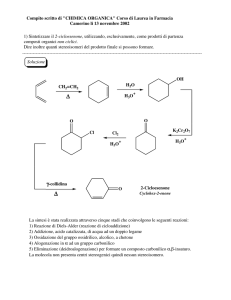
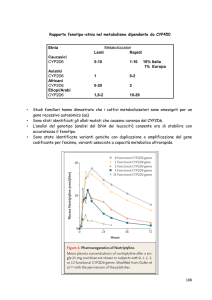
Capitolo 8
METABOLISMO DEI FARMACI
I metabolismo dei farmaci è essenzialmente un meccanismo di difesa di un organismo
vivente contro composti esogeni estranei e non essenziali ai processi vitali: xenobiotici, estranei alla
vita. La finalità è di inattivare 1 dette sostanze (detossificazione) e, contemporaneamente, di
renderle più facilmente eliminabili. Qualche volta la biotrasformazione può aumentare l'attività
biologica della sostanza in circolo: si parla, allora, di profarmaci, qualora venga potenziata
l'attività terapeutica desiderata; o veleni potenziali qualora la nuova attività o l'incremento di
attività così acquisiti non siano desiderati. In qualche caso i metaboliti sono dotati di un quadro
di attività farmacologica parzialmente sovrapponibile al quello del composto di origine; questa
evenienza darà luogo ad una durata d'azione maggiore, che deve suggerire un adeguato
protocollo terapeutico.
Le trasformazioni metaboliche normalmente portano ad un aumento di idrofilia degli
xenobiotici, facilitando così la loro eliminazione per le vie più comuni, come renale, biliare,
fecale, essudativa 2; al contrario la vie meno utilizzate come la polmonare e la traspirativa
richiedono diminuzione della polarità e del peso molecolare in modo da diminuire la tensione di
vapore.
Fra gli xenobiotici che suscitano una risposta di difesa metabolica, non immunitaria, si
possono ricordare:
Farmaci e Sostanze di Sintesi
Polveri e Solventi
Fumo e Bevande
Veleni e Tossine
Precancerogeni e Cancerogeni ...
Data la natura enzimatica delle trasformazioni metaboliche, gli enzimi essere caratterizzati da
una grande adattabilità alla struttura di un substrato così variabile come imprevedibile . Questa
versatilità è ottenuta con uno o più dei seguenti fattori: a) polimorfismo genetico, cioè dotazione
genetica di numerose isoforme; b) polimorfismo inducibile, rapida proliferazione di nuove
isoforme stimolata dalla presenza dello xenobiotico stesso; c) induzione metabolica, aumento
dell'attività enzimatica (dovuta ad aumento della sintesi di enzimi metabolici) in seguito ad
esposizione a xenobiotici ; d) ridotta selettività nei confronti del substrato.
Fattori che influenzano la capacità metabolica:
1) Genetici: differenze di specie e individuali
2) Fisiologici: età, sesso, gravidanza, malattie: insufficienza epatica (riduzione del corredo di enzimi
epatici e digestivi).
3) Farmacodinamici: protocollo terapeutico, distribuzione tissutale, combinazione con proteine,
effetto di primo passaggio (vedi)
4) Ambientali: competizione con altre sostanze, avvelenamento di sistemi enzimatici
5) Attività della Flora Batterica Intestinale: ad. esempio fornisce glucoronidasi che rilibera il farmaco
dall'escreto biliare che così viene riassorbito dall'intestino; bioattivazione di precursori inattivi
(cancerogeni da precancerogeni).
6) Forma farmaceutica: tempo di permanenza nell' intestino...
2
Si consulti un testo di Fisiologia per una sommaria descrizione di queste funzioni escretorie.
75
Fasi Metaboliche
Il metabolismo dei farmaci si attua attraverso due fasi:
REAZIONI DI FASE I o Fase di Funzionalizzazione attuata attraverso i seguenti tipi di reazioni:
1. Reazioni di ossidazione: che introducono nella molecola del farmaco nuovi gruppi
idrofili e/o protici (come, OH NH2 COOH SH ...)
2. Reazioni di idrolisi: che liberano detti gruppi idrofili da eteri,esteri, ammidi, acetali,
emiacetali. epossidi ...
3. Reazioni di riduzione: meno diffuse, che possono generare alcuni di detti gruppi idrofili
o protici
Scopo funzionale:
rendere più idrofilo il composto estraneo per facilitarne l'eliminazione
inattivare il composto
preparare il substrato per la Fase II
REAZIONI DI FASE II o Fase di Coniugazione, attuata attraverso i seguenti tipi di reazioni:
1. Reazioni di coniugazione: accoppiamento con piccole biomolecole ionizzabili o
altamente idrofili, come: Ac. Glucuronico, Ac. Solforico, Aminoacidi, Glutatione ...
2. Reazioni di coniugazione con bioreagenti apolari: Metilazioni Acilazioni
Scopo funzionale:
1. Inattivare e rendere nettamente idrofilo per una completa eliminazione
2. Inattivare e/o aumentare il peso molecolare per facilitare l'eliminazione biliare
Reazioni Enzimatiche di Ossidazione
Il sistema enzimatico più diffuso per l'ossidazione di xenobiotici è il Citocromo P450 3,
chiamato anche CYP450 , monoossigenasi, ossidasi o idrossilasi. É localizzato nella frazione
microsomiale di fegato, rene, polmone, intestino ...
Costituito da:
Eme-Proteina: responsabile del trasporto di e- e attivazione di O2
NADPH associato a NADPH-citocromo reduttasi
e altri cofattori
Differenziatosi geneticamente per metabolismo di steroidi e acidi biliari, è in tutte le specie
viventi caratterizzato da spiccato polimorfismo genetico e polimorfismo inducibile e costituisce
così uno dei più versatili sistema di difesa da sostanze estranee alla biologia cellulare.
Famiglie di CYP450
Sono state identificate più di 110 famiglie di CYP450 per le quali viene adottata la seguente
nomenclatura:
CYP seguito da:
1° Numero, che indica la famiglia con membri presentanti circa il 35% di omologia
Lettera maiuscola, indica la sottofamiglia caratterizzati dal presentare il 40-60% di omologia
2° Numero, sottotipo con omologia ancora più elevata
Il nome deriva dal fatto che la forma ridotta con Fe 2+ lega monossido di carbonio per dare un complesso con un
massimo di assorbimento a 450 nm (nel blu)
3
76
Alcuni Tipi di CYP450, e relativo substrato (da non memorizzare):
CYP1A1
Idrocarburi Policiclici Aromatici, Arilamine, Estrogeni
Indotta da fumo di tabacco e da detti idrocarburi aromatici
Arilamine, Nitrosamine, Idrocarburi Aromatici
Indotta da fumo di tabacco
Cumarine, Aflatossina B1
Testosterone (nel Ratto)
Numerosi farmaci: amine lipofile binding a coppia ionica
Stereoselettiva, non inducibile
Inibita dalla chinina
Alogenoderivati e piccole molecole (etanolo, benzene, acetonitrile, DMF ...
Molto numerosa …
CYP1A2
CYP2A6
CYP2D6
CYP2E1
CYP3A...
N.B. La sequenza aminoacidica dell'eme-proteina determina l'affinità dell'enzima per una
deteminata classe chimica di substrato: la presenza di AA apolari come leucina isoleucina e valina
in prossimità del centro catalitico può formare una sacca lipofilica in grado di legare sostanze
apolari; la presenza di fenilalanina determina affinità per idrocarburi aromatici, residui di AA
bicarbossilici (glutamato e aspartato) tendeno a legare composti con centri cationici, al contrario
residui di AA tipo lisina e arginina determineranno affinità per i composti carichi negativamente
...
