CHIMICA ORGANICA III
(Modulo A)
LM CHIMICA
A.A. 2008-09
L’univers est dissymmetrique
Louis Pasteur, 1860
1
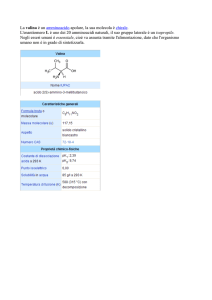
La natura ha una destra ed una sinistra ed è in grado di distinguerle.
O
O
R-(+)-limonene
S-(-)-limonene
(odore di arance)
(odore di limone)
S-(+)-carvone
R-(-)-carvone
(odore di menta)
(odore di cumino)
Anche i batteri sono capaci di distinguere la destra dalla sinistra.
Br
Br
Pseudomonas putida
OH
OH
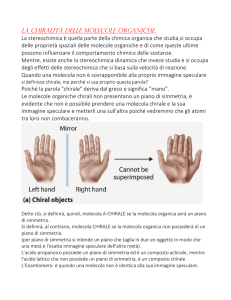
Gli enantiomeri sono chimicamente identici:
come può il nostro naso distinguerli?
come possono i batteri produrli selettivamente?
2
1
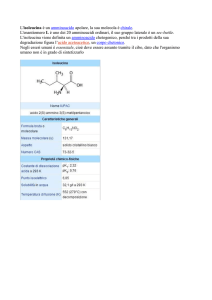
Gli enantiomeri sono chimicamente identici
FINO A CHE NON SONO POSTI IN UN INTORNO CHIRALE
R
CO2H
R
NH2
CO2H
NH2
(S)-α-amminoacido
(R)-α-amminoacido
Alcuni batteri costruiscono la loro parete cellulare con amminoacidi R, in modo da
renderle resistenti agli enzimi usati dagli esseri superiori per idrolizzare i peptidi
Il problema della sintesi asimmetrica diventa vitale quando si tratta di farmaci.
HO
HO
NH2
CO2H
HO
NH2
CO2H
HO
D-dopa
L-dopa
3-(3,4-diidrossifenil)alanina
commercializzato come enantiomero puro
tossico
HN
3
HN
F3C
F3C
fenflurammina racemica
ha effetti collaterali indesiderati
dexfenflurammina
farmaco antifame
Il problema della sintesi asimmetrica non riguarda solo i farmaci.
Z
R O
O
feromone del coleottero giapponese
Popilia Japonica
isomero Z: bastano 25 μg per catturare migliaia di coleotteri
isomero E: solo 10% attività
enantiomero S: inefficace nell’attrazione dei coleotteri
inibitore dell’R (basta 1% S per distruggere
l’attività del feromone)
Negli ultimi 20 anni il problema del controllo della stereochimica assoluta ha
impegnato i chimici organici
4
2
E’ ora possibile (e, con le nuove leggi, necessario) preparare:
farmaci enantiomericamente puri
sostanze “naturali” in modo meno costoso rispetto all’estrazione
SELETTIVITA’ NELLA SINTESI ORGANICA
La SELETTIVITA’ si può definire come la discriminazione osservata in
una reazione
-che comporta attacco competitivo su due o più substrati
-che comporta attacco competitivo su due o più posizioni,
gruppi o facce nello stesso substrato.
Si possono identificare diversi tipi di selettività e, di conseguenza, diversi
livelli di controllo sull’esito delle reazioni organiche
Per prima cosa, consideriamo due situazioni in cui, in alternativa, ha origine la selettività
SELETTIVITA' DI SUBSTRATO
SELETTIVITA' DI PRODOTTO
5
1.
Reazioni che discriminano tra substrati diversi
Si ha selettività di substrato quando un
reagente trasforma due diversi substrati, A
e B, nelle stesse condizioni, nei prodotti C
e D con velocità diverse.
A
B
reagente
k1
reagente
k2
C
D
k1 =
/ k2
I due substrati possono essere isomeri strutturali:
1 equiv H2
+
+
catalizzatore
OH
OH +
HCl conc. freddo
OH +
Cl
I due substrati possono essere diastereomeri:
OH
CrO3
k1
O
CrO3
OH
k2
k1 > k2
6
3
Br
I
-
k1
Br
Br
I
k1 > k2
-
k2
Br
La discriminazione tra enantiomeri porta alla risoluzione cinetica:
NH2
HN
HN
papaina
Ar
CO2H
+
H
N
Ar
HN
Ar
O
OH
O
racemico
OH
OH
t-BuOOH
N
Ti(iPrO)4
(-)-DIPT
N
+
37%
95% ee
59%
63% ee
racemico
2.
OH
N+
O-
7
Reazioni che discriminano tra siti diversi nello stesso substrato
Si ha selettività di prodotto quando in una reazione, in cui si possono formare più
prodotti, questi si formano in rapporto diverso da quello statistico.
A
reagente
[B] =/ [C] =/ [D] .....
B + C + D +.....
La discriminazione tra le diverse posizioni all’interno di una molecola può portare alla
formazione preferenziale di uno o più isomeri
REGIOSELETTIVITA'
CH3
H2SO4, SO3
CH3
35°C
32%
SO3H
+
CH3
CH3
+
SO3H
6%
SO3H
62%
8
4
In certe situazioni la discriminazione tra diversI gruppi o facce porta alla formazione
preferenziale di un enantiomero
ENANTIOSELETTIVITA'
HO2C
OH
H 2O
CO2H
HO2C
fumarasi
CO2H
OMe
O
C
Me2N
H
OH
NMe2
OMe
+ BuLi
40% ee
DEFINIZIONI DI SELETTIVITA’
La CHEMIOSELETTIVITA’ è la reazione preferenziale di un gruppo funzionale
rispetto ad un altro, nelle condizioni di reazione usate.
O
NaBH4
CO2Et
CO2Et
OH
CO2Et
9
CO2Et
H2
Pd-C
Siamo abituati a dare per scontata la chemioselettività. Però il nostro controllo della
chemioselettività è imperfetto, come dimostrato dall’uso esteso dei gruppi protettori
per esempio, non è possibile ridurre direttamente l’estere in presenza del chetone
O
HO
CO2Et
H2O,
H+
O
OH
O
O
1. LiAlH4
CO2Et 2. H2O
H+
O
O
CH2OH
CH2OH
La REGIOSELETTIVITA’ è la reazione preferenziale su uno (o più) dei possibili
siti in una molecola, con conseguente formazione preferenziale di uno (o più)
isomeri strutturali.
può dipendere dalle condizioni di reazione (meccanismi diversi)
Br
HBr, H2O2
HBr, H2O
Br
10
5
più spesso i regioisomeri si formano con lo stesso meccanismo
R'
R'
+
Diels-Alder
R
R
β-eliminazione
+
base
R
R
+
R
Br
R'
R
R
R
R
R'
R
trasposizione pinacolica
H+
R'
R
OH
R'
HO
O
O
SN2 su ossaciclopropani
R'
R
-O
Nu-
+
O
R'
O-
Nu
R'
+
Nu
R
R
R'
R
R'
R
R
R'
R
in questi casi il controllo della regiochimica è più difficile, anche se si può provare cambiando le
condizioni di reazione
11
Il grado di selettività può variare in seguito a variazioni strutturali
O
O
H
O
O
S
H
Ar
H
+
CH2OH
O
H
H
O
PhO2S
51
86
88
O
Ar
SO2Ph
Ar
Ar = fenile
Ar = 3,4-dimetossifenile
Ar = 3,4-metilenediossifenil
H
+
:
:
:
49
14
12
Il controllo della regiochimica più gruppi funzionali identici ma distinguibili va fatto
con l’aiuto dei gruppi protettori.
1. MeOH
HO
O
O
2.
OH
OH
3. LiAlH4
4. H2O,
OH
, H+ HO
O
HO
Br
3. H2O, H+
, H+
HO
HO
O
O
H+
OH
+
O
separato per cristallizzazione
1. NaH, DMF
2.
