Appunti di Cardiochirurgia Capitolo 4 Cardiopatie Congenite
Appunti di Cardiochirurgia
Capitolo 4
Cardiopatie congenite
1
pagina- 1 -
Appunti di Cardiochirurgia Capitolo 4 Cardiopatie Congenite
pagina- 2 -
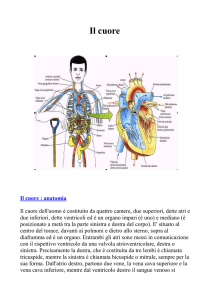
Le malattie cardiovascolari congenite consistono in anomalie strutturali o funzionali del sistema
cardiocircolatorio presenti sin dalla nascita.
Saranno descritte le cardiopatie congenite che possono più frequentemente interessare l’età adulta e/o
l’adolescenza.
Le cardiopatie di pertinenza specificamente pediatrica saranno brevemente accennate.
Epidemiologia: L’incidenza è di 8 – 10 ogni 1000 nati vivi. In Italia ogni anno nascono 3000-3500
nuovi cardiopatici congeniti.
Il sesso maschile è più frequentemente interessato rispetto al femminile, ma possono sussistere notevoli
differenze nel rapporto maschi/femmine tra una malformazione e l’altra.
Eziologia
Le cause delle cardiopatie congenite non sono note nella maggior parte dei casi.
Sono tuttavia conosciuti dei fattori di rischio che nello stato di gravidanza che aumentano la probabilità di
sviluppare cardiopatie congenite quali:
-
cause infettive, come la rosolia contratta da una gestante al I trimestre di gravidanza, che aumenta
il rischio di pervietà del dotto di Botallo,
-
fattori comportamentali quali l’abuso alcool, l’assunzione di alcuni farmaci (talidomide,
difenilidantoina, barbiturici, farmaci antitumorali) costituisce un fattore di rischio generale per lo
sviluppo di cardiopatie congenite del nascituro.
Altri fattori di rischio sono:
-
Fattori ereditari: le persone che nascono in famiglie in cui vi sono stati casi di cardiopatie congenite
hanno maggiore probabilità di esserne affette.
-
Fattori cromosomici: l’aneuploidia (come le sindromi di Down, Turner, Williams) è un fattore che
aumenta il rischio di cardiopatie congenite.
-Classificazione:
Dal funto di vista fisiopatologico si possono distinguere le cardiopatie cianogene (in cui vi è uno shunt
destro>sinistro e quindi cianosi) e quelle non cianogene.
I vizi non cianogeni a loro volta sono distinti tra quelli con normoflusso polmonare e quelli che provocano
un iperafflusso polmonare.
I vizi cianogeni sono invece distinti tra quelli con ipoafflusso e iperafflusso polmonare.
2
Appunti di Cardiochirurgia Capitolo 4 Cardiopatie Congenite
pagina- 3 -
Tabella4; 1: classificazione delle cardiopatie congenite.
Cardiopatie non cianogene
Flusso polmonare aumentato
Flusso polmonare normale o
ridotto
Coartazione aortica
Stenosi polmonare
Stenosi aortica
Ventricolo sinistro ipoplasico
Difetti del setto interatriale
Ritorno venoso polmonare
anomalo
Difetti del setto interventricolare
Pervietà del dotto arterioso di
Botallo;
Cardiopatie cianogene
Flusso polmonare aumentato
Trasposizione dei grossi vasi
Ventricolo destro a doppia uscita
Flusso polmonare ridotto
Tetralogia di Fallot
Atresia della tricuspide
Malattia di Ebstein
CARDIOPATIE NON CIANOGENE CON FLUSSO POLMONARE AUMENTATO.
Difetti del setto interatriale e ritorno venoso anomalo.
I difetti del setto interatriale (DIA) sono la cardiopatia congenita più frequente.
Rappresentano il 15% circa delle cardiopatie congenite nei pazienti che arrivano vivi al primo anno di vita
Capita spesso che la diagnosi di difetto interatriale venga posta nell’età adulta.
Le pazienti di sesso femminile sono circa il doppio rispetto ai maschi.
Nella circolazione fetale il circolo venoso e arterioso sono in comunicazione a livello del setto interatriale
tramite il forame ovale.
Dopo la nascita la pressione nell’atrio sinistro aumenta rispetto al destro, e il forame ovale si chiude,
facendo si che il circolo venoso e il circolo arterioso siano completamente separati.
In caso di presenza di un DIA vi è una comunicazione tra atrio sinistro e atrio destro e, per la maggior
pressione in atrio sinistro, si ha l’immissione di sangue da sinistra a destra (shunt sinistro - destro).
A seconda della loro posizione a livello del setto interatriale si possono genericamente distinguere tre
tipologie principali:
-
difetto tipo ostium secundum, localizzato a livello della fossa ovale. È la variante più comune,
rappresentando circa l’ 80% dei DIA;
-
il difetto tipo ostium primum, localizzato a livello del pavimento del setto in prossimità del piano
atrio-ventricolare. È raramente isolato, bensì è spesso associato a vizi, tipo canale atrioventricolare,
con coinvolgimento delle valvole atrioventricolari;
-
il difetto del seno venoso, è localizzato verso la parte alta del setto e spesso si associa ad un ritorno
venoso anomalo.
3
Appunti di Cardiochirurgia Capitolo 4 Cardiopatie Congenite
pagina- 4 -
Figura 4;1 tipologie di DIA
Il ritorno venoso polmonare anomalo parziale (RVPAP) consiste nel drenaggio di una o più vene polmonari
nell’atrio destro. È solitamente accompagnato da un difetto interatriale tipo seno venoso. Da un punto di
vista fisiopatologico un DIA con associato un ritorno venoso anomalo parziale non cambia molto rispetto ad
un DIA isolato. L’aspetto più importante è che, nel caso di un ritorno venoso anomalo, vi sarà un
iperafflusso polmonare di maggiori dimensioni, e quindi una sintomatologia più spiccata ed una evoluzione
più rapida rispetto al DIA semplice.
figura 4;2: ritorno venoso polmonare anomalo
1 difetto interatriale, 2 vena polmonare anomala
www.pediatriccardiacinquest.mb.
