")
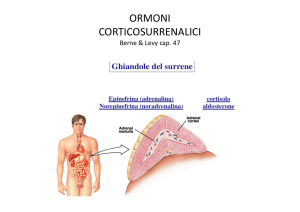
Sindromi corticosurrenaliche: clinica e laboratorio
Biochimica e fisiologia della corticale surrenalica (Rassegna)
Antonello La Brocca
Divisione di Medicina Interna e Ambulatorio di Endocrinologia, Ospedale Civile, ASL 5 Giaveno (TO)
CORTISOLO ED ANDROGENI SURRENALICI:
BIOSINTESI E METABOLISMO
Sebbene le vie biosintetiche siano comuni per i diversi
steroidi corticosurrenalici, la specificità della sintesi nelle
varie zone del corticosurrene è ottenuta mediante la presenza di recettori specifici per l’ACTH o per l’angiotensina,
oltre che con la distribuzione dei sistemi enzimatici specifici.1
In tal modo, le cellule della zona glomerulare del corticosurrene che non posseggono la 17-α-idrossilasi, non
partecipano alla sintesi del cortisolo e degli androgeni, per
i quali questo enzima è essenziale. Viceversa, le cellule
della zona fascicolata e reticolare non posseggono l’enzima 18-idrossilasi e quindi non possono sintetizzare l’aldosterone. Comunque, tutte le cellule possono sintetizzare
desossicorticosterone, in quanto le fasi iniziali di sintesi
sono comuni ad entrambe le vie enzimatiche (Fig. 1).
I principali metaboliti del cortisolo si generano per riduzione del doppio legame e dei gruppi chetonici con formazione di tetraidrocortisolo, tetraidrocortisone, cortolo e cortolone. Questi metaboliti vengono secreti con le urine
coniugati con acido glicuronico e costituiscono circa dal 60
al 70% del cortisolo totale prodotto (Fig. 2). Una piccola
parte del cortisolo prodotto (fino a circa 150 µg al giorno)
viene escreta come tale e rappresenta il cortisolo circolante in forma libera che viene filtrato dal rene (cortisolo libero
urinario).
I principali androgeni prodotti dalla corticale del surrene
(zona fascicolata e reticolare) sono il DEA, il DEAS, l’androstenedione e, in piccole quantità il testosterone2.
L’androstenedione ed il testosterone vengono secreti
anche dall’ovaio e dal testicolo, e una piccola quantità di
DEA è prodotta dall’ovaio.
I processi enzimatici compresi nelle principali vie metaboliche determinano la riduzione dell’anello A ed i metaboliti risultanti vengono coniugati ed escreti nelle urine come
solfati e glicuronidi (Fig. 3).
FISIOLOGIA DELL’ASSE IPOTALAMO-IPOFISISURRENE
Fisiologicamente (Fig. 4), attraverso gli organi di senso,
viene colto uno stimolo aspecifico, quale lo stress, il quale
determina l’attivazione e la modulazione dei neuroni aminergici sopraipotalamici secernenti noradrenalina, acetilco10
lina e serotonina che, a loro volta, influenzano i neuroni
peptidergici ipotalamici. Da parte di questi ultimi viene
immesso nella circolazione portale-ipofisaria il CRH
(Corticotropin Releasing Hormone) il quale arriva all’ipofisi
e ivi stimola la secrezione di ACTH che va ad agire sulle
cellule della zona reticolare e fascicolata del corticosurrene
inducendo la secrezione di cortisolo e di androgeni surrenalici. A sua volta il cortisolo esercita un effetto di feedback negativo sia sull’ipofisi (feed-back corto) che sull’ipotalamo (feed-back lungo).3 Eserciterebbe anche un effetto
di feed-back negativo su sé stesso: feed-back extracorto.
Anche l’ACTH esercita un effetto di feed-back sui neuroni
peptidergici ipotalamici che secernono CRH: esso è denominato feed-back ultracorto. Inoltre l’ACTH eserciterebbe
anche un effetto di feed-back su sé stesso: si parla di auto
feed-back (Fig. 4).
Il CRH è un neuro-ormone prodotto dai nuclei sopraottico e paraventricolare dell’ipotalamo. Tra le azioni biologiche più importanti svolte dal CRH vi è quella di favorire la
steroidogenesi surrenalica inducendo la liberazione, da
parte dell’ipofisi, dell’ACTH.
L’ACTH è un ormone proteico costituito da 39 aminoacidi, dei quali i primi 23(1-23) costituiscono il nucleo attivo
specifico della molecola, mentre i restanti 16 (24-39) costituiscono la coda variabile della molecola, con funzione stabilizzatrice. La sua emivita è di circa 10 minuti. L’ACTH
esercita sia degli effetti sul surrene che extrasurrenalici.