O , H+
O
1. MeSO2Cl, py
2. base (Triton B)
O
OH
10%
O
O
12
63%
6
La STEREOSELETTIVITA’ è la formazione preferenziale di uno (o più) prodotti, che
differiscono solo per la configurazione. Si può a sua volta suddividere in
enantioselettività e diastereoselettività
Si ha ENANTIOSELETTIVITA’ quando i prodotti stereomerici che si possono formare
sono enantiomeri
Alpine-borano
O
HO H
HO3C
H
Ar
N..
90% ee
H
+
Ar
O
N..
66% ee
HO3C
Si ha DIASTEREOSELETTIVITA’ quando i prodotti stereomerici che si possono formare
sono diastereomeri.
La diastereoselettività può essere di due tipi, che si indicano con “diastereoselettività
semplice” e “diastereoselettività assoluta”.
Si può avere diastereoselettività semplice in una reazione in cui si formano due o
più nuovi centri stereogenici (anche con substrato achirale e reagente achirale)
CH3
CH3
H2
H
niente meso
H
Ni
H3C
13
CH3
racemico
R
OH
OLi
O
H
+
O
niente anti
R'
R
R'
CH3
Si può avere diastereoselettività assoluta nella reazione di un substrato
chirale con un reagente achirale (Se il substrato è non racemico, anche il
prodotto può essere non racemico).
CH3
1. BH3/THF
HO
H2
HO
2. H2O2, HO-
CH3
Ni
H
α-pinene
OH H
E’ possibile che una reazione comporti sia enantioselettività che diastereoselettività
O
CH2OH
H
t-BuOOH
Ti(i-PrO)4
(+)-DIPT
OH
OH
t-BuOOH
Ti(i-PrO)4
(+)-DIPT
H
CH2OH
O
14
7
L’enantioselettività viene espressa come eccesso enantiomerico
e.e. =
frazione molare R - frazione molare S
frazione molare R + frazione molare S
x 100 =
[α]oss
[α]max
x 100
La diastereoselettività viene espressa come eccesso diastereomerico
d.e. =
frazione molare D1 - frazione molare D2
frazione molare D1 + frazione molare D2
x 100
Una decina di anni fa c’è stata una discussione
sull’opportunità di abbandonare l’uso di e.e.,
Kagan, 1996
“Is there a preferred expression for the composition of a mixture of
enantiomers? The use of enantiomeric ratio should be encouraged”
Il rapporto enantiomerico è stato espresso sia come numero: q
(rapporto relativo, cioè con denominatore 1), sia come rapporto
normalizzato a percentuale, detto composizione enantiomerica, e.c.
15
composizione enantiomerica =
R
(R+S)
Usare q può avere degli svantaggi. Per esempio, enantiomeri o diastereomeri
potrebbero formarsi in rapporti che variano da 20:80 a 80:20. Se non normalizzati,
i q sarebbero, rispettivamente 0.25 e 4.0.
Se q = R/S e
R≥S
1≤q≤∞
ma se
R≤S
0≤q≤1
L’intervallo per la formazione selettiva dell’enantiomero R va da 1 a ∞, mentre la
selettività per l’enantiomero S va da 0 a 1.
In questo caso è preferibile esprimere la selettività come una percentuale che
abbia come riferimento 50:50
Il modo più conveniente è esprimere il rapporto degli enantiomeri (e.r.)
come percentuale o come frazione molare
L’eccesso enantiomerico è stato introdotto perché la polarimetria era praticamente il solo modo
per determinare la composizione enantiomerica. La sua utilità è svanita con lo sviluppo delle
tecniche spettroscopiche e cromatografiche come medoti proncipali di determinazione degli
enantiomeri.
16
Oggi i termini e.e. e d.e. non sono appropriati per la descrizione della stereoselettività.
8
La stereoselettività di una reazione si riflette sul rapporto dei prodotti.
In condizioni di controllo cinetico, il rapporto dei prodotti è determinato dalle velocità
relative. In condizioni di controllo termodinamico, è determinato dalle costanti di
equilibrio.
Il rapporto dei prodotti (e.r. o d.r.) è il descrittore migliore della
stereoselettività, perché riflette direttamente le costanti relative
di velocità o di equilibrio.
STEREOSPECIFICITA’
è un termine riservato al caso in cui reagenti
stereodifferenziati danno prodotti stereodifferenziati
e/o hanno reattività diversa.
La reazione SN2 è stereospecifica
A
Nu
H
X
B
A
Nu
B
A
H
H
A
Nu
X
B
B
H
Nu
17
SELETTIVITA’ STEREOTOPICA E STEREOFACCIALE
Una reazione comporta “selettività facciale” quando una delle facce
di una molecola è attaccata in modo preferenziale.
Gruppi o atomi che si possono interscambiare per rotazione attorno ad un
asse di simmetria si dicono omotopici.
Le due facce di un doppio legame sono omotopiche se il piano che le divide
contiene un asse di simmetria.
La trasformazione di un gruppo omotopico o dell’altro porta allo stesso prodotto, così
come l’addizione a ciascuna delle due facce omotopiche di un doppio legame:
O
Br2
O
NaBH4
O
Br
HO H
Due gruppi sono enantiotopici se sono fra loro in relazione mediante un
piano o un centro di simmetria.
Due facce sono enantiotopiche se il piano che le divide è un piano di
simmetria che non contiene un asse di simmetria coplanare.
18
9
Si dice che reazioni che danno attacco preferenziale su uno dei due
gruppi enantiotopici o una delle due facce enantiotopiche hanno,
rispettivamente, selettività enantiotopica o selettività enantiofacciale.
Reazioni con questo tipo di selettività si possono avere usando reagenti chirali o
catalizzatori chirali
HO
CH2OH 1 equiv EtCO H
2
H
+
H
CH2OH
HO
HO
CH2OH
H
CH2OCOEt
H
H
CO3H
H
CH2OCOEt
H
+
CH2OH
O
O
+
Due gruppi che non si possono interscambiare con nessuna operazione di
simmetria si dicono diastereotopici.
Due facce sono diastereotopiche se il piano che le divide non è un piano
di simmetria e non contiene un asse di simmetria.
Le trasformazioni di gruppi diastereotopici o l’addizione a facce
diastereotopiche danno diastereomeri.
Si dice che le reazioni che comportano attacco preferenziale di un reagente
ad uno dei due gruppi diastereotopici o ad una delle due facce
diastereotopiche hanno, rispettivamente, selettività diastereotopica o
19
selettività diastereofacciale.
H3C
CO2H
CO2H
H3C
H3C
Δ
H3C
CO2
O
NaBH4
H3C
H3C
H3C
CO2H
+
H
H3C
OH
H +
H
CO2H
H
OH
H3C
La relazione stereochimica fra I vari termini si può riassumere con lo schema:
A
B
A
B
X
X
A
B*
Y
A
B*
X
gruppi
enantiotopici
HZ
A
B
A
B
A
B
HZ
facce
enantiotopiche
A
B
Y
X
X
X
A
B*
gruppi
diastereotopici
H
Z
Z
H
HZ
X
Y
Y
X
A
B*
A
B*
facce
diastereotopiche
HZ
A
B*
H
Z
Z
H
20
10
PROCHIRALITA’: ENANTIOFACCE
Un doppio legame prochirale è quello che in una reazione di addizione dà un prodotto
in cui si formano uno o due centri chirali.
In una reazione con un doppio legame prochirale, un reagente
achirale non è in grado di distinguere tra enantiofacce e perciò
il prodotto è sempre racemico.
Le enantiofacce si identificano con le regole di Cahn-Ingold-Prelog di assegnazione
della configurazione assoluta: ai sostituenti si assegna la priorità nel solito modo.
senso orario
faccia re
senso antiorario
faccia si
re
3
H
Et
O1
2
si
esempi:
H
Et
1. MeMgBr
O
Me
2. H2O
N
OH
H
Et
Me
OH
H
Me
R 1. LiAlH
4
Me
H
+ Et
NHR
2. H2O
Me
+
NHR
H
21
L’addizione al legame doppio C=N dà un prodotto di addizione che di solito
non ha una configurazione assegnabile all’N amminico piramidale (inversione
di configurazione rapida a temperatura ambiente).
Solo quando l’inversione all’N è sufficientemente rallentata, si formano anche qui due
centri chirali.
O2N
O
RCO3H
N :
..