4
Appunti di Cardiochirurgia Capitolo 4 Cardiopatie Congenite
pagina- 5 -
Fisiopatologia
Considerando un DIA semplice senza altri vizi associati, la comunicazione tra i due atri causa uno shunt
sinistro – destro, che provoca un iperafflusso di sangue nel circolo polmonare, la cui entità varia a seconda
delle dimensioni del difetto stesso.
Quando il difetto è sufficientemente ampio da causare un iperafflusso polmonare significativo, avvengono
degli adattamenti a livello del piccolo circolo noti come fenomeno di Eisenmenger:
infatti il continuo incremento delle pressioni nel circolo arterioso polmonare induce delle alterazioni
anatomiche irreversibili sulle arterie polmonari. Aumentano quindi le resistenze vascolari polmonari, che
possono superare le resistenze del circolo sistemico. La pressione nel piccolo circolo può divenire molto
alta, maggiore addirittura di quella sistemica.
Quando questo avviene, la direzione del flusso a livello della malformazione che mette in comunicazione i
due circoli si inverte.
A questo punto il sangue (non ancora ossigenato) si immette nel circolo sistemico (shunt destro-sinistro).
Dal punto di vista clinico l’aspetto peculiare è l’instaurarsi della cianosi.
Tale quadro clinico è noto come sindrome di Eisenmenger.
Si parla di sindrome di Eisenmenger, in quanto il “fenomeno” di Eisenmenger si può osservare in tutte le
cardiopatie congenite con iperafflusso polmonare quali: persistenza del dotto di Botallo, difetti
interventricolari e comunicazioni interatriali. Una volta che la reazione di Eisenmenger si è verificata, il
quadro clinico è simile a prescindere dal vizio cardiaco alla base.
Sintomatologia
Nei casi in cui il DIA sia di piccole dimensioni, il paziente può essere asintomatico. Invece nei casi in cui sia di
maggiori dimensioni, quindi con un importante shunt sinistro- destro, si potranno osservare segni iniziali di
scompenso cardiaco quali dispnea da sforzo e facile affaticabilità.
A questi fanno seguito i sintomi dell’insufficienza ventricolare sinistra in assenza di correzione della
patologia. Qualora si sviluppi la sindrome di Eisenmenger per il sovraccarico di lavoro del ventricolo destro,
si potrà anche sviluppare scompenso cardiaco di tipo destro.
Obiettività clinica:
I reperti auscultatori che possono essere riscontrati in caso di DIA sono:
-
un soffio sistolico eiettivo a livello del focolaio della valvola polmonare, per l’aumento del flusso di
sangue in sistole attraverso la valvola polmonare stessa;
-
un rumore mesodiastolico sul focolaio di auscultazione tricuspidale, per l’iperafflusso diastolico
attraverso la valvola tricuspide;
-
lo sdoppiamento fisso del II tono.
5
Appunti di Cardiochirurgia Capitolo 4 Cardiopatie Congenite
pagina- 6 -
Diagnostica strumentale.
Elettrocardiogramma: è tipico di tutti i vizi con iperafflusso polmonare il riscontro di segni
elettrocardiografici dell’ipertrofia del ventricolo destro. Inoltre per la dilatazione degli atri è frequente il
riscontro di fibrillazione atriale.
Radiografia del torace: potranno essere osservati i segni di dilatazione dell’atrio destro,
del ventricolo destro, dell’arteria polmonare e dei suoi rami. La trama della vascolarizzazione polmonare
sarà incrementata (quadro di iperafflusso polmonare).
Alla radioscopia i rami principali dell’arteria polmonare possono apparire marcatamente pulsanti (danza
ilare).
Ecocardiografia: in alcuni casi, ma non sempre, può visualizzare direttamente il DIA e con tecnica doppler
valutarne il flusso transatriale, inoltre individuerà l’ipertrofia e la dilatazione del ventricolo destro.
Cateterismo cardiaco destro:
si esegue mediante catetere di Swan-Ganz che viene introdotto in vena giugulare (di solito dx) da quì
attraversa la vena cava superiore, l’atrio destro, il ventricolo destro fino all’arteria polmonare.
Tale catetere è dotato di un palloncino gonfiabile in punta che quando è gonfiato, incuneandosi in un ramo
dell’arteria polmonare, misura la pressione di incuneamento polmonare. È inoltre dotato di un sistema in
grado di misurare direttamente la portata cardiaca del circolo polmonare. Inoltre si possono prendere vari
campioni di sangue dalla vena cava superiore, atrio e ventricolo destro e arteria polmonare per misurare
l’ossimetria nel sangue di questi distretti.
L’ossimetria in caso di DIA mostrerà un gradiente di pressione parziale di ossigeno tra il sangue prelevato
nell’atrio destro e quello prelevato nella vena cava.
Il cateterismo mostra inoltre l’incremento delle pressioni nel circolo polmonare.
In particolare, mediante il rilievo delle saturimetrie, si può risalire al valore del rapporto tra la portata del
circolo polmonare (Qp) e quella sistemica (Qs). In condizioni normali le due portate sono uguali e tale
rapporto è uguale a 1.
Nei casi di patologie con shunt sinistro - destro si verifica un aumento della portata polmonare rispetto a
quella sistemica.
Si considera emodinamicamente significativo, e quindi da correggere chirurgicamente, la patologia che
determini un rapporto tra la portata polmonare e quella sistemica maggiore di 1,5.
