")
Le patologie
mitocondriali
Simone Vespa
Edoardo Vespa
Stefano Spagna
Dario Zuppardo
Prof.
Cinzia di Pietro
Polo D 2011/2012
I mitocondri ed il mtDNA
• I mitocondri sono organuli semiautonomi presenti in tutte le cellule
animali e vegetali, aventi la ben nota
funzione di “produttori di energia” da
immagazzinare sottoforma di ATP.
• La peculiarità che li contraddistingue
rispetto agli altri organelli cellulari è la
presenza all’interno della matrice di un
proprio DNA circolare (mtDNA),
probabilmente residuo di un organismo
procariota originariamente a sé stante.
• Questa ipotesi sostiene la teoria
dell’endosimbiosi, secondo la quale un
protoeucariota avrebbe inglobato al suo
interno il protomitocondrio, in cambio di
reciproci benefici (accumulo di energia
tramite metabolismo ossidativo).
• Il genoma mitocondriale codifica per il 93% delle
sue sequenze: ne risulta quindi una struttura
genica compatta, policistronica e quasi totalmente
priva di introni.
• Esso contiene 37 geni, 13 dei quali codificano per
polipeptidi propri del mitocondrio (11 subunità
della catena respiratoria, e 2 subunità della ATPsintetasi), 22 per i tRNA e 2 per gli rRNA, che
esprimono il genoma mitocondriale a livello
dell’0rganismo umano.
• Ovviamente il genoma mitocondriale, in quanto
tale, può andare incontro a mutazioni, più o
meno estese, ma che (così come avviene nel
genoma nucleare) sono il fattore scatenante di
numerose malattie genetiche.
Le patologie
• Le malattie mitocondriali rappresentano un
gruppo eterogeneo di sindromi cliniche
accomunate da un deficit energetico del
metabolismo mitocondriale. [Wallace, 1999]
• Nonostante il mitocondrio sia sede di varie
vie metaboliche fondamentali, per malattie
mitocondriali in senso stretto si intendono
le sindromi associate al deficit della
fosforilazione ossidativa (OXPHOS).
Le normali funzioni mitocondriali dipendono dalla relazione simbiotica
di DNA nucleare (frecce blu e nere) e DNA mitocondriale (frecce rosse).
L’omeostasi cellulare è sotto il doppio controllo di questi due genomi. Le
patologie mitocondriali possono sorgere da disfunzioni che scaturiscono
dall’uno o dall’altro genoma (testo in rosso: malattie del DNA
mitocondriale; testo in blu: malattie del DNA nucleare).
• Poiché i mitocondri sono presenti in tutti i tessuti,
le malattie mitocondriali possono teoricamente
colpire qualsiasi organo. Più spesso, però,
interessano le cellule muscolari e quelle nervose,
data la maggiore richiesta di ATP di questi tessuti,
specie durante lo sviluppo.
• Per la localizzazione specifica che spesso va ad
assumere, le malattie mitocondriali sono spesso
definite come neuro-mio-patie mitocondriali.
Caratteristiche generali
• Le malattie mitocondriali possono essere
causate da mutazioni del DNA
mitocondriale. Alcune di esse colpiscono
un determinato organo (ad esempio la
LHON, Neuropatia ottica ereditaria di
Leber, che coinvolge sostanzialmente
l’occhio), ma molte altre riguardano un
intero sistema e spesso si presentano con
associate disfunzioni neuro-miopatiche.
• Le malattie mitocondriali possono manifestarsi a qualsiasi
età, con una maggiore incidenza nell’età dello sviluppo
puberale, dovuta al maggior consumo energetico.
• In alcuni individui, il quadro clinico è
caratteristico di una specifica disfunzione
mitocondriale e la diagnosi può essere confermata
tramite il test del DNA.
• In molti individui è necessario invece ricorrere ad
un approccio più complesso, che comprende:
1. l’analisi dell’albero genealogico (con particolare
attenzione ai soggetti di sesso femminile, visto che il
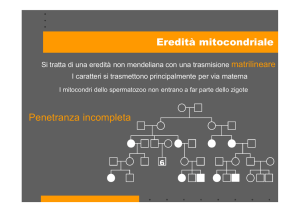
DNA mitocondriale è di esclusiva eredità materna);
2. la misurazione della concentrazione di acido lattico
(che si va gradualmente ad accumulare nei tessuti, che
passano alla fermentazione in quanto risulta inefficiente
la riduzione dell’ossigeno);
3. la biopsia muscolare per analisi istologiche;
4. l’analisi molecolare delle basi del mtDNA, qualora sia
sospettabile una mutazione a livello del genoma
mitocondriale.
