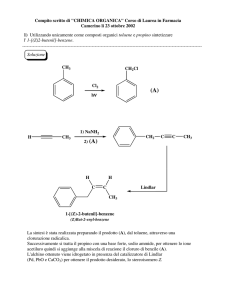
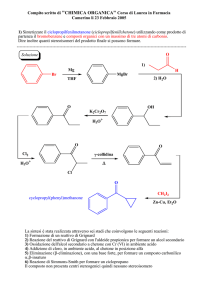
FARMACI ANTIINFIAMMATORI
NON STEROIDEI
- FANS -
Membrana della cellula
TRAUMA
Interno
della
cellula
Istamina
Risposta infiammatoria
Precursori della Bradichinina
Acido Arachidonico
Fosfolipasi A2
PG sintetasi
Cascata
PGE2
Bradichinina
Recettori del dolore
Effetti dei mediatori infiammogeni sui
vasi e sulla migrazione cellulare
Stimolo
(ormonale, immunologico, neuronale)
CELLULE
AGGREGAZIONE
SISTEMA DI
MEDIATORI CASCATA DEL
LIPIDICI COMPLEMENTO COAGULAZIONE MASTOCITARIE PIASTRINICA
Membrana
fosfolipidica
Acido
Arachidonico
LTB4
PGG2
LTC4
PGE2
C3a, C5a
(Attivazione
cellulare)
Fattore di Hageman
PAF
Chinine
Istamina
PGI2
Permeabiltà
vascolare
Vasodilatazione
Edema
Migrazione dei leucociti
5-HT
Cascata dell’Acido Arachidonico
Biosintesi e Metabolismo delle Prostaglandine e dei Trombossani
Membrana fosfolipidica
COX
-1/C
OX2
Attività biologiche dei metaboliti
dell'acido arachidonico
PRODOTTI DELLA CICLOOSSIGENASI
PGE2, PGF2D, PGI2
Vasodilatazione, Antiaggregazione, Riassorbimento osseo,
Regolazione delle risposte immunitarie
TXA2
Aggregazione piastrinica, Vasocostrizione,
Broncocostrizione
PRODOTTI DELLA LIPOOSSIGENASI
LTB4
Chemotassi dei leucociti, adesione e degranulazione
Leucotrieni solfuropeptidici LTC4, LTD4
Permeabilità vascolare, Vasocostrizione,
Broncocostrizione
CLASSIFICAZIONE DEI FANS
NON SELETTIVI COX-1/COX-2
Derivati di acidi carbossilici
Derivati acidici Vari
(enoli)
di acidi aromatici
pirazolici
salicilati
fenamati
Fenilbutazone
Proquazone
Ac. salicilico
Ac. mefenamico
Ossifenilbutazone
Ciproquazone
Diflunisal
Ac. flufenamico
Azapropazone
Benzidamina
Benorilato
Ac. niflumico
Ac. meclofenamico
di acidi aril ed eteroaril
Diftalone
Paracetamolo
Oxicams
acetici
propionici
Piroxicam
Diclofenac
Ibuprofen
Isoxicam
Alclofenac
Naprossene
Sudoxicam
Fenclofenac
Flurbiprofen
Indometacina
Sulindac
Tolmetin
Fentiazac
CLASSIFICAZIONE DEI FANS
IN BASE ALL'EFFICACIA CLINICA
FANS maggiori
Salicilati
Pirazolici
Indometacina
FANS minori
Fenamati
Acidi aril ed eteroarilpropionici
Acidi aril ed eteroarilacetici
Composti vari
CLASSIFICAZIONE DEI FANS
IN BASE ALL'EFFICACIA ED
ALLA TOLLERABILITA'
TOLLERABILITA'
Pirazolici
(Fenilbutazone)
Indolici
(Indometacina)
Salicilici
(Aspirina)
Arilpropionici
Arilpropionici
decrescente
EFFICACIA
Arilacetici
Fenamati
Indolici
Arilacetici
Salicilici
Fenamati
Pirazolici
a. Sia la COX-1 che la COX-2
esistono come dimeri. Il sustrato
acido arachidonico arriva al sito
catalitico
della
cicloossigenasi
attraverso un canale idrofobico.
L’Aspirina acetila irreversi-bilmente
un residuo di serina, vicino, ma
distinto dal sito catalitico. Questo
previene l’accesso del substrato al sito
catalitico dell’enzima.
Modello del sito attivo della cicloossigenasi
con legata la PGG2
"Sito recettoriale" ipotetico della cicloossigenasi
A
H COOH
H
H COOH
H
CH3
B
O O
5A°
8
15
C
11
H 12
H3CO
N
5A°
CH3
O
9
13
Cl
H
7A°
7A°
A = Cavità cationica
B = Superficie idrofoba
Acido Arachidonico
Indometacina
C = Tasca idrofoba
COOH
OH
H3C
NH
H3C
Acido Mefenamico
N
N
COOH
C4H9-n
O CO CH3
O
Fenilbutazone
Acido Acetilsalicilico
FARMACI ANTIINFIAMMATORI NON-STEROIDEI
(FANS)
I principali gruppi di sostanze che appartengono ai FANS
sono:
- Derivati dell'Acido Salicilico
- Derivati Pirazolidin-3,5-dionici
- Derivati dell'Acido Antranilico
- Derivati degli Acidi Arilacetici e Arilpropionici
Queste sostanze hanno in comune le seguenti proprietà:
1. Agiscono per via sintomatica
2. Presentano oltre all'attività antiinfiammatoria, attività
analgesica, antipiretica, antiaggregante piastrinica e
azione sulla dismenorrea primaria (mestruazione
dolorosa).
Gli effetti collaterali più comuni sono: nausea, vomito,
dispepsia e ulcerazioni della mucosa gastrica.
Quest'ultimo effetto è connesso al meccanismo d'azione.
