lez
LE STRATEGIE CATALITICHE
I principi catalitici di base
La funzione principale di un enzima è quella di accelerare una reazione,
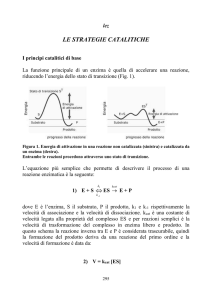
riducendo l’energia dello stato di transizione (Fig. 1).
Figura 1. Energia di attivazione in una reazione non catalizzata (sinistra) e catalizzata da
un enzima (destra).
Entrambe le reazioni procedono attraverso uno stato di transizione.
L’equazione più semplice che permette di descrivere il processo di una
reazione enzimatica è la seguente:
k1
kcat
1) E + S ES E + P
k 1
dove E è l’enzima, S il substrato, P il prodotto, k1 e k-1 rispettivamente la
velocità di associazione e la velocità di dissociazione. kcat è una costante di
velocità legata alla proprietà del complesso ES e per reazioni semplici è la
velocità di trasformazione del complesso in enzima libero e prodotto. In
questo schema la reazione inversa tra E e P è considerata trascurabile, quindi
la formazione del prodotto deriva da una reazione del primo ordine e la
velocità di formazione è data da:
2) V = kcat [ES]
295
Per esprimere la stessa equazione in termini di concentrazione di substrato e di
enzima dobbiamo esprimere ES in funzione di queste due variabili e ciò può
essere ottenuto seguendo il procedimento descritto in figura 2. La figura 2
rappresenta lo schema di analisi di cinetica enzimatica eseguito da Michaelis e
Menten, nel quale si assume che la velocità di dissociazione del complesso
enzima substrato kcat nella direzione del prodotto sia molto bassa in rapporto
alla velocità di formazione e dissociazione (k1 e k-1) del complesso stesso.
Questo implica che ES si troverà in equilibrio con E e con S.
Figura 2. Schema di analisi della cinetica enzimatica nelle condizioni in cui la costante di
velocità kcat sia molto bassa.
L’equazione finale che si ottiene è l’equazione di Michaelis-Menten:
3) V = kcat [E]t [S] / [S]+ KS = Vmax [S] / [S]+ KS
essa permette di riportare la velocità V in funzione della concentrazione del
substrato S e di ottenere il valore di kcat da misure della velocità massima Vmax.
L’equazione si basa sulla condizione che la velocità di formazione del
prodotto sia molto più lenta rispetto alla velocità di formazione e di
dissociazione del complesso. Tuttavia, ciò non è sempre vero, in quanto si
verificano casi in cui kcat è più alta di k-1. Occorre perciò utilizzare un modello
più ampio, il quale si basa sull’assunto che la velocità di dissociazione del
complesso ES sia correlata alla sua velocità di formazione. Inizialmente la
velocità è elevata e la concentrazione di ES aumenta mentre in seguito viene
raggiunto uno stato stazionario in cui la concentrazione del complesso diviene
296
quasi costante (Fig. 3). Tale complesso permarrà fino a quando l’intero
substrato non si sarà consumato.
Figura 3. Rappresentazione dello stato stazionario nella cinetica enzimatica. In questo
stato ES viene consumato con la stessa rapidità con cui è prodotto così che la sua
variazione nel tempo d[ES]/dt = 0.
Considerando il suddetto modello, è possibile affermare che la variazione della
concentrazione di ES nel tempo è data dalla velocità di formazione del
complesso stesso controbilanciata dalla velocità di dissociazione, seguendo
due diverse modalità.
Lo schema rappresentato in figura 4 permette di determinare un’equazione
formalmente identica all’equazione 3) ovvero:
4) V = kcat [E]t [S] / [S]+ KM = Vmax [S] / [S]+ KM
Figura 4. Schema di analisi della cinetica enzimatica nella
situazione di stato stazionario.
297
La differenza tra l’equazione 3) e 4) è nella costante K, che nella prima è
rappresentata dalla costante di equilibrio Ks, mentre nella seconda è data da
KM che è il rapporto fra le tre costanti di velocità k-1, k1 e kcat:
5) KM = k-1 + kcat / k1
Ne deriva che la costante di Michaelis KM, pur non essendo una semplice
costante di equilibrio, si approssima ad essa quando kcat è di molto inferiore di
k-1. La costante di Michaelis può quindi essere associata alla forza di legame
tra enzima e substrato unicamente quando kcat è molto piccolo. Di
conseguenza, un alto valore di KM non implica necessariamente un’ elevata
costante di dissociazione tra enzima e substrato, in quanto potrebbe derivare
dall’elevato valore di kcat. Secondo una definizione operativa, KM è quella
concentrazione del substrato per la quale la velocità enzimatica risulta ½ della
velocità massima (Fig. 5).
Figura 5. Grafico della vecità di reazione in funzione della concentrazione di substrato.
L’altro parametro importante in una reazione enzimatica è kcat , indicato anche
come numero di turnover, che viene misurato in sec-1 e rappresenta il numero
di molecole di substrato trasformate da una molecola di enzima in un secondo.
Maggiore è il valore di kcat, più rapidi sono gli eventi catalitici nel sito attivo.
