della malattia variano in maniera considerevole secondo lo specifico sottotipo e si possono avere da forme di linfoproliferazione
policlonale, a scarsissimo potenziale maligno ma suscettibili di progressione
(Early Lesions), a forme linfomatose estremamente maligne paragonabili a
quelle che insorgono nel paziente non trapiantato [Tabella 1].
In età pediatrica la maggioranza (80%) e la quasi totalità delle forme precoci
(entro i sei mesi dal trapianto) sono a partenza da cellule B infettate ed immortalizzate da Epstein Barr virus (EBV). Le forme non EBV-correlate, in genere
più tardive e più aggressive (2), sono rappresentate principalmente da PTLD
T-cellulari.
L’incidenza è variabile in base all’età del paziente ed all’organo trapiantato:
inferiore al 5% dopo trapianto di rene, inferiore al 10% dopo trapianto di fegato, raggiunge il 10-32% dopo trapianto di polmone ed intestino, sia per l’utilizzo di schemi immunosoppressivi più impegnativi sia per la presenza di abbondante tessuto linfoide intrinseco nel graft (3).
Le forme “Early” possono essere completamente asintomatiche o presentare
una sintomatologia sfumata come ipertrofia adenotonsillare con ostruzione nasale, microadenomegalia diffusa, diarrea recidivante, astenia, calo ponderale,
febbricola; nelle forme monoclonali, al contrario, prevale la focalità per cui ai
sintomi generali si associano i segni dovuti alla presenza di una massa infiltrante in maniera non dissimile ai linfomi del paziente non trapiantato [Figura 1].
Epstein Barr virus (EBV)
is frequently related
to post-transplant
lymphoproliferative
disorder (PTLD). Early
diagnosis and treatment of
polyclonal variants could
probably avoid progression
toward malignant disease.
Treatment strategies for
PTLD
D include reduction
of immunosuppression,
targeting of B-cells with
monoclonal antibodies, or
chemotherapy. Adoptiv
A
Adoptive
doptive
doptive
immunotherapy with EBVspecific C
CTLs to restore a
cellular immune response to
EBV is an innovative and safe
treatment option.
Tabella 1 Classificazione OMS 2008 dei PTLD
Key Words
Post-transplant lymphoproliferative disorder,
EBV-specific cytotoxic T-cell response,
early detection and diagnostics, risk factors,
adoptive T-cell therapy
CATEGORIA
CLONALITÀ
STATO EBV
Lesioni precoci
(iperplasia plasmacitica - mononucleosi-like)
Policlonale
Sempre EBV positivo
PTLD Polimorfico
Monoclonale
Sempre EBV positivo
PTLD Monomorfico
Linfoma B cellulare
Linfoma T cellulare
Monoclonale
Monoclonale
Frequentemente EBV positivo
Raramente EBV positivo
PTLD Linfoma di Hodgkin like
Monoclonale
Sempre EBV positivo
EBV, Epstein-Barr virus; PTLD, disordine linfoproliferativo post trapiant. Modificato da [1]
Giorn Gastr Epatol Nutr Ped 2013; Volume V(1):27-30
27
News in Pediatric Gastroenterology Pharmacology
EBV - MECCANISMO DI INFEZIONE E PATOGENESI
DEL PTLD
EBV è un virus oncogeno, che appartiene alla famiglia herpes virus e ne condivide la caratteristica costante di determinare un’infezione latente che fa seguito ad una fase acuta, replicativa. L’infezione primaria nell’individuo immunocompetente avviene
nell’orofaringe mediante l’interazione tra la glicoproteina virale
GP340 e il recettore di superficie cellulare dei linfociti B: comporta
l'inizio di un ciclo replicativo virale e la morte della cellula infettata
per lisi cui segue il rilascio di nuove particelle virali complete ed
infettive. Al termine della fase replicativa EBV cambia strategia: il
genoma virale inizia la sintesi di alcune proteine nucleari (EBNA-1,
EBNA-2, EBNA-3A, EBNA-3B, EBNA-3C) e di membrana
(LMP-1, LMP-2A e LMP-2B) che governano il ciclo di latenza e
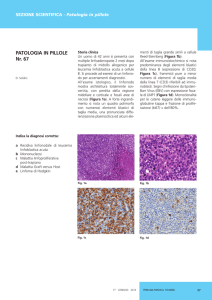
determinano un'attivazione permanente dei linfociti B (immortaFigura 1 Localizzazione sinusale con invasione
lizzazione cellulare). Alcuni di questi blasti entrano in fasi di latenza
intraorbitaria di PTLD B monomorfo (linfoma B a grandi
non proliferativa come cellule memoria capaci di brevi riattivazioni
cellule) a partenza dal tessuto adenoideo, insorto dopo 6
periodiche del ciclo litico. La maggior parte, tuttavia si replica indemesi dal trapianto di polmone in una bambina di 7 anni.