Tra gli effetti che l’ACTH esercita sul surrene (in particolare sulla zona fascicolata e reticolare) occorre ricordare:
- stimolazione della crescita del corticosurrene;
- mantenimento delle dimensioni e dell’integrità del corticosurrene;
- mantenimento dell’attività enzimatica sintetica steroidea;
- aumento del flusso plasmatico surrenalico;
- attivazione dell’AMP ciclico: essa rappresenta lo stimolo e l’avvio alla steroidogenesi;
- accumulo di colesterolo nelle cellule cortisolo-secernenti;
- stimolazione della tappa di conversione del colesterolo
in pregnenolone.
Tra gli effetti extrasurrenalici realizzati dall’ACTH il più
importante è rappresentato dall’azione melanocitostimolante. Essa deriva dal fatto che l’ACTH trae origine da un
precursore di grosse dimensioni, la pro-opiomelanocortina,
la quale genera tre molecole che contengono la sequenza
LigandAssay 11 (1) 2006
Sindromi corticosurrenaliche: clinica e laboratorio
dell’ormone stimolante i melanociti (MSH): il frammento
NH2 terminale che contiene la γ-MSH, l’ACTH con l’α-MSH
e la β-lipotropina (β-LPH) con la β-MSH.
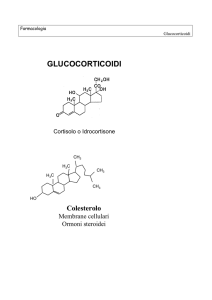
Il Cortisolo, il più importante glucocorticoide nell’uomo,
è sintetizzato, a partire dal colesterolo, dalle cellule della
zona fascicolata e reticolare del corticosurrene sotto l’influenza dell’ACTH. Nell’uomo normale, in assenza di
importanti stress, vengono secreti giornalmente circa 20
mg di cortisolo. Le quantità secrete variano in rapporto ad
un ritmo circadiano di produzione4 (Fig. 5). Generalmente
il picco massimo di secrezione del cortisolo si verifica al
momento del risveglio. Dal risveglio fino a quando non si
va a dormire alla sera la caratteristica di questa secrezione cortisolemica è rappresentata dalla progressiva e
costante riduzione verso valori < a 5 µg/dL. Questa riduzione però non è mai semplice, lineare, progressiva, ma è
frammezzata da picchi di secrezione che sono quanto mai
variabili. Picchi di secrezione costanti e fissi sono invece
quelli legati al pasto e che si realizzano dopo circa due ore
da esso; l’assenza del pasto abolisce tali picchi. Nelle
Figura 1
Principali tappe della steroidogenesi
Figura 2
Principali metaboliti del cortisolo
Figura 3
I principali androgeni cortico surrenalici ed i loro metaboliti
LigandAssay 11 (1) 2006
11
Sindromi corticosurrenaliche: clinica e laboratorio
re in alcune condizioni patologiche ed in particolare in caso
di inappropriata secrezione di cortisolo (malattia e sindrome di Cushing) o in corso di depressione psicogena.
Nel plasma il cortisolo circola legato a delle proteine di
trasporto. La più importante è la transcortina o
Corticosteroid-Binding Globulin (CBG), una α-2-globulina
prodotta a livello epatico, che lega il 95% del cortisolo circolante in condizioni normali. Il rimanente 5% costituisce la
frazione libera e metabolicamente attiva dell’ormone.
Allorquando il cortisolo viene prodotto in eccesso, la CBG
viene saturata e la quantità eccedente dell’ormone viene
legata labilmente all’albumina. L’emivita del cortisolo circolante è di circa 90-100 minuti; esso viene catabolizzato
principalmente dal fegato.
Effetti biologici di interesse clinico del cortisolo
Vengono di seguito descritti i più importanti effetti biologici di interesse clinico esercitati dal cortisolo.
Figura 4
Rappresentazione dell’asse ipolamo-ipofisi-surrene (cortisolo)
Figura 5
Ritmo circadiano di secrezione del cortisolo
prime due ore di sonno (periodo questo nel quale si collocano le fasi più profonde del sonno, e cioè la fase 3 e 4 del
sonno REM) si assiste ad una assoluta mancanza di attività del sistema ipotalamo-ipofisi-surrene, per cui esso è funzionalmente completamente “azzerato”. Trascorse le
prime due ore del sonno, l’asse surrenalico riprende a funzionare per cui compaiono dei picchi di secrezione di cortisolo piccoli ma progressivamente crescenti; successivamente essi si fanno sempre più ampi fino al risveglio, allorquando si manifesta il picco secretorio quantitativamente
maggiore. Occorre tenere presente che la secrezione
dell’ACTH è speculare rispetto a quella del cortisolo, solo
che i picchi di secrezione dell’ACTH sono anticipati di 1520 minuti rispetto a quelli del cortisolo.