6
Appunti di Cardiochirurgia Capitolo 4 Cardiopatie Congenite
pagina- 7 -
Fig4.3 catetere di Swan-Ganz
Storia naturale: la maggior parte dei pazienti portatori di DIA diviene sintomatica generalmente dalla terza
decade. Solitamente la morte avviene per insufficienza cardiaca verso la quinta decade nei pazienti in cui il
difetto non viene corretto prima.
Si pone indicazione all’intervento chirurgico quando il rapporto tra la portata polmonare e la portata
sistemica(Qp/Qs) è maggiore o uguale a 1,5.
Metodologie di correzione.
L’intervento chirurgico avviene mediante l’ausilio della circolazione extracorporea. La tecnica di accesso alla
cavità atriale sinistra è quella descritta da Sondegaard (vedi patologia mitralica).
A seconda delle dimensioni del DIA questo potrà essere chiuso o con una sutura diretta o mediante il
posizionamento di un patch.
In alcuni casi è possibile eseguire la correzione mediante l’impianto transcatetere di dispositivo di
occlusione per via percutanea.
In caso di concomitante DIA e ritorno venoso anomalo l’intervento chirurgico mira a correggere entrambi i
difetti. Il patch dovrà essere posizionato in modo tale da far proseguire lo sbocco della vena polmonare
anomala attraverso il difetto interatriale preesistente nell’atrio sinistro.
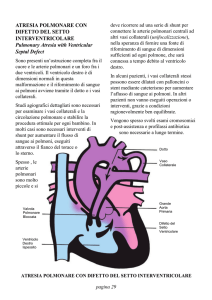
Difetti del setto interventricolare.
Il difetto del setto interventricolare (DIV) origina nella costituzione del setto che sepimenta i due ventricoli.
Dunque in presenza di un DIV sussiste una comunicazione diretta tra il ventricolo destro e il sinistro, con
conseguente shunt sinistro - destro.
I DIV sono abbastanza frequenti e raramente vengono diagnosticati in età adulta, bensì entro la prima
decade e possono presentarsi isolati o associati ad altri vizi cardiaci congeniti.
7
Appunti di Cardiochirurgia Capitolo 4 Cardiopatie Congenite
pagina- 8 -
I DIV possono essere classificati in base a tre aspetti fondamentali: le dimensioni, la sede e il numero in cui
si presentano:
• la loro superficie può essere da pochi mm2 fino ad alcuni cm2;
• perciò che riguarda la sede, la maggior parte dei DIV sono di solito riscontrati a livello della parte
prossimale membranosa del setto.
I difetti della pars membranacea vengono poi distinti a seconda della localizzazione rispetto alla crista
ventricularis, una trabecola muscolare che attraversa la parte posteriore del tratto di efflusso ventricolare
destro: si distinguono DIV sopracristali e sottocristali e la maggior parte dei DIV membranosi è in sede
sottocristale.
I DIV sottocristali hanno sede nelle vicinanze della valvola aortica e possono associarsi a malformazioni di
tale valvola. I difetti sopracristali sono ubicati subito al di sotto della valvola polmonare, e spesso sono
associati ad alterazioni che la coinvolgono.
I difetti settali della pars membranacea sono in genere singoli mentre quelli muscolari sono più
spesso multipli (Swiss cheese).
Figura 4; 4: tipologie di DIV : 1 del setto membranoso, , 2-3 sottocristale, 4 -5 sopracristale, 6-7-8 del setto
muscolare.
Fisiopatologia: la fisiopatologia dei difetti interventricolari non è dissimile da quella dei difetti interatriali.
Tuttavia essendo il ventricolo sinistro una cavità a elevata pressione, di solito a parità di dimensioni lo shunt
di un DIV sarà maggiore rispetto a quello di un DIA.
Dunque generalmente l’evoluzione clinica nei DIV è più rapida e più grave che non in caso di DIA.
Il difetto interventricolare, mettendo in diretta comunicazione i due ventricoli, determina uno shunt
sinistro-destro per la differenza di pressione che sussiste tra le due camere.
L’entità dello shunt dipende dalle dimensioni del DIV e dal gradiente pressorio tra i ventricoli sinistro e
destro.
Quando difetto è piccolo, lo shunt sarà trascurabile e le pressioni nel ventricolo destro e nel piccolo circolo
non ne risentiranno. Tale condizione è definita malattia di Roger.
8
Appunti di Cardiochirurgia Capitolo 4 Cardiopatie Congenite
pagina- 9 -
In caso di difetto di maggiori dimensioni, con un significativo flusso di sangue da sinistra a destra, vi sarà un
incremento delle pressioni del piccolo circolo. Come è già stato precedentemente detto per i DIA, tale
aumento pressorio sarà inizialmente reversibile, ma nel tempo l’ipertensione polmonare diverrà
irreversibile per l’adattamento dei vasi del circolo polmonare all’aumento del flusso (reazione di
Eisenmenger).
L’incremento pressorio del circolo polmonare si riverbera anche a livello del ventricolo destro.
Con un aumento di pressione del ventricolo destro lo shunt sinistra-destra attraverso il DIV si riduce.
Qualora la pressione ventricolare destra divenga simile a quella nel sinistro, il flusso attraverso il DIV può
invertirsi: a questo punto avremo uno shunt destra – sinistra, e sangue venoso si immetterà nel circolo
sistemico arterioso con comparsa di cianosi (sindrome di Eisenmenger).
Sintomatologia: in caso di piccolo difetto il paziente è asintomatico.
Difetti di dimensioni maggiori, tali da condizionare un iperafflusso e un’ipertensione polmonare, causano
facile affaticabilità e dispnea da sforzo, sintomi tipici della congestione polmonare, ed
eventualmente edema polmonare acuto. L’iperafflusso polmonare favorisce (come nel DIA) l’insorgenza di
infezioni polmonari. La cianosi può verificarsi in caso si instauri una severa ipertensione polmonare, che
determini l’inversione dello shunt attraverso il difetto.