Un elemento utile nella descrizione di una reazione enzimatica è kcat/KM,
ovvero il rapporto tra le due costanti sopra descritte. Il suo significato si palesa
soprattutto quando la concentrazione del substrato è molto più piccola di KM,
per cui quasi tutto l’enzima si trova in forma libera. In tal caso l’equazione 4)
si riduce a:
6) V = [E] [S] kcat /KM
298
Il rapporto kcat/KM rappresenta quindi una costante di velocità al secondo
ordine per la reazione tra l’enzima ed il substrato indicando il valore di
efficienza di specificità dell’enzima per un particolare substrato. Quanto più
alto è questo valore tanto più l’enzima è specifico per quel definito substrato.
Le unità di misura della costante sono M-1 s-1.
Un caso interessante è quello in cui kcat >> k-1, ovvero quando la dissociazione
del complesso ES si sposta verso la formazione del prodotto il rapporto kcat/KM
si riduce a:
7) kcat/KM = kcat k1 / k-1 + kcat ~ = kcat k1 / kcat ~ = k1
In questo caso l’efficienza dell’enzima dipende unicamente dalla velocità di
associazione tra l’enzima ed il substrato. Il limite superiore dell’efficienza
catalitica è determinato quindi dalla velocità di collisione tra il substrato ed il
sito attivo dell’enzima. Questi enzimi sono detti perfetti e la loro efficienza è
limitata dalla diffusione. Il fattore che limita la reazione enzimatica è di tipo
fisico e non chimico.
Di seguito verranno descritte in modo dettagliato due classi di enzimi: le
proteasi a serina e le superossido dismutasi a rame-zinco (SOD).
Le proteasi a serina
Le proteasi a serina costituiscono una particolare classe di proteasi, il cui
meccanismo catalitico si basa sulla fondamentale presenza di un residuo di
serina nel sito attivo. Esse sono in grado di idrolizzare il legame peptidico
all’interno di una catena peptidica. In particolare, le proteasi a serina
eucariotiche, (quali la tripsina, la chimotripsina, l'elastasi), sono molto simili
sia relativamente al proprio sito catalitico che alla struttura tridimensionale.
Sono sintetizzate sotto forma di zimogeni inattivi e vengono attivate a seguito
di tagli proteolitici. La tripsina e la chimotripsina, ad esempio, vengono
sintetizzate nel pancreas ed attivate nell'intestino ad opera di altre proteasi.
Entrambe sono delle endopeptidasi (idrolizzano cioè i legami peptidici interni
della proteina, dando origine a frammenti più corti).
Un’altra proteasi a serina è la subtilisina, isolata dal batterio Bacillus subtilis.
Essa possiede un sito attivo identico agli enzimi sopra menzionati, ma una
struttura tridimensionale del tutto differente e non correlata. La principale
forza motrice della catalisi delle proteasi a serina è rappresentata da un insieme
di tre aminoacidi, denominati la “triade catalitica”, una serina, un’istidina ed
un aspartico, conservata in tutte le proteasi in una posizione spaziale idonea al
taglio di specifici legami peptidici.
299
Formazione della chimotripsina attiva
La forma attiva normale della chimotripsina è costituita da tre catene
peptidiche che traggono origine da una singola catena iniziale denominata
chimotripsinogeno. Come si può osservare nella figura 6, il chimotripsinogeno
possiede 245 residui e cinque ponti disolfuro.
Figura 6. Attivazione della chimo tripsina.
Il processo di maturazione avviene in seguito a quattro tagli sequenziali che
avvengono sui legami peptidici che seguono i residui 13 e 15 e 146 e 148. I
due dipeptidi costituiti dai residui 14-15 e 147-148 vengono rilasciati, mentre i
tre nuovi polipeptidi sono tenuti assieme dai ponti disolfuro (Fig.7), oltre che
dalle interazioni non covalenti. L’eliminazione dei due dipeptidi porta ad un
riarrangiamento tridimensionale ed, in particolare, l’eliminazione del dipeptide
14-15 conduce alla formazione di un ponte salino tra il nuovo N-terminale del
residuo 16 e l’aspartico 194, il quale provoca un riarrangiamento della serina
195, (facente parte della triade), indispensabile per l’attivazione dell’enzima.
Figura 7. Schema dell’eliminazione dei due dipeptidi nell’attivazione della chimo
tripsina.
300
La triade è presente in più proteasi a serina, tra queste vi è la subtilisina di
origine batterica, non correlata da un punto di vista strutturale con le proteasi a
serina digestive, come la tripsina e la chimotripsina. La triade catalitica è
conservata anche nella carbossipeptidasi II, un’esopeptidasi isolata da germe
di grano. Nei tre tipi di proteasi, l’ordine delle sequenze aminoacidiche
primarie dei residui localizzati nei corrispondenti siti attivi è molto diverso
(Fig. 8), così come differenti sono le strutture tridimensionali delle proteine.
Conseguentemente, remota è la possibilità che tali proteasi siano state
originate da una proteina ancestrale comune. Le proteasi a serina sembrano
piuttosto rappresentare un esempio di evoluzione convergente1, ossia, partendo
da geni diversi e quindi da strutture proteiche diverse, la natura ha scoperto e
selezionato per almeno tre volte il sito attivo formato dalla triade, denotandone
l’estrema efficienza.