H
N
Le enantiofacce si possono avere anche con i dieni, come in:
EtO2C
EtO2C
CO2Et
CO2Et
H
Me
Me
CO2Et
S
Me
H
CO2Et
H
EtO2C
CO2Et
CO2Et
Me
Me
EtO2C
R
CO2Et
H Me
CO2Et
22
11
CENTRI PROCHIRALI (PROSTEREOGENICI): ATOMI O GRUPPI
ENANTIOTOPICI
Due qualsiasi sostituenti identici, legati ad un atomo (generalmente C) sp3 che lega
due altri gruppi diversi sono prochirali (enantiotopici).
Me
OH
S
H
Me
D
Se i due diversi H del metilene
OH
dell’etanolo vengono sostituiti,
pro R
H
H
per esempio con deuterio, si
Me
pro S
formano enantiomeri.
R
OH
D
H
L’identificazione dei singoli atomi o gruppi enantiotopici usa un’estensione
della convenzione di Cahn-Ingold-Prelog.
La configurazione risultante (R o S) definisce i gruppi come, rispettivamente,
pro-R e pro-S
CH2CO2Me
HO
pro S
HO
CH2CO2H
H
CH2CO2H
H
CH2CO2H
MeOH, H2SO4
HO
pro R
H
CH2CO2H
CH2CO2Me
23
Gli atomi (o i gruppi) enantiotopici non sono necessariamente legati allo stesso atomo:
HO2C
H
CO2H
H
meso
HO2C
H
CO2H
D
HO2C
D
CO2H
H
PROCHIRALITA’: DIASTEREOFACCE
Un alchene con un centro stereogenico direttamente legato ha due
facce diastereotopiche.
L’addizione di un reagente achirale ad un enantiomero singolo passa
attraverso stati di transizione diastereomerici.
L’attacco sulle due facce del doppio legame avviene con velocità diverse.
La reazione procede in modo diastereoselettivo.
b
c
b
c
a
[O]
b
c
a
O
a
O
diastereomeri
24
12
a [O]
b
c
ΔG
b
c
a
[O]
ΔΔG =/
=/
/ ΔG (1)
ΔG =
(2)
b
c
b
c
a
+ [O]
b
c
O
(1)
a
(2)
O
coordinata di reazione
In una reazione controllata cineticamente, le velocità di formazione dei prodotti sono legate alle loro
energie di attivazione
ΔΔG =/ = [ΔG(1)=/ - ΔG(2)=/ ] = RT ln (k2/k1)
- ΔG =/ = RT ln k
dove k1 e k2 sono le costanti di velocità legate agli stati di transizione che portano rispettivamente a
(1) e (2).
Affinché l’ossaciclopropano (2) si formi 100 volte più velocemente
dell’(1) a temperatura ambiente (300K):
ΔΔG =/ = 8.314 x 300 x 2.303 log 100 J mol-1
= 11.5 kJ mol-1 (2.74 kcal mol-1)
25
Un rapporto 100:1 è normale per una reazione mediata da enzimi, ma non per una in cui
gli enzimi non siano coinvolti.
Un valore generalmente utile è 95:5. Una miscela in questo rapporto è spesso
utilizzabile senza ulteriore purificazione.
EFFETTO DELLA TEMPERATURA SULLA DIASTEREOSELETTIVITA’
Applicando l’equazione di Arrhenius ed assumendo che A sia uguale per la formazione
di (1) e di (2):
/ /RT
A e-ΔG =(2)
k1 = A e-ΔG=/(1)/RT
k2
=
= e [-ΔG=/(2) + ΔG =/(1)]/RT
/ /RT
-ΔG=
k
A e-ΔG=/ /RT
k2 = A e
(2)
1
(1)
k2/k1 è una misura della diastereoselettività
poiché
ΔG=/(2) < ΔG =/(1)
e [-ΔG=/(2) + ΔG =/(1)]/RT è massima a valori piccoli di T
In generale, la diastereoselettività aumenta con il diminuire
della temperatura a cui viene eseguita la reazione.
26
13
METODI PIU’ COMUNI PER AVERE UN SINGOLO ENANTIOMERO
RISOLUZIONE DELLA MISCELA RACEMICA
1.
Qualunque sbilanciamento nella formazione di enantiomeri deriva, in ultima analisi, dalla
Natura.
Una sintesi di laboratorio, a meno che non comporti substrati o reagenti
enantiomericamente puri, darà sempre una miscela racemica.
Esempio: sintesi del feromone del coleottero giapponese
Li
R
CN
R
CN
O
R
H2, cat
OH
CN
Z
OH
R
1. KOH
2. HCl
O
O
R=
feromone racemico
E’ relativamente facile avere il controllo sulla configurazione del doppio legame, ma non
c’è controllo stereochimico sulla formazione del centro stereogenico.
Se vogliamo solo il feromone R, dobbiamo tentare la risoluzione della miscela
racemica.
La risoluzione è stata effettuata sull’alcool precursore del feromone.
R
CN
27
(S), non
desiderato
O
N
H
O
R
R
CN
CN + O C N
Z
O
OH
O
alcool racemico
N
H
i diastereomeri si separano per cromatografia
R
R
R
1. KOH
O
O 2. HCl
feromone R
enantiomericamente puro
CN
CN
Cl3SiH
OH
alcool R
enantiomericamente puro
O
O
N
H
Per una sintesi industriale non è neppure presa in considerazione la
possibilità di buttare l’altro enantiomero (spese di smaltimento).
28
14
STRATEGIA DELLA “RISERVA CHIRALE”
2.
Un modo più economico di preparare enantiomeri puri è farli da materiali di partenza
enantiomericamente puri.
LA “RISERVA CHIRALE”: I CENTRI CHIRALI “PRONTI PER L’USO” DELLA
NATURA
Si basa sulla possibilità di trovare un un composto naturale enantiomericamente puro,
adatto ad essere trasformato nel prodotto desiderato.
zuccheri, amminoacidi
esempio:
O
O
O
O
N
H
NH2
+
CO2H
HO
NH2
estere metilico
dell'(S)-fenilalanina
aspartame
O
OMe
NH2
CO2H
acido (S)-aspartico
La maggior parte delle sintesi asimmetriche richiede più di uno-due passaggi dai
29
composti della “riserva chirale”.
esempio:
Feromone prodotto dal maschio del coleottero della corteccia del genere
Ips: miscela di prodotti enantiomericamente puri, tra I quali l’(S)-(-)ipsenolo
CO2H
NH2
OH
(S)-(-)-leucina
(S)-(-)-ipsenolo
il gruppo amminico si deve convertire in OH, mantenendo la configurazione
: OH
CO2H
O
HONO, H2O
N H
+
N
NH2
(S)-(-)-leucina
H
O
inversione
..
H2O
seconda
inversione
O
CO2H
OH
30
15
CO2H
CO2H
H+
+
O
OH
O
OH
LiAlH4
O
CO2R
ROH
H+
O
O
O
OTs
TsCl, py
O
1. acido
O
O
2. base
O
BrMg
1.
OH
2. H+
(S)-(-)-ipsenolo
esempio:
Feromone dell’aggregazione del coleottero dell’ambrosia: sulcatolo in miscela 65:35
degli enantiomeri.
il chimico deve preparare entrambi gli enantiomeri
separatamente e mescolarli nella giusta proporzione
OH
OH
(R)-sulcatolo
(S)-sulcatolo
O
31
OH
OH
HO
il feromone naturale li contiene
in miscela 65:35
?
OH
CHO
OH OH
HO
(R)-sulcatolo
2-desossi-D-ribosio
questi OH devono essere rimossi
O
OH MeOH
HO
H+
HO
O
HO
OMe
MsCl
O
MsO
OMe
MsO
HO
Ms = metansolfonile, CH3SO2-
KI
O
I
OMe
Ni Raney
O
OMe
H2O
O
OH
H+
I
Ph3P
OH
CHO
Wittig
OH
(R)-sulcatolo
32
16
L’(S)-sulcatolo non si può preparare con questo metodo, perché lo zucchero L non è
disponibile (e anche il D è piuttosto costoso).
Soluzione ?