Inoltre presentano tossicità a livello renale ed epatico e
disordini nel quadro ematico.
Fra le varie proposte di meccanismo d'azione quella che
trova maggior credito prende in considerazione
l'inibizione della CICLOOSSIGENASI.
DERIVATI DI ACIDI AROMATICI
Derivati dell'Acido Salicilico (Ac. 2-Idrossi Benzoico)
Derivati dell'Acido Fenamico (Ac. N-FenilAntranilico)
Derivati dell'Acido Salicilico
Lo sviluppo di farmaci derivanti dall'Acido salicilico ha avuto
inizio dalla scoperta del glicoside SALICILINA presente nella
corteccia di Salix Alba che presentava attività antipiretica.
CH2OH
CH2OH
Idrolisi
COOH
Ox
OH
O(C6H11O5)
OH
Alcool Salicilico
Salicilina
Acido Salicilico
L'Acido Salicilico viene sintetizzato secondo la sequenza
di KOLBE.
OH
O- Na+
CO2
OH
COO- Na+
+
COOH
H
4.4 bar, 135°C
E' molto irritante (come tale viene somministrato solo per
uso esterno) per cui si usano i sali di sodio, magnesio, colina
e trietanolamina.
In terapia si usano anche gli esteri di metile e propile.
ACIDO ACETILSALICILICO (Aspirina)
OCOCH3
COOH
Sintesi
OH
+
OCOCH3
(CH3CO)2O / H
' , toluene
COOH
COOH
L'Aspirina è il composto più diffuso e largamente
utilizzato. Viene impiegato anche come antiaggregante
piastrinico.
Si usa alla dose di 1gr per 24h come
analgesico-antipiretico e alla dose di 4 gr per 24h
come antireumatico.
Irrita la mucosa gastrica. Per limitare i danni
gastrointestinali sono state
preparate varie
formulazioni contenenti i sali di calcio o magnesio o
preparando
compresse
a
rivestimento
gastroresistente.
L'Aspirina inibisce la cicloossigenasi mediante una
reazione di transesterificazione, pertanto è un
inibitore irreversibile della cicloossigenasi.
COOH
+
CICLOOSSIGENASI
Nu
OCOCH3
Nu = OH di una Serina529
COOH
+
OH
CH3
CO
Nu
CICLOOSSIGENASI
METABOLISMO DELL'ACIDO SALICILICO
Salicilato libero
HO
COOH
Ossidazione
OH
OH
COOH
Coniugazione con
Ac. glucoronico
Glucoronato estereo
Glucoronato etereo
Coniugazione
con glicina
CONHCH2COOH
OH
Acido
Salicilurico
DIFLUNISAL
COOH
OH
F
F
Sintesi
OH
1. NaOH
2. CO2
+
Diflunisal
3. H
F
F
E' quattro volte più attivo dell'Aspirina, presenta una
minore attività gastroirritante e possiede una durata
d'azione più lunga. Ha scarsa attività antipiretica e
antiaggregante piastrinica. Viene somministrato in
compresse da 250-500 mgr, 2-3 volte al dì.
BENORILATO
COO
NHCOCH3
OCOCH3
Unisce con un legame labile l'Aspirina e il
Paracetamolo. E' meno irritante dell'Aspirina.
Derivati dell'Acido Fenamico
Sono derivati dell'Acido N-fenilantranilico ed agiscono in
modo analogo ai salicilati, inibendo la cicloossigenasi.
COOH
COOH
CF3
NH
R3
N
R1
NH
Ac. Niflumico
R2
R1 = R2 = CH3; R3 = H
Ac. Mefenamico
R1 = R3 = Cl; R2 = CH3
Ac. Meclofenamico (Lenidolor)
R1 = R3 = H; R2 = CF3
Ac. Flufenamico
Le relazioni struttura-attività possono essere così riassunte:
1. La funzione amminica è essenziale in quanto la sua
sostituzione con O, CH2, SO2, ..... N-COCH3 determina
una significativa diminuzione di attività.
2. I sostituenti R1, R2, R3 aumentano l'attività. La ragione
potrebbe essere la mancanza di coplanarità dei due
anelli.
3. I due anelli aromatici non devono essere coplanari.
HOOC
H
N
CH3
CH3
Inattivo
4. Il gruppo carbossilico deve essere in posizione orto.
ACIDO MEFENAMICO
COOH
-
COO K
+
+
H2N
Br
(AcO)2Cu
H3C
N
H
CH3
H3C
CH3
E' il capostipite di questa classe e viene utilizzato principalmente
come analgesico. Viene metabolizzato rapidamente per
ossidazione a COOH del gruppo CH 3 in meta. Il tempo
emivita è circa 2h.
ACIDO FLUFENAMICO
CF3
COOH
+
COOH
Cu
K2CO3
H2N
Cl
CF3
2'
3'
N
H
4'
6'
5'
Possiede buona attività antiinfimmatoria. Viene metabolizzato
per ossidrilazione in 4' ed ha un'emivita di circa 3h.
ACIDO NIFLUMICO
CF3
COOH
+
N
Cl
H2N
COOH
N
CF3
N
H
E' un bioisostero dell'acido flufenamico. Ha mostrato buona
attività antiinfiammatoria.
DERIVATI DI ACIDI ARIL- ED ETEROARILACETICI E PROPIONICI
Inibitori competitivi e reversibili della cicloossigenasi.
- I derivati Arilacetici sono identificati dal suffisso
FENAC (R = H)
- I derivati D-Arilpropionici sono identificati dal
suffisso PROFEN (R = CH3)
D
CH
X
COOH
R
Y
Î Sostituenti in D più voluminosi del CH3 causano una
netta diminuzione di attività.
Î X è un gruppo con carattere prevalentemente
idrofobico.
Î Y può essere un alogeno.