Inibizione del CYP450 :
Inibitori reversibili: monossido di carbonio, fluorochinoloni, cimetidina, …
Inibitori complessanti: complessano gli intermedi del ciclo catalitico del CYP450 e sono
nitrosoalcani, antibiotici macrolidici, …
Inibitori irreversibili: Inibizione “suicida” o inibiz. basata sul metabolismo, cioè l'enzima
produce dei metaboliti che lo inattivano. Questi metaboliti sono radicali formati formati da
alcheni, alchini, da aloalcani ..., da ormoni androgeni e progestinici, epossidi e acilalogenuri da
polialogenoderivati (vedi avanti).
Centro Catalitico Ferro(III) eme tiolato:
R S
N
3
N
Fe
N
N
OH2
La carica del ferro trivalente (N° di coordinazione 6) è
neutralizzata da due carica negative su due atomi di azoto pirrolici
del nucleo porfirinico (dove per brevità le catene laterali sono
omesse) e la terza carica negativa è sullo zolfo di un residuo di
cisteina della porzione proteica del citocromo. Il sesto legando è
una molecola di acqua, debolmente legata, che appunto nel ciclo
catalitico viene sostituita dall'ossigeno.
Ciclo Ossidativo
a) Il substrato R-H viene adsorbito su una regione proteica dal CYP450-[Fe(III)]
77
b) Il Fe(III) del gruppo eme, dopo riduzione 4 a Fe(II) da parte del NADPH, lega una
molecola di ossigeno che prende il posto della molecola di acqua. Dopo una serie di
reazioni in sequenza nelle quali interviene altro NADPH si genera la forma del CYP450
cataliticamente attiva per l'ossigenazione: Fe(V)=O, ossene perferrile con Fe a numero di
ossidazione 5+ e ossigeno con caratteristiche di biradicale ·O· ovvero di ossigeno
atomico.
c) L'ossene perferrile strappa un atomo di idrogeno dal substrato in modo da formare il
radicale R· ; la probabilità e le percentuali relative dei vari possibili radicali dipende dalla
rispettiva stabilità e dalla libertà sterica. Si ricorda che i radicali, come i carbocationi, sono
stabilizzati da effetti a rilascio elettronico. Il Fe si riduce a 4+ fornendo il perferrilidrossido, Fe(IV)-OH, dove ·OH ha carattere di radicale
d) Accoppiamento radicalico fra R· e ·OH . Il Fe ritorna a numero di ossidazione 3+.
e) Rilascio del prodotto ossidrilato R-OH e rigenerazione del CYP450.
SCHEMA 1 Ciclo ossidativo del CYP450
RH
P450[Fe(II)H2O
]
RO
H
P450[Fe(II) RO
H]
P450[Fe(II)RH]
+2e
O
22H
H2O
P450[Fe(V)=ORH]
P450[Fe(IV)O
HR]
I passaggi sopra descritti possono essere riassunti dalle seguenti equazioni, dove ·O·
sostituisce il perferril-ossene e ·OH il perferril-idrossido:
O2 + 2H+ + 2 e·O· + R-H
·O· + H2O
[ R· + ·OH] R-OH
O2 + 2H+ + 2e-
R-OH + H2O
Ossidazione di Catene Alchiliche:
Le catene alchiliche sono più reattive dei residui arilici nelle reazioni radicaliche: il centro
radicalico viene stabilizzato da effetti di risonanza (coniugazione con legami ), iperconiugativi
(coniugazione con legami C-H) ed induttivi, come dal seguente prospetto:
Stabilità del radicale carbocatione
Il Ferro-eme, analogamente a quello della emoglobina, per poter legare l'ossigeno molecolare deve essere ridotto a
Fe (II), cioè ferro a numero di ossidazione 2+.
4
78
allilico > benzilico > CIII > CII > CI > arile
Effetto iperconiugativo potenziato da vicinanza di metili:
Me3C∙ > Me2CH∙ > MeCH2∙ > RCH2CH2∙ > R2CHCH2∙ > R3CCH2∙
Reattività per Impedimento Sterico
CI > arile > CII > CIII (più deboli ma opposti agli effetti precedenti)
Meccanismo dell'ossidazione enzimatica di radicali alchilici
Lo schema sotto riportato illustra una tipica ossidrilazione di una catena alchilica che
procede con un meccanismo radicalico come già descritto in precedenza. Un eventuale prodotto
secondario può derivare dalla deidrogenazione ossidativa con introduzione di un doppio legame;
questa via metabolica è facilitata da carbocationi intermedi stabili e impediti stericamente ad
avvicinarsi al sito catalitico.
SCHEMA 2
Fe(III)
Fe(IV) OH
CH2
CH
CH2
CH2
OH
CH
(Principale)
CH2
Fe(V)=O Fe(IV) OH
Fe(IV) OH
CH
CH
CH2
CH
Fe(III) OH
Fe(III) OH
Fe(III)
Ad es. nei barbiturici la resistenza dei residui idrocarburici in C5 è determinante per la durata
del sonno da questi indotto: raggiunge una durata di 12 h con il fenobarbitale e di 4-6 h con gli
altri in accordo con la rispettiva emivita come sotto riportato.
SCHEMA 3
H 3
O
H
N
O
CH2 CH3
N
H
1
O
Fenobarbitale
t1/2 = 80-100 h
H
O
Pentobarbitale
t1/2 = 15-50 h
O
1'
CH2
N
CH2 CH3
N
H
4'
CH2 CH3
CH CH2
O
1' 2'
N
5
O
O CH3
3'
O
CH3
Aprobarbitale
t1/2 = 14-34 h
O
N
CH3
3'
1'
CH2 CH
CH2
CH3
N
CH CH3
O
CH CH3
N
H
CH
H
3'
CH2
H
O
Amobarbitale
t1/2 = 10-30 h
Nel fenobarbitale il gruppo CH2 legato al sistema eterociclico dà radicali poco stabili per
l'effetto elettronattrattore di detto anello; più disponibili per l'ossidrilazione da parte del CYP450
rimangono il CH3 e il fenile, relativamente poco reattivi.
Nel Pentobarbitale è presente un radicale ramificato, 1'-metilbutile; tuttavia il carbonio
terziario 1', oltre che risentire del risucchio di elettroni da parte dell'eterociclo, è stericamente più
impedito dei C secondari 2' e 3', dei quali è preferito 3' per il minor ingombro e per
l'iperconiugazione con il metile 4'.
Nell'aprobarbile il carbonio più reattivo verso l'ossidrilazione radicalica è l'1' in posizione
allilica: l'efficace stabilizzazione per risonanza compensa e supera l'inattivazione da parte
dell'eterociclo.
Nell'amobarbitale il centro più attivo per l'ossidrilazione è il C3' del radicale isoamilico: è
terziario, è inoltre lontano dall'eterociclo ed adiacente a due metili.
79
Si può concludere che nei barbiturici, come in altri composti, l'introduzione di ramificazioni
o di insaturazioni nei sostituenti in C5 riduce sensibilmente la loro durata d'azione per essere
metabolizzati più velocemente ed estensivamente.
