Rivista Italiana di Genetica e Immunologia Pediatrica - Italian Journal of Genetic and Pediatric Immunology
Anno IV numero 2 - aprile 2012 | direttore scientifico: Carmelo Salpietro - direttore responsabile: Giuseppe Micali
Diagnostica per immagini nella Sclerosi Tuberosa
Cicero G, Pizzino MR, Crapanzano M, Costa A, Alterio T, Centorrino R, Morabito G, Arrigo T
Dipartimento di Scienze Pediatriche, UOC di Genetica ed Immunologia Pediatrica - Università di Messina
La Sclerosi Tuberosa, conosciuta anche come sindrome di “Bourneville-Pringle”, è una
malattia a trasmissione autosomica dominante, classificata tra le facomatosi. Essa è
caratterizzata da lesioni cutanee, nervose e splancniche, dovute ad iperplasia delle
cellule ectodermiche e mesodermiche. Clinicamente si evidenziano: adenoma sebaceo,
epilessia e ritardo mentale. A diffusione mondiale, ha la stessa frequenza di incidenza in
tutte le razze ed in entrambi i sessi. Solo in un terzo dei casi è familiare ed imputabile ad
una trasmissione autosomica dominante, la cui prevalenza è stimata tra 1/50.000 e
1/300.000; nei rimanenti casi risulta sporadica. In genere la malattia progredisce
lentamente. Tra i casi gravi, circa il 30% muore prima del quinto anno, e il 50-75% prima
di raggiungere l'età adulta. Il peggioramento è soprattutto a carico della sfera intellettiva
(1).
La patologia è causata da mutazioni in uno dei due geni oncosoppressori TSC1 (20%
dei casi) e TSC2 (80%).
Il gene TSC1, costituito da 23 esoni, è localizzato sul braccio lungo del cromosoma 9
(9q34) e codifica per una proteina di 130 kDa chiamata Amartina. Il gene TSC2, invece,
localizzato sul braccio corto del cromosoma 16 (16p13), consiste di 41 esoni e codifica
per una proteina di 200-kDa, la Tuberina. La posizione del gene TSC2 è adiacente a
quella del gene PKD1, coinvolto nella malattia del rene policistico, e a questa vicinanza
può imputata la presenza delle cisti renali in pazienti affetti da TSC. Sono state
segnalate più di 200 varianti alleliche della mutazione di TSC1 e quasi 700 per TSC2.
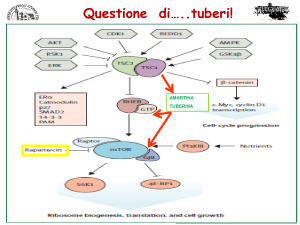
Il complesso Amartina-tuberina interviene nel processo di integrazione dei segnali
cellulari, come la formazione di ATP, i fattori di crescita, l'integrità genomica. Se uno dei
due fattori è assente, come nella TSC, manca l’inibizione nei confronti della proteina
Rheb, GTP-dipendente, che a sua volta andrà a stimolare l’enzima mTOR,
determinando anomala crescita e proliferazione cellulare. Gli elementi cellulari all'interno
delle lesioni sono alterati sia per numero che per dimensioni (2).
Le manifestazioni cliniche della malattia sono estremamente variabili e, in alcuni casi,
assenti.
I pazienti con mutazioni a carico del gene TSC1 hanno generalmente un fenotipo clinico
più blando, caratterizzato da una minore frequenza di crisi epilettiche, un ritardo
cognitivo di grado moderato-severo, un minor numero di noduli subependimali e di tuberi
corticali, un minor coinvolgimento renale e cutaneo e assenza di amartomi retinici. Al
contrario, le mutazioni del TSC2 sono associate ad una più precoce comparsa di crisi
epilettiche e ad una maggior frequenza di spasmi infantili. A livello cutaneo i reperti più
frequentemente individuati sono: le macchie ipomelanotiche e gli angiofibromi. Le prime
possono essere già presenti alla nascita, accrescendosi lentamente di numero e
dimensione con la pubertà. Gli angiofibromi risultano tipicamente distribuiti in maniera
simmetrica sul solco naso-labiale, sulle guance, sul mento e meno comunemente sulla
fronte e sul cuoio capelluto.