La piccola aveva manifestato a 2 mesi dal trapianto la
finitamente, sebbene a basso ritmo (4). Il controllo dell’infezione riprima infezione EBV con viremie elevatissime. Al momento
chiede entrambi i meccanismi di risposta immunitaria, cellulare ed
della diagnosi di PTLD la carica viremica era minima
umorale: la produzione di anticorpi limita la diffusione del virus nella forma infettiva, mentre l'attivazione del meccanismo di sorveglianza T cellulare agisce sulla proliferazione delle cellule B-memoria EBV infette, dimostrandosi efficace nel controllare la diffusione delle
cellule trasformate. Nel paziente immunosoppresso la funzione T-cellulare è deficitaria (i farmaci inibitori delle calcineurine limitano principalmente la funzione dei linfociti T) consentendo, quindi, ai linfociti B immortalizzati dal virus di andare incontro ad una proliferazione incontrollata ed allo sviluppo di
malattia linfoproliferativa (5)
MONITORAGGIO EBV E DIAGNOSI PRECOCE
Il fattore di rischio più significativo per PTLD è l’età al trapianto, per cui i bambini sono ipso facto una
popolazione a rischio. Tale rilievo è chiaramente un epifenomeno che rimanda a quanto detto circa
il ruolo patogeno dell’EBV. Il reale fattore di rischio, molto comune nei bambini, è infatti lo status di
sieronegatività per EBV al momento del trapianto. Tale condizione minimizza la possibilità di sviluppo di un'efficace risposta immune e, in ultima analisi, la citolisi dei blasti proliferanti EBV+. Anche il
tipo di trapianto effettuato porta con sè un potenziale di rischio: quello di organi con una maggiore
quantità di tessuto linfoide o che richiedono un’immunosoppressione più profonda (es. polmone, cuore o intestino) implica un rischio di PTLD notevolmente maggiore. Altri fattori di rischio minori, non
universalmente riconosciuti, sono il matching tra un ricevente EBV- ed un organo EBV+ (la prima
infezione nei riceventi di organi EBV- è solo ritardata, non evitata), la concomitante infezione da
CMV ed il tipo di immunosoppressione (tacrolimus più di ciclosporina) (1,3).