Acido (S)-lattico
CO2Et 1. LiAlH4
protezione
di OH
OTs
2. TsCl
OR
CO2Et
OR
O
1. deprotezione
2. base
dall'(S)-lattato di etile si possono ottenere entrambi
gli enantiomeri del metilossaciclopropano
OH
TsCl
(S)-lattato di etile
(S)-2-idrossipropanoato di etile
CO2Et LiAlH
4
OH
OTs
base
O
OTs
OH
(S)
BrMg
O
(S)-sulcatolo
OH
BrMg
O
(R)
33
(R)-sulcatolo
Per sintetizzare molecole con più di un centro chirale, basta prenderne uno solo dalla “riserva chirale”,
purché per introdurre gli altri si possano usare reazioni diastereoselettive.
Poiché il primo centro chirale ha configurazione assoluta definita, qualsiasi reazione diastereoselettiva
che controlli la stereochimica relativa di un nuovo centro chirale ne definisce anche la configurazione
assoluta.
esempio:
Me
Me
OH
HO
OH
NH2
acido (S)-lattico
Me
OH
OMe
metil mycamminoside
CO2H
Me
O
Me
OAc
OMe
COCl
CO2H
CO2H
OAc
OMe
BrMg
acido (S)-lattico
Me
O
OAc
H2
OMe
cat
Me
O
H
OAc
OMe
OMe
Me
O
OMe
O
H
OMe
Questo raro amminozucchero è stato sintetizzato dall’acido (S)-lattico acetilato: la
ciclizzazione introduce il secondo centro chirale in modo selettivo, perché il metile va nella
posizione pseudoequatoriale ed il metossile in quella pseudoassiale (effetto anomerico).
34
17
Il terzo centro chirale è stato controllato dalla riduzione assiale del chetone, che dà
l’alcool equatoriale. Questo poi indirizza il quarto ed il quinto centro stereogenico
mediante epossidazione. Infine il nucleofilo amminico attacca l’ossaciclopropano con
inversione di configurazione.
O
HO
O
HO
H
O
H
OMe
OMe
OH
H 2N
Me
OMe
O
HO
OH
NH2
..
metil mycamminoside
HNMe2
Problemi della strategia della “riserva chirale”
il composto desiderato deve essere strutturalmente abbastanza vicino ad
uno dei composti della “riserva naturale” (una sintesi con troppi passaggi
dà più scarti della risoluzione racemica)
mancata disponibilità di entrambi gli enantiomeri per la maggior parte dei
composti naturali sinteticamente utili (amminoacidi, zuccheri).
Esempio:
HO2C
feromone del coleottero giapponese
NH2
HONO
CO2H
H2O
HO2C
+
N
CO2H
N
acido (S)-glutammico
35
+
H2 O
Ph3P
1. SOCl2
HO2C
O
O
2. H2, Pd
BaSO4
OHC
O
O
-
C8H17
Wittig
C8H17
O
O
enantiomero
sbagliato!
(+ 10-15% di E)
36
18
3.
STRATEGIA DELLA SINTESI ASIMMETRICA
Quando si crea un nuovo centro stereogenico in una molecola non chirale usando
reagenti achirali si ha una miscela racemica.
δ-
O
.
.. . .
. . δR R' Nu
=/
δ-
stati di
transizione
enantiomerici
δ-
O..
.. .
HO
Nu
R
R'
=/
..
Nu R R'
E
O
HO Nu
R
R
R'
R'
Li
O
HO
esempio
37
La sintesi diastereoselettiva si basa sul rendere il più diversi possibile degli stati di
transizione diastereomerici.
Li
OH
Me
Me
N
Me
N
Me
N
Me
Li
O
attacco equatoriale
favorito
OH
Me
N
Me
Me
attacco assiale
sfavorito
O
Possiamo usare il principio alla base della risoluzione per trasformare stati di
transizione enantiomerici in stati di transizione diastereomerici?
Sì, se una molecola (o parte di molecola) enantiomericamente pura è
presente nel corso della reazione ed interagisce con lo stato di
transizione, in modo da controllare la formazione del nuovo centro
stereogenico.
38
19
attacco nucleofilo su un chetone in un ambiente chirale
δ-
δ-
E
O
.
.. . .
. . δR R' Nu
=/
stati di
transizione
diastereomerici
R
R'
R
=/
..
Nu R R'
δ-
O
HO Nu
O..
.. .
R'
HO
Nu
R
R'
composti enantiomerici prodotti in quantità diverse
Questa molecola può essere:
-un reagente
-un catalizzatore
-legata al substrato in modo covalente
39
AUSILIARI CHIRALI
☺ CHE COSA SI INTENDE PER STRATEGIA DELL’AUSILIARIO CHIRALE
⇒ Un composto enantiomericamente puro (di solito derivato di un prodotto
naturale semplice), chiamato ausiliario chirale viene legato al substrato.
⇒ Si esegue una reazione diastereoselettiva che, a causa della purezza
enantiomerica dell’ausiliario chirale, dà un solo enantiomero del prodotto.
⇒ L’ausiliario chirale viene rimosso (per esempio, per idrolisi), lasciando il
prodotto di reazione come enantiomero singolo.
Gli ausiliari chirali migliori si possono riciclare: anche se servono quantità
stechiometriche, non c’è scarto.
Il prodotto della reazione di Diels-Alder tra ciclopentadiene ed acrilato (=propenoato) di
benzile dà solo il diastereomero endo, ma in miscela racemica.
O
OH
+
Cl
Bn = benzile
O
O
+
O
dienofilo
diene
achirale
achirale
OBn
O
OBn
miscela 50:50 dei due enantiomeri
40
20
Se l’estere benzilico (achirale) si sostituisce con un’ammide derivata da un’amminoacido naturale (valina), la diastereoselettività rimane la stessa, ma l’ambiente chirale fa
sì che si formi un solo enantiomero del prodotto.
Cl
O
O
O
O
+ HN
O
base
N
O
O
Et2AlCl
O
N
O
diene achirale
enantiomero singolo
derivato da (S)-valina
enantiomero singolo
del dienofilo
O
OLi
HN
O
+
O
Bn = benzile
OBn
solo questo enantiomero
L’ausiliario chirale è enantiomericamente puro e lo stereocentro non viene coinvolto
nella reazione di Diels-Alder.
Il prodotto è diastereomericamente ED enantiomericamente puro.
L’introduzione dell’ausiliario chirale e la sua successiva rimozione hanno dato
lo stesso prodotto della reazione senza l’ausiliario chirale, ma come
41
enantiomero singolo.
L’ausiliario chirale dell’esempio è uno dei più usati tra I derivati dell’ossazolidinone.
Si forma facilmente ed in modo economico dall’(S)-valina e si può riciclare.
Nell’ultimo passaggio, la transesterificazione con alcool benzilico rigenera
l’ausiliario chirale.
O
NH2
CO2H
Me2S.BH3
O
NH2
OH
EtO
OEt
HN
O
K2CO3
(S)-valina
Dovrebbero essere disponibili entrambi gli enantiomeri degli ausiliari chirali più utili.
la (R)-valina non è naturale e quindi è costosa
H2 N
OH
O
+ EtO
K2CO3
OEt
O
HN
O
norefedrina
ausiliario chirale derivato
dalla norefedrina
Utilizzato nella stessa reazione di Diels-Alder dà l’enantiomero
42
21
O
HN
2.
O
O
1. NaH
O
N
O
O
O
Et2AlCl
O
N
O
Cl
ausiliario chirale
dalla norefedrina
unico diastereomero
O
OLi
HN
O
+
O
ausiliario chirale
recuperato: si può riutilizzare
OBn
solo questo enantiomero
come funziona l’ausiliario chirale?
L’isopropile scherma una faccia del doppio legame del dienofilo coordinato all’acido
di Lewis: quando il ciclopentadiene si avvicina, lo deve fare dalla faccia opposta.
Et
O
O
N
O
Et 2Al Cl
O
Et
Al
N
Et2
.. O Al O
.
.. H
N
O
..
..
..
O
O
faccia
schermata
43
ruolo importante dell’acido di Lewis Et2AlCl nel fissare la conformazione scis del legame singolo del dienofilo
Et
s-cis
O
Et2
O Al O
Et
Al
N
O
HN
O
s-trans
Et2
O Al O
HN
O
O
sfavorito per
ingombro sterico
L’8-fenilmentolo è un ausiliario chirale preparato dal composto naturale pulegone, in
cui il fenile scherma una faccia del dienofilo.