IBUFENAC
H3C
CH
CH2
CH2 COOH
H3C
E' il capostipite degli acidi Fenilacetici. E' stato introdotto
in terapia nel 1963 ed è stato ritirato dal commercio nel
1968 a causa della sua tossicità (epatotossico).
IBUPROFEN (Brufen)
CH3
H3C
CH
CH2
CH
COOH
*
H3C
E' il capostipite degli acidi Arilpropionici. E' utilizzato
(300 mg 3-4 volte al dì) nell'artrite reumatoide,
nell'osteoartrite, etc......
E' un farmaco ben tollerato e viene eliminato per via
renale prevalentemente inalterato.
La somministrazione del racemo porta alla escrezione di
una miscela dei due enantiomeri in cui prevale la forma
(S)-(+).
Uno studio approfondito del problema ha permesso di
scoprire che in vivo si verifica una inversione d
configurazione (R)-(-) Î (S)-(+).
Enantiomero
Somministrazione
Rapporto (S)/(R)
Escrezione
Rapporto (S)/(R)
(S)-(+)
95:5
95:5
(R)-(-)
6:94
80:20
Racemo
50:50
70:30
In vitro l'enantiomero (S)-(+) è circa 60 volte più
attivo dell'enantiomero (R)-(-).
In vivo questa differenza di attività fra i due
enantiomeri è decisamente meno pronunciata.
Meccanismo di inversione
COOH
Ar
Acil-CoA transferasi
CoA-SH
C
CH3
H
COSCoA
Ar
C
CH3
H
Idrolasi
(R)
Racemasi
COOH
Ar
C
H
CH3
(S)
Acil-CoA transferasi
CoA-SH
Idrolasi
COSCoA
Ar
C
H
CH3
Metodi generali di sintesi dei racemati
O
1-
Ar
OH
NaBH4
C
Ar
Br
PBr3
CH
CH3
CH3
Ar
CH3
NaOH
CH
Ar
CH
CN
COOH
CH3
2-
Ar
CH2
COOR
1. NaH
2. CH3I
CH3
NaOH
Ar
CH
COOH
Ar
CH
CH3
CH3
NaCN
Ar
CH
COOH
RO
3-
Ar
OR
C
X
shift 1,2
del gruppo Ar
CH
CH3
Ar
CH
CH3
COOR
Per la trasposizione si usano acidi di Lewis e solventi
polari protici. Con tale metodologia sono stati
sintetizzati Ibuprofen e Naproxen.
Br
O
O
CH3
CH3
Br2
EtOH
H+
EtO
Br
CH3
EtO
shift 1,2
ZnBr2
EtO
O
Ibuprofen
Et
Br
Br
Zn
CH3
YO
OY
Br
YOOC
H
X
R
Ar
H
R
Ar
Metodi generali di sintesi
dei singoli enantiomeri
1. Classica risoluzione con Cinconidina.
2. Risoluzione cinetica degli esteri con enzimi (lipasi).
3. Idrogenazione asimmetrica di precursori insaturi.
4. Trasposizione asimmetrica di chetali derivanti da
D-alogeno chetoni.
1. Classica risoluzione con Cinconidina.
H2C H2C
H
H
COOH
CH3
N
HO
.
H
H3CO
(S)-(+)-Naproxene
N
Cinconidina
Il Naproxene è il primo FANS commercializzato come
singolo enantiomero. Ha un tempo di emivita lungo
(12-15h) rispetto agli altri componenti della classe (2-4h).
L'enantiomero (S)-(+) è molto più attivo dell'enantiomero
(R)-(-).
2. Risoluzione cinetica degli esteri con enzimi (lipasi).
COOH
H
CH3
CH
COOCH3
Carboxylesterasi NP
CH3
Resa: <50%
H3CO
H3CO
+- Naproxene
100% e.e.
COOH
H
CH3
CH3
CH
COOCH3 Carboxylesterasi NP
Resa: <50%
H3C
+- Ibuprofen
CH3
CH3
H3C
95% e.e.
3. Idrogenazione asimmetrica di precursori insaturi.
Processo MONSANTO
(elettrocarbossilazione + idrogenazione asimmetrica)
O
O
.
eH3CO
e-, 2CO2
H3CO
OOC
OCOO
HO
COOH
H+
H3CO
-H2O
H3CO
H CH3
COOH
H2, cat.*
COOH
MeOH, 135 atm
sub/cat. = 215 H3CO
12h
H3CO
PPh2
cat.*
O
Ru
PPh2
92% resa
97% e.e.
O
Atropisomerismo
O O
Ru (-S-BINAP) (OAc)2
H3C
H
O
COOH
COOH
H2
cat.*
96% e.e.
4. Trasposizione asimmetrica di chetali derivanti da
D-alogeno chetoni.
H3C
Ar
H
Cl
AlCl3
H +
ClOC
MeOH
C
C
CH3
ZnCl2
CH3
COOCH3
C
C
OMe H Cl
Ar
H+
Ar
CO
H
MeO
H
Ar
Cl
H
H2O
CH3
Ar
+
H
COOH
C
CH3
Ibuprofen: 82% e.e.
Naproxen: 96% e.e.
Processo ZAMBON
H3COOC
O
O
COOCH3
O
1a fase
H3CO
H3CO
H3COOC
O
COOCH3
O
H3COOC
O
+
COOCH3
O
2a fase
Br2
Br
H3CO
Br
H3CO
Br
Br
1
94%
>>
2
6%
2
1
3a fase
H2O/H+
HOOC
HOOC
COOH
O
COOH
O
O
Br
H3CO
H2O/H+
O
Br
H3CO
Br
Br
4a fase
100 °C Trasposizione
+
H2O/H
Sostituzione
HOOC
COOH
CH3
COOH
O
COO
O
OH
H3CO
HOOC
OH
H3CO
Br
Br
1. MeOH
2. H2 - Pd/C
3. CH3SO2Cl / NEt3
H3COOC
CH3
O
COOH
COOCH3
O
H3CO
Br
OSO2CH3
H3CO
H2 - Pd/C
5a fase
H
Trasposizione
+
H2O/H
CH3
COOH
H3CO
99% e.e.