Il bioritmo di secrezione del cortisolo e dell’ACTH può
subire variazioni in conseguenza di un’alterazione del
ritmo sonno-veglia, come può accadere in seguito a cambiamenti di fuso orario o di turno nell’attività lavorativa, e
sono in genere necessari da cinque a sette giorni affinché
la secrezione circadiana riacquisti il normale ritmo.
Alterazioni del ritmo circadiano si possono inoltre verifica12
Effetti sul metabolismo glucidico
Il cortisolo aumenta i livelli ematici di glucosio attivando
la neoglucogenesi 5. La neoglucogenesi epatica è fisiologicamente importante per il mantenimento della glicemia in
condizioni di digiuno e quindi per la salvaguardia del metabolismo glucosio-dipendente del sistema nervoso, nonché
per il mantenimento delle riserve di glicogeno. Dopo un
digiuno notturno, il fegato è la sola fonte di glucosio. Il
fegato utilizza substrati del catabolismo muscolare e adiposo (alanina, lattato, glicerolo) e produce glucosio-6fosfato, cha a sua volta può dare origine a glucosio circolante oppure a glicogeno immagazzinato negli epatociti. I
glucocorticoidi facilitano l’afflusso di substrati attivando il
catabolismo proteico muscolare e la lipolisi, aumentano la
captazione di aminoacidi da parte del fegato, inducono la
sintesi di enzimi-chiave del processo neoglucogenetico. Il
cortisolo favorisce poi l’azione di altri ormoni direttamente
o indirettamente neoglucogenetici, come il glucagone e le
catecolamine. La facilitazione di questi effetti ormonali
rientra nel generico concetto di azione permissiva dei glucocorticoidi.
Il cortisolo, inoltre, riduce l’utilizzazione del glucosio inibendone la captazione da parte delle cellule insulino-sensibili. In sostanza, svolge un’azione antiinsulinica che
rende ragione dell’insulino-resistenza osservabile in corso
di stress e nella sindrome di Cushing, nonché dell’ipersensibilità all’insulina osservabile nella malattia di Addison. I
glucocorticoidi non modificano l’affinità di legame dell’insulina per i recettori di membrana e non riducono significativamente il numero dei siti recettoriali. L’azione antiinsulinica è post-recettoriale, ossia il cortisolo interferisce significativamente, diminuendone a più livelli l’efficienza funzionale, sulla traslocazione indotta dall’insulina di proteine trasportatrici (carrier) preposte al trasporto transmembrana
del glucosio dal compartimento microsomiale alla membraLigandAssay 11 (1) 2006
Sindromi corticosurrenaliche: clinica e laboratorio
na plasmatica cellulare. Fra le proteine carrier quella ritenuta più insulino-dipendente è quella indicata con la sigla
GLUT-4, espressa in modo privilegiato nel tessuto adiposo
e muscolare.
Effetti sul metabolismo lipidico
Il cortisolo inibisce l’assorbimento del glucosio da parte
del tessuto adiposo, riducendo perciò la disponibilità di glicerofosfato per la riesterificazione degli acidi grassi e quindi limitando la lipogenesi. Contemporaneamente sembra
che aumenti la liberazione degli acidi grassi liberi. I glucocorticoidi inoltre controllano indirettamente il metabolismo
dei grassi potenziando la risposta lipolitica ad agenti come
l’ormone della crescita o le catecolamine.
Effetti sul metabolismo proteico
Il cortisolo aumenta il catabolismo proteico e rende
negativo il bilancio azotato. L’azione ormonale si esplica in
sede extraepatica (tessuto muscolare, connettivo, osso)
ed è seguita dall’aumentata captazione di aminoacidi circolanti da parte del fegato, oltre che da aumentata escrezione renale. I muscoli costituiscono la più larga riserva
corporea di aminoacidi; se si prescinde dal contenuto idrico, oltre l’80% della massa muscolare è costituita da proteine. L’eccesso di glucocorticoidi, soprattutto se prolungato nel tempo, comporta una riduzione anche vistosa della
muscolatura scheletrica per impoverimento delle proteine
strutturali. L’ipotrofia muscolare comporta una diminuzione
della forza muscolare.