Obbiettività clinica: il reperto auscultatorio tipico è un soffio sistolico tra i quattro e i sei sesti,
maggiormente udibile al quarto spazio intercostale sulla linea parasternale destra, eventualmente
accompagnato da un fremito.
Diagnostica strumentale.
Elettrocardiogramma: l’ECG sarà normale nei casi di piccoli difetti. Nei casi di vizi di dimensioni maggiori,
poiché entrambi i ventricoli sono sottoposti a un sovraccarico di lavoro, si potrà riscontrare ipertrofia
ventricolare destra, sinistra o di entrambi i ventricoli.
Radiografia del torace: si potranno osservare i segni radiologici relativi alla dilatazione ventricolare sia
destra che sinistra, alla dilatazione dell’arteria polmonare e
dei suoi rami principali, e all’aumento della trama vasale polmonare.
Ecocardiogramma: può direttamente visualizzare il difetto ed evidenziare lo shunt mediante il color
doppler. Saranno inoltre visualizzati i ventricoli ipertrofici e dilatati.
Cateterismo cardiaco: si potrà rilevare una maggiore ossigenazione del sangue prelevato dal ventricolo
destro rispetto a quello prelevato nell’atrio destro. Il cateterismo consente anche la misurazione diretta
delle pressioni nell’arteria polmonare.
L’ossimetria e la misurazione delle pressioni polmonari consentono una stima indiretta dell’entità del
difetto, mediante valutazione comparativa della portata (gittata/min) nel circolo polmonare e nel circolo
sistemico (QP/QS).
9
Appunti di Cardiochirurgia Capitolo 4 Cardiopatie Congenite
pagina- 10 -
Storia naturale: i DIV di piccole dimensioni possono chiudersi spontaneamente, e quelli che rimangono
pervi, se non danno complicanze emodinamiche, non vanno operati (malattia di Roger). Al contrario i DIV di
dimensioni maggiori con shunt significativo (Qp/Qs>1.5) se non operati determinano la morte del 40% dei
pazienti entro la seconda decade di vita, e del’80% entro la quarta.
Il flusso turbolento provocato dal difetto interventricolare facilita l’impianto di batteri nei pressi del difetto
e tale rischio sussiste anche se il difetto è piccolo.
Indicazioni alla correzione chirurgica: nei difetti di piccole dimensioni non c’è indicazione alla correzione.
Nei difetti più grandi l’indicazione si pone quando il rapporto tra la portata polmonare e la portata
sistemica(Qp/Qs) è maggiore o uguale a 1,5.
Tecnica di correzione.
L’intervento chirurgico avviene mediante l’ausilio della circolazione extracorporea. La tecnica di accesso al
DIV è in genere transatriale destra. Una volta eseguita l’atriotomia destra si esegue la retrazione del lembo
settale della tricuspide o l’eventuale sua disinserzione (con successivo reimpianto) per visualizzare
correttamente il setto interventricolare. Dopo la visualizzazione del difetto si posiziona un patch di dacron o
di pericardio.
Per quanto in alcuni casi eseguire la correzione per via percutanea sia tecnicamente possibile, non la si
consiglia in prima battuta poiché è gravata da un notevole rischio di causare blocco atrioventricolare con
necessità di impianto di un pace maker.
Pervietà del dotto arterioso di Botallo.
Il dotto arterioso di Botallo durante la vita fetale connette l’arteria polmonare con l’aorta in sede
immediatamente distale all’ostio dell’arteria succlavia sinistra.
Nella circolazione fetale il dotto di Botallo permette il passaggio di sangue dall’arteria polmonare in aorta.
Alla nascita il dotto si restringe e poi si chiude per l’aumento della pressione parziale di ossigeno nel sangue
arterioso dopo l’inizio della respirazione. Nella maggior parte dei casi il dotto arterioso si chiude
fisiologicamente dopo le prime 24 ore dalla nascita, ma in alcuni casi anche dopo un periodo di tempo
maggiore
Si considera patologica la persistenza della pervietà del dotto ai tre mesi di vita.
Il dotto arterioso pervio può essere isolato o eventualmente associato ad altre cardiopatie congenite,
la maggior parte dei pazienti con portatori del difetto è di sesso femminile.
10
Appunti di Cardiochirurgia Capitolo 4 Cardiopatie Congenite
pagina- 11 -
www.slideshare.net
Figura 4; 5 pervietà del dotto di Botallo
Fisiopatologia.
Dopo la nascita si verifica l’aumento di pressione nell’aorta, che supera quella della polmonare facendo si
che il flusso di sangue all’interno del dotto arterioso sia diretto dall’aorta alla polmonare
(shunt sinistro - destro).
Il calibro del dotto può variare da pochi millimetri a circa 15 mm; se il dotto è di dimensioni ampie, il flusso
di sangue nel circolo polmonare aumenterà in maniera importante.
Si avrà quindi un aumento delle pressioni del circolo polmonare che saranno inizialmente reversibili, quindi
con la possibilità di ritornare a valori normali se il dotto viene chiuso.
Se questo non succede, e le pressioni del piccolo circolo rimangono elevate nel tempo a tale livello, si
instaura il fenomeno di Eisenmenger.
Sintomatologia. Nei casi in cui il dotto arterioso è di piccole dimensioni, il paziente può essere asintomatico.
Invece nei casi in cui il dotto sia di maggiori dimensioni, quindi con un importante shunt sinistro destro si
potranno osservare segni iniziali di scompenso cardiaco quali dispnea da sforzo e facile affaticabilità. In
assenza di correzione della patologia fanno seguito i sintomi dell’insufficienza ventricolare sinistra. Inoltre,
a livello del dotto arterioso pervio vi è maggior rischio che si sviluppi un’endoarterite infettiva.