Il monitoraggio per PTLD del bambino trapiantato, in mancanza di un singolo parametro efficace
ed affidabile, si basa su un insieme di parametri clinici e bioumorali. In primo luogo vi è necessità di
seguire l’andamento dell’infezione da EBV, specie dopo l’avvenuta prima infezione o riattivazione del
virus. Lo strumento più utile e quello su cui vi è maggiore esperienza è la viremia EBV. È piuttosto
forte infatti l’evidenza che, al momento della fase replicativa, quindi in genere precocemente dopo il
trapianto, alte viremie EBV rappresentino un importante fattore di rischio. Nel tempo la tecnica di
rilevazione del DNA si è evoluta e si è passati dalla semplice PCR su plasma, che misura principalmente le copie di DNA “libero” provenienti dalla lisi cellulare, alla misurazione delle copie presenti
all’interno delle cellule mononucleate (PBMC, peripheral blood mononuclear cells). Quest’ultima
tecnica offrirebbe un quadro molto più preciso del numero di linfociti B EBV-carrier in fase di latenza (6). Tuttavia non sempre è documentata una correlazione fra PTLD e “viral load”. La recente introduzione del test di valutazione della risposta linfocitotossica EBV-specifica appare promettente. La
rilevazione di una risposta cellulo-mediata ridotta o assente, espressa come numero di linfociti T del
paziente producenti Interferon-gamma dopo attivazione con antigeni di EBV (ELISPOT), indica
28
Giorn Gastr Epatol Nutr Ped 2013; Volume V(1):27-30
TIPO DI PTLD
TERAPIA DI PRIMA SCELTA
Riduzione
dell'immunosoppressione (RI)b
Lesioni precoci
Sistemico
Polimorfico
Localizzato
RI, se possibileb e:
• Solo Rituximab
o
• Chemioimmunoterapiae
RI, se possibileb e:
• RT ± Rituximab
o
• Chirurgia ± Rituximab
o
• Solo Rituximab
RI, se possibileb e/o:
Monomorficoa
• Solo Rituximabd
o
• Chemioimmunoterapiac
RISPOSTA INIZIALE
TERAPIA DI SECONDA SCELTA
Risposta completa
Gestione immunosoppressionee
e monitoraggio EBV PCR
Persistenza o progressione
della malattia
Rituximab and monitor EBV PCR
Risposta completa
Persistenza o progressione
della malattia
Risposta completa
Persistenza o progressione
della malattia
Si consideri la profilassi per la sindrome lisi tumorale (vedi NHODG-B)
Vedi anticorpo monoclonale e riattivazione virale (NHODG-B)
aIl trattamento si basa unicamente sull’istologia
altamente sintomatici o che non tollerano la chemioterapia a
causa di comorbidità
e
strettamente monitorati; la RI deve essere coordinata con Il reincremento dell’immunosoppressione deve essere
individualizzato, tenendo in considerazione il livello di RI
l'equipe di trapianto
iniziale ed il tipo di trapianto d'organo. Tali decisioni
cChemioimmunoterapia concomitante o sequenziale
devono essere prese in collaborazione con il team di
dCome parte di un approccio graduale in pazienti che non sono
trapianto.
bLa risposta alla RI è variabile ed i pazienti devono essere
Monitoraggio EBV PCR e:
• Osservazione
o
• Continuare RI, se possibile
± mantenimento Rituximab
Chemioimmunoterapiac
o
Sperimentazione clinica
o
Immunoterapia cellulare
con linfociti T citotossici EBV
specifici (se EBV correlate)
Vedere le linee guida
istologiche appropriate
per il follow-up
Se RI era la terapia di prima
scelta, poi Rituximab o
Chemioimmunoterapiac
o
Se Rituximab era la
monoterapia di prima scelta,
poi Chemioimmunoterapiac
o
Sperimentazione clinica
o
Immunoterapia cellulare
con linfociti T citotossici EBV
specifici (se EBV correlate)
Note: Tutte le raccomandazioni sono categoria 2A, se non diversamente indicato.
Sperimentazioni cliniche: NCCN crede che la migliore gestione di qualsiasi malato di cancro sia la sperimentazione clinica. La partecipazione a sperimentazioni cliniche
è particolarmente incoraggiata.
Figura 2 Trattamento del PTLD. Linee guida 2013 del National Comprehensive Cancer Network
infatti un deficit del paziente a sviluppare una risposta citotossica nei confronti di cellule EBV-positive.