L’azione catabolica sulle proteine strutturali si rende
evidente anche a livello cutaneo. Nell’ipercortisolismo cronico la cute è sottile, fragile, poco elastica, facilmente vulnerabile, il derma è impoverito di connettivo, e ciò perché i
glucocorticoidi inibiscono la sintesi del collagene e l’attività
dei fibroblasti.
Effetti sul metabolismo osseo
L’osteoporosi da eccesso di glucocorticoidi è conseguenza della diminuzione del bilancio calcico in quanto
determinano una diminuzione dell’assorbimento intestinale
di calcio per una diminuzione dell’idrossilazione della 25OH-vitamina D3 in 1-α-25-diidrossi-vitamina D3, un aumento dell’ecrezione renale di calcio e di fosforo, un aumento
della secrezione di Paratormone (PTH), un aumento dell’attività degli osteoclasti ma soprattutto una riduzione del
numero e dell’attività degli osteoblasti per probabile blocco
maturativo e differenziativo. 6
Effetti sul metabolismo idrico ed elettrolitico
In condizioni di normale funzione renale, il cortisolo, a
concentrazioni fisiologiche o parafisiologiche, ha azione
diuretica, in quanto è in grado di esercitare un effetto perLigandAssay 11 (1) 2006
missivo sulla capacità di eliminare un carico idrico, ciò per
il mantenimento di un adeguato flusso glomerulare e per
riduzione del riassorbimento tubulare. E’ inoltre ostacolata
l’azione della Vasopressiona (ADH). Il cortisolo, però, è
potenzialmente dotato di attività mineralcorticoide, nel
senso che può legarsi ai recettori per l’aldosterone e favorire quindi la ritenzione di sodio e l’eliminazione di potassio. Con la ritenzione di sodio si ha allora ritenzione di
acqua.
Il cortisolo favorisce l’escrezione renale di calcio e di
fosforo. E’ diminuito il riassorbimento tubulare mentre è
aumentata la filtrazione glomerulare. Il cortisolo inibisce
anche l’assorbimento intestinale di calcio, espletando un’azione opposta a quella della 1,25-diidrossicolecalciferolo
(vitamina D3). L’effetto ormonale globale è ipocalcemizzante; a seguito della riduzione dei livelli calcemici, è stimolata la secrezione di PTH, con conseguente riassorbimento della componente minerale dell’osso che, insieme
alla riduzione dell’attività appositiva degli osteoblasti, è
responsabile dell’osteoporosi anche notevole che caratterizza le sindromi da eccesso di glucocorticoidi. Anche l’assorbimento intestinale di fosforo è ostacolato dai glucocorticoidi.
Effetti sull’apparato cardiovascolare
Il cortisolo partecipa al controllo della pressione arteriosa. Lo testimoniano da un lato l’ipotensione della malattia
di Addison e dall’altro l’ipertensione della sindrome di
Cushing. Nel soggetto normoteso, comunque, l’iniezione
singola anche di notevoli quantità di cortisolo non causa
ipertensione. L’ipertensione arteriosa è, invece, la regola
se l’eccesso di glucocorticoidi si prolunga nel tempo.
L’azione ormonale sulla pressione arteriosa riconosce
diverse cause: dall’attività mineralcorticoide all’effetto “permissivo” sulla azione vasocostrittrice delle catecolamine
endogene, ai complessi effetti sul sistema renina-angiotensina-aldosterone.
A livello cardiaco, l’azione dei glucocorticoidi può quindi definirsi inotropa positiva, cronotropa positiva e dromotropa positiva. In particolare, risulta facilitata la conduzione
atrio-ventricolare (accorciamento del tratto P-Q dell’elettrocardiogramma sotto carico corticoide; allungamento dello
stesso nelle crisi da iposurrenalismo acuto).
Effetti sull’apparato respiratorio
Il cortisolo favorisce l’interazione catecolamine/β-recettori nell’albero bronchiale. In particolare esercita un effetto
di “sblocco” delle cellule con recettori beta-adrenergici presenti a livello bronchiale, con “facilitazione” dell’azione di
beta-stimolazione (con conseguente broncodilatazione)
delle catecolamine endogene: effetto di modulazione
recettoriale che determina quindi un potenziamento d’azione tra cortisolo-glicocorticoidi e sostanze beta2-stimolanti.
Altri effetti dei glicocorticoidi riguardano la sintesi e la
13
Sindromi corticosurrenaliche: clinica e laboratorio
secrezione del surfactante alveolare da parte degli pneumociti di II tipo.