Qualora si sviluppi la sindrome di Eisenmenger per il sovraccarico di lavoro del ventricolo destro, si potrà
anche sviluppare scompenso cardiaco di tipo destro.
Obbiettività clinica.
Il segno clinico più importante è un soffio sisto-diastolico, che ha la sua massima
intensità sul focolaio di auscultazione della polmonare.
Esso è tipico della pervietà del dotto, in quanto la pressione sia sistolica che diastolica in aorta supera
quella in arteria polmonare, per cui il flusso ematico attraverso il dotto è sistolico e diastolico. Quando il
soffio è molto intenso si può palpare un fremito.
Nel caso in cui la pressione del piccolo circolo aumenti in modo significativo, il gradiente pressorio tra aorta
e polmonare si riduce con conseguente riduzione di intensità del soffio.
11
Appunti di Cardiochirurgia Capitolo 4 Cardiopatie Congenite
pagina- 12 -
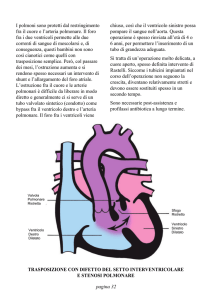
La cianosi si presenta solo quando, a causa dell’ipertensione polmonare, si inverte la direzione del flusso
attraverso il dotto (shunt destro-sinistro).
Diagnostica strumentale.
Elettrocardiogramma: può essere normale se il dotto è piccolo. Se invece è ampio, il ventricolo sinistro è
sottoposto a un sovraccarico di lavoro, con riscontro all’elettrocardiogramma di segni di ipertrofia del
ventricolo sinistro. In caso di sindrome di Eisenmenger potranno evidenziarsi i segni di ipertrofia
ventricolare destra, eventualmente associati ai segni di ipertrofia sinistra.
Radiografia del torace: evidenzia soprattutto i segni dell’iperafflusso polmonare con distensione e
dilatazione dell’arteria polmonare e dei suoi rami.
Nel quadro radiologico della sindrome di Eisenmenger si osserverà la arteria polmonare e suoi rami
principali dilatati, mentre la periferia polmonare risulta particolarmente povera di trama vascolare.
Ecocardiografia: visualizza direttamente il dotto arterioso e con la funzione color doppler misura il flusso al
suo interno. A livello cardiaco si potrà evidenziare l’ipertrofia ventricolare.
Cateterismo cardiaco: in quanto il vizio fa si che sangue arterioso proveniente dall’aorta si immetta
nell’arteria polmonare, l’ossimetria proverà una differenza di ossigenazione fra il sangue prelevato
dall’arteria polmonare e quello prelevato dal ventricolo destro.
Il cateterismo inoltre misura direttamente le pressioni e le resistenze del circolo polmonare.
Anche in questo caso la stima del rapporto QP/QS>1,5 fa porre indicazione per la correzione del difetto.
Storia naturale.
Nei casi in cui il dotto sia di dimensioni ridotte in pazienti asintomatici, il vizio non modiifica le prospettive
di vita. Se invece il dotto è molto grande, sussiste il rischio di morte entro i primi mesi di vita per
scompenso cardiaco. Nei casi di dimensioni intermedie si potrà avere assenza di sintomi fino
al’adolescenza, con dispnea da sforzo via via ingravescente, fino allo scompenso conclamato verso la terza
o quarta decade.
Terapia: consiste nella chiusura diretta del dotto. L’intervento è eseguito senza circolazione extracorporea.
12
Appunti di Cardiochirurgia Capitolo 4 Cardiopatie Congenite
pagina- 13 -
CARDIOPATIE NON CIANOGENE CON FLUSSO POLMONARE NORMALE
Stenosi aortica
Nei casi di stenosi valvolare aortica congenita la valvola è in genere bicuspide.
La bicuspidia aortica è la cardiopatia congenita più frequente: peraltro c’è da dire che non sempre è causa
di una stenosi . Nei casi di stenosi il quadro clinico è sovrapponibile a quello della stenosi acquisita (si veda
in seguito).
La bicuspidia, se non genera stenosi è di solito asintomatica. Si ricorda che tale malformazione può essere
associata ad altri vizi congeniti, in particolare la coartazione aortica, inoltre i pazienti con valvola aortica
bicuspide sono più soggetti a sviluppare nel tempo aneurismi dell’aorta ascendente, con un rischio di
rottura maggiore rispetto quello della popolazione generale.
Coartazione dell’aorta.
La coartazione aortica consiste in un restringimento congenito del vaso, che può interessare il decorso
dell’aorta per una lunghezza variabile. Di solito la coartazione è localizzata a livello dell’istmo aortico, e a
seconda della sua estensione può essere distinta in postduttale (dell’adulto) e preduttale (infantile).
La coartazione postduttale è anche chiamata dell’adulto in quanto permette di sopravvivere fino all’età
adulta.
La forma preduttale è in genere più grave: raramente il paziente sopravvive fino all’età adulta senza
correzione chirurgica.
La prognosi dei due tipi di coartazione è diversa in funzione delle diverse caratteristiche anatomiche.
Nella forma postduttale il segmento stenotico del vaso è solitamente circoscritto e localizzato a livello
dell’aorta discendente (distalmente all’istmo aortico e al legamento arterioso, che è il residuo fibroso del
dotto di Botallo), senza interessare l’arco aortico.
La forma infantile interessa l’aorta nel segmento prossimale al dotto di Botallo, e solitamente determina
ipoplasia di una parte dell’arco aortico.
In tutti i casi la coartazione aortica può essere associata ad altri difetti congeniti quali, bicuspidia aortica e
pervietà del dotto di Botallo.
13
Appunti di Cardiochirurgia Capitolo 4 Cardiopatie Congenite
Figura 4;6 coartazione aortica preduttale
pagina- 14 -
figura 4;7 coartazione aortica postduttale
La coartazione aortica ostacola il normale flusso di sangue attraverso il segmento vasale interessato.