• (A) Eredità materna nella Leber hereditary optic neuropathy
(LHON): la donna trasmette la mutazione ad ogni nascituro,
mentre l’uomo non trasmette mai alcuna mutazione;
• (B) Eredità autosomica dominante nella dominant optic
atrophy (DOA): uomo e donna hanno la medesima possibilità
del 50% di trasmettere la mutazione ad ogni nascituro;
• (C) Eredità autosomica recessiva nella Friedreich ataxia
(FRDA): ogni allele mutato del genitore ha un possibilità del
50% di essere trasmesso alla generazione successiva, ma la
malattia si manifesta sono quando il nascituro riceve l’allele
mutato da entrambi i genitori.
Concentrazione di acido lattico nel siero di
pazienti affetti da miopatie mitocondriali
Variazioni istopatologiche nelle miopatie
mitocondriali
Neuropatia ottica ereditaria di Leber
• La neuropatia ottica ereditaria di Leber (LHON)
è la più frequente delle patologie mitocondriali:
comporta perdita della vista (in genere tra i 15
ed i 35 anni di età) a causa della progressiva
demielinizzazione del nervo ottico, che nella
gran parte dei casi va incontro a morte.
• La causa, identificata per la prima volta nel 1988
dal ricercatore statunitense Douglas Wallace,
consiste in una mutazione puntiforme, a carico
di una specifica coppia di basi: le mutazioni
primarie più comuni sono la G11778A, la
G14484A o la T3460C.
• La prima mutazione scoperta (G11778A)
nonché la più frequente e pericolosa,
produce la sostituzione di istidina al
posto di arginina (CAC anziché CGC) in
una subunità del complesso I della catena
di trasportatori di elettroni (NADH
deidrogenasi IV), che risentirà così di un
funzionamento anomalo.
• Simile è il procedimento che segue alle
altre due mutazioni(G14484A e T3460C),
ma esse riguardano rispettivamente la
ND6 e la ND1).
Proportion of
Leber
Hereditary
Optic
Gene Symbols
Neuropathy
Attributed to
Mutations in
This Gene
Test Method
~90%
Targeted
mutation
analysis
m.14484T>C
m.3460G>A
MT-ND1
Select
mitochondrial
genes (ND6)
Test
Availability
m.11778G>A
MT-ND4
MT-ND6
Mutations
Detected
~10%
Sequence
analysis
Other mtDNA
sequence
variants
Clinical
• Queste anomalie riducono
la capacità dei mitocondri
di compiere correttamente
la fosforilazione ossidativa,
e quindi di produrre ATP,
anche se è stato provato
che nell’eziologia della
malattia sono compresi
altri svariati fattori,
genetici ed ambientali.
• In ogni caso, della carenza di energia risente
immediatamente il nervo ottico, in quanto fortemente
dipendente dal metabolismo ossidativo, producendo le
manifestazioni cliniche della malattia come la rapida
degenerazione del nervo e quindi la perdita della vista.
Patogenesi e sviluppo della malattia
•
•
(A) Forma acuta: iperemia, pseudoedema, micro-teleangectasia
(dilatazioni e tortuosità dei vasi papillari);
(B) Forma cronica: a tre anni dalla comparsa, atrofia ottica, specialmente a
carico del settore temporale; perdita delle fibre nervose papillari.
Epidemiologia
• L’incidenza della malattia è stata stimata, con
variazioni a seconda dei luoghi, tra 1/25.00050.000. Ricordiamo che la LHON è una malattia
ad eredità materna. Tuttavia non tutti gli
individui che presentano il difetto a livello del
genotipo presentano anche un fenotipo
patologico: solo il 10% delle femmine ed il 50%
dei maschi che posseggono il gene mutato vanno
incontro allo sviluppo della malattia.
• Quest’ultimo dato ci permette di introdurre la caratteristica
di penetranza incompleta del gene mutato della LHON e
l’evidente superiorità di frequenza nei maschi anziché nelle
femmine (come abbiamo visto a causa di fattori ambientali,
fisiologici e genomici particolari).