Effetti sul sistema endocrino
I glucocorticoidi influenzano la funzione tiroidea. In condizioni di ipercortisolismo cronico i livelli basali di TSH sono
normali, ma è inibita la risposta al TRH. In genere, è compromessa la sintesi dell’α-subunità degli ormoni glicoproteici ipofisari e quindi, così come per il TSH, anche per le
gonadotropine FSH ed LH si ha torpidità di risposta allo stimolo con lo specifico releasing hormone (RH) e ridotta pulsatilità nella secrezione di base. I livelli circolanti degli
ormoni tiroidei totali (T4 e T3) sono tendenzialmente bassi;
i glucocorticoidi riducono inoltre la sintesi epatica di thyroxine binding globulin (TBG) e frenano la conversione periferica di T4 a T3 favorendo, invece, la via alternativa a
reverse T3 (rT3). Le concentrazioni degli ormoni liberi (FT4
ed FT3) sono di regola normali e i segni clinici di ipotiroidismo sono molto sfumati o del tutto assenti.
Per quanto attiene alla funzione gonadica, oltre agli
effetti sulle gonadotropine sopra menzionati, nel sesso
maschile l’eccesso di glucocorticoidi diminuisce la secrezione di testosterone agendo direttamente sulle cellule di
Leydig. La risposta testicolare allo stimolo gonadotropinico
risulta inibita. I livelli circolanti di testosterone sono bassi
anche per la ridotta sintesi epatica di SHBG (Sex Hormone
Binding Globulin).
Nella donna la modulazione negativa glucocorticoide
coinvolge più il complesso ipotalamo-ipofisario che non l’ovaio. La pulsatilità spontanea ad alta frequenza della gonadotropina LH è compromessa. L’amenorrea ipogonadotropa è molto frequente nelle pazienti con sindrome di
Cushing.
I glucocorticoidi influenzano negativamente la secrezione e l’azione dell’ormone dell’accrescimento (GH).
L’eccesso di cortisolo nell’età infantile si accompagna di
regola a rallentamento o arresto della crescita. La risposta
del GH plasmatico a test provocativi è spesso bloccata. Vi
sono evidenze consolidate che i glucocorticoidi inibiscano
l’espressione genica per l’ormone (ridotto mRNA specifico). I glucocorticoidi si oppongono anche all’azione dell’ormone della crescita (GH), limitando la sintesi dell’IGF-1 e/o
quella dei recettori per il fattore di crescita GH-dipendente.
I glucocorticoidi, come già in precedenza riportato,
aumentano in via indiretta la secrezione di PTH, insulina,
glucagone. Modulano negativamente sintesi e azione della
vasopressina, positivamente sintesi e azione dell’adrenalina.
Effetti sul sistema nervoso centrale
Il cervello è estremamente sensibile ai glucocorticoidi;
le fluttuazioni all’interno dell’intervallo fisiologico producono modificazioni dei livelli di soglia di certe sensazioni e di
alcune funzioni superiori, come la capacità di concentra14
zione, la memoria e la capacità intellettiva. In generale, l’insufficienza dei glucocorticoidi deprime l’attività cerebrale
causando apatia e facile stancabilità, mentre il loro eccesso causa uno stato di euforia, insonnia ed iperattività. Nel
secondo caso possono anche manifestarsi chiari segni di
psicosi, quasi sempre reversibili con il ritorno alla norma
dei livelli ormonali. Nell’insufficienza corticosurrenale il
flusso ematico cerebrale risulta ridotto. Questa è probabilmente una manifestazione locale dell’insufficienza circolatoria generalizzata ed è reversibile con la somministrazione di cortisolo. In queste condizioni l’insufficienza vascolare può essere responsabile della depressione della funzione cerebrale.
Effetti sul sistema emocoagulativo
L’ecceso di glucocorticoidi che si verifica ad esempio
nella sindrome di Cushing è responsabile di una ipercoagulabilità ematica e quindi di una spiccata trombofilia per 7:
(a) aumento dei livelli plasmatici di alcuni fattori della
coagulazione: fattore II, V, IX e soprattutto fattore VIII
di von Willebrand;
(b) alterazione del sistema fibrinolitico: aumento della sintesi e del rilascio di plasminogeno, di α2-antiplasmina e
dell’inibitore dell’attivatore del plasminogeno (PAI-1) e
diminuzione del rilascio dell’attivatore tissutale del plasminogeno (tPA).