Figura 8. Rappresentazione schematica dei residui del sito attivo in tre proteasi a
struttura tridimensionale diversa.
Le quattro caratteristiche strutturali delle proteasi a serina necessarie a conferirne la
funzione.
Le proteasi a serina contengono quattro caratteristiche strutturali conservate
necessarie al mantenimento ed all’ottimizzazione del proprio meccanismo
catalitico (Fig. 9): la triade catalitica, la buca dell’ossianione, la tasca di
specificità e la presenza di interazioni deboli aspecifiche.
1
Evoluzione convergente: processo dovuto a selezione naturale, per cui geni diversi che danno
luogo a proteine con strutture originariamente diverse evolvono verso una medesima funzione
biochimica, che si traduce ad esempio nella medesima configurazione del sito attivo.
301
Figura 9. Le quattro caratteristiche strutturali delle proteasi a serina.
1) Triade catalitica
La triade catalitica è costituita da tre aminoacidi serina, istidina e aspartico che
nella chimotripsina si trovano in posizione Ser195-His57-Asp102 (Fig. 10) e
nella subtilisina in posizione Ser221-His64-Asp32. La triade è fortemente
conservata e permette l’attacco nucleofilo della serina sul carbonio del legame
peptidico che deve essere tagliato.
Figura 10. La triade catalitica della tripsina.
2) Buca dell’ossianione
La buca dell’ossianione nel processo catalitico delle proteasi è l’elemento che
consente l’accelerazione della reazione enzimatica. Serve infatti a stabilizzare
il complesso di transizione e quindi ad abbassarne l’energia di attivazione. La
stabilizzazione avviene attraverso legami idrogeno tra gruppi presenti in
questa cavità e l’ossigeno legato al carbonio, a sua volta legato dalla catena
laterale della serina. Nella chimotripsina, ad esempio, si ha la formazione di 2
legami idrogeno tra l’ossigeno parzialmente negativo del carbonile e 2 NH
della catena principale di Ser195 e Gly193. La capacità di legare in modo
efficace il complesso di transizione tetraedrico velocizza la reazione.
302
3) Tasca di specificità
Le proteasi a serina manifestano una parziale specificità nella proteolisi di
legami peptidici prossimi ad alcune catene laterali. Considerando come punto
di partenza il gruppo N-terminale della proteina substrato, la catena laterale di
riconoscimento precede il legame che deve essere tagliato. La specificità è a
carico di una tasca i cui elementi principali sono 3 residui, la cui dimensione e
carica varia a seconda della catena laterale della proteina substrato con cui
devono interagire.
4) Interazioni aspecifiche
Il riconoscimento tra la proteasi e la proteina substrato avviene attraverso una
serie di legami idrogeno tra le due catene principali. Nello specifico, essi
avvengono con un “loop” presente nella proteasi e la proteina substrato e
portano alla formazione di un breve foglietto β antiparallelo con la proteina
substrato. Peculiare è il fatto che uno di questi legami a idrogeno si fortifichi
(riducendo la sua distanza) nel passaggio dal complesso enzima-substrato alla
formazione dello stato di transizione. Si rileva, in tal modo, la sua utilità ai fini
della stabilizzazione dello stato di transizione e, quindi, dell’accelerazione
della reazione.
Reazione catalitica
Le proteasi a serina sono così denominate perchè un residuo di serina è
responsabile dell'attacco nucleofilo al carbonile del legame peptidico.
Normalmente l'ossidrile della serina non è un buon nucleofilo, in quanto il pK
di dissociazione del protone è estremamente elevato e quindi il suo protone
non si stacca. Nelle proteasi a serina, però, è presente la "triade catalitica", che
coinvolge i residui aminoacidici di Asp-102, His-57 e Ser-195 la cui funzione
è quello di abbassare il pKa dell'ossidrile della serina permettendone la
ionizzazione. Il primo evento del meccanismo catalitico è l’interazione
enzima-substrato. Questo riconoscimento permette la cessione del protone
dell’ossidrile della catena laterale della serina 195 all’istidina 57 (Fig. 11) che
diventa doppiamente protonata ed è stabilizzata dalla carica negativa dell’Asp102. L’Asp 102 non si protona durante il processo di catalisi avendo un pKa <
2, bensì orienta His-57 in modo tale che possa prendere il protone da Ser-195 e
stabilizza la carica positiva di His-57 protonata.
303
Figura 11. Attacco nucleofilo della catena laterale della serina 195 sul carbonile della
catena principale da proteolizzare.
La serina sarà così in grado di effettuare l’attacco nucleofilo del carbonio
carbonilico del legame peptidico, generando un intermedio tetraedrico (Fig.
12):
Figura 12. L’intermedio tetraedrico.
L'intermedio tetraedrico è stabilizzato dalla formazione di legami idrogeno tra
l'ossigeno del carbonile interagente con la serina ed i gruppi NH di Gly-193 e
Ser-195.
Successivamente, l'intermedio tetraedrico collassa nell’intermedio acil-enzima
(Fig. 13), a seguito della catalisi acida esercitata da His-57, avviene cioè la
donazione di un protone da parte del N3 dell’istidina. L’acil-enzima è
estremamente instabile ma è stato caratterizzato strutturalmente, attraverso
diffrazione a raggi X, nell’elastasi bloccandolo a basse temperature.