I limiti di tale tecnica sono la scarsa standardizzazione (diversi tipi di melange antigenici) e l’assenza
di una reale validazione “sul campo” come predittiva di sviluppo di PTLD (7). Accanto alla sorveglianza virologica, l’attento follow-up clinico riveste un ruolo cruciale. È necessario valorizzare prontamente quei segni e sintomi, spesso sfumati o aspecifici, non riconducibili ad una causa nota ed alternativa. L’ipertofia adenotonsillare, comune nel bambino, nel paziente trapiantato assume
un'importanza completamente diversa. È infatti noto che l’anello linfatico orofaringeo rappresenta il
principale serbatoio di replicazione del virus EBV ed è precocemente sede di fenomeni di iperplasia
linfoide (8) istologicamente non dissimili da quello che si osserva nella mononucleosi infettiva, salvo il
fatto che non tendono alla autolimitazione spontanea. Il tessuto adenotonsillare, infatti, è la sede più
frequente di PTLD focali monoclonali nel bambino (3), verosimile evoluzione di quadri inizialmente
policlonali e benigni.
L’obiettivo di una diagnosi precoce non può che essere perseguito tramite un campionamento istologico dei tessuti accessibili in maniera mini-invasiva (adenoidi, tonsille, tessuto linfoide associato alla
mucosa intestinale) o la biopsia di una lesione focale sospetta.
L’individuazione di un paziente con sintomi compatibili, in assenza di chiare lesioni focali, offre la
preziosa opportunità di evidenziare un processo linfoproliferativo in stadio precoce e scarsamente
aggressivo. Presso il nostro Istituto è operativo un programma di screening istologico precoce che,
in pazienti selezionati sulla base di elementi clinici, ha permesso di dimostrare un'incidenza superiore all'80% di forme “Early” (9) e di avviare precocemente un'adeguata presa in carico terapeutica. Tale modus operandi riteniamo sia il principale determinante della completa assenza di diagnosi di PTLD monomorfi nella nostra coorte di pazienti trapiantati che ormai approssima i 5
anni di follow-up mediano.
Giorn Gastr Epatol Nutr Ped 2013; Volume V(1):27-30
29
News in Pediatric Gastroenterology Pharmacology
Key Points
• I disordini linfoproliferativi post trapianto rappresentano un gruppo
clinicamente ed istologicamente eterogeneo di malattie, con
ampio spettro clinico: da lesioni
benigne a forme linfomatose.
• l principale fattore di rischio è
rappresentato dall'infezione primaria da EBV che avviene dopo
il trapianto, in quanto nel paziente immunosoppresso la funzione
T-cellulare è deficitaria consentendo ai linfociti B immortalizzati
dal virus di andare incontro ad
una proliferazione incontrollata
ed allo sviluppo di malattia linfoproliferativa.
• Il monitoraggio dell'infezione si
avvale della determinazione
della viremia EBV mediante PCR
e del test di valutazione della
risposta linfocitotossica EBVspecifica.
• La diagnosi precoce richiede
un’attenta sorveglianza clinica
ed il campionamento istologico
dei tessuti accessibili in maniera
mini-invasiva (adenoidi, tonsille,
tessuto linfoide associato alla
mucosa intestinale) o la biopsia
di una lesione focale sospetta.
• L'approccio terapeutico dipende soprattutto dal sottotipo di
disordine: tuttavia la terapia più
innovativa, clinicamente validata, è rappresentata dall’uso di
linee cellulari T-linfocitarie autologhe addestrate in vitro ad aggredire i linfoblasti portatori di EBV e
ripristinare l’immunosorveglianza
virus specifica.
30
TERAPIE INNOVATIVE
Il trattamento delle diverse forme di PTLD è schematizzato in Figura 2. Le forme linfomatose si avvalgono della combinazione di Rituximab (anticorpo monoclonale antiCD 20), capace di eliminare la maggioranza dei linfociti B, con cicli brevi di chemioterapia che limita le popolazioni cellulari rapidamente proliferanti. Con questa
combinazione la frequenza di remissione completa supera l’80%. Il novero di presidi
terapeutici tradizionalmente adoperati nel caso di forme “Early” policlonali è al contrario molto limitato. La relativa benignità del quadro non giustifica infatti l’uso del
Rituximab (che non presenta un profilo di sicurezza ideale) e l’unico approccio storicamente adoperato è stato la riduzione o la sospensione della immunosoppressione.