Nel SNC le lesioni tipiche sono: i tuberi corticali, i noduli subependimali periventricolari e
gli astrocitomi gigantocellulari.
I tuberi sono noduli in crescita localizzati nella corteccia cerebrale e responsabili dei
disturbi neurologici focali. Sono spesso multipli, di grandezza variabile e tendono
progressivamente alla calcificazione.
I noduli subependimali e gli astrocitomi gigantocellulari generalmente sono silenti; solo
nel 5% dei casi possono arrivare ad ostruire il forame di Monro provocando ipertensione
endocranica.
Clinicamente, l’epilessia rappresenta il segno più precoce di malattia e, qualora non
trattata o resistente ai farmaci, può portare a ritardo di sviluppo intellettivo. I tuberi
corticali rappresentano i foci epilettogeni della Sclerosi Tuberosa ed esiste una relazione
topografica tra le anomalie elettroencefalografiche e le lesioni rilevabili in Risonanza
Magnetica, con una manifestazione più precoce per le lesioni temporo-occipitali e più
tardiva per quelle frontali.
Il ritardo mentale è stato osservato in circa il 50% dei bambini con ST. Non è ancora del
tutto chiaro se le crisi siano responsabili del deficit cognitivo o se entrambi siano aspetti
differenti della stessa disfunzione cerebrale. (4)
L’interessamento renale è testimoniato dalla individuazione di angiomiolipomi e dalla
malattia renale cistica. Gli angiomiolipomi, costituiti da quantità variabili di elementi
vascolari, cellule muscolari lisce immature, epitelioidi e adipose. Le cisti renali,
solitamente multiple e bilaterali, possono originare da tutti i segmenti del nefrone e
presentano uno sviluppo età-correlato.
Le lesione cardiaca maggiormente rappresentata è il rabdomioma, frequentemente
identificato durante la vita fetale e nell’età neonatale; tende a regredire nel 70% dei casi
entro il quarto anno di età.
Le tre principali lesioni polmonari sono la linfangioleiomiomatosi, l’iperplasia multifocale
micro nodulare degli pneumociti e il tumore a cellule chiare.
La linfangioleiomiomatosi è la lesione polmonare più comune nelle giovani donne, con
età media di insorgenza di 33 anni.
E’ caratterizzata da diffusa proliferazione interstiziale dei fasci di cellule muscolari lisce e
lesioni cistiche nel parenchima polmonare.
Dispnea e ricorrenti pneumotorace spontaneo sono le presentazioni più comuni, con
una progressione lenta e costante verso l’insufficienza respiratoria.
L’iperplasia multifocale micronodulare degli pneumociti è raramente associata a TSC. Si
manifesta sottoforma di noduli amartomatosi multicentrici e delimitati, con proliferazione
di pneumociti di tipo II lungo i setti alveolari.
I tumori a cellule chiare in genere non sono capsulati e costituite da cellule rotonde,
ovali o affusolate, chiare con abbondante componente granulare eosinofila
intracitoplasmatica. (2)
Altre manifestazioni splancniche possono essere i linfangioleiomiomi epatici e
retroperitoneali, gli amartomi splenici e colo-rettali.
Diagnosi
Le indagini strumentali più utili nel confermare la diagnosi sono la TC, che evidenzia con
maggiore accuratezza le lesioni calcifiche periventricolari, e la Risonanza Magnetica, più
sensibile nell'evidenziare lesioni corticali.
I progressi nel campo del “Neuroimaging” stanno ulteriormente migliorando la diagnosi
della Sclerosi Tuberosa, con positive ricadute sul trattamento dei pazienti.
La Diffusion MRI, la PET e la Magnetoencefalografia svolgono già un ruolo importante
nella localizzazione non invasiva dei tuberi ai fini della resezione chirurgica.