Mutations
Number of
analyzed
pedigrees
Median onset
age
Male:female
ratio
Visual recovery
G3460A
17
24,5
3,3:1
24%
G11778A
115
26
4:1
16%
T14484C
40
23
4,7:1
45%
Approcci terapeutici
• Anche se la perdita della vista è di solito il sintomo
più evidente, sono state riportate associazioni della
LHON con anomalie cardiache, neurologiche o
scheletriche.
• Un approccio terapeutico a questa malattia prevede
l’intensificazione del metabolismo ossidativo
tramite la somministrazione di cofattori della via
del trasporto degli elettroni, quali il succinato o il
coenzima- Q, anche se, ad oggi, i risultati sono
sempre stati poco influenti ai fini del decorso.
• Un’altra via risolutiva potrebbe consistere nella
terapia genica, in pratica la sostituzione, nei
mitocondri, del gene mutato con un gene normale.
Atassia di Friedreich
(FRDA)
CARATTERI GENERALI
• Malattia genetica degenerativa, rara (1:50000), che si
manifesta come atassia (perdita di coordinazione)
cerebellare
• L’origine è una mutazione a carico del DNA nucleare
(primo introne del gene FXN del cromosoma 9): la
tripletta GAA subisce un numero eccessivo di ripetizioni
• Riduzione della sintesi di frataxina mediante il
silenziamento di FXN, che codifica la proteina
• Trasmissione autosomica recessiva
FRATAXINA
• Proteina di 210 amminoacidi,
ubiquitaria, principalmente localizzata
nella matrice mitocondriale.
Funzioni:
• Regolazione fosforilazione ossidativa
(produzione di ATP)
• Assemblamento di ISC (complesso
contenente ferro-zolfo appartenente alla
catena respiratoria)
• Biogenesi dell’eme (come chaperonina)
• Deposito e omeostasi del ferro
TRIPLETTA GAA
• Si ripete per slippage mispairing
• L’espansione può essere sia meiotica che mitotica, e
risulta fortemente instabile; è età-dipendente; e
cellularmente selettiva (principalmente neuroni gangli
dorsali, ad alto dispendio energetico)
• Lunghi tratti ripetuti causano la formazione di strutture
secondarie anomale (sticky DNA), che possono dar luogo
a:
– Strutture auto-associanti, che rallentano
meccanicamente lo scorrimento della RNA polimerasi.
– Formazione di eterocromatina, che silenzia il gene.
Formazione dell’eterocromatina dovuta a ripetizione di GAA
• Il meccanismo di regolazione (patologica) del gene è
quindi epigenetico.
• La FRDA infatti viene definita come “malattia da
cromatina”, poiché è la modalità di compattamento della
cromatina che determina l’inibizione del gene e la
disfunzione proteica.
• Perché la disfunzione genica si manifesti sul fenotipo, è
necessario che entrambi gli alleli di FXN presentino
l’espansione di GAA (omozigosi).
• In FXN può verificarsi anche una eterozigosi “allele con
GAA ripetuto-allele con mutazione puntiforme”
Silenziamento del gene
• La completezza dell’mRNA trascritto da FXN
dipenderà dall’estensione dell’espansione
eterocromatica.
• Anche la variabilità nella presentazione clinica si
può spiegare col numero di nucleotidi ripetuti.
• Maggiore è il numero di ripetizioni di GAA
(quindi la lunghezza del tratto eterocromatico),
maggiore sarà l’inibizione della sintesi di frataxina.
Differenze tra mRNA di FXN in sani e affetti da FRDA
I principali effetti patologici del deficit di frataxina sono:
• Accumulo mitocondriale del ferro dovuto al
decadimento degli ISC, che in breve tempo ad uno
squilibrio delle difese antiossidanti e alla formazione
di radicali liberi.
• Aumento della sensibilità a stress ossidativi.
• Mancata funzionalità della catena respiratoria e di
conseguenza carenza di energia in quelle cellule che
ne abbisognano maggiormente:
– Cellule nervose dei gangli dorsali (cervelletto)
– Fibre di tessuto miocardico.
ASPETTO CLINICO
Sono proprio i danni da stress ossidativo a queste cellule
che determinano l’aggravarsi della patologia. I primi
sintomi sono:
• Progressiva perdita del coordinamento motorio e
della sensibilità
• Danni neurologici come atrofia ottica e perdita
d’udito (20 % dei casi),
• Diabete mellito (10 %).
• Il danno più grave riguarda le fibre miocardiche, il
cui malfunzionamento porta a cardiomiopatia
ipertrofica, che è la causa di morte principale per gli
affetti da FRDA.
