Metabolismo
dei
composti azotati:
aminoacidi e nucleotidi
Metabolismo degli aminoacidi
I composti azotati
• L’azoto contenuto nella cellula è presente
soprattutto negli AMINOACIDI delle
proteine e nelle BASI AZOTATE degli acidi
nucleici.
• Queste sostanze sono sottoposte ad un
continuo ricambio che vede un flusso
costante in entrata attraverso gli alimenti
e la sintesi endocellulare e in uscita
attraverso il loro catabolismo in modo tale
che il bilancio dell’azoto sia zero.
Metabolismo degli
aminoacidi
• A differenza dei
carboidrati e dei lipidi, la
cui principale funzione è
quella di fornire energia, il
ruolo principale degli
aminoacidi è quello di
partecipare a reazioni
biosintetiche, in
particolare la sintesi delle
proteine.
• Le proteine (gli aminoacidi)
sono usate come fonte
secondaria di energia.
Infatti forniscono solo il
15% del fabbisogno
energetico giornaliero
complessivo nell’uomo
adulto.
Pool di aminoacidi
• E’ di circa 100g, distribuito nell’organismo.
• E’ piccolo a confronto con l’ammontare delle
proteine dell’organismo (circa 18kg per un uomo
di 70kg).
• Il 75% degli aminoacidi attenuti dall’idrolisi delle
proteine endogene (=dell’organismo) è riutilizzato
nella biosintesi di nuove proteine.
• L’azoto presente negli aminoacidi non viene
utilizzato per scopi energetici e solo in piccola
parte per la biosintesi delle basi azotate dei
nucleotidi.
Turnover delle proteine
• Ammonta a circa 400g al giorno di proteine
dell’organismo che vengono costantemente degradate e
risintetizzate.
• La vita media (EMIVITA) varia molto da proteina a
proteina.
• Per es. le proteine che funzionano fuori dalle cellule
(enzimi digestivi, proteine plasmatiche…) ed enzimi
regolatori delle vie metaboliche hanno vite medie molto
brevi (ore o giorni), mentre proteine strutturali
(collagene, cheratina..) sono metabolicamente stabili ed
hanno emivite lunghe (mesi o anni).
• Proteine ricche in sequenze dette PEST (prolina,
glutammato, serina e treonina) hanno vite medie brevi.
• Proteine danneggiate per es. da ossidazioni o marcate
con ubiquitina (piccola proteina intracellulare) sono
preferenzialmente degradate.
Catabolismo delle proteine
• Gli aminoacidi derivano dalla demolizione delle
proteine cellulari, dalla degradazione delle
proteine ingerite e dalla demolizione delle proteine
del corpo in mancanza di sostanze nutrienti (come
nel digiuno o nel diabete non controllato).
• Le proteine assunte con la dieta sono digerite da
parte degli ENZIMI PROTEOLITICI del tubo
gastro-enterico. La maggior parte delle proteasi
vengono sintetizzate sotto forma di zimogeni
inattivi e successivamente attivati nello stomaco o
nell’intestino mediante la rimozione proteolitica di
parti della loro catena polipeptidica. Altre proteasi
idrolizzano enzimi ed altre proteine presenti nella
cellula.
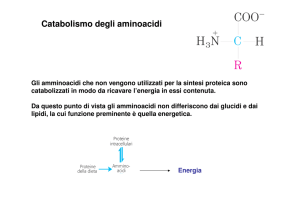
Catabolismo degli aminoacidi
• Non esistono forme di deposito nell’organismo per gli
aminoacidi.
• Gli aminoacidi in eccesso rispetto alle necessità
biosintetiche delle cellule vengono immediatamente
degradati.
• Gli aminoacidi, oltre a C,H,O, contengono N che non può
essere conservato nell’organismo e deve essere eliminato in
forma non tossica. L’N viene eliminato dall’organismo
attraverso le urine come:
•
•
•
•
UREA (86%)
AMMONIACA (3%)
CREATININA (5%)
ALTRI PRODOTTI del metabolismo degli aminoacidi (6%)
• L’N viene assunto dall’organismo in vari composti presenti
nella dieta, ma i più importanti sono proprio gli aminoacidi
contenuti nelle proteine della dieta.
• Si distinguono 2 fasi nel catabolismo degli aa:
• 1a fase: RIMOZIONE DELL’α-AMINOGRUPPO mediante
reazioni di transaminazione e di deaminazione ossidativa che
trasformano l’aminoacido nel corrispondente α-chetoacido.
• 2a fase: TRASFORMAZIONE DELLO SCHELETRO
CARBONIOSO dell’α-chetoacido in intermedi comuni delle vie
metaboliche. Questi intermedi possono essere poi
completamente degradati a CO2 e H2O con produzione di energia
(ATP) oppure essere usati per produrre glucosio
(→AMINOACIDI GLUCOGENETICI) o acidi grassi e corpi
chetonici (da acetil-CoA: →AMINOACIDI CHETOGENETICI).