Diagnosi prenatale e prevenzione
L'aspetto prenatale delle lesioni della sclerosi tuberosa alla Risonanza Magnetica è
identico a quello delle immagini neonatali. Noduli iperintensi nelle sequenze pesate T1
correlano con tuberosità periventricolari e subcorticali nei reperti patologici posteriori.
Tuttavia, una normale Risonanza Magnetica non esclude la diagnosi di sclerosi
tuberosa, mentre una certa percentuale di lesioni rimane, comunque, non evidenziata
(1).
Tuberi corticali
La RM è la tecnica più sensibile nell’identificazione di tuberi corticali, comunemente
situati alla giunzione tra sostanza grigia e bianca, ma che interessano anche la corteccia
sovrastante e la sostanza bianca sottocorticale.
Spesso la corteccia sovrastante un tubero è displasica e può mostrare pachigiria o
polimicrogiria. Mentre negli adulti i tuberi risultano ipointensi nelle sequenze T1-pesate
ed iperintensi nelle immagini pesate in T2 e FLAIR, nei neonati e nei bambini il segnale è
iperintenso nelle T1 pesate e ipointenso nelle T2-pesate. MRI, elettroencefalografia
(EEG), tecniche di medicina nucleare e imaging come la tomografia ad emissione di
positroni (PET) e SPECT sono utilizzati per localizzare i tuberi che fungono da foci
epilettogeni prima dell'intervento chirurgico. La SPECT, in particolare, è in grado di
dimostrare un aumento dell'attività radio tracciante in corrispondenza della regione
epilettogena. (2)
Il 90% dei tuberi è più frequentemente riscontrabile nel parenchima cerebrale,
solitamente sono molteplici, anche se in rari casi sono stati repertati tuberi corticali
solitari. Meno comunemente rispetto al cervello, tuberi sono presenti nel cervelletto,
dove può risultare evidente solo all'esame istologico ma, a differenza dei tuberi corticali,
i tuberi cerebellari sono di solito a forma di cuneo e non epilettogeni. Rara la presenza
nel tronco cerebrale e nel midollo spinale. (3)
I noduli subependimali sono presenti in circa 80% dei pazienti TS e sono situati attorno
alla parete del ventricolo laterale ed il terzo ventricolo, solitamente in prossimità del
forame di Monro. Dal punto di vista istologico sono costituiti cellule di grandi dimensioni
e cellule gliali, sono coperte da un sottile strato di ependima. Alla RM, queste lesioni
sono isointense o iperintense nelle sequenze T2-pesate ed iperintensa in sequenze T1
pesate, sebbene in T2 possano mostrare ipointensità centrale circondata da orletto
iperintenso. Enhancement variabile è dimostrato dopo somministrazione di contrasto. I
noduli sono più inclini alla calcificazione rispetto ai tuberi corticali e, pertanto, più
facilmente rilevabili in TC.
Gli astrocitomi subependimali a cellule giganti, anche se rari, sono i tumori cerebrali più
comuni, che si verificano in circa il 26% dei pazienti TSC. Si tratta di tumori di grado 1
con crescita lenta, minima invasione del parenchima, assenza di edema cerebrale.