Ossidazione Anelli Aromatici:
Come si è accennato in precedenza i residui aromatici sono più resistenti all'ossidazione
radicalica rispetto a residui alifatici. In ogni caso la loro reattività e alquanto variabile, risentendo
sensibilmente della presenza di sostituenti e degli ingombri sterici quasi in maniera parallela alle
stesse reazioni condotte in ambiente chimico. I sostituenti a rilascio elettronico stabilizzano i
radicali e, particolarmente, i carbocationi intermedi della reazione enzimatica di ossidrilazione e
così aumentano la reattività orientando l'OH in para e orto; quest'ultima posizione è meno
favorita per la compressione sterica e per l' effetto orto. La presenza su strutture aromatiche di
sostituenti a risucchio elettronico rendono praticamente impossibile l'ossidazione da parte del
CYP450. Allo stesso modo gli anelli eterociclici (es. anelli pyrrolici, piridinici, pirimidinici ...), a
causa dell'aromaticità e dell'effetto elettronattrattore dell'eteroatomo, praticamente sono molto
più resistenti alla ossidazione enzimatica così come lo sono a quella chimica.
Ad es. l'ossidazione della fenacetina, un vecchio analgesico antipiretico ormai in disuso,
procede secondo il seguente schema:
NHCOCH3
CYP450
HO
NHCOCH3
Il prodotto di ossidazione in para, cioè la p-idrossi-acetanilide o paracetamolo, è nettamente
il principale, come avverrebbe in una sostituzione elettrofila aromatica essendo il sostituente RCONH orto/para orientante. Come si vedrà nel Cap. 22, il paracetamolo è più attivo e meno
tossico dell'acetanilide ( da ritenere un pro-drug ): la biotrasformazione è quindi favorevole.
Meccanismo dell'ossidazione enzimatica di residui aromatici attivati
Un possibile meccanismo dell'ossidazione dei sistemi aromatici da parte del CYP450 viene
illustrato nello schema seguente. Gli ossidanti, come gli elettrofili, forniscono dapprima un
complesso che può dar luogo al complesso radicalico o ad un comlesso ionico, comune
alle sostituzioni elettrofile aromatiche; nel primo caso il Fe(V)=O/eme si appropria di uno dei
due elettroni del legame riducendo il suo numero di ossidazione da 5+ a 4+, nel secondo
acquista entrambi gli elettroni riducendosi a Fe3+. Dal complesso radicalico si distacca Fe3+
lasciando un elettrone spaiato sull'ossigeno: l'accoppiamento dei due centri radicalici formati,
sull'ossigenio e sul carbonio, fornisce l'epossido. Gli epossidi sono agenti alchilanti molto efficaci
alchilando gli azoti del gruppo eme del citocromo, residui lisinici della parte proteica del stesso
citocromo (catalisi suicida) o di altri enzimi producendo varie manifestazioni tossiche. Esistono,
tuttavia numerosi meccanismi di difesa come l'idrolisi a cicloesadien.glicole o la coniugazione con
il glutatione (vedi avanti) per dare composti meno reattivi.
80
SCHEMA 4
R
R
H
Fe(V)=O
(Compl. )
R
R
Fe(III)
(Epossido)
Idrolasi
H
R
GSH
(Enzima)
Fe(III)
R
OH
HO
O
Fe(IV) O
(Compl. rad.)
R
R
HO
NIH shift
H
H
S G
H
Fe(III)
Fe(III) O
O
H
HO
Principale
(Compl. )
Nel complesso ionico il distacco di Fe 3+ lascia una carica negativa sull'ossigeno che si
accoppia con il C+ per dare l'epossido come sopra. Tuttavia, preferibilmente, si ha migrazione di
ione idrogeno negativo o idruro (NIH shift) con distacco di Fe3+, l'ossigeno negativo fornisce gli
elettroni per il legame con il C vicinale; la forma carbonilica così ottenuta tautomerizza
completamente a fenolo. Il fenolo è molto più stabile dell'epossido perchè quest'ultimo ha
tensioni di legame e perde la stabilizzazione aromatica. Il fenolo si può formare anche per
dismutazione 5 dell'epossido.
Il -glicol del cicloesadiene prodotto dalla epossidoidrolasi per autoossidazione o per
deidrogenazione aromatizza facilmente per dare l' o-difenolo corrispondente o catecolo, che poi a
sua volta è soggetto ad autossidazione per dare un o-chinone:
R
R
ossidazione o
R
O O
O2
R
O O
O2
deidrogenazione
HO
HO
OH
H
OH
O
H
OH
O
radicale idrochinone
catecolo
O
chinone
Il radicale idrochinone esercita azione citotossica combinamdosi con proteine e/o DNA. Anche
il chinone, per addizione nucleofilica 1,4 di gruppi amminici, produce l'effetto citotossico tipico
di queste sosttanze (vedi Cap. ??? , metabolismo delle catecolamine). Il glutatione, G-SH, quando
presente, evita l'azione tossica dei chinoni, comportandosi lui stesso da ottimo nucleofilo:
H
R
R
4
R
+
H
3
H
-
G S
R
GS
R
O
HO
OH
GS
2
1
O
O
H O
O
HO
O
HO
Schema della dismutazione. Nell'epossido i due carboni legati all'ossigeno hanno numro di ossidazione zero, dopo
il riassestamento di elettroni rappresentato con le frecce, il C legato all'OH di ossida a 1+ e l'altro si riduce ad 15
R
H
0
0
O
R
H
R
H
R
H
O
R
R
1+
1-
HO
H
81
L'anione radicale superossido, –O2· , produce direttamente danni cellulari ossidativi o
indirettamente producendo H2O2 per opera della superossidodismutasi (SOD) e radicali OH·
con altri processi enzimatici:
O O +
+
+ 2H
O O
SOD
O2 + HO OH
Analogamente la cancerogenità del benzene e degli idrocarburi aromatici in genere ( fenoli e
idrochinoni e di tutti gli intermedi di ossidazione a chinone) può almeno in parte essere attribuita
alla formazione di idrochinone radicalico e chinone in seguito a doppia ossidrilazione da parte
del CYP450, successiva formazione del radicale idrochinonico per opera di perossidasi e infine
autossidazione a chinone con produzione di anione radicale superossido, come visto in
precedenza:
CYP450
O
OH
OH
CYP450
O
O2
Peroxid.
Autoss.
OH
OH
O
L'epossido idrolasi (Schema 4) catalizza il processo di trans addizione di acqua agli epossidi per dare i
corrispondenti trans-1,2-dioli (-glicoli) e così svolge un importante ruolo nella detossificazione di
epossidi elettrofili altamente reattivi per la notevole tensione degli angoli di legame nel ciclo a tre termini.
Gli epossidi, in assenza di idrolasi oppure di glutatione e glutatione transferasi che fornisce un addotto
nettamente più idrofilo inerte e facilmente coniugabile con acido glucuronico come l'-glicole, potrebbero
attaccare proteine e DNA innescando processi di cancerogenesi e di mutazione. L'alta attività
cancerogena del benzo[a]pirene ( (1) Schema 5, dove le lettere 'b' indicano anelli benzenoidi a tre legami ;
le 'c' anelli chinonoidi con soli due legami ) è appunto dovuto alla formazione di una epossido nella
"regione di baia" cioè nella parte convessa della molecola. I doppi legami ossidabile dal CYP 450 sono
quelli esterni alla regione di baia: il legame p 4-5 è quello più ossidabile e forma il trans-benzo[a]pirene4,5-diidrodiolo (2), questo viene poi eliminato come solfato o come glucoronide senza alcun problema.