304
Figura 13. L’intermedio acil-enzima
In tal modo, si verifica la liberazione della porzione del polipeptide con NH2
terminale, ma il resto del polipeptide risulta legato con legame estere a Ser195.
Il nuovo gruppo aminico viene rilasciato e sostituito da una molecola di acqua
che interagisce con l’N3 dell’istidina (Fig. 14).
Figura 14. Liberazione del nuovo N-terminale e interazione di una molecola di acqua con
l’istidina 57.
Il residuo di His-57 preleva un protone dalla molecola di acqua generando un
gruppo ossidrilico che attacca il carbonio carbonilico: si genera così un nuovo
intermedio tetraedrico (Fig. 15). L'intermedio tetraedrico viene stabilizzato
dalla formazione di legami idrogeno tra l'ossianione ed i gruppi NH di Gly193 e Ser-195.
305
Figura 15. Formazione del nuovo intermedio tetraedrico.
L'intermedio tetraedrico collassa a causa della catalisi acida esercitata da His57, ovvero His 57 cede un protone alla catena laterale di Ser 195. La seconda
parte del polipeptide viene rilasciata e la proteasi torna nello stato iniziale (Fig.
16), pronta per un nuovo ciclo di catalisi.
Figura 16. Rigenerazione dell’enzima nelle condizioni iniziali.
In questa parte della reazione l’acqua è il gruppo nucleofilo e la serina è il
gruppo uscente.
306
La buca dell’ossianione
Come in parte anticipato, la buca dell’ossianione ha un ruolo determinante
nello stabilizzare lo stato di transizione e quindi nell’accelerare la reazione
catalitica.
Figura 17. La buca dell’ossianione prima e dopo interazione con l’intermedio
tetraedrico.
L’importanza della buca dell’ossianione è stata messa in evidenza dall’analisi
comparata di alcune strutture di proteasi a serina e loro inibitori ottenute per
diffrazione X. Lo schema della proteina in prossimità della buca, riportato in
figura 17, mostra l’interazione tra la proteasi ed il substrasto prima e dopo la
formazione dello stato di transizione. Prima della formazione dello stato di
transizione, il carbonio carbonilico del legame peptidico che deve essere
proteolizzato, viene distorto per entrare nella tasca dell’ossianione, ma non
riesce ad effettuare nessuna interazione non covalente con i residui della
proteasi. Successivamente alla formazione del complesso tetraedrico (ovvero
dello stato di transizione), l’ossigeno del gruppo carbonilico, provvisto di
carica negativa (ossianione), interagisce e forma un legame idrogeno con il
gruppo NH della catena principale sia di Ser 195 che di Gly 193. Inoltre, la
distorsione avvenuta a causa della formazione del complesso tetraedrico, porta
alla creazione di un ulteriore legame idrogeno tra il gruppo NH del residuo,
che precede il legame peptidico ed il carbonile della Gly 193.
Il complesso tetraedrico ha, rispetto al legame enzima-substrato, un legame
preferenziale con la proteasi, che conferisce gran parte dell’efficienza della
proteasi stessa.
La tripsina ed il suo inibitore
Una dimostrazione dell’esistenza del complesso tetraedrico è data dalla
struttura derivante dalla diffrazione X del cristallo della tripsina coordinata
307
con il suo inibitore naturale, l’inibitore della tripsina di pancreas bovino
(BPTI). Il BPTI è una piccola proteina di 58 residui che forma con la tripsina
un complesso molto stabile, grazie ad una notevole complementarità di
superficie ed alla presenza di numerose interazioni deboli. La costante di
dissociazione tra queste due proteine è 10-13 M e questa forte interazione
impedisce a qualsiasi molecola di tripsina, che sia stata attivata
prematuramente nel pancreas, di svolgere una qualsiasi attività proteolitica di
proteine presenti nell’organo.
Il BPTI interagisce con la proteasi tramite un’interazione simil-substrato (Fig.
18).
Figura 18. Interazione della tripsina con l’inibitore BPTI.
La catena laterale di una lisina si introduce nella tasca di specificità ed il
legame peptidico Lys-Ala si posiziona con il carbonio carbonilico pronto ad
essere attaccato dalla catena laterale della serina del sito attivo. Il complesso
ha una conformazione che si trova lungo le coordinate di reazione, che
conduce alla formazione del complesso tetraedrico. L’ossigeno della serina
crea contatti van der Waals con il carbonio carbonilico del legame peptidico
del BPTI, tuttavia la reazione non procede perché, essendo il complesso
sigillato, il gruppo non può essere allontanato e l’acqua non può entrare. Di
conseguenza, il sistema non si deforma, non si ottimizza la formazione del
complesso tetraedrico ed il BPTI funge da inibitore perfetto. Quindi, affinchè
la reazione possa proseguire, occorre che si ottimizzi il legame che porta alla
formazione dello stato di transizione e che il sistema sia flessibile, cioè in
308
grado di cambiare orientazione. Questo complesso, invece, è estremamente
rigido e sigillato, per cui non permette alla reazione di procedere.