DERIVATI DI ACIDI ARILACETICI
DICLOFENAC (Voltaren)
CH2COOH
NH
Cl
Cl
E' un ibrido tra la serie degli acidi Arilacetici e quella dei
Fenamati. Ne è risultato un composto molto attivo che si
utilizza alla dose di 25mg 3-4 volte al dì.
Sintesi
COCl
NH
Cl
+
COCl
N
COCl
Cl
Cl
C
O
Cl
O
NH2
AlCl3
N
Cl
1. KOH
2. HCl
Diclofenac
O
NH2 , KOH
Cl
'
W.K.
N
Cl
O
Cl
ALCLOFENAC
Cl
H2C
CH CH2 O
CH2 COOH
Sintesi
OH
Cl
Cl
OCH2 CH CH2
+ H2C
base
CH CH2Br
Cl
OCH2 CH CH2
CH2O, HCl
1. NaCN
2. KOH
3. HCl
ClH2C
Cl
OCH2 CH CH2
HOOC
H2C
FENCLOFENAC
CH2 COOH
O
Cl
Cl
INDOMETACINA
CH2
H3CO
N
C
COOH
CH3
O
Cl
Viene usata nel trattamento della gotta,
dell'artrite reumatoide, osteoartrite e spondilite.
Si somministra in capsule (200 mgr) o in supposte
(100 mgr).
Possiede un'emivita di circa 2h ed il 20% viene
escreto inalterato. I metaboliti derivano da:
demetilazione, N-deacilazione e glucoronazione.
I principali effetti collaterali sono disturbi a
carico del tratto gastrointestinale e del sistema
emopoietico e reazioni di sensibilizzazione.
Sintesi dell'Indometacina
1. Processo MERCK
NH NH2
+
HCl
H3C CO CH2 CH2 COOCH3
CH3OH
Fisher
OCH3
CH2 COOH
CH2 COOCH3
H3CO
1. NaOH
H3CO
2. HCl
N
N
CH3
H
H
t-C4H9OH
CH3
COCl
CH2 COOC4H9
H3CO
NaH
+
ZnCl2
N
CH3
H
Cl
CH2 COOC4H9
H3CO
N
CO
Cl
CH3
'
210 °C
Indometacina
Sintesi Indolica di Fisher
CH3
NH NH2
NH N C
CH3
CH3
+ C O
CH3
+
CH2
H
NH NH2
H H
C
H
C
C
+
CH3
N
H
NH2
CH
-H
C
CH3
NH2
NH NH
2
CH3
CH3
NH2
- NH3
N
H
CH3
2. Processo SUMITOMO
NH N
NH NH2 . HCl
CH CH3
CH3CHO
+
OCH3
OCH3
N
N
Piridina
COCl
Cl
CH CH3
CO
HCl
Cl
C2H5OH
H3CO
NH2 . HCl
N
CH3
CO
Cl
+
H3CO
CO
CH2
CH2 COOH
H3CO
N
N C
CO
Cl
HCl
Fisher
Indometacina
CH2 CH2 COOH
CH3
Sono stati preparati e saggiati più di 350 composti con
struttura simile all'Indometacina.
L'introduzione di un gruppo CH3 sulla catena laterale
non aumenta la potenza, ma l'enantiomero (S)-(+) è il
più attivo.
CH3
CH COOH
H3CO
CH3
N
C O
Cl
Da misure chimico-fisiche si è osservato che la
conformazione più stabile dell'Indometacina è cisoide
non planare.
CH2 COOH
H3CO
N
CH3
C O
Cl
Infatti l'analogo rigido è inattivo.
CH2 COOH
H3CO
N
CH3
O
Cl
Relazioni struttura-attività
nell'Indometacina
CH2
COOH
R3
R2
N
R1
R1
VANTAGGIOSO
SFAVOREVOLE
X
Acile alifatico
CO
(X = Alog., CF3, SCH3)
CH2 ;
S
Alchile
CO
R2
CH3
H,
R3
5-CH3O-; 5-CH3CO-;
5-Cl, 5-H
5-F; 5-(CH3)2N-;
Sostituzione
nelle altre
posizioni
5-CH3-
Nel corrispondente derivato indenico dell'Indometacina
la forma cisoide A è circa 5 volte più attiva della
corrispondente forma transoide B, anche se l'attività di
A è circa la metà di quella dell'Indometacina.
CH2COOH
H3CO
CH2COOH
H3CO
CH3
CH3
H
H
Cl
Cl
A
B
Da questi composti si è arrivati al Sulindac.
SULINDAC
CH2 COOH
F
CH3
C
H
H3C
S
O
Il Sulindac è meno attivo dell'Indometacina, però è meglio
tollerato. E' un profarmaco in quanto il prodotto attivo è
il corrispondente tioetere che deriva dalla riduzione
enzimatica del Sulindac.
Il corrispondente solfone ed il suo glucoronato
costituiscono i principali metaboliti.
CH2 COOH
F
CH3
C
H
H3C S
Metabolita attivo
CH2 COOH
F
Rid.
enzimatica
CH3
C
H
CH2 COOH
F
H3C S
Ox.
enzimatica
O
CH3
C
H
H3C O2S
Prodotto di escrezione
Sintesi
H2C
O
F
CN
CN
F
COOH
COOH
H3C COO-NH4+
CH3
CH3
CH2 CN
F
- CO2
CH2 COOH
F
1. KOH
2. HCl
CH3
CH3
CHO
CH2 COOH
F
H3CS
CH3
- H2O
H
H3CS
NaIO4
Sulindac
TOLMETIN
CH2 COOH
N CH3
O
H3C
Ottimo analgesico, antipiretico e antiinfiammatorio.
E' ben tollerato.