Sebbene il tasso di regressione sia molto elevato, tale approccio comporta un rischio
elevato e non accettabile di rigetto acuto o cronico e di perdita del graft. Un possibile
approccio alternativo può essere l’uso di farmaci immunosoppressori differenti dal
tacrolimus senza ridurre significativamente l’impegno immunosoppressivo globale.
Esistono in letteratura diversi dati che indicano negli inibitori delle m-TOR delle valide alternative al tacrolimus e la nostra esperienza preliminare con la rapamicina è
stata più che positiva. L’approccio più innovativo, tuttavia, è l’uso di linee cellulari Tlinfocitarie autologhe addestrate in vitro ad aggredire i linfoblasti portatori di EBV che
presentano un profilo di immunogenicità particolarmente favorevole. L’esperienza
con queste cellule, dette CTL (Cytotoxic T Lymphocytes) EBV specifiche è ormai
consolidata ed i risultati sono eccellenti (10). Il principale ostacolo al loro utilizzo è la
disponibilità di una Cell-Factory capace di prepararle per ogni singolo paziente.
CORRESPONDING AUTHOR
SILVIA RIVA
Epatologia Pediatrica e Trapianto di Fegato
ISMETT - University of Pittsburgh Medical Center di Palermo
Via Tricomi, 1 - 90127 Palermo
Tel. + 39 091 2192111
Fax + 39 091 2192201
E-mail: [email protected]
BIBLIOGRAFIA
1. Kamdar KY, Rooney CM, Heslop HE. Posttransplant lymphoproliferative disease following liver
transplantation. Curr Opin Organ Transplant 2011 Jun;16(3):274-80.
2. Pinho-Apezzato ML, Tannuri U, Tannuri AC et al. Multiple clinical presentations of lymphoproliferative
disorders in pediatric liver transplant recipients: a single-center experience. Transplant Proc 2010
Jun;42(5):1763-8.
3. Allen UD, Farkas G, Hébert D et al. Risk factors for post-transplant lymphoproliferative disorder in
pediatric patients: a case-control study. Pediatr Transplant 2005 Aug;9:450-5.
4. Snow AL, Martinez OM. Epstein-Barr virus: evasive maneuvers in the development of PTLD. Am
J Transplant 2007 Feb;7(2):271-7.
5. Martinez OM, de Gruijl FR. Molecular and immunologic mechanisms of cancer pathogenesis in
solid organ transplant recipients. Am J Transplant 2008 Nov;8:2205-11.
6. Baldanti F, Grossi P, Furione M et al. High levels of Epstein-Barr virus DNA in blood of solid-organ
transplant recipients and their value in predicting posttransplant lymphoproliferative disorders. J Clin
Microbiol. 2000 Feb;38(2):613-9.
7. Davis JE, Sherritt MA, Bharadwaj M et al. Determining virological, serological and immunological
parameters of EBV infection in the development of PTLD. Int Immunol. 2004;16:983-9.
8. Shapiro NL, Strocker AM. Adenotonsillar hypertrophy and Epstein-Barr virus in pediatric organ
transplant recipients. Laryngoscope. 2001;111:997-1001.
9. Sciveres M, Vitulo P, Riva S et al. Early Detection Of Lymphoproliferative Disorders (Ptld) In
Paucisymptomatic Pediatric Liver Transplant Recipients By Adenotonsillar Histology. J Pediatr
Gastroenterol Nutr 2011;52 S1:E19 (abstract).
10. Heslop HE, Slobod KS, Pule MA et al. Long-term outcome of EBV-specific T-cell infusions to
prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood.
2010;115:925-35.
Giorn Gastr Epatol Nutr Ped 2013; Volume V(1):27-30