O
O
+
HO
(S)-pulegone
8-fenilmentolo
Cl
base
O
O
dienofilo chirale
Una reazione di Diels-Alder catalizzata da acido di Lewis con un ciclopentadiene
sostituito ma achirale dà un solo enantiomero dell’addotto.
44
22
BnO
+
O
BnO
O
diene achirale
BnO
O
AlCl3
O
CO2R*
dienofilo chirale
R* = ausiliario chirale
Il fenile scherma una faccia del dienofilo ed il diene si deve avvicinare dalla faccia opposta,
dando uno solo dei possibili enantiomeri endo.
Corey ha usato i quattro centri chirali creati nella reazione per ottenere i
centri chirali attorno all’anello del ciclopentanone delle prostaglandine.
Dopo ossidrilazione dell’enolato dell’estere, l’ausiliario chirale è stato rimosso per
riduzione. Il ciclopentanone ottenuto (scissione del diolo con periodato, ossidazione
di Baeyer-Villiger e iodolattonizzazione) è servito a Corey come punto di partenza per
la sintesi di molte prostaglandine.
1. LDA
2. O2, (EtO)3P
3. LiAlH4
BnO
CO2R*
BnO
BnO
BnO
H2O2
NaIO4
(Baeyer-Villiger)
OH
R*OH
O
O
OH
O
45
L’uso degli ausiliari chirali è stato sviluppato soprattutto per derivati chirali di enolati,
con ausiliari chirali facilmente disponibili e facilmente recuperabili.
H X
c
XC = chiral auxiliary
R
OH
X
introduzione
dell’ausiliario
R *
N
R
R
R'
condensazione
X
c reazione
E
diastereoselettiva
H Xc
O
O
O
O
O
R
riciclo dell’ausiliario
R *
c
idrolisi
OH
E
riciclo dell’ausiliario
Xc
R'
reazione
diastereoselettiva
N
R *
E
O
X
c
R'
R *
idrolisi
R'
E
L’ausiliario chirale deve:
- essere facile da introdurre
- predisporre la molecola ad un’enolizzazione altamente selettiva
- indirizzare la costruzione diastereoselettiva del nuovo legame
- essere staccato in condizioni blande, non distruttive, senza racemizzazione del prodotto
46
23
Ausiliari chirali usati con più successo nella sintesi asimmetrica
O
R
N
OH
N
H
Me
Meyers
OMe
O
N
H
Me
OH
Yamada
N
H
R
Me
N
N
MeO
Whitesell
R'
Schöllkopf
O
Me
OMe
N
R
OR
N
OH
O
OR
SONR2
Kunz
Oppolzer
Seebach
OR
O
NH2
OR
47
N
NH2 OMe
Enders
OH
Corey
OH
OH
SO2Ph
N
OH
Hemlechen
Hoffmann
48
24
Gli ausiliari chirali sono stati usati soprattutto nella reazione di Diels-Alder asimmetrica,
nell’alchilazione asimmetrica e nella condensazione aldolica asimmetrica
esempio:
ALCHILAZIONE DI ENOLATI CHIRALI
N Li+
1) R'' X
N SO Ph
2
H
O
THF/HMPT
H R''
HO
R'
2) LiAlH4
R'
LiO
enolato E
N SO Ph
2
OCOCH2R
N Li+
R'
1) R'' X
N SO Ph
2
R'
O
THF
R''
HO
H
2) LiAlH4
H
LiO
enolato Z
La deprotonazione selettiva porta al corrispondente enolato dell’estere E o Z semplicemente cambiando
il solvente e questo porta ai due diversi diastereomeri a partire dallo stesso ausiliario chirale
49
Tra i più usati
O
O
O
N
O
S
S
O
N
O
ossazolidinoni chirali
R
Evans
(1981)
O
N
R'
O
R'
R
R
Fujiata/Nagao (1985)
Crimmins (1997)
N
R'
Me N
N
Me
O
O
N
O
N
O
O
R'
S
Helmchen (1984)
O
O
O
N
R'
Yan (1991)
R'
N
O
N
O
Oppolzer (1983)
O
N
R'
R'
OR
OR
Gosh (1998)
R'
Me
Me
R
O
O
O
O
Davies (1995)
Seebach (1998)
Sibi (1995)
O
O
O
Davies (1991)
OR
Kunz (1992)
50
25
Primi impieghi degli ossazolidinoni di Evans come ausiliari chirali
. Li
.. ...
O
O
O
O
O
N
O
O
R
O
N
R''
R'' X
R
O
N
Alchilazione asimmetrica
(1982)
R
O BBu2
O
O
N
O
O
R
O
OH
N
R'' CHO
“Evans syn”
R''
R
Condensazione aldolica
asimmetrica syn (1982)
O
O
X
X
.M
.. ...
O
O
O
N
R
O
N
O
R''
O
O
R
N
R''
R
Diels-Alder asimmetrica
(1984)
51
ALCHILAZIONE DI ENOLATI CHIRALI
esempio:
Una delle reazioni in cui gli ausiliari chirali (soprattutto gli ossazolidinoni di Evans,
che si possono facilmente trasformare in derivati enolizzabili di acidi carbossilici)
sono stati più applicati è l’alchilazione degli enolati.
+
Cl
HN
O
O
O
O
N
O
O
LDA
O
Li..
.
O
N
O
Il trattamento con una base (LDA) a bassa temperatura produce un enolato, che
può essere attaccato solo da una faccia. Inoltre, l’ausiliario voluminoso permette la
formazione solo del’enolato Z. Infine, la struttura è resa rigida dalla chelazione del
litio.
O
Li..
.
O
N
I
O
O
Li..
.
O
N
elettrofilo
O
rapporto
diastereomeri
PhCH2I
> 99:1
bromuro di allile 98:2
EtI
94:6
52
26
E+
l'elettrofilo attacca da sopra
..Li. .
O
O
H N
O
la faccia inferiore è schermata dall'isopropile
Come si può vedere, la reazione è diastereoselettiva al 100%. Il problema è che, quando
si rimuove l’ausiliario chirale, il prodotto finale può essere contaminato da un po’
dell’altro enantiomero.
ECCESSO ENANTIOMERICO
OLi
O
O
N
O
O
O
O
miscela 98:2 di
diastereomeri
+
HN
O
miscela 98:2 di enantiomeri
96% e.e.
53
Il modo più semplice di determinare l’eccesso enantiomerico è misurare l’angolo di rotazione del
piano della luce polarizzata. Però non sempre si conosce il potere rotatorio dell’enantiomero puro.
Le misure al polarimetro dipendono
dalla temperatura,
dal solvente,
dalla concentrazione
e possono essere affette da errori grossi se ci sono piccole quantità di impurezze
con elevata attività ottica.
HPLC: usando una fase stazionaria chirale. Gli enantiomeri si separano e si
determinano quantitativamente (uv o indice di rifrazione).
GC: si usano colonne impaccate con
una fase stazionaria chirale come
questo derivato dell’isoleucina
O
CF3
N
H
O
O
NMR: per separare gli enantiomeri spettroscopicamente bisogna metterli in un
intorno chirale, per esempio legandoli ad un reagente enantiomericamente puro.
Uno dei più usati è quello noto come alogenuro acilico di Mosher, che permette di
determinare l’eccesso enantiomerico dall’integrazione dei segnali sia nello spettro
1H NMR, sia in quello 19F.
54
27
OH
O
OH
R
+
R
MeO
O
base
Cl
F3C
MeO
F3C
miscela di enantiomeri
Ph
R
O
O +
MeO
F3C
O
Ph
R
miscela diastereomerica degli esteri di Mosher
Un altro sistema per discriminare tra gli enantiomeri è aggiungere nel tubo NMR un
composto enantiomericamente puro, che complessi il campione in esame: i complessi
sono diastereomerici e perciò hanno chemical shifts diversi e si possono integrare.
reagenti di shift chirali (sali di lantanidi)
F3C
H
OH
(S)-(+)-TFAE
2,2,2-trifluoro-1-(9-antril)etanolo
può formare legame
idrogeno e dare π-stacking
(1H e 19F NMR)
55
COME MIGLIORARE L’e.e.