Sintesi
(CH2O)X
N
CH3
NH(CH3)2.HCl
Mannich
N
CH3
CH2 N
N
CH3
CH3I
CH3
CH3
+ CH3
CH2 N CH3
I- CH3
NaCN
'
N
CH2 CN
CH3
CH2 CN
H3C
COCl
N
AlCl3
CH3
O
H3C
1. NaOH
2. HCl
Tolmetin
DERIVATI DI ACIDI ARILPROPIONICI
IBUPROFEN (Brufen)
CH3
CH
COOH
S = R per inversione in vivo.
Dose di mantenimento: 1.2 gr/dì.
NAPROXEN (Xenar, Naprosin)
CH3
CH
COOH
S>R
Dose di mantenimento: 500 mgr/dì.
H3CO
FLURBIPROFEN (Froben)
CH3
CH
COOH
S>R
Dose di mantenimento: 150 mgr/dì.
F
FENOPROFEN (Fepron)
CH3
CH
O
COOH
S=R
Dose di mantenimento: 1.8 gr/dì.
CHETOPROFEN (Orudis, Ketangel)
O
CH3
CH
COOH
S>R
Dose di mantenimento: 150 mgr/dì.
BENOXAPROFEN (Coxigon)
CH3
CH
O
N
Cl
COOH
Dose di mantenimento: 600 mgr/dì.
DERIVATI ENOLICI
FENILBUTAZONE (Butazolidina)
Il Fenilbutazone è il primo rappresentante della classe
di antiinfiammatori a struttura Pirazolidin-3,5-dionica.
H3C
H
O
nC4H9
C6H5
N
N
O
C6H5
Fenilbutazone
H3C
H3C
CH3
N
N
N
O
C6H5
Aminofenazone
Nel 1949 il Fenilbutazone fu immesso sul mercato come
solubilizzante (componente acido) dell'Aminofenazone che
è un composto con attività analgesica e antipiretica.
Successivamente si è osservato che anche il
Fenilbutazone
possiede
ottime
caratteristiche
antiflogistiche e può essere impiegato nel trattamento di
forme reumatiche e nella gotta.
Viene somministrato alle dosi di 300-400 mgr/dì, però a
causa dell'emivita particolarmente lunga (72h) si ha
pericolo di accumulo.
I principali effetti collaterali sono danni epatici, ulcera,
variazioni del quadro ematologico con anemia aplastica e
agranulocitosi.
Relazioni struttura-attività
nel Fenilbutazone
H
O
nC4H9
5
N1
2
N
4
3
O
La funzione E -dicarbonilica enolizzabile è
essenziale. Tale funzione ha carattere acido.
Per eliminazione (sostituzione) dell'atomo di H
in 4 si ha perdita di attività.
Il radicale butile può essere sostituito con un
radicale a tre atomi di carbonio anche insaturo.
E' possibile, ma comunque non migliora
l'attività, introdurre in para all'anello aromatico
in posizione 1 un sostituente (CH3, Cl, NO2).
Permane l'attività (minore) se si elimina il fenile
in posizione 1 all'eterociclo; quello in posizione
2 è essenziale.
I due anelli aromatici non sono coplanari, ma formano
un angolo di circa 45°.
Il radicale butilico si dirige verso la stessa direzione
del fenile in 2.
OSSIFENBUTAZONE (Tanderil)
H
O
C4H9
N
O
N
OH
E' fra i metaboliti del Fenilbutazone quello che si è
dimostrato attivo quanto il Fenilbutazone stesso e viene
impiegato alla dose di 300 mgr/dì.
SUXIBUTAZONE
FECLOBUZONE
BUMADIZONE CALCICO
C4H9 O
RCO2
O
COO
C4H9
H2C
. 1/2 Ca
N
N
NH
O
C6H5
N
C6H5
R = HOOC CH2 CH2
Suxibuzone
R = Cl
Feclobuzone
Bumadizone calcico
Questi composti sono dei profarmaci in quanto la funzione
E -dicarbonilica enolizzabile essenziale non è presente, ma
si forma in vivo.
C4H9 O
C4H9 O
R
C
O
O
HO
O
N
O
N
N
N
-CH2O
Retroaldolica
C4H9
O
O
N
N
O
C4H9
O
O
H
N
N
C6H5
. 1/2 Ca
C6H5
PRODOTTI COMMERCIALI
FENILBUTAZONE (Butazolidina)
O
C4H9
N
O
N
300/400 mgr/dì
C6H5
C6H5
Sintesi
C6H5
COOC2H5
CH
C4H9
HN
RO-Na+
HN
'
+
COOC2H5
Fenilbutazone
C6H5
OXIFENBUTAZONE (Tanderil)
O
C4H9
O
300 mgr/dì
N
N
OH
SUXIBUTAZONE (Solurol)
O
C4H9
C
HOOC
O
O
800 mgr/dì
N
O
C6H5
N
C6H5
FECLOBUZONE
O
C4H9
Cl
C
O
O
N
O
N
1 gr/dì
C6H5
C6H5
BUMADIZONE CALCICO (Eumotol)
C4H9
COO
. 1/2 Ca
NH
O
N
C6H5
C6H5
300 mgr/dì
FEPRAZONE (Zepelin)
O
H3C
H3C
N
O
C6H5
N
300-400 mgr/dì
C6H5
SULFINPIRAZONE (Enturen)
O
C6H5
O
S
N
O
N
300-400 mgr/dì
C6H5
C6H5
Sintesi
COOC2H5
CH CH2 CH2 S C6H5 +
COOC2H5
C6H5
1. RO-Na+
HN
2. H2O2
HN
C6H5
Sulfinpirazone
AZAPROPAZONE (Prolixan)
C3H7
O
N
O
CH3
N
600-900 mgr/dì
H3C
N
N
CH3
Sintesi
O
O2N
CH3
+
CH3
N
N
H3C
H2N
N
CN
CH3
H3C
N
N
CH3
H
rid.