Ma quando viene ossidato il legame 7-8 per dare il trans-benzopirene[a]pirene-7,8-diidrodiolo (3), questo
viene rapidamente riossidato in 9-10, perché il legame p pur essendo nella regione di baia, ora è
completamente non aromatico. Sul benzo[a]pirene-7,8-diidrodiolo-9,10-diidroossido (4) non possono
intervenire né la epossido idrolasi né la glutatione transferasi perché impedite stericamente; tuttavia
l'epossido (4) è in grado di legarsi al DNA per produrre tumori cutanei o polmonari:
Regione
di baia
12
1
b
b
2
11
10
P450
epossido
idrolasi
3
9
b
b
c
7
6
5
8
4
HO
O
OH
(1)
(3)
P450
(Perossidasi)
P450
epossido
O
idrolasi
OH
O
(2)
OH
HO
OH
(4) Cancerogeno
N- O- S-Dealchilazioni ossidative:
82
Le dealchialzioni procedono secondo i seguenti schemi, producendo da una parte una
ammina o un alcol o un tiolo e dall'altra sempre un aldeide. Quando presente il gruppo metilico è
quello che preferenzialmente subisce l'ossidrilazioe e poi il distacco.
R NH CH3
R NH CH2 OH
gem.diolo
R NH2 +
CH2O
R O CH3
R O CH2 OH
gem.diolo
emiacetale
R OH
+
CH2O
R S CH2CH3
R S CH CH3
OH
emitioacetale
R SH
+ O CH CH3
Nel caso di assenza di gruppi metilici direttamente legati all'eteroatomo (come
nell'etilalchilsolfuro sopra riportato), l'ossidazione attacca il carbonio direttamente legato
all'eteroatomo stesso.
Meccanismo delle N- O- S-dealchilazioni
La via principale è quella che conduce ad aldeide e all'ammina dealchilata. Come prodotto
decondari si può avere l'N-ossido, secondo il seguente schema:
Fe(V)O
R N
R
Fe(IV)O
H
C R'
R'
R N
R
H
C R'
R'
Fe(IV)O
Fe(III)
R N
R
C R'
R'
Fe(IV)O
O
R N
R
Fe(III)
H
C R'
R'
(Secondario)
OH
O
R N H
R
+
C R'
R'
R N
R
C R'
R'
H2O
H+
R N
R
C R'
R'
R N
R
C R'
R'
Lo stesso schema è valido per le ossigeno- e tio-dealchialzioni.
Deamminazione Ossidativa
Simile alle N-dealchilazioni procede con lo stesso meccanismo, con la differenza che
richiede un altro sottotipo di CYP450 e libera ammoniaca invece di una ammina secondaria o
primaria
83
NH2
NH2
CH2
HC OH
HO CH
CYP450
HC=O
NH3
HO CH
HO CH
HO
HO
HO
OH
OH
OH
3,4-diidrossi.glicolaldeide
nor-adrenalina
Dealogenazione ossidativa (e riduttiva):
Gli alogenoderivati, per la loro stessa natura quasi sempre xenobiotici, sono molto diffusi: si
usano come insetticidi, pesticidi, anestetici generali, plasticizzanti, isolanti in trasformatori
elettrici, antiincendio e solventi commerciali. Solo gli alogenuri alchilici 6 prendono parte
facilmente a sostituzioni nucleofiliche (SN1 o SN2) o a dealogenazioni ossidoriduttive. Le
sostituzioni da parte di un nuclefilo dell'alogeno è tipica dei monoalogeno derivati e dei 1,2dialoderivati, mentre il cumulo di due o più atomi di alogeno su un atomo di carbonio riduce la
mobilità dell'alogeno stesso: gli atomi di alogeno con il loro effetto elettron attattore riducono la
stabilità del sia del carbocatione che del radicale intermedi [ R-(X)2C+ R-(X)2C∙]. Di qui la
possibilità di impiego del CCl4 come antiincendio e del teflon (perfluoroidrocarburo) nelle
protesi. La mobilità degli alogenuri dipende dalla forza di legame C-X e dalla polarizzabilità (cioè
dal volume) di X- come gruppo uscente; presenta, quindi, il seguente ordine: I > Br > Cl >> F.
Gli alogenoderivati e i polialogenoderivati non vicinali risultano tossici alchilando centri
nucleofilici di enzini, acidi nucleinici e altre biomolecole di interesse vitale. Il nostro organismo
di difende coniugando questi agenti alchilanti con glutatione (in presenza di glutatione transferasi)
7:
-
R-X + SG
→ R-S-G + Cl
-
→ ... derivati mercapturici stabili e idrofilici
La deidroalogenazione ossidativa catalizzata dal citocromo P450 è la via metabolica più
comune per numerosi idrocarburi gem-polialogenati. L'ossidazione produce una gem-aloidrina
intermedia che può eliminare acido alogenidrico e formare un derivato carbonilico (aldeidi,
chetoni, alogenuri acilici, fosgene [diclouro dell'ac. carbonico], ... ). La sequenza di reazioni,
inizialmente radicaliche e poi ioniche, è illustata nello schema sotto riportato. Per l'attacco iniziale
è necessaria la presenza di un -idrogeno.
Gli arilalogenuri ed i vinil alogenuri sono stabilizzati per risonanza che porta anche a rafforzamento e diminuzione
della polarità del legame C-X. L'effetto induttivo +I dell'alogeno è in direzione contraria all'effetto mesomerico +M
del radicale e pur prevalendo ne risulta indebolito:
6
Cl
Cl
Cl
Cl
Cl
Cl
Purtroppo gli 1,2-dialogeno derivati diventano mutageni perchè trasformati dal GSH in ioni episulfonio che sono
ancora più reattivi del composto di partenza e alchilano il DNA:
7
S
X
C
+ G-SH
C
+
X
-
-H -X
G
C
C
SN interna
C
X
-X
S
G
C
84
CF3
Fe(IV)OH
Fe(V)=O
Cl
Fe(IV)OH
Cl
C H
CF3
Br
Fe(III)
C
CF3
Br
Br
gem.aloidrina
alotano
C OH
H2O
Cl
CF3
HBr
Cl
CF3
COOH + HCl
idrolisi
C O
NH Proteina
acil cloruro
CF3
C O
acila NH2 di proteine (lisina)
Cl
Cl3C H
Cl3C OH
Proteina NH
C O
cloroformio
C=O
Proteina NH
Cl
fosgene
Gli alogenuri di acile e di carbonile sono tra gli intermedi più reattivi e più tossici: possono
reagire con l'acqua per formare acidi carbossilici e ioni alogenuri meno tossici, ma possono
reagire anche con molecole tissutali con conseguenze dannose. Le proteine acilate si comportano
da apteni stimolando risposte immunologiche e di ipersensibilizzazione (vedi). Il cloramfenicolo
(RNHCOCHCl2) è biotrasformato nell'alogenuro acilico (RNHCOCOCI) che acila selettivamente l'apoproteina del CYP450 rendendolo così inattivo. Fortunatamente, data la relativa
inerzia dei gem-polialogenuri, questa attivazione metabolica avviene su una bassa percentuale di
composto.
Ossidazioni Catalizzate da FMO
Un altro complesso enzimatico, la flavina monoossigenasi (FMO), la quale, analogamente al
CYP450 sebbene con meccanismo differente, attiva l'ossigeno molecolare. Anche la FMO è
R3N:
R2NH
2 R SH
R3N
R2N OH
R S
N-ossido di amine III
R3N+ O-
O
Idrossilamine con amine II
S R
Disolfuri da tioli
O
R S R
Solfuro
R S R
R S R
O
O
Solfossido
Solfone
microsomiale, ma è meno diffusa, presenta poche isoforme e minor inducibilità rispetto al
CYP450; è quindi dotata di minor specificità di substrato; tipicamente catalizza l'ossigenazione di
atoni di N e S, ma non le reazioni di dealchilazione di detti eteroatomi come viene brevemente
riportato nello schema successivo. I gruppi funzionali ossidati dalla FMO sono: Ammine 2ª e 3ª
acicliche cicliche aromatiche ed eteroaromatiche, le idrossilamine , le idrazine, tioli, solfuri aciclici
ciclici e teroaromatici, polisolfuri; i prodotti di ossidazione per la maggior polarità vengono
escrete dai reni.