Si determina quindi un gradiente pressorio a monte e a valle della coartazione, che causa
un’importante ipertensione a carico di tutte le strutture vascolari che stanno a monte della coartazione, ed
una ipoperfusione di tutte le struttura a valle dove invece la pressione arteriosa è ridotta.
Si ha inoltre lo sviluppo di circoli collaterali. Il più importante si instaura a livello delle le arterie succlavie,
che portano sangue alle arterie mammarie interne, e delle arterie intercostali le quali sono variamente
connesse col circolo arterioso collegato all’aorta distale.
Sintomatologia: nelle forme preduttali di solito si hanno manifestazioni precoci di scompenso cardiaco.
Nelle forme postduttali, che in genere si manifestano in adolescenza, i sintomi tipici sono quelli dello
scompenso. Spesso viene riferita facile affaticabilità agli arti inferiori.
Inoltre a livello della coartazione vi è maggior rischio di infezioni batteriche (arteriti) che a volte possono
essere il primo segno della patologia.
Obbiettività clinica:
si potrà riscontrare un soffio sistolico in crescendo decrescendo sulla linea parasternale sinistra e sul dorso
a livello della colonna vertebrale toracica.
Si potrà inoltre apprezzare una riduzione dei polsi femorali, e una differenza di pressione arteriosa sistolica
fra gli arti superiori e gli arti inferiori, in genere superiore ai 30 mmHg.
Nella forma infantile questa differenza è dovuta alla riduzione della pressione arteriosa negli arti inferiori.
Al contrario nell’adulto è dovuta in genere ad incremento progressivo della pressione negli arti superiori.
14
Appunti di Cardiochirurgia Capitolo 4 Cardiopatie Congenite
pagina- 15 -
Diagnostica strumentale:
Elettrocardiogramma: segni di ipertrofia ventricolare sinistra.
Radiografia del torace:a livello dell’ombra cardiaca si potrà riscontare ipertrofia del ventricolo sinistro.
Inoltre si potranno osservare delle irregolarità a livello dei margini inferiori della parte posteriore delle
coste. Queste sono causate dall’erosione ossea provocata dalla dilatazione delle arterie intercostali che
fanno parte del circolo collaterale prima descritto.
Le incisure costali compaiono di solito dopo i 12 anni di età.
Ecocardiogramma: può evidenziare direttamente la coartazione aortica, ed evidenziare con il color doppler
il flusso turbolento da essa provocato. La velocità massima permette di stimare il gradiente di pressione a
cavallo della stenosi.
Storia naturale: senza correzione chirurgica, nella forma infantile si ha la morte per scompenso entro i primi
anni di vita.
Nella forma adulta, in assenza di correzione, il paziente vive in genere fino a 30-35 anni.
Terapia. Per le forme di coartazione preduttali l’intervento è in genere eseguito con l’ausilio della
circolazione extracorporea e deve mirare a ricostruire l’arco aortico.
Nelle forme postduttali in genere l’intervento può essere eseguito senza CEC, e consiste nel resecare il
segmento stenotico e ripristinare la continuità del vaso o con una sutura termino-terminale tra i due
monconi aortici, o mediante interposizione di una protesi vascolare.
In alcuni casi è possibile eseguire la dilatazione della coartazione anche per via percutanea.
Stenosi polmonare
In tale condizione vi è un ostacolo allo svuotamento del ventricolo destro. Le malformazioni che lo
determinano possono essere a livello della valvola polmonare oppure nell’infundibolo sottovalvolare.
Quando l’ostruzione è a livello della valvola polmonare questa si presenta come un diaframma con
un’apertura al centro.
L’ostacolo nell’infundibolo può essere rappresentato dalla presenza di una struttura fibrosa al di sotto del
piano valvolare, oppure da una diffusa ipertrofia muscolare del tratto di efflusso del ventricolo destro.
In tutti i casi sul ventricolo destro grava un sovraccarico di lavoro.
Quando la pressione sistolica del ventricolo destro raggiunga o superi i 60 mmHg il ventricolo si adatta
ipertrofizzandosi.
Per vincere l’ostruzione all’efflusso dal ventricolo destro, la pressione intraventricolare aumenta,
producendo un gradiente pressorio fra la camera ventricolare e l’arteria polmonare, necessario per
mantenere il flusso attraverso l’ostruzione.
15
Appunti di Cardiochirurgia Capitolo 4 Cardiopatie Congenite
pagina- 16 -
Sintomatologia: dipende dalla severità della stenosi. Se è lieve, il paziente è asintomatico.
Nei casi di stenosi di media gravità di solito rimane asintomatica fino all’età adulta.
Le ostruzioni gravi sono sintomatiche già nell’infanzia.
I sintomi principali sono dispnea da sforzo e facile affaticabilità.
Nel caso di elevata pressione intraventricolare che si riverberi in atrio destro, è possibile l’apertura del
forame ovale e uno shunt destro-sinistro con possibile comparsa di cianosi.
Obbiettività clinica: il reperto auscultatorio principale consiste nel riscontro di un soffio sistolico, udibile in
particolare sul focolaio auscultatorio della valvola polmonare, determinato dal sangue che viene spinto dal
ventricolo destro attraverso l’ostruzione. Il soffio è in genere intenso, rude con intensità in crescendo
decrescendo, definito “a diamante”.
Diagnostica strumentale
ECG: evidenzia i segni di ipertrofia del ventricolo destro: onda R maggiore di 7 mm in V1 e V2,deviazione a
destra dell’asse del QRS e T negativa in V1-V2 e in D2, D3, aVF.
Radiografia del torace: si può rilevare una dilatazione dell’arteria polmonare e la dilatazione del ventricolo
destro.