Effetti sulla risposta immune
Sul sistema immunitario i glicocorticoidi esercitano i
seguenti effetti:
(1) forte inibizione dell’ ipersensibilità di tipo III (ad esempio la malattia da siero che coinvolge vasi, membrane,
cellule, con l’interessamento del complemento);
(2) inibizione dell’attacco dell’anticorpo all’antigene di
membrana nelle cellule del sistema reticolo-endoteliale
nell’ipersensibilità di tipo II (trombocitopenia autoimmune, anemia emolitica);
(3) inibizione dell’ ipersensibilità di tipo I mediante:
(a) blocco della proliferazione dei cloni di cellule produttrici di IgE;
(b) inibizione della funzione delle cellule T-helper;
(4) inibizione dell’ ipersensibilità ritardata di IV tipo per
effetto inibente sui linfociti T attivati dalle linfochine
(tant’è che la intradermoreazione alla tubercolina viene
totalmente bloccata dalla somministrazione di glucocorticoidi);
(5) effetto antinfiammatorio (inibizione della flogosi) in
quanto determinano:
(a) inibizione del reclutamento dei neutrofili e dei monociti-macrofagi a livello della sede della flogosi;
(b) diminuzione del numero dei linfociti circolanti, specie dei linfociti T;
(c) diminuzione della produzione di un attivatore del
plasminogeno, con ostacolo alla migrazione dei leucoLigandAssay 11 (1) 2006
Sindromi corticosurrenaliche: clinica e laboratorio
citi nella sede della flogosi;
(d) diminuzione della depurazione da parte del sistema
reticolo-endoteliale di cellule ricoperte di anticorpo (inibizione dell’ipersensibilità di tipo II);
(e) diminuzione della sintesi di prostaglandine, trombossano, leucotrieni, per blocco della fosfolipasi A2.
Effetti di rilievo ai fini di interferenze con i
processi infettivi
(1) diminuzione della produzione di monociti a livello
midollare;
(2) diminuzione del numero delle cellule fagocitarie miste;
(3) aumento dei neutrofili circolanti per aumento dell’afflusso dal midollo e diminuzione della clearance periferica;
(4) diminuzione delle attività specifiche dei neutrofili per:
(a) diminuzione della fagocitosi;
(b) diminuzione delle proprietà depuranti del sistema
reticolo-endoteliale.
realizza la idrossilazione in posizione 17-α: ciò differenzia
i mineralcorticoidi dai glucocorticoidi nei quali tale idrossilazione si realizza (Fig. 6) Determinanti per l’attività biologica dell’aldosterone sono la presenza nella sua molecola
del doppio legame in posizione 4 e del gruppo chetonico in
posizione 3 (Fig. 7).
Nel corso delle tappe biosintetiche del passaggio da
progesterone ad aldosterone si formano anche due composti intermedi che sono: il desossicorticosterone ed il 18idrossicorticosterone (Fig. 6). Essi sono da considerare dei
mineralcorticoidi, anche se “meno potenti” dell’aldosterone.
Il ritmo medio di secrezione dell’aldosterone è di 100200 µg/die. Nel sangue per il 30% si trova alla stato libero
e per il 70% si lega debolmente alle seguenti proteine plasmatiche di trasporto: CBG e albumina, e di conseguenza
Effetti sui meccanismi di difesa dell’ospite
(1) soppressione della risposta immunitaria alle infiammazioni acute e croniche;
(2) alterazione della risposta immunologia, in particolare
dell’ipersensibilità ritardata, come già visto in precedenza;
(3) disturbo dei normali meccanismi intracellulari di processamento dei materiali captati dalle cellule fagocitarie;
(4) diminuzione della produzione di interferone;
(5) interferenza con i processi di guarigione dei tessuti lesi;
(6) effetto antipiretico che maschera infezioni anche gravi.
Effetti biologici di interesse clinico degli
androgeni surrenalici
Le più importanti azioni biologiche di interesse clinico
esercitate dagli androgeni surrenalici sono sostanzialmente analoghe a quelle effettuate dagli androgeni di provenienza testicolare, e consistono in un aumento della sintesi proteica, della massa muscolare e della forza muscolare per incremento del glicogeno muscolare (effetti di tipo
anabolico), incremento della deposizione di matrice ossea,
sviluppo dei caratteri sessuali secondari (voce, distribuzione della peluria corporea), arretramento dell’attaccatura
dei capelli, ipertrofia delle ghiandole sebacee con induzione dell’acne, oltre ad un modesto contributo all’effetto
gonadico sullo sviluppo del pene e del clitoride alla pubertà (surrenarca).