Ossidazioni non Microsomiali
85
Nella frazioni mitrocondriale e nella frazione solubile di omogenati di tessuti esistono altri
tipi di ossidasi, fra le quali le più diffuse sono le seguenti.
Ossidazioni di Alcoli:
Le alcoldeidrogenasi, enzimi non specifici NAD dipendenti, ossidano la maggior parte
degli alcoli 1ª a corrispondenti aldeidi, mentre solo alcuni dei secondari sono convertiti a chetoni;
i rimanenti alcoli 2ª, unitamente agli alcoli 3ª, rimangono immodificati nelle biotrasformazioni di I
Fase e sono eliminati come tali o come coniugati.
CH3-CH2-O-H
CH3-CHOH-CH3
CH3-CHO
CH3-CO-CH3
L'etanolo per circa 1/3 viene metabolizzato anche da una determinata isoforma di CYP450
(CYP2E1) la cui attività viene fortemente incrementata dal consumo delle bevande alcoliche.
L'induzione di questa isoforma contribuisce all'attivazione metabolica di molte sostanze, come
anestetici generali, analgesici, benzodiazepine ... la cui assunzione contemporanea all'alcol o da
parte di forti bevitori deve essere vivamente sconsigliata. L'acetaldeide contribuisce alla tossicità
epatica, cardiovascolare e di altro tipo. Inoltre, l'acetaldeide combinandosi con triptofano e
derivati produce carboline (vedi) che si comportano da inibitori inversi del GABA; l'inibizione
del GABA, che è essenzialmente un neurotrasmettitore di sinapsi inibitorie , produce
iperecittabilità, responsabile dell'iniziale azione disinibente de euforica e del perdita di controllo
motorio attraverso le vie extrapiramidali, a sua volta responsabile del tremore (delirium tremens)
degli alcolizzati.
Ossidazioni di Aldeidi:
Le aldeidi sono composti dotati di buona reattività anche in vivo, perciò vengono
prontamente trasformate in composti più stabili e meno pericolosi per i processi vitali: possono
essere ridotte ad alcoli 1ª o ossidate ad acidi carbossilici. L'ossidazione delle aldeidi viene
catalizzata da aldeide deidrogenasi NAD dipendente o da enzimi metalloflavoproteici come la
xantina ossidasi e l'aldeide ossidasi.
CH3-CHO
CH3-COOH
Deamminazione Ossidativa:
La deamminazione di ammine 1ª catalizzata dalle le MAO, Mono Ammino Ossidasi, procede
in modo analogo a quella catalizzata dal CYP450 riportata in precedenza: di formano gli stessi
intermedi, dei quali nello schema sotto riportato si riportano per brevità l'immina e
l'ammonaldeide prodotta per idrolisi di quest'ultima.
RCH2NH2
MAO
RCH=NH
+ H2O
OH
RCH
- NH3
RCHO
NH2
Le DAO o Di Ammino Ossidasi catalizzano la stessa reazione nelle sostanze provviste di
due centri basici, come istamina, cadaverina e putrescina prodotte per decarbossilazione
della istidina, lisina e ornitina rispettivamente:
NH2
H
N
H2N
(CH2)5
NH2
H2N
(CH2)4
NH2
N
86
Ossidazione delle Purine:
Catalizzate da xantinossidasi, che sono metallo flavoproteine. Ad es., la 6-mercaptupurina
fornisce l'acido mercapturico, secondo la seguente equazione:
SH
SH
H
N
N
N
N
N
HO
6-mercaptopurina
H
N
OH
N
N
6-mercapturico
-Ossidazioni:
Farmaci che contengano residui di acidi carbossilici lineari, a numero pari di atomi di C ed
eventualmente con uno o più doppi legami alternati e a struttura cis, vengono rapidamente e
completamente biotrassformati dalle -ossidasi, secondo un meccanismo esaurientemente
trattato nei testi di Chimica Biologica. Gli acidi alifatici ramificati o a numero dispari di atomi di
C o a struttura trans vengono metabolizzati più lentamente e solo parzialmente, interrompendosi
la sequenza ossidativa in prossimità di una ramificazione, di un ciclo e di una struttura non
riconosciuta.
L'efficacia dalla -ossidazione metabolica degli acidi alifatici spiega la grande difficoltà
incontrata nell'usare in terapia i prostanodi naturali o sintetici per il fatto che la loro struttura
conserva parte dell'acido arachidonico dal quale derivano e perciò viene rapidamente attaccata
dalle -ossidasi.
Riduzioni
Le reazioni di riduzioni sono rese possibili da reduttasi più o meno specifiche alle quali
molto spesso è associato come coenzima il NADPH. I substrati più comuni ed i relativi prodotti
di riduzione sono brevemente elencati nella seguente Tabella
Substrato
Aldeidi
Chetoni
Ar-N=N-Ar
Azocomposti
Ar-NO2
Nitroderivati
R-S-S-R
Disolfuri
R-SO2-R
Solfoni
Prodotti di riduzione
Acoli I
Alcoli II
Ar-NH-NH-Ar
Idrazocomposti
Ar-NO
Nitrosoderivati
2 R-SH
Tioli o mercaptani
R-SO-R
Solfossidi
2 Ar-NH2
Arilammine
Ar-NHOH
Arilidrossilamine
ArNH2
R-S-R
Solfuri
Idrolisi
I gruppi funzionali suscettibili di idrolisi (SNAcilica) in ordine di reattività decrescente sono:
Epossidi > Esteri > Lattoni > Amidi > Imidi > Lattami> Uretani > Uree > Barbiturici.
87
Gli enzimi che attivano le reazione di idrolisi vengono dette genericamente, idrolasi, e sono
praticamente ubiquitari, presentando la più alta attività nel fegato e nel plasma
Carbossiesterasi o Carbossilesterasi.
Sono presenti in molti tessuti e nel plasma (tubo digerente ??) ed agiscono non solo su composti
endogeni come diacil- e monoacil-gliceroli, acil-CoA, ma anche su molti xenobiotici. Quindi sono
dotate di scarsa specificità e possono idrolizzare non solo esteri (es.: procaina) ma anche amidi
(es.: procainamide), tioesteri (es.: spironolattone), esteri organofosforici (es.: paraoxon), anidridi
(es.: diisopropilfluorofosfato), epossidi, … Nella Tabella successiva vengono riportati tipici
esempi di idrolisi metaboliche, che alle quali si può applicare i principi che regolano la reattività in
vitro: forza basica e stabilità del gruppo uscente, energia dei legami che si devono rompere e
impedimenti sterici. Inoltre, la velocità delle reazioni di idrolisi, come sostituzione nucleofile
aciliche, dipendono dalla carica positiva sul carbonio carbonilico: effetti induttivi e mesomerici
elettron attrattori che la incrementano aumentano la reattività e viceversa, come risulta dai
seguenti esempi.
Uree: R2N-CO-NR2 hanno i più bassi valori di carica positiva sul carbonio carbonilico e
quindi più bassa reattività verso i nucleofili per il doppio effetto mesomero elettrondonatore
dei due atomi di azoto adiacenti. Ancora più resistenti all'idrolisi sono le uree cicliche tipo
barbiturici
Uretani: R2N-CO-O-R' hanno rettività leggermente superiore avendo l'ossigeno del gruppo
O-R' un effetto mesomerico elettrondonatore inferiore all'azoto.