Se con l’ausiliario chirale si ha ancora l’1-2% dell’altro diastereoisomero, si può ricorrere
alla cristallizzazione, anche se si perde qualche % di prodotto.
esempio
Durante la sintesi dell’antibiotico complesso X206, Evans aveva bisogno di grandi quantità di
questa semplice molecola.
N
O
O
O
O
O
N
1. NaN(SiMe3)2
2.
O
OSiMe2t-Bu
1. LiAlH4
2. t-BuMe2SiCl
OSiMe2t-Bu
I
> 99% ee
diastereomeri 98:2
frammento di X-206
cristallizzazione
diastereomeri >99:1
56
28
Tornando all’addizione di Diels-Alder discussa in precedenza, i diastereomeri
si formano in rapporto 93:7, ma basta una ricristallizzazione per avere 81% di
prodotto diastereomericamente puro > 99%.
O
O
N
O
O
Et2AlCl
O
N
O
O
O
N
O
prodotto principale
si forma anche 7% di questo addotto
☺
Uno dei grandi vantaggi degli ausiliari chirali è di rendere più facile anche
la purificazione finale.
Ci sono anche degli svantaggi:
-Devono essere legati durante la costruzione della molecola e devono
essere rimossi alla fine della sintesi. I migliori ausiliari chirali si possono
riciclare, ma nella sintesi ci sono almeno due passaggi “improduttivi”.
- Scoprire un ausiliario chirale che funzioni richiede laboriose ricerche.
spesso ausiliari chirali potenzialmente promettenti danno in realtà bassi
57
ee.
REAGENTI CHIRALI E CATALIZZATORI CHIRALI
Una delle reazioni più semplici per trasformare un’unità prochirale in una chirale è la
riduzione di un chetone.
NaBH4
OH
O
o LiAlH4
R
R
chirale ma
racemico
prochirale
E’ stata usata la strategia dell’ausiliario chirale, ma è concettualmente più semplice
tentare di ottenere un singolo enantiomero usando un reagente chirale: in altre parole,
legare “l’influenza” chirale al reagente e non al substrato.
Uno dei primi tentativi di realizzare
questa strategia è stato di legare un
alcool chirale (alcool del Darvon: il
suo estere è il medicinale Darvon) al
riducente LiAlH4.
Sfortunatamente, questo reagente
non ha dato grandi risultati:
funziona discretamente solo per
avere alcooli propargilici.
OH NMe2
LiAlH4
Ph Ph
Ph Ph
alcool del Darvon
O
H - H
Al +
O NMe2
agente riducente chirale
agente riducente chirale
OH
R
R
R'
R'
70-80%ee
58
29
Più efficace è il “reagente CBS”, derivato chirale del boroidruro sviluppato da
Corey, Bakshi e Shibita, che si basa su un eterociclo stabile del B ottenuto da un
alcool derivato dalla prolina.
O
O
Cl
H
H
N
H
CO2H
N
CO2H
CO2Bn
NaOH, H2O
(S)-(-)-prolina
H
1. HCl
2. NaOH
N
H
H
MeOH, H+
N
CO2Bn
2. PhMgCl
OH
H
MeB(OH)2
N
B O
OH
Me
reagente CBS
L’agente riducente attivo si fa complessando l’eterociclo con il borano.
H
BH3
N
B O
Me
H
+
- N
H3B
B O
Me
agente riducente attivo
catalizzatore
O
59
OH
10% catalizzatore
BH3
chetone prochirale
resa 99%, 97% ee
Bastano quantità catalitiche del boro-eterociclo, perché il borano è in grado di ridurre
I chetoni solo se è coordinato all’N.
Le riduzioni con CBS funzionano meglio quando i due gruppi legati al CO sono
stericamente differenziati.
Solo quando l’O del CO è complessato con il B dell’eterociclo il C è
abbastanza elettrofilo da venire attaccato dalla debole fonte di idruro.
+
H
O
Me B N+
H
- N
H 3B
B O
Me
O
sostituente
più grande
O
RL R
S
H
RL R
S
sostituente
più piccolo
BH2
H
O
R
L
- N+
O
B
H
B H
Me
RS
H
HO H
RL R
S
il gruppo grande
pseudoequatoriale
L’idruro viene trasferito in uno stato di transizione ciclico a sei termini, dove il 60
gruppo più voluminoso preferisce disporsi in posizione pseudoequatoriale.
30
Il CBS è uno dei migliori agenti di riduzione asimmetrica ideato dai chimici.
La Natura effettua riduzioni in continuazione, ogni volta con 100%
ee, usando gli enzimi.
L’uso di enzimi come reagenti chimici ha il problema che gli enzimi di solito
sono “substrato-specifici”.
Si usano sistemi multienzimatici: le cellule viventi.
Particolarmente efficace per ridurre i chetoni è il lievito, soprattutto quanto si tratta di
β-chetoesteri. La reazione si effettua agitando il chetone con una sospensione
acquosa di lievito vivo, che perciò deve essere alimentato con zucchero.
O
lievito di birra
CO2Et
OH
resa 55%
fino a 97% ee
CO2Et
glucosio
Per quanto riguarda i sostituenti grande e piccolo del chetone, la selettività del lievito di birra è
opposta a quella del reagente CBS.
Un’importante applicazione della riduzione con lievito di birra è nella sintesi del
citronellolo.
OH
CO2Et
1. TsCl
2. LiAlH4
3. NaH,
1.
OTs
Br
OBn
CuLn
2. Na, NH3
OH
sostituzione
con inversione
citronellolo
88% ee
Dopo riduzione dell’estere, protezione e sostituzione nucleofila ad opera dell’opportuno
cuprato, si ottiene citronellolo con ee migliore di quello naturale, la cui purezza
61
enantiomerica varia notevolmente a seconda della pianta da cui è stato estratto.
Il modo più studiato di effettuare una riduzione enantioselettiva è l’idrogenazione in
presenza di un catalizzatore chirale.
L’idrogenazione catalitica di un carbonile non dà grandi risultati.
Migliori sono le enantio-selettività nell’idrogenazione catalitica di doppi legami C=C,
soprattutto quelli con nelle vicinanze eteroatomi in grado di coordinare il metallo del
catalizzatore (OH, NHR).
sintesi del farmaco analgesico Naprossene
esempio:
CO2H
H2
[(S)-BINAP]Ru(OAc)2
MeO
H
H
CO2H
MeO
(S)-naprossene
Il principio è semplice: il catalizzatore sceglie una delle facce enantiotopiche
del doppio legame e a quella addiziona idrogeno.
Il catalizzatore contiene un metallo (Ru) ed un legante.
PPh2
PPh2
(R)-BINAP
PPh2
PPh2
(S)-BINAP
62
31
Come molti altri leganti per l’idrogenazione asimmetrica, BINAP è una difosfina
chelante: il metallo sta tra i due atomi di fosforo, in un intorno chirale.
Il BINAP ha “chiralità assiale” per l’impossibilità di rotazione intorno al legame C-C
che unisce le due unità naftaleniche.
H
Ph2P H
PPh2
PPh2
H
H
PPh2
Il BINAP si sintetizza in laboratorio e si risolve.
HO2C
O
1.
PPh2
1. Mg
Br 2. Ph2POCl
Br
PPh2
O
dibromuro racemico
OCOPh
CO2H
OCOPh
PPh2
PPh2
2. cristallizzazione
3. base
4. riduzione (HSiCl3)
(S)-BINAP
bis-fosfinossido racemico
Questo processo rende il BINAP
piuttosto caro, ma ne basta molto
poco:
CBS
BINAP
10% mol
0.0002% mol
uso industriale
63
Composti derivati dal binaftile, per esempio i BINOLs sono oggi disponibili
commercialmente, per essere usati come leganti chirali
esempio:
O
O
OH
OH
(R)-BINOL; (S)-BINOL
(R); (S)
O
O
O
O
O
S
S
O
O
O
(R); (S)
(R); (S)
O
O
CF3
S
O
O
O
O
S
CF3
O
O
O
Br
Br
O
O
(R); (S)
(R); (S)
OH
OH
OH
OH
OH
OH
Br
(R); (S)
O
Br
(R); (S)
Br
Br
O
O
O
O
Br
Br
(R); (S)
(R); (S)
O - K+
O - K+
OH
OH
OH
OH
(R); (S)
(R); (S)
64
J. M. Brunel, Chem. Rev. (2005), 105, 857
32
BINAP-Ru(II) funziona particolarmente bene nell’idrogenazione di alcooli allilici e di
acidi carbossilici α,β-insaturi.