Figura 6
Tappe biosintetiche dell’aldosterone e dei mineralcorticoidi
Aldosterone e mineralcorticoidi: biosintesi,
metabolismo e fisiologia
Le cellule della zona glomerulare del surrene non posseggono l’enzima 17-α-idrossilasi per cui in esse non si
LigandAssay 11 (1) 2006
Figura 7
Molecola dell’aldosterone
15
Sindromi corticosurrenaliche: clinica e laboratorio
ha un’emivita relativamente breve (circa 20 minuti) ed
un’elevata velocità di clearance metabolica.
Due sono i composti che vengono prodotti in quantità
importanti dal catabolismo dell’aldosterone, e che sono
privi di attività mineralcorticoide: il tetraidroaldosterone che
viene coniugato con acido glicuronico e solforico nel fegato ed eliminato attraverso le urine, e l’aldosterone glicuronide che viene eliminato attraverso le urine (Fig. 8).
Il meccanismo fisiologico fondamentale che regola la
secrezione dell’aldosterone è rappresentato dall’attivazione del sistema renina-angiotensina.8 Infatti la renina prodotta da parte delle cellule dell’apparato iuxtaglomerulare
del rene va ad agire su di un substrato secreto dal fegato,
l’angiotensinogeno, ne idrolizza il legame leucina-leucina
producendo così angiotensina I, che è un decapeptide; sull’angiotensina I agisce un enzima, l’enzima di conversione
(ACE) sintetizzato nel polmone, nel rene ed in altri distretti vascolari, il quale stacca dall’angiotensina I due aminoacidi (la leucina e l’istidina) dando così origine all’angiotensina II, che è un octapeptide. L’angiotensina II va a sua
volta a stimolare la biosintesi ed il rilascio dalle cellule della
zona glomerulare del corticosurrene di aldosterone (Fig.
9).
Anche la secrezione di aldosterone avviene in maniera
episodica, con un ritmo molto simile a quello del cortisolo.
Non sono comunque noti né il meccanismo né la finalità
fisiologica di tale modalità di secrezione. E’ tuttavia noto
che essa non è dovuta né all’ACTH, in quanto il ritmo di
secrezione dell’aldosterone è mantenuto anche in assenza
di ACTH, né alle modificazioni plasmatiche di potassio e di
sodio. Non è stato inoltre completamente stabilito se tale
ritmo di secrezione sia dovuto alle modificazioni plasmatiche dell’angiotensina.
Effetti biologici dell’aldosterone
L’Aldosterone ha un ruolo fondamentale nella regola-
zione del volume extracellulare e del metabolismo del
potassio, mediato dal classico meccanismo di azione
genomico attraverso il legame con il suo recettore localizzato nei classici organi bersaglio. Infatti l’ormone stimola il
riassorbimento di sodio attraverso varie strutture epiteliali:
tubulo renale, ghiandole sudoripare, salivari e gastrointestinali, agisce a livello della parte distale del nefrone dove
favorisce il riassorbimento di sodio ed acqua ed aumenta
l’escrezione degli ioni potassio ed idrogeno. Da ciò ne deriva di conseguenza un aumento dei liquidi extracellulari
(volemia) e quindi una azione potenziatrice sull’incremento della pressione sanguigna.9,10 Queste azioni sono sicuramente coinvolte nella fisiopatologia degli iperaldosteronismi primitivi e secondari (Fig. 10).
Sono stati inoltre descritti effetti non genomici rapidi
che hanno luogo tramite meccanismi coinvolgenti vie di
trasduzione di segnale. Un loro ruolo in situazione patologiche umane non è stato ancora dimostrato, ma potrebbero essere coinvolti nella regolazione della funzione endoteliale. Più recentemente, il riscontro di recettori di mineralcorticoidi in tessuti non epiteliali come il cuore, i vasi e il
cervello ha aperto nuovi orizzonti nella comprensione dei
molteplici ruoli che l’aldosterone può avere sul sistema cardiovascolare in particolare sul rimodellamento vascolare e
cardiaco, sulla sintesi del collageno e lo sviluppo di fibrosi.