Esteri: R-CO-O-R' carica O=C+ ancora più elevata per il debole effetto induttivo di R che
sostituisce R2N dei precedenti uretani. Gli esteri di acidi aromatici Ar-CO-OR' risultano
ancora più resistenti all'idrolisi dei corrispondenti aromatici per l'efficace effetto mesomerico
elettron donatore dell'anello aromatico:
OR
C
C
C
OR
O
O
O
O
C
OR
OR
Questo effetto elettrondonatore dell'anello aromatico viene accresciuto sensibilmente dalla
presenza di sostituenti elettrondonatori come gruppi NR2 > OR > R specie nelle posizioni
para e orto, aumentando ulteriormente la resistenza all'idrolisi e quindi la durata dell'effetto di
anestetici locali come la procaina (vedi Cap. 11).
Anidridi: R-CO-O-CO-R sono molto più facilmente idrolizzate degli esteri per la maggiore
stabilità del gruppo uscente, essendo R-COO- molto meno basico di R'-O Acilalogenuri: R-CO-X si idrolizzano con estrema facilità sia per il potenziamento della carica
O=C+ per l'effetto induttivo dell'alogeno X sia per la stabilità del gruppo uscente X-
88
TABELLA
CH3
O
H2C
CH2
O
O
N
CH2
CH3
CH3
CH2
OH
H2O
H2C
+
CH2
H2N
N
CH2
HO
H2N
CH3
CH2
Procaina
CH3
O
H2C
CH2
NH
O
N
CH2
CH3
CH3
CH2
OH
H2O
H2C
+
CH2
H2N
N
CH2
H2N
H2N
CH3
CH2
Procainamide (L'idrolisi enzimatica , come in vitro, è sensibilmente più lenta della procaina: a parità di
-
-
ingombri sterici, RNH è un gruppo uscente meno favorevole si RO dell'estere)
O
O
O
H3C
CH3
H2O
H
H
O
H
S
O
H3C
CH3
O
H
H
H
O
CH3
O
+
SH
HO
CH3
Spironolattone ( Si noti che l'anello lattonico - un estere ciclico - rimane integro. Ciò è da attribuire alla
maggiore stabilità dei sistemi ciclici: la struttura aperta è ottenibile solo in ambiente
nettamente alcalino [pH > 8] nella forma HO-(CH2)nCOO- M+ )
NO2
O
O P O
O
H3C
H2O
O
H3C
O P O-H
O
H-O
H3C
H3C
NO2
+
Dietil-p-nitrofenil.fosfato (Paraoxon) ( Il p-nitrofrnolo viene idrolizzato di preferenza sul etanolo, perchè
il p-nitrofenato base molto più debole, quindi gruppo uscente più favorevole dell' alcolato)
O
O
H2O
iPr-O
P
iPr-O
F
iPr-O
P
OH
+
H-F
iPr-O
Diisopropil.fluorofosfato (DFP) (La rimozione idrolitica dell' HF e nettamente favorita di quella dell' alcol
isopropilico. Sia per la minor enegia di legame P-F rispetto a P-O, sia per la stabilità di F- , base
nettamente più debole di iPr-O- )
89
Esteri Fosforici: (RO)2-PO-OR' sono più velocemente idrolizzati degli analoghi esteri di acidi
carbossilici, perché l'anione fosfato [ (RO)2-PO-O- ] ha minor contenuto energetico dell'anione
carbossilato ( R-COO- ) derivando da acido più forte. Il fosfato viene trasferito sull'enzina e
va ad esterificare l' OH di un residuo serinico in prossimità del sito attivo dell'enzima stesso. La
carbossiesterasi viene così inattivata irreversibilmente, ma in questo modo viene protetta
l'acetilcolinesterasi sinaptica del sistema colinergico. L'azione tossica e le proprietà insetticide
degli esteri fosforici sono appunto dovuti all'inibizione per foforilazione dell'acetilcolinesterasi
con meccanismo simile all'inibizione della carbossiesterasi (vedi Cap. 20). Alcuni componenti
di questa classe possono essere inattivati anche da sistemi enzimatici diversi dalle idrolasi, come
il CYP450, le flavino monossigenasi e le glutatione S-transferasi.
Non sempre il metabolismo degli xenobiotici da parte delle carbossiesterasi comporta
detossificazione ad es. le idrolisi di acetato di vinile e di nitroso amidi producono acetaldeide e
metidiazo idrossido rispettivamente che combinandosi con il DNA risultano cancerogeni:
CH3-CO.O-CH=CH2 → CH3-COOH + [ HO-CH=CH2 ] → O=CH-CH3
R-CO.N(NO)CH3 →
RCOOH + [ O=N-NH-CH3 ] → HO-N=N-CH3
Così, la cocaina ed alcuni suoi metaboliti vengono idrolizzati da una carbossiesterasi epatica che,
in presenza di etanolo proveniente da bevande alcoliche, produce transesterificazione
trasformando il gruppo carbossimetilico in carbossietilico ( per la struttura completa della cocaina
si veda il Cap. 11):
COCAINA-COOCH3
→ COCAINA-COOCH2CH3
Gli esteri etilici sono ancora attivi e più lipofili, incrementando l'attività e la tossicità epatica
dell'alcaloide al punto da risultare mortale ad alti dosaggi e in presenza di forti dosi di etanolo.
Peptidasi.
Amidasi
Epossido idrolasi
(riportare da pag156)
Le epossido-idrolasi sono stereoselettive fornendo trans - dioli, come accennato in precedenza in
" Meccanismo dell'ossidazione enzimatica di residui aromatici ". La notevole reattività degli epossidi è
dovuta alla deformazione degli angoli di legame dai normali valori di 109° a circa 60°.
90
REAZIONI DI CONIUGAZIONE
Fase 2
Gli agenti coniuganti più comuni sono: acido glucuronico - acido solforico - glicina.
Le reazioni di coniugazione possono essere precedute da quelle della Fase I, ma per sostanze
provviste già di adatti gruppi la coniugazione è immediata.
I prodotti di coniugazione hanno le seguenti caratteristiche generali:
Nettamente più idrofili, ad esclusione di quelli di metilazione e acilazione
Molto pesso ma non sempre inattivi
Per la maggior parte dei farmaci come degli xenobiotici, la coniugazione rappresenta un
meccanismo di detossificazione anche se è noto che alcuni di questi intermedi risultano
farmacologicamente attivi o sono coinvolti nella carcinogenesi, nelle reazioni allergiche ed in
danni tissutali. Esempi tipici di coniugati attivi sono il 6-glucoronide della morfina che è più
attivo della stessa morfina ed il minoxidil solfato che è il metabolita attivo (antiipertensivo) del
minoxidil.
Coniugazione con glicina
Acil glucuronazione
COOH
OH
O.Solfoconiugazione
O.glucuronazione
NH2
Acetilazione
N.glucuronazione
La sequenzialità delle coniugazioni di una stessa sostanza può dar origine a svariati prodotti
di coniugazione, come nel caso dell'acido p_amminosalicilico (antitubercolare), che può essere il
substrato di più di un enzima metabolizzante, così che processi di coniugazione diversi possono
competere per lo stesso gruppo funzionale. Il risultato è una vasta gamma di metaboliti escreti
con le urine o con le feci.
Gli enzimi di coniugazione, quando un farmaco sia somministrato come racemato, possono
mostrare stereospecificità verso uno degli enantiomeri. Anche la via di somministrazione, orale o
endovenosa, può condizionare il tipo di biotrasformazione per il verificarsi di coniugazioni
presistemiche intestinali.