Caratteristiche generali della struttura delle proteasi a serina
Le strutture tridimensionali delle proteasi a serina eucariotiche sono molto
simili. La prima struttura è stata risolta nel 1967 ed è quella della
chimotripsina. La proteina è costituita da due domini, ognuno di circa 120
residui. Ogni dominio forma un barilotto β composto da sei filamenti di cui i
primi quattro formano un motivo a greca e gli ultimi due un motivo a forcina
antiparallela (Fig. 19 e Fig. 20).
Figura 19. Struttura schematica della chimo
tripsina.
Figura20. Diagramma topologico del dominio della
chimotripsina.
309
Il sito attivo si trova in una cavità compresa tra i due domini ed i residui che
costituiscono la triade catalitica si trovano sui “loops” di connessione dei
filamenti β. Due residui, l’His 57 e l’Asp 102, sono presenti sul primo
dominio, l’altro, la Ser 195 si trova sul secondo. In dettaglio: l’His 57 e la Ser
195 sono all’interno del “loop” 3-4 del dominio 1 e 2 rispettivamente, mentre
l’Asp 102 è nel “loop” 5-6 del dominio 2. Gli altri residui che possono
considerarsi parte del sito attivo, quali i residui che definiscono la tasca di
specificità o che formano legami idrogeno con la catena principale della
proteina substrato, si trovano sui “loops” 3-4 e 5-6 del dominio 2. Anche in
questo caso è rispettata la regola generale secondo cui la parte strutturale e la
parte funzionale sono localizzate in regioni differenti dell’enzima. La parte
strutturale è costituita dai filamenti β che formano il barilotto, mentre la parte
del sito attivo si trova sulle anse di connessione dei filamenti β.
I due domini presentano un basso grado di identità, ma sono molto simili da un
punto di vista tridimensionale e ciò fa supporre che queste proteasi siano frutto
di una duplicazione genica di un unico gene ancestrale. Tuttavia, quest’ ultimo
non avrebbe potuto possedere la triade catalitica, in quanto i residui della
triade si trovano uno su un dominio e due sull’altro, contravvenendo all’ipotesi
formulata. In realtà, vari esperimenti di mutagenesi sito diretta hanno
evidenziato che le proteasi continuano ad avere un’attività catalitica anche
dopo eliminazione di alcuni residui della triade, dimostrando che la triade non
è essenziale ma serve ad ottimizzare l’efficienza enzimatica. E’ plausibile
dunque ritenere che le proteasi derivino da duplicazione di un unico gene
ancestrale.
Ulteriore peculiarità delle proteasi consiste nel fatto che la tasca di specificità,
accomodando la catena laterale del residuo precedente il legame peptidico che
deve essere proteolizzato, determina la specificità di taglio.
In figura 21 sono rappresentate le tasche della chimotripsina, tripsina ed
elastasi. La specificità è essenzialmente determinata dalla tipologia delle
catene laterali degli amionoacidi in posizione 189, 216 e 226. Le loro
caratteristiche chimico-fisiche ne determinano la specificità: per la
chimotripsina è per catene laterali aromatiche, per la tripsina è per catene
laterali cariche positivamente e per l’elastasi è per catene laterali a dimensioni
ridotte.
Figura 21. Le tasche
di specificità di
chimotripsina,
tripsina ed elastasi.
310
La proteasi a serina subtilisina
La subtilisina è una proteasi batterica a serina con caratteristiche strutturali
completamente differenti da quelle eucariotiche. La subtilisina è una proteina
di 275 residui con una struttura tridimensionale complessa, in cui il dominio
principale è rappresentato dal dominio N-terminale, costituito da cinque
filamenti β paralleli circondata da quattro α-eliche due per ogni lato (Fig. 22)
Figura 22. Struttura schematica della subtilisina.
Il motivo è un motivo di tipo α/β in cui è presente un’eccezione strutturale.
Infatti, tutti i motivi di tipo β-α-β presenti nelle proteine sono di tipo destrorso,
mentre nella subtilisina il motivo β2-αB-β3 è di tipo sinistrorso, come si può
osservare nel diagramma topologico di figura 23. Il motivo di questa
eccezione è dovuto al fatto che l’His 64, facente parte della triade catalitica, è
situata nel primo giro dell’elica αB. Se il motivo fosse destrorso, la posizione
tridimensionale dell’istidina sarebbe inadatta a formare la triade catalitica.
Figura 23. Diagramma topologico che mette in evidenza la connessione di tipo sinistrorso
del motivo β2-αB-β3.
311
Nonostante le forti diversità strutturali tra la subtilisina e le altre proteasi a
serina descritte, anche la proteasi batterica possiede le quattro caratteristiche
strutturali tipiche delle proteasi a serina, configurando palesemente un caso di
evoluzione convergente a livello molecolare. La subtilisina è stata ampiamente
studiata al fine di comprendere in profondità il meccanismo di funzionamento
delle proteasi a serina. In particolare, alcuni studi hanno permesso di delineare
bene il ruolo della buca dell’ossianione nella stabilizzazione dello stato di
transizione.
Nella subtilisina, il complesso tetraedrico viene stabilizzato da legami
idrogeno che avvengono con la catena laterale dell’asparagina 155 e non con
la catena principale (come nel caso della tripsina e della chimotripsina) (Fig.
24). Eliminando per mutagenesi sito diretta questo residuo, è stato possibile
verificarne il ruolo da un punto di vista funzionale.