Rimozione dell’azoto
dagli aminoacidi
• Avviene mediante 2 tipi di reazione:
• TRANSAMINAZIONE
• DEAMINAZIONE OSSIDATIVA
Attraverso queste reazioni si ha la
formazione di ammoniaca (NH3) e
di aspartato che forniscono
ciascuna un amino gruppo nella
sintesi dell’urea (H2N-CO-NH2)
α-Transaminazione
• Consiste nel passaggio reversibile del gruppo amminico
legato al Cα da un aminoacido ad un chetoacido.
L’aminoacido si trasforma nel chetoacido corrispondente
e il chetoacido nell’aminoacido relativo.
• Gli enzimi che catalizzano queste reazioni sono detti
AMINOTRANSFERASI O TRANSAMINASI e usano
come coenzima il piridossale-5’-fosfato (derivato della
vitamina B6).
• Queste reazioni sono usate anche per produrre aminoacidi
dai corrispondenti chetoacidi.
• La transaminazione avviene praticamente in tutti gli
organi, nel citoplasma cellulare.
• Le reazioni di transaminazione che portano alla sintesi di urea
spostano il gruppo aminico degli aminoacidi da degradare all’αchetoglutarato che così diventa glutammato:
• Tra le transaminasi la più abbondante e importante è la
GLUTAMMICO-PIRUVICO TRANSAMINASI detta anche
ALANINA AMINOTRANSFERASI (GPT o ALT):
• Parte del glutammato che si produce dalla reazione subisce
un’altra reazione di transaminazione catalizzata dalla
GLUTAMMICO-OSSALACETATO TRANSAMINASI o
ASPARTATO AMMINOTRANSFERASI (GOT o AST) che sposta
l’amino gruppo dal glutammato all’ossalacetato che diventa
aspartato.
Valore diagnostico delle
transaminasi
• Le transaminasi sono enzimi
intracellulari, la loro presenza a
livelli elevati nel plasma indica
un danno a carico di organi
ricchi di questi enzimi (malattie
del fegato come l’epatite,
l’infarto del miocardio o
malattie muscolari).
Deaminazione ossidativa
• Tramite questa reazione un aminoacido perde il gruppo
aminico (-NH2) sotto forma di ammoniaca (NH3) e si
trasforma nel corrispondete α-chetoacido:
• Importantissima è la deaminzione ossidativa reversibile del
glutammato catalizzata dall’enzima glutammato
deidrogenasi che utilizza NAD+ (→NADH) e avviene nei
mitocondri:
Produzione e trasporto dell’ammoniaca
L’NH3 viene costantemente prodotto nei tessuti:
• Per deaminazione ossidativa degli aa
• Per degradazione di purine e pirimidine
• Per degradazione di neurotrasmettitori
• Per degradazione di altri composti azotati della dieta
• Per azione della flora batterica che degrada i composti
azotati presenti nel tubo gastroenterico liberando NH3
• I livelli ematici di NH3 sono però bassi (NH3 è tossica,
soprattutto per il cervello, perché aumenta il pH del
plasma e consuma α-chetoglutarato e glutammato), grazie
al fatto che l’NH3 viene convertita in un composto non
tossico, la glutammina, e che il fegato la rimuove
rapidamente trasformandola in urea.
• Nei tessuti l’NH3 viene ceduta al glutammato che si
converte in glutammina secondo la reazione catalizzata
dalla glutammina sintetasi:
Glutammato + NH3 + ATP → glutammina + ADP + Pi
• La glutammina rappresenta una forma non tossica di
trasporto dall’NH3 verso il fegato (che la trasformerà
in urea) o verso il rene (che la eliminerà nelle urine
come NH3) dove avviene la reazione inversa catalizzata
dalla glutamminasi, presente nei mitocondri.
• Nel muscolo l’NH3 viene ceduta al piruvato per la
sintesi di alanina, utilizzato come trasportatore di
NH3 di piruvato al fegato. L’ NH3 viene escreta e il
piruvato utilizzato per la sintesi di glucosio.
Produzione di ammoniaca
A. Di Giulio, A. Fiorilli, C. Stefanelli Biochimica per le Scienze motorie Copyright 2011 C.E.A. Casa Editrice Ambrosiana Reazioni di assimilazione
dell’ammoniaca e
principali destini dall’azoto.
CICLO DELL’UREA
• Poiché l’ammoniaca è molto tossica per i tessuti, gli
animali uretelici allontanano l’azoto aminico sotto
forma di urea (H2N-CO-NH2).
• L’UREA è prodotta solo nel fegato tramite il ciclo
dell’urea che avviene in parte nella matrice
mitocondriale e in parte nel citoplasma.