GLUCURONAZIONE: Coniugazioni con Acido Glucuronico
É la più diffusa via coniugativa. Il fegato è particolarmente ricco non solo di acido
glucuronico ma anche di UDP-glucuronil-transferasi (*), cioè dell'enzina che trasferisce l'acido
glucuronico sul substrato
Glucosio-1-fosfato + UTP UTP-glucosio (+ 2 NAD + UDPG-deidrogenasi) UDP-glucoronato
91
O
CH 2 OH
O
O
H
-O.P.O. P. O. P.O
O- O- O-
O
COO
1)
OH
O PO3 2OH
Farmaco
nucleofilo
COO
-
O
Uridina-Ribosio
OH
R
.Y.
-
O
OH
O
OH
2) Ossidazione
Y
R
O
(*)
O
OH
-
O
OH
O
P. O. P.O
+
O
-O. P. O. P.O
- OO
OH
-
OH
O
Uridina-Ribosio
Uridina_Ribosio
Glucuronidasi
Ac. glucuronico + R-Y-H
Cioè, il -glucosio-1-fosfato reagisce dapprima con Uridintrifosato (UTP) per dare
uridindidfosfato--glucosio con eliminazione di pirofostato inorganico e con conservazione di
configurazione del C1 del glucosio. Poi il gruppo alcolico 1ª in C6 viene deidrogenato ad
aldeide e quindi ossidato ad acido. Poi il centro nuclefilo , HY: (dove YH = OH, NH, SH) , del
substrato dà una tipica SN2 da retro con inversione di configurazione catalizzata da UDPGtransferasi (*). La sostituzione è resa possibile dal fatto che il gruppo uscente uridindifosfato è
una base molto debole essendo l'anione (fosfato) coniugato con un acido forte (uridin di
fosforico). Si ottiene quindi un -glucoronide. La UDP-glucuronil transferasi ha proprietà
inducibili: efficaci agenti di induzione sono i barbiturici ed il fumo di tabacco.
Nella glucuronazione si ha un grande aumento di idrofilia prodotto dai gruppi ossidrilici
liberi e soprattutto dall'anione carbossilato. Il coniugato dopo filtrazione glomerulare non è
affatto riassorbibile nell'ansa. Quando il glucuronide ha un elevato peso molecolare (superiore a
circa 500 Dalton), la via di eliminazione preferita è quella biliare. Il glucuronide così secreto
nell'intestino non verrebbe affatto riassorbito attraverso la parete intestinale ed andrebbe
incontro a completa eliminazione fecale se non venisse idrolizzato da glucoronidasi ivi presenti
(prodotte anche dalla flora batterica). Questo riassorbimento (effetto di secondo passaggio), che
è proporzionale all'entità della liberazione del farmaco dal glucuronide, è solo parziale.
I glucuronidi sono in relatà dei glucosidi o emiacetali ciclici: la loro suscettibilità all'idrolisi è
quindi intermedia fra esteri e gli eteri ed è promossa dagli acidi ed elettrofili, mentre è insensibile
alle basi ed ai nuclefili. I gruppi funzionali che posssono essere glucuronati sono:
alcoli → O-glucuronidi a carattere etereo, buons stabilità: alcoli Iª > IIª > IIIª
fenoli → O-glucuronidi a carattere etereo c. s., ostacolo da orto-sostituenti ingombranti
acidi → O-acilglucuronidi a carattere estereo, molto sensibili all'drolisi
ammine → N-glucuronidi poco stabili in ambiente acido
tioli → S-glucuronidi poco stabili in ambiente acido
amine IIIª → N-glucuronidi quarternari, minor stabilità dei precedenti
COO-
O
COO-
O OH
OO
(H+)
OH
R
OH
R
+ Proteina-NH2
O
OH
O
C4-O-glucurunide
Proteina-NH-CO.R
(Aptene)
OH
Ac. glucuronico
OH
H+/OH-
Ac. Glucuronico + R-COOH
Contrariamente ai C1-O-glucuronidi eterei, i C1-O-acilglucuronidi, data la natura esteri
acetalici sono sensibili agli alcali ed ai nucleofili e, data la mobilità del gruppo acilico, possono
dar luogo a reazioni di trans-esterificazione, che possono portare al trasferimento dell'acile sugli
92
altri ossidrili della stesso residuo glucuronico 8 o su gruppi amminici di proteine o altre
biomolecole. La proteina così acilata può comportarsi da aptene e causare risposte i
mmunologiche in seguito ad una successiva esposizione alla sostanza acida
COO(vedi precedente cshema). Questa ipersensibilizzazione è responsabile delle
O NR3
reazioni anafilattiche all'acido acetilsalicilico e ad altri FANS. La frequenza di
OH
queste risposte immunotossiche dipendono dalla reattività dell'acil
glucuronide e dalla stabilità della proteina antigenica: antiinfiammatori come
OH
il benoxaprofene, zomepirac, indoprofene ... sono stati ritirati dal commercio.
OH
Questa reattività degli O-acilglucoronidi può essere responsabile di
N.glucuronide IVª
epatotossicità e crcinogenesi. L'induzione di tumori alla vescica da parte di
arilamine sembra legata alla formazione di N-glucuronidi di N-idossiarilamine (prodotti di
ossidrilazione con CYP450, vedi). Questi glucuronidi si concentrano nelle urine, dove, per il pH
acido si idrolizzano a N-idrossiarilamine che possono subire eleiminazione di acqua e convertirsi
in ioni arilnitrenio, capaci di reagire con nuclefili endogeni, come gli acidi nucleinici, inizioando il
processo mutagenetico e precarcinogeno.
% Glucuronazione
O-acil-glucuronide + proteina proteina acilata aptene
Altra somministrazione reazione immunologica (ipersensibilità)
Comune ad aspirina e molti FANS.
Steroidi, bilirubina, tirosina e tiroxina, … elininati come glucuronidi
Esempi di glucuronidi attivi della morfina:
3-O-glucoronide: conc. Plasm. 20 x M ; antagonista
6-O“
“
“
2 x M ; potente - agonista
In competizione con solfoconiugazione: stessi substrati
I C2- , C3- e C4-O-acilglucuronidi ottenuti per trasposizione dei C1-O-acilglucuronidi sono dei regioisomeri non
più riconosciuti dalle glucuronidasi e quindi resitenti all'idrolisi.