Figura 24. Rappresentazione schematica del sito attivo della subtilisina.
Sostituendo l’Asn155 con un’altro aminoacido, la cui catena laterale non è in
grado di effettuare legame idrogeno con il complesso tetraedrico, si riduce il
valore di kcat/KM. Tale diminuzione deriva da una riduzione del valore di kcat,
perché eliminando la possibilità di effettuare il legame idrogeno non si abbassa
l’energia di attivazione del complesso di transizione. La buca dell’ossianione
ha quindi un ruolo determinante nel modulare l’efficienza catalitica di questa
classe di enzimi.
312
Superossido dismutasi a rame-zinco
Le superossido dismutasi sono una classe di metallo enzimi che catalizzano la
dismutazione dello ione superossido in perossido di idrogeno ed ossigeno,
attraverso la riduzione ciclica del rame presente nel sito attivo. La reazione è la
seguente:
202-. +2H+ 02 + H202
La funzione di questo enzima è di proteggere le cellule dall’azione tossica del
radicale superossido che si forma nelle cellule aerobiche, principalmente a
causa della perdita di elettroni dai diversi componenti delle catene di trasporto
elettronico. Le superossido dismutasi sono ubiquitarie, risultando presenti in
quasi tutti i sistemi biologici aerobi. In base alla natura del metallo prostetico
presente nel sito attivo, che consente loro di svolgere l’attività catalitica, sono
distinte in Cu,ZnSOD, FeSOD e MnSOD. Le ultime due sono caratterizzate da
una simile struttura secondaria (ad α-elica) e terziaria e sono presumibilmente
evolute da uno stesso gene ancestrale.
Le superossido dismutasi a rame-zinco, Cu,Zn-SOD, sono caratterizzate da
una struttura secondaria a foglietti β organizzati in motivi strutturali a greca a
formare una struttura terziaria a barile. Generalmente, negli eucarioti si trova
nel citoplasma, ma è stata riscontrata anche nel periplasma dei batteri gramnegativi.
Caratterisitiche strutturali
Le Cu,Zn-SOD eucariotiche sono proteine omodimeriche di peso molecolare
pari a 32 KDa. Ogni subunità (Fig. 25) è composta da 150-153 aminoacidi e
contiene un atomo di rame, essenziale per il processo catalitico, e uno di zinco
che ha principalmente funzione strutturale. La subunità è costituita da otto
filamenti β antiparalleli, disposti in una struttura terziaria a cilindro
leggermente schiacciato da un lato e collegati tra loro da tre lunghe anse non
ordinate in una struttura secondaria definita (“loop” 4,7- “loop” 6,5- “loop”
7,8) e da regioni con inversione di catena (“turns”and “tight turns”). La
struttura è a "β-barrel" ed evidenzia il motivo topologico definito a “greca” che
è comune a molti sistemi biologici di grande interesse, tra cui le
immunoglobuline.
313
Fig. 25. Struttura tridimensionale di una subunità
della Cu,Zn-SOD bovina. I segmenti β sono mostrati
come freccie e il ponte disolfuro è evidenziato in
nero. Due grandi anse si protendono verso il
solvente a formare la parte superiore e inferiore del
canale del sito attivo. Il rame e lo zinco giacciono sul
fondo di questo canale, con il rame accessibile al
solvente dalla parte dell'osservatore. Il diagramma
in basso a destra illustra la tipologia a "greca”
assunta dal cilindro β aperto e disteso su un piano.
Il cilindro è asimmetrico e può essere diviso in due sezioni: una comprende dal
primo al quarto segmento β, l’altra dal quinto all’ottavo. I segmenti β di
quest'ultima sono più corti, instaurano un numero inferiore di legami idrogeno,
possiedono un'alternanza meno regolare delle catene laterali e una maggiore
distorsione (Fig. 26), spiegando così l’asimmetria del cilindro β.
Figura 26. Diagramma topologico di una subunità di Cu,Zn SOD.
314
Sono presenti tre anse principali: la 6,5; la 7,8; la 4,7 (Fig. 26 e Fig. 27).
Fig. 27. Il dimero della Cu,Zn superossido dismutasi.
L'ansa 6,5 può essere divisa in due sottodomini: il primo, che si estende dalla
Gly47 alla Pro60, contribuisce al contatto tra le subunità, forma un lato del
canale del sito attivo ed è stabilizzato dall'unico ponte disolfuro del monomero
(Cys55-Cys144); il secondo, che comprende i residui tra His61 e Leu82, è la
regione che include i quattro ligandi dello zinco che provvedono a mantenere
lo ione completamente nascosto al solvente. L'ansa 7,8 forma una sorta di
coperchio sul sito attivo ed è funzionalmente rilevante, in quanto contiene i
residui che, attraverso un’attrazione elettrostatica, guidano il substrato nel
canale del sito attivo, verso il rame catalitico Cu(II). L'ansa 4,7 è la più piccola
e lega l'Asp99 all'Arg113 dando origine ad una connessione a greca che
attraversa una estremità del cilindro β.
Struttura del sito attivo metallico
Studi di cristallografia e spettroscopia hanno dimostrato che la struttura del
sito attivo della superossido dismutasi a rame e zinco è altamente conservata.