Sn
H
H3C
N
N
N
COOC2H5
CH3
+
CH C3H7
COOC2H5
N
CH3
Azapropazone
PARACETAMOLO
(Femidol, Saridon, Tachipirina)
NHCOCH3
OH
Il Paracetamolo ha buone attività analgesiche e
antipiretiche. L'attività antiinfiammatoria è nulla.
E' un inibitore rapido, reversibile e non-competitivo
della cicloossigenasi. Infatti esplica la sua azione
sull'enzima
attraverso
la
riduzione
della
concentrazione degli idroperossidi che sono essenziali
nell'attività della cicloossigenasi..
E' un metabolita sia dell'Acetanilide che della
Fenacetina.
NHCOCH3
NHCOCH3
OC2H5
Acetanilide (Antifebbrina)
Fenacetina
Questi composti sono stati ritirati dal commercio in
quanto sono nefrotossici. Inoltre la 4-etossianilina, un
metabolita della fenacetina, trasforma l'emoglobina in
meta-emoglobina che non possiede la capacità di
trasportare ossigeno.
NH2
NHCOCH3
0.1%
OC2H5
p-Fenetidina
OC2H5
Fenacetina
NHCOCH3
NHCOCH3
NHCOCH3
Solfato
OH
Glucuronide
Paracetamolo
P-450 Ossidasi
HO N COCH3
OH
Intermedi tossici
NCOCH3
G
Glutatione
NHCOCH3
O
Macromolecole nucleofile
cellulari
NHCOCH3
Glutatione
OH
Acido Mercapturico
OH
Macromolecole
cellulari
Morte cellulare
OXICAMS
Evoluzione delle strutture che hanno portato agli Oxicams.
H
C
C
C
C
C
C
+
O R O- H
O R O
O
H
X
Arile
O
O
H
X
Arile
S
O
O
O
CONHR
N
S
O
CH3
O
OH
CONHR
S
O
N
O
CH3
OXICAMS
OH
CONHR
S
O
N
CH3
O
Piroxicam
R=
N
R=
20-40 mg/dì
in dose unica
t1/2 45h
Isoxicam
N
CH3
O
N
Sudoxicam
R=
t1/2 24h
S
Sono inibitori rapidi e reversibili della cicloossigenasi.
Vengono impiegati nel trattamento della osteoartrite
e dell'artrite reumatoide e possono essere utilizzati
per lunghi periodi. Mostrano effetti collaterali a
carico del tratto gastrointestinale.
La ricerca attuale è orientata alla preparazione di
appropriati profarmaci che migliorano sensibilmente
il profilo farmacocinetico degli Oxicams sopra
riportati.
CH3
O
OCO
CH OCOOEt
CONH
CH CH C6H5
CONH
N
N
S
N
CH3
O
O
N
S
O
O
Ampiroxicam
CH3
Cinoxicam
O
O
N
N
O
N
S
O
CH3
O
Droxicam
Sintesi
O
N
O
ClCH2COOCH3
H
S
O
N
-
OH
CH2
S
O
O
Saccarina
O
OH
O
-
COOCH3
COOCH3
+
RO Na
N
DMSO
CH
COOCH3
S
O
S
O
O
CH3I
R-NH2
OXICAMS
NH
O
COMPOSTI VARI
PROQUAZONE (Biarison)
C6H5
N
900 mgr/dì
H3C
N
O
CH
CH3
H3C
BENZIDAMINA (Tantum)
O (CH2)3 N
N
CH3
CH3
150 mgr/dì
N
CH2 C6H5
DIFTALONE (Aladione)
O
N
500-750 mgr/dì
N
O
Nuovi approcci per lo sviluppo di FANS
con ridotta tossicità gastrointestinale:
FANS che rilasciano NO
FANS inibitori selettivi della COX-2
FANS che rilasciano NO
L'ossido d'azoto (NO) è capace di esercitare effetti
citoprotettivi simili a quelli osservati con le
prostaglandine. Infatti l'NO modula il flusso di
sangue, il rilascio di muco e ripara i danni della
mucosa.
Flurbiprofen nitrossibutilestere (HTC-1026)
CHCOO(CH2)4 ONO2
CH3
F
E' in fase II di sperimentazione clinica come
antiinfiammatorio ma possiede ulteriori indicazioni
terapeutiche per incontinenza urinaria ed
osteoporosi (NO inibisce gli osteoclasti).
Chetoprofen nitrossibutilestere
CH3
CHCOO(CH2)4
O
C
ONO2
NITROASPIRINE
CH2
ONO2
R=
NCX 4016
COOR
OCOCH3
R=H
Acido
Acetilsalicilico
R = (CH2)4
ONO2
NCX 4215
1. NO che si libera può controbilanciare l'effetto
negativo dell'aspirina dovuto all'inibizione
delle prostaglandine a livello gastrointestinale.
2. Le NO-aspirine mostrano un potenziato
effetto antiinfiammatorio dovuto al ruolo che
l'NO
gioca
nei
processi
patologici
infiammatori.
3. Le NO-aspirine sono potenziali nuovi agenti
antitrombotici.
NCX-4016
O
H3C
O
O
O
O
NO2
Il suo profilo farmacologico indica che l'introduzione
del gruppo che rilascia NO non solo elimina la
tossicità gastrointestinale, ma anche aumenta
l'attività analgesica e antitrombotica.
A differenza dell'aspirina l'NCX-4016 non potenzia la
risposta ulcerogenica gastrica allo stress ipotermico
(28-30 °C), ma anzi mostra una protezione dose
dipendente contro le lesioni gastriche indotte da
HCl/Etanolo.
E' in fase clinica I in Inghilterra.
FANS inibitori selettivi della COX-2
Acido Arachidonico
Enzima
COSTITUTIVO
nello stomaco,
piastrine e rene.