L’ecocardiogramma visualizza direttamente sia l’infundibolo del ventricolo destro sia la valvola polmonare.
Il color doppler evidenzia il flusso turbolento presente in sistole nell’arteria polmonare dopo l’ostruzione.
Dalla velocità massima del flusso si calcola il gradiente pressorio trans-valvolare.
Storia naturale: i pazienti con stenosi severa non trattata chirurgicamente vanno incontro a morte per
scompenso destro in giovane età.
Il trattamento consiste nella correzione chirurgica che varia a seconda delle caratteristiche anatomiche
della stenosi, ma che in tutti i casi mira a ripristinare il fisiologico flusso tra ventricolo destro e arteria
polmonare. Nel caso di una stenosi sottovalvolare sarà rimossa la membrana sottovalvolare, o corretta
l’ipertrofia infundibulare. Nel caso di importanti alterazioni anatomiche della valvola questa dovrà essere
sostituita con una protesi.
16
Appunti di Cardiochirurgia Capitolo 4 Cardiopatie Congenite
pagina- 17 -
Ventricolo sinistro ipoplasico
La sindrome del cuore sinistro ipoplasico è rara .
Rappresenta la più comune causa di morte per anomalie cardiache nel primo mese di vita.
La fisiologia del cuore sinistro ipoplasico è definita come l’ incapacità del cuore sinistro a sostenere
un'adeguata gittata cardiaca dopo la nascita, a causa dell’ iposviluppo di una o più strutture sinistre del
cuore.
Alla nascita la presenza di un difetto interatriale o interventricolare, e la pervietà del dotto di Botallo sono
necessari alla sopravvivenza del neonato.
La correzione chirurgica avviene in tre gradi: il primo è definito intervento di Norwood, il secondo consiste
nell’anastomosi cavopolmonare bidirezionale secondo Glenn, e il terzo nella correzione secondo Fontan.
CARDIOPATIE CIANOGENE CON RIDOTTO FLUSSO POLMONARE
Tetralogia di Fallot.
La tetralogia di Fallot è una cardiopatia congenita con quattro aspetti fondamentali:
1 presenza di un difetto del setto interventricolare alto;
2 ostruzione all’efflusso del ventricolo destro (stenosi polmonare valvolare o sottovalvolare);
3 Aorta a cavaliere: l’aorta non nasce dal ventricolo sinistro bensì ha origine a cavallo fra i due ventricoli
sopra il difetto interventricolare .
4 Ipertrofia ventricolare destra.
Epidemiologia: fra i vizi cianogeni la tetralogia di Fallot è la più frequente, costituendo il 6% di tutti i vizi
cardiaci congeniti.
Descrizione morfologica:
1) il difetto interventricolare è solitamente ampio; generalmente ha sede nel setto membranoso sopra la
crista ventricularis;
2) l’ostruzione all’efflusso dal ventricolo destro in circa il 20% dei casi è determinata da una stenosi della
valvola polmonare.
Più frequentemente è causata da una struttura fibrosa sottovalvolare o da un restringimento di tutto il
tratto di efflusso del ventricolo destro;
3) aorta a cavaliere. L’aorta prende origine sia dal ventricolo destro sia dal ventricolo sinistro, è posta a
cavallo del setto interventricolare sopra il difetto interventricolare;
4) l’ipertrofia del ventricolo destro è causata dalle alterazioni emodinamiche, condizionate dai difetti sopra
descritti.
17
Appunti di Cardiochirurgia Capitolo 4 Cardiopatie Congenite
pagina- 18 -
Figura 4;8 : tetralogia di Fallot.
Fisiopatologia. Il sangue pompato dal ventricolo destro entra nell’ arteria polmonare solo parzialmente, per
via dell’ostruzione nel tratto di efflusso. I vasi polmonari vengono quindi ipoperfusi. Il sangue che non passa
in arteria polmonare ha due vie di uscita dal ventricolo destro: l’aorta (che in parte origina dal ventricolo
destro stesso) e il difetto interventricolare. Peraltro, siccome l’aorta nasce a cavallo del DIV, anche il sangue
che lo attraversa entrerà comunque nell’aorta, attraverso la parte del vaso che ha origine dal ventricolo
sinistro.
In questo modo il sangue venoso entra nel circolo sistemico (shunt destra-sinistra) causando ipossiemia
arteriosa e cianosi.
Il ventricolo destro diviene ipertrofico, dovendo lavorare in condizioni di sovraccarico per le elevate
resistenze al flusso, rappresentate dall’ostruzione polmonare e dalle pressioni sistemiche dell’aorta e del
ventricolo sinistro.
La quantità di sangue che riesce ad arrivare nel circolo polmonare dipende dall’ostruzione e dalle resistenze
nel circolo polmonare e sistemico.
Sintomatologia. La diagnosi avviene di solito precocemente. Nel bambino piccolo si osservano irritabilità e
inappetenza. Durante il pianto compare o si accentua la cianosi per l’aumento delle resistenze polmonari,
secondario all’incremento della pressione intratoracica, causata dalla manovra di Valsalva fatta dal
bambino durante il pianto.
Lo stesso si verifica in altre condizioni in cui si effettui una manovra di Valsalva come tosse ed evacuazione
delle feci.
Lo shunt destro-sinistro può causare ipossia cerebrale, con la possibilità che si verifichino sincopi e
convulsioni.
18
Appunti di Cardiochirurgia Capitolo 4 Cardiopatie Congenite
pagina- 19 -
Il bambino affetto da tetralogia di Fallot assume la caratteristica posizione di “squatting”, che consiste
nell’accovacciarsi con le ginocchia piegate e le braccia intorno alle gambe. Assumendo tale posizione si
incrementano le resistenze arteriose periferiche, con ostacolo al flusso di sangue venoso dal ventricolo
destro verso l’aorta e il difetto interventricolare, riducendo pertanto la cianosi.