Il sito catalitico (Fig. 28) di ciascuna subunita' è orientato nella regione
opposta all’interfaccia che le unisce ed è costituito da un lungo canale che ha
per base il foglietto β del cilindro ed è delimitato lateralmente dalle anse 6,5 e
7,8, creando una sorta di coperchio attorno ad esso. In fondo al canale si
collocano il rame e lo zinco, il primo esposto al solvente, mentre il secondo è
inaccessibile ad esso.
315
Figura 28. Monomero della Cu,Zn-SOD bovina:
localizzazione del sito attivo e relativa
disposizione dei due metalli.
Il rame è coordinato agli atomi di azoto di quattro istidine; His44 e His 46
localizzate nel sesto segmento β, His118 del settimo segmento β e His61 del
“loop” 6,5 (Fig. 28 e Fig. 29). La disposizione dei suoi ligandi determina la
geometria quadrato planare con distorsione tetraedrica.
La distorsione, insieme al particolare orientamento degli anelli imidazolici,
rende la posizione del rame più aperta dal lato del solvente, permettendo la
presenza di una molecola di acqua coordinata in posizione assiale. In diversi
enzimi eucariotici si è osservato che l'His61 forma un ponte tra il rame e lo
zinco, per cui i due metalli sono compresi in un piano a circa 6 Å di distanza.
Figura 29. Il cluster metallico della Cu,Zn superossido dismutasi.
I ligandi dello zinco sono collocati sul “loop” 6,5 e sono rispettivamente
l’atomo di azoto delle His61, 69, 78 ed il residuo Asp81; la geometria del sito
è di tipo tetraedrico, con una distorsione a piramide trigonale con l’Asp81
all’apice (Fig. 29). I tre anelli istidinici sono collocati quasi di fronte allo zinco
316
in direzione del sito attivo, l’atomo risulta, così interno alla struttura proteica
ed inaccessibile al solvente (Fig. 28).
I due metalli rappresentano il cuore del sito attivo con l’atomo di rame
direttamente coinvolto nella reazione di ossido riduzione necessaria a
dismutare lo ione superossido. Esistono tuttavia alcuni residui proteici, oltre ai
ligandi dei metalli, che hanno un ruolo fondamentale nel processo catalitico. In
particolare, tutte le superossido dismutasi a rame e zinco analizzate presentano
in posizione 141 un residuo di arginina che è altamente conservato. Questo
residuo si posiziona frontalmente all’atomo di rame e risulta determinante sia
nell’attrazione che nel posizionamento dell’ione superossido.
Meccanismo di catalisi enzimatica
Il ciclo catalitico è costituito da due reazioni in cui lo ione superossido
reagisce alternativamente con il rame alla stato ossidato e allo stato ridotto. Le
due reazioni sono caratterizzate da una medesima efficienza catalitica, essendo
per ambedue kcat/KM identica e molto elevata, dell’ordine di 2x109 M-1 s-1 (Fig.
30).
La catalisi ha inizio con il sopraggiungere di una molecola di ione superossido,
che spiazza la molecola d’acqua assiale, legando un atomo di ossigeno al Cu2+
e formando con l’altro un legame idrogeno con l’azoto del guanidinio di
Arg141, il quale stabilizza il complesso rame-substrato. L’O2-.legato provoca
la riduzione di Cu2+ "rameico" alla forma Cu+ "rameoso”,
contemporaneamente, si rompe il legame tra l’His61 ed il rame, viene
rilasciato O2 e l’azoto di His61 dissociato si protona.
Una seconda molecola di O2-. si lega con il rame nello stato ridotto Cu+. Un
trasferimento elettronico dal Cu+ abbinato ad un trasferimento protonico
dall’His61, con l’aggiunta di un secondo protone, proveniente da una molecola
di acqua del solvente, permette la formazione di una molecola neutra di H2O2,
che viene eliminata. Il rame, allo stato ossidato, ripristina il legame con l’azoto
di His61 deprotonata, con conseguente ritorno allo stato strutturale iniziale del
sito attivo.
In sintesi, il ciclo catalitico è costituito da una reazione di ossidoriduzione
effettuata dal rame che passa da ossidato a ridotto e nuovamente ad ossidato,
una volta dismutando il superossido in ossigeno, l’altra in acqua ossigenata.
317
Figura 30. Schema del ciclo catalitico della superossido dismutasi.
L’ipotesi elettrostatica
Dati sperimentali hanno dimostrato che l’efficienza di dismutazione dello ione
superossido è identica sia per l’enzima con il rame allo stato ossidato che in
quello ridotto. Inoltre, il valore di kcat/KM è molto elevato, dell’ordine di 2x109
M-1 s-1, uno dei maggiori mai riscontrati per qualsiasi famiglia di enzimi.
Tali dati indicano che si tratta di una reazione ai limiti della diffusione, sia
quando il rame è ossidato sia quando il rame è ridotto. La chimica dell’enzima
non risulta in grado di influenzare l’efficienza enzimatica, perciò l’efficienza è
governata da fattori fisici indipendenti dallo stato di ossidazione del rame.