COX-1
COX-2
Enzima
INDOTTO dalle
citochine,
endotossine,
mitogeni
Celecoxib
Rofecoxib
Indometacina
Aspirina
altri FANS
Indometacina
Aspirina
altri FANS
Prostaglandine
COX-1 e COX-2 differiscono nella selettività per
gli inibitori a causa del diverso amminoacido in
posizione 523 che è una ISOLEUCINA nella
COX-1 e una VALINA nella COX-2.
SELETTIVITA’ COX-2
VALINA (COX-2)
COX-2
COX-1 ISOLEUCINA (COX-1)
Br
N
N
H2NO2S
SC 558
CF3
Inibitore specifico della COX-2:
interazione solo con COX-2
FANS legati ai siti attivi della
COX-1 rispetto alla COX-2
A.
Flurbiprofen legato nel canale del sito attivo della COX-1
ovina.
B.
SC-588, inibitore selettivo COX-2, legato nel canale del sito
attivo della COX-2 di topo.
L'analisi della struttura amminoacidica delle
due isoforme COX-1 e COX-2 ha evidenziato:
- una sostanziale analogia per il sito catalitico.
- un numero limitato di amminoacidi differenti
nel canale idrofobico attraverso il quale il
substrato transita per raggiungere il sito attivo
dell'enzima. La presenza però di una valina
nella COX-2 , al posto di una isoleucina nella
struttura della COX-1, rende disponibile una
tasca laterale non accessibile invece nella
COX-1.
GLI INIBITORI SELETTIVI COX-2 HANNO
CARATTERISTICHE STRUTTURALI TALI DA
POTERSI POSIZIONARE STABILMENTE NELLA
TASCA LATERALE CARATTERISTICA DELLA
COX-2,
INIBENDONE
SELETTIVAMENTE
L'ATTIVITA' ENZIMATICA.
NON SONO INVECE IN GRADO DI INTERAGIRE
IN MODO PERSISTENTE ED EFFICACE CON IL
SITO ATTIVO (PIU' RISTRETTO) DELLA COX-1.
CITOCHINE
MITOGENI
Dexametasone
COX-1
COSTITUTIVO
COX-2
INDOTTO
FANS
TXA2
PGI2
PGE2
Piastrine
Mucosa
gastrica
Endotelio
vascolare
Rene
FANS:
EFFETTI
COLLATERALI
PROSTAGLANDINE
INFIAMMAZIONE
DOLORE, GONFIORE
EFFETTI
TERAPEUTICI
INIBITORI SELETTIVI COX-2
Si
possono
considerare
inibitori
COX-2
"tempo-dipendenti". Inizialmente infatti hanno uguale
potenza su COX-1 e COX-2, ma la potenza contro COX-2
aumenta selettivamente dopo 10 minuti di incubazione. La
rimozione del farmaco per dialisi ripristina l'attività
COX-1, ma non COX-2, suggerendo l'instaurarsi di un
cambiamento irreversibile nell'enzima indotto COX-2.
Vengono suddivisi in due classi:
- ARILSOLFONAMMIDI
X
N
H
Y
O
O
H3C
R
S
- DIARILETEROCICLI
YO2S
ANELLO
ETEROCICLICO
X
ARILSOLFONAMMIDI
Derivano dal razionale approccio di sostituzione del gruppo
carbossilico degli acidi arilacetici con il gruppo isostero
solfonammidico.
R
O
NO2
O
O
S
H3C
N
H
NIMESULIDE (Aulin)
R=
NS-398
R=
F
F
O
X
O
O
S
H3C
N
H
CGP 28238
X=O
NS-398
X=S
DIARILETEROCICLI
Derivano dall'Indoxolo uno dei primi FANS non acidi.
H3CO
Indoxolo
N
H
H3CO
Studi di relazione struttura-attività hanno evidenziato
che l'anello eterociclico può essere di tipo diverso e che i
due gruppi arilici devono essere in posizione 1-2
dell'eterociclo che li mantiene in una corretta posizione
per poter interagire con l'enzima.
YO2S
ANELLO
ETEROCICLICO
X
DUP-697
Y
X
CH3
F
ETEROCICLO
Br
S
SC-57666
CH3
F
SC-58125
CH3
F
N N
CF3
CELECOXIB
NH2
CH3
N N
CF3
ROFECOXIB
CH3
H
O
O
SELETTIVITA' COX-1/COX-2
COX-1 (IC50,PM)
COX-2 (IC50,PM)
INIBITORI
NON SELETTIVI
PREFERENZIALI
SELETTIVI
- Naproxene
0,11
- Diclofenac
0,4
- Indometacina
0,2
- Piroxicam
<0,12
- Acido Mefenamico
<0,17
- NS-398
42
- DUP-697
85
- NIMESULIDE (Aulin)
55
- MELOXICAM (Mobic)
3
- JTE-522
>156
- CELECOXIB (Celebrex)
375
- ROFECOXIB (Vioxx)
800
- VALDECOXIB (Bextra)
28000
INIBITORI PREFERENZIALI COX-2
NIMESULIDE (Aulin) 1985
O
NO2
O
O
S
H3C
N
H
MELOXICAM (Mobic) 1996
O
O
S
CH3
N
H
N
S
CH3
OH
O
N
Viene utilizzato da pazienti con artrite reumatoide ed
osteoartrite. Ha un'emivita di circa 20h ed un effetto
terapeutico simile al Piroxicam, al Diclofenac e al
Naproxene, ma possiede maggiore tollerabilità
gastrointestinale probabilmente per la maggiore
selettività verso la COX-2.
INIBITORI SELETTIVI COX-2
INIBITORI SELETTIVI COX-2
CELECOXIB (Celebrex)
O
H2N
O
S
N
N
CF3
H3C
Appartiene alla classe degli 1,5-diarilpirazoli.