• La reazione complessiva del ciclo è:
2NH+4 + HCO-3 + 3ATP + H2O →
→ urea + 2ADP + 4Pi + AMP + 5H+
• La sintesi di una molecola di urea consuma 4 legami
fosforici ad alta energia ed è quindi un processo
irreversibile .
Il ciclo dell’urea parte dal
mitocondrio con una molecola di
ione ammonio che viene attivata
mediante trasformazione in un
composto ad alta energia di
idrolisi: il CARBAMILFOSFATO.
Questa reazione è catalizzata
dalla CARBAMILFOSFATO
SINTETASI 1 e richiede
l’idrolisi di 2 ATP.
Il carbamilfosfato si combina
con l’ornitina per formare
citrullina che reagisce con
l’aspartato (il secondo donatore
di un amino gruppo) per dare
argininosuccinato che rilascia
arginina e fumarato.
L’arginina si decompone in urea
e ornitina.
ornitina
Carbamil
fosfato
citrullina
arginina
Arginina succinato
La regolazione del ciclo
dell’urea avviene proprio a
livello della carbamilfosfato
sintetasi 1. Questo enzima
richiede la presenza di un
attivatore allosterico, l’Nacetilglutammato, che è un
derivato del glutammato che
rappresenta un segnale di
abbondanza di aminoacidi.
Alcuni degli intermedi e
sottoprodotti del ciclo
dell’urea sono anche
intermedi del ciclo di Krebs,
creando un collegamento tra
le 2 vie metaboliche.
Trasformazione dello scheletro carbonioso
• Il catabolismo dello scheletro carbonioso degli aminoacidi converge
nella produzione di 7 composti:
• Acetil-CoA e acetoacetato (→ da aa. chetogenetici: possono essere
utilizzati per la sintesi di corpi chetonici, acidi grassi e colesterolo
ma non per la sintesi del glucosio)
• Piruvato, αchetoglutarato, succinilCoA, fumarato,
ossalacetato (→ da aa
glucogenetici: sono
precursori del glucosio
attraverso la
gluconeogenesi)
• Tutti e 7 i composti
possono dare luogo alla
produzione di energia
(ATP) mediante la loro
completa ossidazione a
CO2 e H2O
• Gli aminoacidi sono precursori nella
sintesi di altri composti:
• Porfirine (gruppo eme
dell’emoglobina e della mioglobina)
• Creatina (deposito di energia nel
muscolo, fosfocreatina)
• Purine e pirimidine (basi azotate dei
nucleotidi)
• Neurotrasmettitori (noradrenalina,
serotonina..) e ormoni (adrenalina…)
Biosintesi degli aminoacidi
• Nell’uomo 8 aminoacidi sono considerati ESSENZIALI:
essi non possono essere sintetizzati dall’organismo o sono
sintetizzati solo in quantità insufficienti. Quindi devono
essere assunti con l’alimentazione. (fenilalanina, isoleucina,
lisina, leucina, metionina, treonina, triptofano e valina)
• Gli aminoacidi NON ESSENZIALI possono invece essere
sintetizzati da intermedi del metabolismo o da aminoacidi
essenziali (alanina, aspartato, asparagina, cisteina,
glutammato, glutammina, glicina, prolina, serina, tirosina).
• Esistono poi reazioni di interconversione attraverso le
quali molti aminoacidi possono essere trasformati uno
nell’altro assicurando che tutti siano presenti nei rapporti
ottimali per essere usati nella biosintesi proteica.
Catabolismo
dei
nucleotidi
Catabolismo degli acidi nucleici
• Gli acidi nucleici DNA ed RNA sono idrolizzati
nei loro nucleotidi da cui vengono rimossi
successivamente il gruppo fosfato e il ribosio.
• Le basi azotate sono sottoposte ad una serie di
reazioni con lo scopo di renderle più idrofiliche
e quindi facilmente eliminabile con le urine
• Le pirimidine sono degradate ad ammoniaca
(urea), anidride carbonica e β-alanina o altri
composti idrosolubili.
• Le basi puriniche sono trasformate attraverso
una serie di reazioni di deaminazione in acido
urico.
Quesiti
1) In quali forme viene eliminato N (azoto) dall’organismo e attraverso quale mezzo?
2) Catabolismo degli aminoacidi: indicare in quali fasi si articola e darne una breve descrizione.
3) Che cosa sono le Transaminasi? Quale e’ la loro funzione? Quali sono le transaminasi di
importanza ai fini diagnostici?
4) Da quali substrati si produce NH3 (ammoniaca)? In che forma viene eliminata dall’organismo?
5) Che cosa costituiscono Glutammina ed Alanina nel metabolismo dell’azoto?
6) Dove avviene il ciclo dell’urea e quale e’ la sua funzione?
7) Quali sono I prodotti del catabolismo dello scheletro carbonioso degli aminoacidi?
8) Elencare gli aminoacidi essenziali
9) Quale e’ il prodotto finale del catabolismo delle basi puriniche?