L’effetto dell’aldosterone che può contribuire al danno
vascolare comprende la disfunzione endoteliale, l’ipertrofia
e l’iperplasia delle cellule muscolari liscie, la fibrosi della
matrice del collageno, la diminuita “complianza” arteriosa e
venosa, l’aumento delle resistenze vascolari periferiche
(causato dall’ingresso del sodio nelle cellule muscolari
liscie della parete vascolare con conseguente aumento
della contrattilità) e l’alterato controllo autonomico da
disfunzione dei baroriflessi, in aggiunta ai ben noti e già
descritti effetti sulla ritenzione di acqua e sale. A livello
miocardico l’aumento del contenuto di collageno e quindi la
fibrosi unite all’ipertrofia indotta dall’aumento dei valori
Figura 8
Principali metaboliti dell’aldosterone
16
LigandAssay 11 (1) 2006
Sindromi corticosurrenaliche: clinica e laboratorio
Figura 9
Rappresentazione del sistema renina-angiotensina-aldosterone
Aumento
Ipotassiemia
Aldosterone
Ritenzione sodica
PRA
e ipervolemia
1 fase
2 fase
Volume dipendente
Volume dipendente
Escape
sodica
Aumento della portata
Aumento del contenuto
cardiaca
di Na+ nelle fibrocellule
muscolari dei vasi
Vasocostrizione
Aumento delle resistenze
periferiche
IPERTENSIONE ARTERIOSA
Figura 10
Meccanismi patogenetici indotti dall’eccesso di aldosterone
LigandAssay 11 (1) 2006
17
Sindromi corticosurrenaliche: clinica e laboratorio
pressori causa una riduzione della “complianza” ed, in ultima analisi, una insufficienza diastolica11.
Alcuni studi hanno dimostrato che il fenomeno dell’aumentata sintesi di collageno e della fibrosi aldosteronedipendente è preceduto da processi di tipo infiammatorio,
prevalentemente perivascolari, caratterizzati da infiltrazioni
di monociti-macrofagi e da un’aumentata espressione e
concentrazione di citochine e chemochine proinfiammatorie, fenomeni bloccati dal trattamento con antagonisti
recettoriali dell’aldosterone. Altri studi segnalano un ruolo
dello stress ossidativo come primo evento di questa cascata infiammatoria. Infine, molto recentemente, è stato dimostrato come questi meccanismi possano aver luogo anche
a carico di cellule mononucleate del sangue periferico circolanti, attivate prima che invadano il circolo coronarico
intramurale, tramite un aumentato ingresso di ioni-Calcio
intracellulare. Anche questo fenomeno di interfaccia neuroendocrino-immunologico può essere bersaglio di farmaci antialdosteronici.
4.
Weitzmann Ed, Fukushima D, Nogeire C. Twentyfour
hour pattern of episodic secretion of cortisol in normal
subjects. J. Clin. Endocrinol. Metab. 1971; 33: 14-22.
5.
Mcmahon M, Gerich J, Rizza R. Effects of
glucocorticoid on carbohydrate metabolism. Diabetes
Metab. Rev. 1988; 4: 17-30.
6.
Lukert Bp, Raisz Lg. Glucocorticoid-induced
osteoporosis: pathogenesis and management. Ann. Inter.
Med. 1990; 112: 352-364.
7.
La Brocca A, Terzolo M, Pia A, Paccotti P, De Giuli P,
Angeli A. Recurrent thromboembolism as a hallmark of
Cushing’s sindrome. J. Endocrinol. Invest. 1997; 20: 211214.
8.
Quinn Sj, Williams Gm. Regulation of aldosterone
secretion. Ann. Rev. Physiol. 1988; 50: 409-426.
BIBLIOGRAFIA
9.
Funder Jw, Pearle Pt, Smith R, Smith Ai.
Mineralcorticoid action: target tissue specificit is enzyme,
not receptor, mediated. Science, 1988; 242: 583-585.
10.
Weber Kt. Aldosteronism revisited: perspectives on less
well-recognized actions of aldosterone. J. Lab. Clin. Med.
2003; 142: 71-80.
11.
Muiesan Ml, Rizzoni D, Salvetti M, et Al. Structural
changes in small resistance arteries and left ventricular
geometry in patients with primary and secondary
hypertension. J. Hypertens. 2002; 20: 1295-1304.
1.
Curnow Km, White Pc, Pascoe L. Adrenal steroid
biosynthesis. Curr. Opin. Endocr. Diab. 1994; 1: 10-15.
2.
Parker Ln. Control of adrenal androgen secretion.
Endocrinol. Metab. Clin. North Am. 1991; 20: 401-421.
3.
Dallman M, Akana S, Scribner K. Stress, feedback and
facilitation in the hypothalamo-pituitary-adrenal axis. J.
Neuroendocrinol. 1992; 4: 517-526.
Per corrispondenza:
Dr. Antonello La Brocca
Via Mons. Carlo Bovero n. 54 - 10094 Giaveno (TO)
Tel.: 011/9349042 - Fax: 011/9349042
e-mail: [email protected]
18
LigandAssay 11 (1) 2006
")