8
93
Solfoconiugazione
Preferenziale per fenoli: Catecolamine
Acidi Biliari
Ormoni steroidei
Farmaci Fenolici e Derivati tirosinici
Solfonati anche: alcoli amine >> tioli
Limitata dalla disponibilità di solfato:
A basse concentrazioni di substrato prevale su glucuronazione
Ad alte
“
“
cede alla
“
Alcuni solfoconiugati possono risultare attivi:
Minoxidil solfato
Morfina 6-solfato
Altri tossici per spiccate proprietà alchilanti
Sedi principali: fegato e intestino
SO42- + 2 ATP (ATP-solforilasi + Chinasi) 3-Fosfoadenosina5-fosfosolfato
PPi
NH2
+ 2 ADP +
N
N
O
N
N
O
OH
O
CH2 O P O S OO-
+
O
Ar OH
O
Ar
O S OO
OPO32Anidride mista
legame debole
CONIUGAZIONE con AMINOACIDI
Sostanze con gruppi COOH ramificate, alicicliche e aromatiche
Lineari: beta-ossidazione e acetato
AA principale: Glicina (taurina per ac. biliari)
Coniugati sempre non tossici
R-COOH + HS-CoA + ATP
ADP + R-CO-S-CoA
(acilsintetasi)
94
R-CO-S-CoA + H2N-CH2-COO- HS-CoA + R-CO-NH-CH2-COO(aciltransferasi)
O
COOH
+H N
3
CH2 COO-
N
COO-
H
Acido ippurico
95
ACETILAZIONE
Avviene principalmente su gruppi amminici:
ammine 1ª aromatiche e alifatiche
aminoacidi
idrazine
sulfanilamidi: N1 e/o N4
Alcuni coniugati conservano attività: N-acetil-procainamide
Polimorfismo ereditario:
Individui acetilatori lenti più predisposti alla tossicità acuta
“
“
Ac
RNHSO2
NH2
veloci: più predisposti alla tossicità cronica
tumore alla vescica e fegato
S CoA
RNHSO2
NH COCH3
acetiltransferasi
(*) Ossidrilazione catalizzata da CYP 450
[Ar-NH+ ]: ione nitrenio, fornisce legami covalenti con ac. nucleinici e proteine
cancerogeno (tumore alla vescica)
Ar NH2
O
AcCoA
Ar NH
C CH3
O
CYP450
O
Ar NH C CH3
Acetil N_ossido
O
Ar NH O C CH3
Ar NH+
Acetilidrossilammina
arilnitrenio
96
Coniugazione con Glutatione : Sintesi Mercapturica
Coinvolge sostanze alchilanti: suscettibili all'attacco nucleofilico
Protegge dall'alchilazione proteine, enzimi, ac. nucleinici
METILAZIONE
O- e N-Metilazioni : più attive su composti endogeni
Molto spesso si osserva incremento di attività
S-METILAZIONE:
DETOSSIFICANTE
disulfiram, captopril, penicilamina
97
6-propiltiouracile, 6-mercaptopurina
R-SH e H2S
98
% Metabolismo:
Coniugazione del Cianuro
Complessasione con Fe dei Citocromi ed Emoglobina
Programma di Tossicologia
Polimorfismo Genetico
Grande variabilità nel metabolismo dei farmaci CYP 450-dipendenti:
polimorfismo genetico , maggiore causa
induzione enzimatica
funzionalità epatica e renale, malattie
sesso, età, attività fisica, rischi professionali, bioritmi, dieta …
Pol. Gen. :
diversità genetica nell' espressione naturale di isoforme di CYP 450
Isoforme: diversa capacità di catalizzare le biotrasformazioni
risposte insolite o esagerate a normali dosaggi di un farmaco
CYP 450 che catalizza ossidrilazioni e demetilaziono ossidative (CY2D6):
Fenotipo Attivo Metabolizzatori estensivi:
alta capacità di detossificazione
ma maggior rischio per .estensiva attivazione di percancerogeni
Fenotipo Poco Attivo Metabolizzatori scadenti (5-10% caucasici)
Risposte esagerate sia terapeutiche che tossiche
Insensibili alla codeina per mancata O-demetilazione
Polimorfismo è stato associato anche aad altri processi enzimatici:
Acetilasi di idrazine, amine aromatiche, benzodiazepine …
Colinesterasi serica
Alcool deidrogenasi, aldeide deidrogenasi
Epossido idrolasi
Xantinossidasi
Ad es.: 50 % degli orientali mancano di aldeide deidrogenasi …
99
Metabolismo Extraepatico
FEGATO :
SEDE PRIMARIA
Molti Enzimi metabolizzanti gli xenobiotici sono ubiquitari
Superfici gastrointestinale e polmomare: relativamente più ricche:
Presenti: varie famiglie di CYP 450 (ossidazioni e demetilazioni)
Enzimi di coniugazione, acetilazione, idrolisi …
Per os sono particolarmente evidenti interazioni fra farmaci e farnaci/dieta:
Induzioni e Inibizioni
Induttori (es. fumo): biodisponibilità di altri farmaci
Inibitori ( es. eritromicina, steroidi ): “
“
“
Cavoletti di Bruxelles: rallentano la 2-ossidrilazione del testosterone
Vit B6 dietaria aumenta l'attività della L-AA-decarbossilasi intestinale
Tiramina (formaggi, vino rosso, banane ) come substrato inibisce le MAO intestinali e sistemiche
Paracetamolo (fenoli) co-somministrato a etinilestradiolo ne aumenta del 48%
la conc. Ematica competendo per la solfoconiugazione.
produce -glucuronidasi, solfatasi e varie glucosidasi
Ruolo importante nel metabolismo presistemico
-glucoronidasi, solfatasi, …
riciclo entero-epatico
Farmaci: digossina, contraccettivi, cloramfenicolo
Endogeni: H. Tiroidei, Ac. biliari, ac. folico, colesterolo
Microflora intestinale:
Riduttasi:
NO2
Ar-N=N-Ar
R2SO
nitroimidazoli
sulfalazina, prontosil rosso, … (prodrugs)
sulfinpirazone
100
Metabolismo a livello Polmonare
Polmone possiede tipiche attività CYP450 , FMO, Epossido idrolasi e
coniugazioni confrontabili a quelle epatiche
Polmone sede di 1° Passaggio dopo somministrazioni: Endovenosa
2° Passaggio
altre vie
Intramuscolare
Sottocutanea
Dermica
Si accumulano nel polmone per interazione con fosfolipidi tissutali:
-bloccanti
oppioidi
antidepressivi triciclici
NUCOSA NASALE HA UNA ATTIVITÀ CYP450 PIÙ ALTA CHE IN OGNI ALTRO TESSUTO
ATTIVA SU : DECONGESTIONANTI, ESSENZE, ANESTETICI, ALCOLI, NICOTINA, COCAINA,
…
101
Stereochimica e Metabolismo
Enzimi chirali
buona stereoselettività di substrato
stereospecificità di prodotto
Stereoselettività: azione preferenziale su un isomero (> velocità)
Es.:
decarbossilazione di S-a-metildopa a S-a-metildopamina
i relativi enantiomeri D inattaccati
HO
HO
COOH
OH
HO
(S)--metildopa
(R)- -metildopa
(S)--metildopamina
STEREOSPECIFICITÀ: PRODUZIONE
NH2
H2N
H2N
OH
H3C
CH3
HO
Es.:
HOOC
H
CH3
PREVALENTE DI UNO DEI POSSIBILI ISOMERI
riduzione del metadone produce prevalentemente un isomero
Ossidrilazione fenitoina: idem
Riduzione del naltrexone solo 6-a-isomero
Stereoselettività di substrato-prodotto, es.: R--metildopamina
-idrossilata selettivamente
prodotto un solo isonero : (1R, 2S)-a-metilnoradrenalina
HO
HO
COOH
H
HO
COOH
CH3
CH3
H2N
HO
H2N
HO
(S)--metildopa
102
Tossicità da Metabolismo Ossidativo
Ossidazioni
Bioattivazione
(*) stericamente impediti
Talvolta
Sost. Nucleofiliche: epossidi (*)
chinoni (*)
radicali (*)
non attaccati da enzimi detossificanti
epossido idratasi o glutatione S-transfer.
coniugati reattivi dove coniugante e buon grppo uscente
103
104
Interazione fra Farmaci
Spesso un farmaco modifica il decorso metabolico di altri farmaci:
Sinergismo
Antagonismo
Reazioni tossiche
Evitare somministrazioni simultanee specie di farmaci molto attivi.
Es.
fenobarbitale: potente induttore di CYP450
diminuisce attività: fenitoina, anticoagulanti, …
anti-MAO potenziano azione adrenergici e antidepressivi
allopurinolo usato come antigottoso, inibitore xantinossidasi
porta ad accumulo di 6-mercaptopurina, immunosoppressore
Differenze di sesso nel metabolismo
Poche notizie: probabilmente dovute ai diversi ormoni sessuali
Es. N-demetilazione dell'eritromicina è più alta nella Donna
Differenze di velocità di metabolizzazione:
propranololo: 50% più elevata mel maschio
ossidrilazione di anelli aromatici: simili
N-demetilazione di meperidina nella gravidanza e con contraccettivi
differenze anche nella velocità di glucuronazione
105