Un risultato sperimentale interessante riguarda la misura dell’efficienza
enzimatica kcat/KM in funzione del pH e della forza ionica. Questa misura,
riportata nel grafico di figura 31, indica che ad ogni singola forza ionica
l’efficienza è pH indipendente per valori di pH compresi tra 7 e 9. Per valori
superiori l’efficienza diminuisce, ma in percentuale inferiore se i valori di
forza ionica sono alti. In particolare, mentre per valori di pH tra 7 e 10
l’efficienza diminuisce all’aumentare della forza ionica, per valori di pH tra 11
e 12 l’efficienza catalitica è quasi costante qualsiasi valore assuma la forza
ionica. La dipendenza del valore di kcat/KM dalla forza ionica indica che tale
parametro è condizionato dall’elettrostatica, inoltre, la diminuzione di kcat/KM
all’aumentare del pH presuppone la presenza di uno o più gruppi con un pK
intorno a 10.5, la cui deprotonazione produce una riduzione dell’efficienza
catalitica.
318
Figura 31. Grafico di kcat/KM in funzione del pH a cinque diversi valori di forza ionica.
Le informazioni raccolte hanno portato alla formulazione della così detta
“ipotesi elettrostatica”, secondo la quale il meccanismo guida per l'interazione
enzima-substrato si basa sulla presenza di cariche positive nell'area circostante
il sito attivo. In linea con questa ipotesi la distribuzione del potenziale
nell’intorno della proteina (Fig. 32) mostra un peculiare andamento.
Figura 32. Andamento delle linee
equipotenziali
nell’intorno della
proteina.
La proteina è infatti circondata da valori di potenziale negativi, tranne che in
due regioni definite corrispondenti alla regione occupata dal sito attivo. Lo
ione superossido, a carica negativa, viene respinto da ogni regione della
proteina, ad eccezione delle regioni che contengono il sito attivo. Le cariche
presenti sulla proteina creano un imbuto elettrostatico che convoglia il
superossido verso la regione del sito attivo. Vi è quindi un’agevolazione
elettrostatica alla diffusione del superossido verso il sito attivo, che risulta
identica sia per la proteina ossidata che per quella ridotta, spiegando
l’identicità del valore di kcat/KM.
319
L’ipotesi elettrostatica giustifica anche l’alto valore di kcat/KM, infatti la
superficie occupata dal sito attivo è una piccola percentuale della superficie
totale dell’enzima. Se la collisione dello ione superossido con il sito attivo
fosse determinata unicamente dalla diffusione, la sua probabilità sarebbe bassa
e la superossido dismutasi non potrebbe avere gli alti valori di kcat/KM misurati
sperimentalmente. Poiché la collisione è agevolata dall’imbuto elettrostatico,
l’incontro sito attivo-substrato viene facilitato dal potenziale elettrostatico
prodotto dalle cariche presenti sull’enzima stesso. E’ stato inoltre dimostrato
che la distribuzione del potenziale elettrostatico è invariata nel corso
dell’evoluzione, essendo indipendente sia dalla carica netta delle proteine, che
dalla conservazione di singoli residui (Fig. 33).
Figura 33. Distribuzione del potenziale elettrostatico in superossido dismutasi di diversa
specie.
L’ipotesi elettrostatica conferma che l’efficienza catalitica non è modulata
dalla chimica dell’enzima bensì dalla fisica, o meglio, dalla distribuzione del
potenziale elettrostatico. Le superossido dismutasi a rame e zinco sono quindi
enzimi caratterizzati da una kcat/KM molto elevata. Ciò è dovuto al fatto che il
sito attivo è preformato in modo ottimale per reagire con il suo substrato e che
il limite al processamento del substrato è determinato unicamente dalla
capacità del substrato di raggiungere il sito attivo. Si tratta di una classe di
enzimi che viene definita perfetta, per cui considerando la più semplice
equazione per il procedere di una reazione enzimatica:
320
k1
kcat
1) E + S ES E + P
k 1
si propone la situazione in cui kcat >> k-1, ovvero in cui la dissociazione del
complesso ES è spostata verso la formazione del prodotto. In questo caso, il
rapporto kcat/KM si riduce:
2) kcat/KM = kcat k1 / k-1 + kcat ~ kcat k1 / kcat ~ k1
l’efficienza enzimatica è assimilabile in prima approssimazione alla velocità di
associazione enzima-substrato k1.
L’unica via per migliorare l’efficienza di questo enzima è di attrarre in
maniera ottimale il substrato verso il sito attivo. Ciò si ottiene migliorando la
distribuzione del potenziale elettrostatico e rendendo l’imbuto più efficiente
nel processo di attrazione del substrato carico negativo. Tale approccio è stato
perseguito introducendo aminoacidi a carica positiva ed eliminando
aminoacidi a carica negativa in prossimità del sito attivo, dando luogo ad
enzimi superefficienti con kcat/KM dell’ordine di 1 x 1010 M-1 s-1 (Fig. 34).
Questi esperimenti, oltre ad aver originato mutanti super-efficienti, hanno
dimostrato definitivamente che lo step limitante della reazione enzimatica non
si trova all’interno della chimica dell’enzima bensì al di fuori dell’enzima
stesso: lo step limitante consiste nella capacità dell’enzima di attrarre il
substrato verso se stesso.
Figura 34. Valori di kcat/KM per alcuni
mutanti di superossido dismutasi.
321
322