H2
OH
H
OH
[(S)-BINAP]Ru(OAc)2
(R)-citronellolo
geraniolo
R'
R
H
R"
H
R'
H2
CO2H
[(R)-BINAP]Ru(OAc)2 R
R"
H2
CO2H [(S)-BINAP]Ru(OAc)2
H
OAc
H2
CO2H
NHAc
[DIPAMP]RhL2+
(L = solvente)
MeO
H H
OAc
H
R
H
R"
CO2H
OMe
Se il doppio legame porta anche un gruppo amminico, si
formano amminoacidi.
In questo caso è meglio un catalizzatore di Rh.
MeO
R'
P. P
. ..
MeO
CO2H
(R,R)-DIPAMP
H NHAc
95% ee
CO2H
HO
NH2
HO
65
L-dopa
Le idrogenazioni catalizzate da Rh sono di enorme importanza industriale per la
domanda di amminoacidi (naturali e non).
CO2H
NHAc
CO2H
H2
[PNNP]RhL2+
NHAc
N-acetil-L-fenilalanina
83% ee, che sale a 97%
per ricristallizzazione
N
N
PPh2 PPh2
DNNP
Il processo industriale usa la difosfina DNNP. Il prodotto inizialmente ottenuto ha
ee 83%, che sale a 97% dopo ricristallizzazione.
Nella manifattura dell’aspartame, l’accoppiamento con l’acido aspartico naturale
(100% ee) trasforma l’1.5% dell’enantiomero minore in un’impurezza
diastereomerica rimuovibile per cristallizzazione.
La ricristallizzazione di campioni di circa 85% ee ha buone probabilità
di migliorare l’ee. Campioni con ee molto minori tendono a diminuire
l’ee per ricristallizzazione. Molto dipende dalla struttura del cristallo.
La difficoltà di aumentare bassi ee per ricristallizzazione è uno degli
svantaggi della tecnica dei reagenti chirali.
66
33
Un altro processo collegato alla riduzione ha acquistato notevole importanza,
per la sua applicazione industriale.
La compagnia giapponese Takasago prepara circa il 30% delle 3500 tonnellate/anno
di L-mentolo dal citronellale.
Me
Me
Me
CHO
Me
ZnCl2
=
H2, cat.
OH
O
H
OH
L-mentolo
(R)-citronellale
nella reazione di trasposizione intramolecolare il metile nello stato di transizione ciclico
preferisce essere equatoriale ed indirizza la formazione dei due nuovi centri chirali. La
reazione è accelerata dall’acido di Lewis con l’ossigeno.
Me
ZnCl2
Me
+
O
H
O
H
Me
Me
ZnCl2
OH =
OH
67
Quello che rende notevole la sintesi, però, è un altro passaggio.
Il pinene (un terpene prodotto con e.e. bassi dagli alberi di pino) è usato come substrato
economico, enantiomericamente impuro, per formare mircene (terpene achirale), da cui si
ottiene un’ammina allilica.
Li
NEt2
Et2NLi
β-pinene
H2O
NEt2
mircene
Nel passaggio chiave, il BINAP di Rh catalizza la trasposizione dell’ammina allilica ad
enammina, creando un nuovo centro chirale con 98% ee.
H H
H H
CHO
NEt2 Rh[(S)-BINAP] +
H2O
2
NEt2
1 kg
7 tonnellate
98% ee
(R)-citronellale
La reazione richiede solo 0.01 % mol
Come funzioni esattamente questa reazione e che cosa renda di successo il
catalizzatore non è chiaro: non siamo in grado di dire come la chiralità del 68
legante indirizzi la formazione del nuovo centro stereogenico.
34
Rh richiede
carbonile (base
di Lewis) in β al
doppio legame
O
R
R'
N
H
gruppo in grado di coniugare
o ad attrazione elettronica
Ru richiede
OH in α al
doppio legame
R
HO
R
R'
R"
HO
R'
R"
O
69
RIEPILOGO DEI METODI DI SINTESI ASIMMETRICA
Metodo
risoluzione
chiral pool
Vantaggi ☺
entrambi gli
enantiomeri
disponibili
100% ee garantito
ausiliario chirale spesso ee eccellenti,
migliorabili per
cristallizzazione
reagente chirale spesso ee eccellenti,
migliorabili per
cristallizzazione
catalizzatore
economico: usate
chirale
solo piccole quantità
di materiale riciclabile
Svantaggi
massima resa 50%
Esempi
sintesi di BINAP
spesso disponibile un
solo enantiomero
sintesi derivate
da amminoacidi
e zuccheri
ossazolidinoni
passaggi extra per
introdurre e rimuovere
l’ausiliario
solo pochi reagenti hanno
successo e spesso per
pochi substrati
solo poche reazioni sono
veramente di successo; la
ricristallizzazione può
aumentare ee solo già alti
enzimi, agente
riducente CBS
idrogenazione
asimmetrica,
epossidazione,
diossidrilazione
70
35
MEMORIA DI CHIRALITA’: una strategia emergente per la sintesi
asimmetrica
Il termine “memoria di chiralità” (memory of chirality, MOC), è stato coniato nel 1991,
da Fuji, il primo a lavorare su questo principio.
MOC ha attratto l’attenzione, perché sembra realizzare l’impossibile: un solo
centro chirale viene distrutto in un processo, ma il suo ricordo si mantiene!
Ci sono varie definizioni per descrivere la “memoria della chiralità”
“La chiralità centrale su un C α ad un carbonile viene mantenuta come chiralità assiale
transiente dell’enolato intermedio e poi rigenerata come chiralità centrale nel prodotto
di reazione” (Fuji, 1991)
“La chiralità del materiale di partenza si conserva, per un periodo di tempo
limitato, in un intermedio reattivo” (Fuji, 1998)
“La chiralità di un materiale di partenza che ha un C sp3 chirale viene mantenuta
nel prodotto di reazione, anche se la reazione procede attraverso un intermedio in
cui quel C non è più chirale (carbabioni, monoradicali singoletto, carbocationi)”
(Matsumura, 2002)
“Una reazione con “memoria di chiralità” si può definire come una sostituzione
formale su un centro stereogenico sp3, che procede stereospecificamente, anche
se la reazione procede per trigonalizzazione di quel centro e nonostante nel
71
sistema non siano presenti altri elementi permanentemente chirali”(Carlier, 2005)
La rotazione attorno a legami Csp3-Csp3 richiede di solito meno di 7 kcal/mole
Per la rotazione attorno a legami Csp2-Csp2 si raggiungono facilmente le 16 kcal/mole
MEMORIA DELLA CHIRALITA’ NELLA CHIMICA DEGLI ENOLATI
Fuji ha considerato che la deprotonazione del centro stereogenico in α al carbonile in
un chetone non necessariamente porta all’enolato achirale: nelle opportune condizioni
si possono formare enolati conformazionalmente chirali:
O
R
H R'
R"
base
OR
R"
R'
achirale
R
R'
R
R'
OM
A
MO
A
B
M
B
M
O
R"
O
R"
R
R'
chiralità assiale
R
R'
chiralità planare
Perché la reazione MOC abbia successo, questi enolati chirali si dovrebbero formare in modo
72
enantioselettivo e non dovrebbero racemizzare rapidamente nella scala dei tempi della alchilazione.
36
esempio:
MeO
KO
O
KH, 18-corona-6
OEt
Me
OMe
MeI
EtO
O
MeO
OEt
THF, da -78°C a -20°C
EtO
OEt
93% e.e.