• Le principali terapie per FRDA, anche se fino adesso non
hanno mostrato grande efficacia, riguardano:
– Antiossidanti per combattere la formazione di
radicali liberi, contrastando la perdita di elettroni
durante la respirazione cellulare.
– Chelanti del ferro per ridurre gli accumuli
mitocondriali.
– Eritropoietina, che sembra agire a livello posttrascrizionale.
– Inibitori delle deacetilasi istoniche, in grado
ripristinare l’espressione del gene silenziato
(eterocromatico).
– Cellule staminali mesenchimali, che aumentano
l’efficacia di enzimi antiossidanti (catalasi e glutationeperossidasi), o autologhe adulte, il cui gene per la
frataxina è stato modificato in vitro.
MERRF
Myoclonic Epilepsy with Ragged Red Fibers
• L’epilessia mioclonica a fibre rosse
sfilacciate(nota anche come sindrome di
Fukuhara) è dovuta ad una mutazione del
mtDna (A8344G).
CARATTERISTICHE PRINCIPALI:
1.Mioclono
2.Epilessia generalizzata
3.Atassia
4.Fibre rosse sfilacciate
MANIFESTAZIONI FREQUENTI:
•Perdita dell’udito neurosensoriale
•Miopatia
•Neuropatia Periferica
•Demenza
•Bassa statura
•Atrofia ottica
NOTA: <50% presenta
cardiomiopatie,retinopatie
pigmentose,oftalmoparesi e
lipomi multipli.
ANALISI CLINICA
Il paziente presenta:
• Acido lattico nel sangue e nel liquido cerebrospinale.
• Aumento della proteina CSF.
• EEG mostra picchi e scariche d’onda,rilevate scariche
epilettiformi focali.
• RM cerebrale mostra atrofia cerebrale e calcificazione dei
gangli della base.
• La biopsia muscolare mostra le tipiche fibre rosse
sfilacciate(RRF).(tricromica di Gomori).
• Analisi biochimiche di enzimi della catena respiratoria in
estratti muscolari di solito mostrano una diminuita attività del
complesso della catena repiratoria contenenti subunità
codificate dal mtDna,soprattutto carenza di citocromo c
ossidasi.
(A) Le fibre muscolari mostrano
eteroplasmia , con lievi
proliferazioni di mitocondri
mutanti, che manifestano delle
fibre " rosse sfilacciate “nel
momento in cui sono trattate
con coloranti.
(B) Proliferazione
contrassegnata là dove la
maggior parte dei mitocondri
ha il gene mutato.
Mutazioni del mtDNA
MERRF può essere causata da più di una mutazione
del mtDNA,anche su diversi geni.
Gene Symbol
Test Method
MutationsDetected
Mutation Detection
Frequency by Test
Method 1
m.8344A>G
>80%
Test Availability
m.8356T>C
MT-TK
Targeted mutation
analysis
m.8363G>A
~10%
m.8361G>A
MT-TF
MT-TP
Clinical
m.611G>A
Mutation
scanning/sequence
analysis
<5%
2
m.15967G>A
mtDNA
Mutation
scanning/sequence
analysis
Sequence variants
90%-95% 3
Cloverleaf structures of human mitochondrial tRNALys from wild-type cells (left) and from MERRF patientderived cells with the A8344G mutation (right). sU* indicates the modified uridine, and U on a round black
background, unmodified uridine. G on a square black background represents the point mutation. The
other modified nucleosides were determined previously: 1-methyladenosine (m1A), 2-methylguanosine
(m2G), pseudouridine ( ) and N6-threonino carbonyladenosine (t6A) (Helm et al., 1998; Yasukawa et al.,
2000a).
Ereditarietà
• La malattia si trasmette con
ereditarietà materna.
Il padre di un probando non è a
rischio di avere la malattia.
La madre invece,che possiede
sicuramente la mutazione,può
presentare o meno i sintomi.
La diagnosi prenatale è
possibile solo se la malattia è
già stata rilevata nella madre.
Tuttavia non si può prevedere il
fenotipo.
Sindrome di Kears-Sayre
• La KSS è un disturbo multisistemico di origine
mitocondriale: esordisce entro i 20 anni, con oftalmoplegia
esterna progressiva e degenerazione pigmentaria della
retina, ed in aggiunta almeno uno dei seguenti danni: blocco
cardiaco completo, proteine nel CSF maggiori di 100 mg/dl e
atassia cerebellare.