Eterogenei alla RM (T1 iso / ipointenso, T2/FLAIR iso / iperintenso), contengono
calcificazioni e dimostrano enhancement disomogeneo. In considerazione della loro
natura vascolare, TC e RM può essere utile nella valutazione non invasiva prima
dell'intervento chirurgico. La RM spettroscopica può essere utile per attestare l’aumento
del rapporto Cho / Cr e la riduzione di NAA/Cr. Gli astrocitomi vengono monitorati di
solito con MRI per eventuali cambiamenti nelle dimensioni e per l'insorgenza di
idrocefalo. (2)
Nell’ambito della sostanza bianca si trovano molto spesso aree di alterato segnale. Esse
consistono per lo più in strie tenuemente iperintense nelle sequenze dipendenti dal T2,
a forma di nastro o di cuneo con apice diretto verso la profondità: esse spesso collegano
la superficie ventricolare alla corteccia. (5) Sono più frequenti nei lobi frontale e parietale
e, occasionalmente, si verificano nel cervelletto. Anomalie superficiali della sostanza
bianca è presente in quasi tutti i tuberi corticali e mostra iperintensità T2, riflettendo gliosi
o riduzione del contenuto di mielina. L' angiomiolipoma renale in TC si presenta come
una massa corticale non calcifica, sebbene anche il tessuto renale radiologicamente
normale può contenere LAM microscopici. Caratteristiche radiografiche che possono
aiutare a differenziare l’ angiomiolipoma dal carcinoma renale sono: l’enhancement
omogeneo in TC con mezzo di contrasto, la mancanza di calcificazioni e l’isoecogenicità
omogenea all’indagine ultrasonografica. L'imaging con Risonanza Magnetica può
contribuire ad identificare la componente lipidica delle lesioni, ma per una diagnosi di
certezza è auspicabile il ricorso alla biopsia. La linfoangioleiomiomatosi polmonare alla
TC è riscontrabile sottoforma di cisti, a pareti sottili e di dimensioni variabili, in
comunicazione con le vie respiratorie. Talvolta vengono identificate opacità reticolari,
causate da edema interstiziale conseguente ad ostruzione linfatica. Chilotorace e
pneumotorace sono complicanze ricorrenti che possono essere identificate
all’imaging. La biopsia, in casi dubbi, risulterà risolutiva. Il tumore a cellule chiare del
polmone viene, invece, identificato all’imaging sottoforma di nodulo parenchimale
periferico o di massa, senza evidenza di cavitazione o calcificazione. La visualizzazione
viene accresciuta dalla somministrazione di MdC in TC, probabilmente per la ricca
componente vascolare presente. (2)
Terapia
Nei pazienti con sclerosi tuberosa ed epilessia, la lamotrigina risulta efficace e ben
tollerata, mentre l'ormone adrenocorticotropo (ACTH) sopprime gli spasmi flessori e
tende a normalizzare le anomali dell'EEG. L'escissione chirurgica dei tumori è
considerata inutile, specialmente negli individui gravemente compromessi (con
l'eccezione degli amartomi renali che causano IRC). Tuttavia alcuni pazienti possono
beneficiare della dermoabrasione delle lesioni del volto (sebbene queste recidivino
frequentemente) e della resezione dei tumori cerebrali responsabili di epilessie intrattabili
o di ipertensione endocranica (1).
Bibliografia
1) http://malattierare.regione.veneto.it/cerca_it/dettaglio.php?lang=ita&id=1567
2) Clinically Relevant Imaging in Tuberous Sclerosis Radhakrishnan R, Verma S.. J Clin
Imaging Sci 2011;1:39
3) Neuroradiology: Pictorial Essay: Neuroimaging of Tuberous Sclerosis: Spectrum of
Pathologic Findings and Frontiers in Imaging Babak N. Kalantari, Noriko Salamon AJR
May 2008 190:W304-W309; doi:10.2214/AJR.07.2928
4) http://nuke.sioh.it/Portals/0/bibilioteca/congresso%20Genova
/OK%20REALE%202.pdf
5) La valutazione RM della sclerosi tuberosa C. UGGETTI Rivista di Neuroradiologia 16:
547-550, 2003
www.geneticapediatrica.it trimestrale di divulgazione scientifica dell'Associazione Pediatrica di Immunologia e Genetica
Legge 7 marzo 2001, n. 62 - Registro della Stampa Tribunale di Messina n. 3/09 - 11 maggio 2009
Direttore scientifico
Carmelo Salpietro - Direttore responsabile
Giuseppe Micali - Segreteria redazione
Basilia Piraino - Piera Vicchio
Direzione-Redazione: UOC Genetica e Immunologia Pediatrica - AOU Policlicnico Messina