Viene inoltre riferita dispnea da sforzo, secondaria all’ipossigenazione del sangue arterioso, e
all’ipoperfusione del circolo polmonare.
Obbiettività clinica. Ispettivamente il paziente può mostrare una cianosi che si aggrava (o compare) in corso
di manovra di Valsalva.
Il reperto auscultatorio principale è un soffio sistolico udibile in particolare sul focolaio auscultatorio della
valvola polmonare, determinato dal sangue che viene spinto dal ventricolo destro attraverso l’ostruzione. Il
soffio è in genere intenso, rude con intensità in crescendo decrescendo definito “a diamante”.
Diagnostica strumentale.
Elettrocardiogramma. Mette in evidenza un’importante ipertrofia e sovraccarico del ventricolo destro.
Radiografia del torace. Si riscontano i segni dell’ ipertrofia del ventricolo destro. L’aorta ascendente è in
genere dilatata. I campi polmonari appaiono ipoperfusi.
Ecocardiogramma. Mette in evidenza l’ipertrofia ventricolare destra e l’aorta a cavaliere; può individuare
pure il difetto interventricolare.
Cateterismo cardiaco. Misura l’aumento di pressione del ventricolo destro, il passaggio diretto del catetere
dal ventricolo destro nell’aorta, la ridotta ossigenazione del sangue arterioso.
Storia naturale. In assenza di correzione chirurgica la morte avviene tra la prima e la seconda decade. Circa
il 10% dei pazienti sopravvive fino all’età adulta.
Terapia: la correzione chirurgica viene eseguita con la circolazione extracorporea. Consiste sia nella
correzione dell’ostruzione del ventricolo destro che nella applicazione di un patch, che deve essere
posizionato in modo tale da chiudere il difetto interventricolare e anche la comunicazione tra l’aorta e il
ventricolo destro .
Malattia di Ebstein
È rara (1% dei vizi cardiaci congeniti).
Consiste in un abbassamento del piano tricuspidalico verso l’apice del ventricolo sinistro.
Frequentemente la valvola tricuspide e l’apparato sottovalvolare sono alterati nella loro morfologia e
funzionalità.
Spesso è associata ad un difetto interatriale.
19
Appunti di Cardiochirurgia Capitolo 4 Cardiopatie Congenite
L’alterazione emodinamica
pagina- 20 -
principale della malattia di Ebstein è la ridotta capacità contrattile del
ventricolo destro, che causa una riduzione di flusso del piccolo circolo, e ipertensione nelle strutture a
monte cioè l’ atrio destro e il circolo venoso sistemico.
La sintomatologia è essenzialmente rappresentata da dispnea da sforzo e facile affaticabilità.
Spesso il paziente può essere cianotico per uno shunt destra-sinistra attraverso il difetto interatriale, che si
instaura per l’elevata pressione nell’atrio destro.
Diagnostica strumentale
Elettrocardiogramma: mostra onde P di tipo polmonare. Spesso si rilevano aritmie sopraventricolari per la
dilatazione atriale .
Radiografia del torace: si riscontrano l’atrio destro dilatato e la ridotta vascolarizzazione polmonare.
Ecocardiogramma: conferma la diagnosi potendo visualizzare direttamente la sede d’impianto anomalo
della tricuspide.
La malattia di Ebstein senza difetto interatriale ha generalmente un decorso benigno,
per cui non è necessaria una terapia chirurgica.
Atresia della tricuspide.
Si tratta d una grave cardiopatia congenita, in cui l’orifizio della tricuspide rimane occluso, e
il ventricolo destro è solitamente rudimentale.
Affinché sia compatibile con la vita è necessario che sussista una comunicazione interatriale o
interventricolare, e la pervietà del dotto di Botallo. Pressoché la totalità del flusso cardiaco è indirizzato a
livello sistemico, e la perfusione polmonare è dipendente dalla pervietà del dotto di Botallo.
La diagnosi è in genere ecocardiografica.
La storia naturale ha una prognosi severa con mortalità del 70% entro i primi sei mesi di vita per i pazienti
non trattati chirurgicamente.
La terapia medica punta a mantenere pervio il dotto di Botallo mediante la somministrazione di
prostaglandine.
La terapia chirurgica prevede tre gradi di correzione. Il primo è la derivazione succlavio-polmonare di
Blalock-Taussing, il secondo è la correzione secondo Glenn. La correzione definitiva è denominata di
Fontan.
20
Appunti di Cardiochirurgia Capitolo 4 Cardiopatie Congenite
pagina- 21 -
CARDIOPATIE CIANOGENE CON FLUSSO POLMONARE AUMENTATO
Trasposizione dei grossi vasi
Nella trasposizione dei grossi vasi si verifica che l’aorta prende origine dal ventricolo destro e l’arteria
polmonare dal ventricolo sinistro. Tale situazione è compatibile con la vita alla nascita, se sussistono un
difetto interatriale o interventricolare, e la pervietà del forame ovale.
La sopravvivenza è dipendente da un precoce intervento chirurgico di correzione del difetto.
Storicamente ricordiamo l’ inversione del ritorno atriale secondo Senning o Mustard.
Attualmente la tecnica utilizzata consiste nella deconnessione dei grossi vasi dai ventricoli, e il loro
reimpianto nella posizione corretta con successivo reimpianto degli osti coronarici.
Ventricolo destro a doppia uscita
Consiste nell’emergenza dal ventricolo destro sia dell’arteria polmonare che dell’aorta, è di solito associato
a un difetto interventricolare subaortico o subpolmonare.
La correzione consiste nel posizionare un patch, che oltre a chiudere il difetto, ripristini il flusso di sangue
del ventricolo sinistro verso l’infundibolo sinistro e la valvola aortica.
Altrimenti si chiude il difetto e si esegue una inversione delle grandi arterie come nella trasposizione dei
grossi vasi.
21