• Delezioni su ampia scala del DNA mitocondriale sono spesso
evidenziate a livello dei muscoli scheletrici. Caratteristiche
supplementari associate con la KSS possono includere
miopatia, distonia, anomalie endocrine, sordità bilaterale,
demenza, cataratta e acidosi renale tubulare prossimale.
• Si tratta di una miopatia
progressiva che coinvolge
primariamente i muscoli
oculari estrinseci. Di solito i
sollevatori delle palpebre
sono i primi ad essere colpiti,
andando in ptosi, seguita da
oftalmoparesi.
• Una volta cominciata,
progredisce fino a che gli
occhi si paralizzano. Il
coinvolgimento simultaneo
di tutti i muscoli extraoculari
fa si che gli occhi rimangano
in una posizione centrale,
cosi che strabismo e diplopia
non siano comuni.
Quando il paziente tenta di sollevare le sue palpebre e di
guardare verso il basso, il muscolo frontale viene
contratto, corrugando la fronte. Le palpebre sono
anormalmente sottili a causa dell'atrofia dei muscoli
sollevatori. I muscoli orbicolari dell'occhio sono spesso
coinvolti insieme a quelli extraoculari-
Diagnosi
• La diagnosi è essenzialmente clinica. I livelli ematici di lattato
e piruvato di solito sono elevati in presenza della KSS. Nel
CSF, il livello di lattato è elevato, anche se i livelli del medesimo
nel sangue sono nella norma. Inoltre la KSS eleva il livello delle
proteine nel CSF. Anche se un test di PCR eseguito su DNA,da
campioni ematici può portare alla scoperta di delezioni
nell'mtDNA, il modo migliore per raggiungere la diagnosi
definitiva è l'analisi di un campione bioptico muscolare.
• Si raccomanda uno screening per escludere le anomalie
endocrine, che si presentano in molti pazienti. I metodi di
screening possono includere test per misurare il glucosio
sierico, la funzione tiroidea, calcio e magnesio.
•
L'MRI del cervello ha limitato uso diagnostico infatti i rilievi dell’ MRI
possono essere normali o mostrare atrofia cerebrale e cerebellare ma
correlano ben poco con la KSS. Utili ad una corretta diagnosi sono
l’elettrocardiogramma che rivela difetti nella conduzione cardiaca e
l‘ elettroretinografia che aiuta a stimare la degenerazione retinica. Nei
pazienti con KSS cambiamenti degenerativi spongiformi si hanno sia
nella sostanza grigia che bianca del cervello.
•
La KSS compare in seguito a
specifiche delezioni nel
DNA mitocondriale in
grado di indurre un
particolare fenotipo
patologico, la maggior parte
delle quali sono sporadiche
e si crede che accadano
come mutazioni della
cellula germinale o a livello
dello sviluppo embrionale.
• Le delezioni variano per
dimensioni (1.3-8 kb) e
posizione all'interno del
genoma mitocondriale;
comunque, il singolo sito
più comune e tra le
posizioni 8469 e 13147
(deletion hotspots).
• Questa mutazione di 4.9-kb incide per un
terzo dei casi di KSS. Delezioni avvengono
in tutti i tessuti; tuttavia al posto di esse si
riscontrano talvolta duplicazioni in tandem
del DNA.
Com'e possibile che un gruppo eterogeneo di delezioni
mitocondriali possa condurre ad un fenotipo simile?
• Il meccanismo proposto è basato sul fatto che la
trascrizione dell'mtDNA è policistronica, cioè che
tutti i geni codificanti situati sul filamento H e su
quello L sono trascritti come due soli grandi filamenti
precursori di RNA.
• Questi poi vengono tagliati in filamenti di RNA
separati, che comprendono al loro interno filamenti
di RNA transfer. Così, una delezione in qualsiasi
punto del genoma mitocondriale può alterare la
trascrizione o la traduzione di geni che non erano
stati interessati direttamente dalla mutazione.
Terapia
• Non esiste alcuna terapia in grado di
modificare la storia della malattia in KSS.
In futuro, un potenziale trattamento nei
pazienti con KSS può tentare di inibire la
replicazione dell'mtDNA mutante o
stimolare la replicazione dell'mtDNA
wilde-type. La prognosi per i pazienti che
ne soffrono è infausta infatti la morte è
comune nella terza o quarta decade di vita
")