In vitro ha mostrato eccellente selettività per la COX-2 (IC50
COX-2/COX-1 : 0.04/15).
In vivo possiede potente attività antiinfiammatoria dopo
somministrazione per os:
ED50 7,1 mg/Kg per infiammazione acuta nel saggio
dell'edema indotto da carragenina;
ED50 0.37 mg/Kg/die per infiammazione cronica nel modello
di artrite.
Non produce nei topi tossicità gastrointestinale nè acuta, nè
cronica.
Nel saggio dell'edema indotto da carragenina nella zampa di
ratto inibisce la produzione di PGE2 alla dose di 0,1-2 mg/Kg,
mentre non tocca la PGE2 gastrica e renale fino alla dose di
600 mg/Kg.
Presenta un buon profilo farmacocinetico con emivita di 12h.
Il Celecoxib è il primo inibitore selettivo COX-2
approvato dalla FDA alla fine del 1998 ed è entrato
in commercio in America nel 1999 e
successivamente in altri paesi, tra cui l'Italia.
Gli studi clinici hanno mostrato che non produce
erosioni gastriche o ulcere, mentre hanno provato la
sua efficacia come analgesico ed antiinfiammatorio
in pazienti con osteoartriti ed artrite reumatoide.
Rappresenta una valida opzione nel trattamento del
dolore provocato dalle patologie infiammatorie delle
ossa.
Sintesi del Celecoxib
O
O
NaOCH3
CH3 +
H3C
MeOH
'
CF3
O
H3C
O
O
O
O
CF3 +
H2N
S
EtOH
N
H
H3C
O
H2N
NH2
O
S
N
N
H3C
CF3
'
ROFECOXIB (Vioxx)
O
O
S
H3C
O
O
Ha la stessa efficacia dell'Ibuprofen sia come
analgesico che come antipiretico, indicando che le
prostaglandine derivate dalla COX-2 sono
importanti mediatori del dolore e contribuiscono alla
febbre.
Studi clinici effettuati su pazienti affetti da artrite
reumatoide hanno evidenziato che il Rofecoxib ha
provocato infiammazioni gastriche nel 4% dei casi
rispetto al 26% del Naproxene.
Il Rofecoxib inoltre non ha effetto sul citocromo
P450.
Nel 1999 la Merck ha ottenuto dall'FDA
l'approvazione del Vioxx per il trattamento
dell'osteoartrite e del dolore con un'unica
somministrazione al giorno.
Nel 2004 il Vioxx è stato ritirato dal commercio
perchè cardiotossico. Sono stati registrati numerosi
casi di morte per infarto del miocardio.
Sintesi del Rofecoxib
H3C
S
CH3
Cl
AlCl3
+
H3C
S
CH3
O
O
O
O
H2O2
H3C
O
O
S
Br2
H3C
S
CH3
Br
O
O
O
O
O-Na+
H3C
S
O
O
DMF
O
O
O
O
S
H3C
O
O
(i-Pr)2NH
DMF
JTE-522
O
O
S
H2N
F
O
CH3
N
Il JTE-522 è attualmente in fase II di sperimentazione
clinica per il trattamento di artrite reumatoide ed
osteoartrite.
Inoltre questo composto ha mostrato attività
antiipertensiva in uno studio condotto su ratti,
indicando che le prostaglandine prodotte per azione
della COX-2 possono essere coinvolte nell'ipertensione
renovascolare.
Il JTE-522 è stato testato in vitro e in vivo per chiarire
un'eventuale relazione tra l'espressione della COX-2 e
le metastasi al fegato del cancro al colon e valutare il
potenziale effetto terapeutico. Gli effetti inibitori
mostrati dal JTE-522 in vivo sulle metastasi al fegato
ha permesso di concludere che gli inibitori COX-2 sono
potenzialmente utili nel prevenire tumorogenesi
colorettali ed anche metastasi ematogene del cancro al
colon.
VALDECOXIB (Bextra)
N
O
CH3
H2N
S
O
O
Il Valdecoxib è un inibitore COX-2 altamente selettivo ed ha
mostrato, dopo somministrazione orale, un'elevata potenza in
modelli animali sia di infiammazione acuta che cronica. E'
entrato in commercio nel 2004 per il trattamento di artrite e
dolore. Sono stati osservati effetti collaterali: eruzioni
cutanee, disturbi epatici e renali.
ATTIVITA' In Vitro (IC50, PM)
Enzimi ricombinanti umani
COX-1
COX-2
COX-1/COX-2
Valdecoxib
140 + 19
0.005 + 0.001
28000
Celecoxib
15 + 3.4
0.04 + 0.01
375
ATTIVITA' In Vivo (EC50, mg/Kg)
Edema indotto
da carragenina
nel ratto
Artrite
nel ratto
Bolla d'aria
nel ratto
Valdecoxib
10.2 + 1.4
0.032 + 0.002
0.05 + 0.02
Celecoxib
7.13 + 0.79
0.373 + 0.163
0.33 + 0.08
PARECOXIB SODIUM
N
O
Na
H3C
CH3
N
CH2
O
S
O
O
E' un potente e selettivo inibitore COX-2 somministrabile
per via parenterale. Si tratta infatti di un 'prodrug' che in
vivo rigenera il Valdecoxib. Si è dimostrato molto efficace
nelle forme di dolore acuto (ED 50 5mg/Kg) ed è in fase di
sperimentazione clinica per il trattamento di queste
patologie.
Sintesi
N
O
N
1. (RCO)2O, Et3N
O
2. NaOH
CH3
H2N
O
S
O
Na
N
R
O
O
CH3
S
O
R = CH3
R = CH2-CH3 Parecoxib Sodium
R = CH2-CH2-CH3
Profilo farmacocinetico della conversione del
PARECOXIB SODIUM a VALDECOXIB
Inversione dell’iperalgesia indotta da Carragenina dopo
somministrazione endovena di PARECOXIB SODIUM
Carragenina