OEt
enolato con chiralità
assiale dinamica
66% e.e.
resa 48%
A sostegno dell’ipotesi che l’enantioselettività è legata alla MOC a causa della
rotazione ristretta attorno al legame Csp2-Csp2:
MeO
Il prodotto di O-alchilazione racemizza a 21°C con un tempo di dimezzamento
di 53 min, corrispondente a un’energia di attivazione di 22.6 kcal/mole
OMe
OEt
Me
OEt
MeO
O
OEt
Con una barriera di energia
inferiore per la rotazione, si
ha racemizzazione
MeI
KH, 18-corona-6
O
MeO
OEt
THF, da -78°C a -20°C
OEt
OEt
0 % e.e.
resa 51%
96% e.e.
73
La prima cosa che si impara studiando la chimica organica è che un centro stereogenico
enantiopuro sp3, se diventa trigonale e poi di nuovo tetraedrico, dà una miscela racemica
Y
X
R
R'
H
R +
R'
H
Y-
R'
H
H
R'
R
R
X-
Y
substrato
enantiopuro
intermedio
achirale
prodotto
racemico
In assenza di altri controllori della chiralità, un risultato non racemico sarebbe possibile
solo se l’intermedio possedesse qualche forma di chiralità conformazionale.
Per sua natura, la chiralità conformazionale sarà di breve durata. Il fenomeno è stato
denominato “chiralità dinamica”, perché la purezza enantiomerica del prodotto
dipende dal tempo e dalla temperatura.
Consideriamo la fenilalanina e l’acido fenilpropanoico:
H H
H NH2
CO2H
H CO2H
CO2H
H
achirale
CO2H
H
g-
g+
chiralità centrale
statica
H
chiralità dinamica
In circostanze opportune, la chiralità conformazionale potrebbe
influenzare il destino stereochimico di un intermedio reattivo
74
37
La formazione di un intermedio conformazionalmente chirale non è una condizione
sufficiente per la MOC: questo intermedio si deve formare enantioselettivamente
I requisiti essenziali per la memoria di chiralità sono illustrati nella seguente ipotetica reazione di
deprotonazione/metilazione:
MeI
base
(S)-A-H
veloce
(M)-A-
MeI
molto lento
molto
lento
molto
lento
(S)-A-Me
veloce
MeI
(P)-A-
(R)-A-Me
veloce
1. La deprotonazione del centro stereogenico deve generare un intermedio reattivo
conformazionalmente chirale, con elevata enantioselettività (M e P sono descrittori
arbitrari)
2. Questo intermedio conformazionalmente chirale non deve racemizzare rapidamente
(almeno non nella scala dei tempi della successiva reazione).
3. L’intermedio conformazionalmente chirale deve reagire con MeI con elevata
stereospecificità.
Basta che uno di questi requisiti non sia soddifsatto, per non avere enantioselettività
Il processo MOC comporta trasferimento di chiralità: (1) da chiralità centrale
statica a chiralità conformazionale transitoria e (2) da chiralità conformazionale
75
transitoria a chiralità centrale
Come assicurare un efficiente trasferimento di chiralità in entrambi i passaggi
rappresenta la sfida principale della strategia MOC.
CHIRALITA’ DINAMICA
Visto che la chiralità conformazionale è, per definizione, transitoria, che tempo di
vita deve avere l’intermedio reattivo conformazionalmente chirale?
Assumendo che la racemizzazione dell’intermedio sia unimolecolare, si può calcolare il
tempo di dimezzamento a varie temperature
Dipendenza di t1/2 di racemizzazione dalla energia di attivazione e dalla
temperatura
t1/2 di racemizzazione
a -78°C
t1/2 di racemizzazione
a 25°C
12
2.4 sec
3.5 x 10-5 sec
14
7 min
1.0 x 10-3 sec
16
20 h
3.0 x 10-2 sec
18
148 giorni
0.9 sec
20
70 anni
26 sec
Barriera per la
racemizzazione
ΔG≠ (kcal/mole)
A -78°C, una barriera di 16 kcal/mole dà ad un intermedio reattivo il tempo sufficiente
per dare una reazione intermolecolare lenta, senza racemizzazione significativa.
A temperatura ambiente, invece, la racemizzazione è 2 milioni di volte più veloce: in
questo caso si potrebbe ottenere una reazione enantioselettiva solo per reazione
76
intramolecolare o per intrappolamento con il solvente.
38
REAZIONI STEREOSELETTIVE DI COMPOSTI CARBONILICI
1. ADDIZIONE NUCLEOFILA A COMPOSTI CARBONILICI
Ci sono tre modi per controllare la stereoselettività dell’addizione a composti carbonilici:
-uso di substrato chirale
-uso di reagente chirale
-uso di catalizzatore chirale
USO DI SUBSTRATO CHIRALE
Se il substrato è chirale, gli stati di transizione sono diastereomerici, con il risultato
che si formano due prodotti diastereomerici in quantità diversa.
esempio
H3C
H
O
H
1. RMgBr
H3C
H
OH
H
R
2. H3O+
H3C
H
O
R
H3C
H
1. LiAlH4
2.
R = Me
R = Et
R = Ph
40% d.e.
50% d.e.
60% d.e.
OH
H
R
H3O+
R = Me
R = Et
R = i-Pr
R = t-Bu
50% d.e.
50% d.e.
66% d.e.
96% d.e.
77
Sono state sviluppate diverse regole per spiegare la stereoselettività di queste reazioni.
Regola di Cram.
Con il centro chirale adicacente al carbonile, considerando la conformazione in
cui il gruppo più grande è coplanare anti al carbonile, il diastereomero prevalente
corrisponde all’addizione del nucleofilo dal lato del gruppo più piccolo.
Cram
R
O
S
Nu-
H
RM
Nu-
R
RL
O
CH3
H
Regola empirica: non cerca di spiegare i fatti sperimentali, ma di correlarli.
Non rappresenta accuratamente né lo stato fondamentale, né quello di transizione, ma
fornisce un metodo di previsione.
Regola di Felkin.
Lo stato di transizione preferito si basa su una conformazione in cui il più voluminoso
dei gruppi legati in a (L) si mette perpendicolare al piano del carbonile in anti rispetto
al Nu che si avvicina ed il secondo gruppo più voluminoso (M) è gauche rispetto al
carbonile.
Felkin
OR
RM O
RL
Nu-
RS R
S
RL
R RM
Nu78
39
Queste regole funzionano bene quando sono coinvolti solo fattori sterici (selettività
relativamente modesta).
M
..
... ..
.
OX
Se c’è un eteroatomo e possibilità di chelazione, il
risultato della reazione si spiega assumendo che il
gruppo contenente l’eteroatomo venga tenuto
coplanare sin con il carbonile (modello chelato di
Cram).
RS
Di solito si hanno selettività migliori.
esempi
Mg
..
.. . O
..
O
H3C
H3C
OH
OH
MgBr
O
H3C
H3C
+
O
H3C
H3C
CH3
O
C
H2 H
CH3
2. H3O+
86
O
RL
R
1.
CH3
modello chelato
di Cram
OH
1. RMgBr
H
C10H21
O
14
OH
+
H
R
C10H21
C
H2 H
2. H3O+
:
O
C
H2 H
R=
94
:
6
R=
95
:
5
R
H
C10H21
79
O
O
C
H2 H
OH
1. LiAlH4
R
C10H21
O
C
H2 H
2. H3O+
R=
R=
H
R
C10H21
OH
+
O
C
H2 H
76
:
24
80
:
20
R
H
C10H21
Sono stati usati anche gli ausiliari chirali.
O
Mg
O
O
RMgBr
OH
O
-78°C
O
H
R = Me 98% d.e.
R H
O
O
OH
O 1. KBH(O-iPr)3
O
H
2. H3O+
O
R = Me 90% d.e.
H R
80
40
Questo metodo è stato utilizato per la sintesi di entrambi gli enantiomeri del feromone frontalina.
O
O
RMgBr
O
O
O
2. H3O+
-78°C
CH3
OH
CH3
1. LiAlH4 HO
OH
CH3
unico diastereomero
O
O3
-78°C
(-)-frontalina
100% e.e.
O
O
O
RMgBr
O
O
O
1. PDC
OH
H
2. MeMgBr
-78°C
H
unico diastereomero
O
OH
CH3
O
LiAlH4
HO
OH
CH3
O
O3
O
(+)-frontalina
100% e.e. 81
USO DI REAGENTE CHIRALE
Sono stati sviluppati diversi riducenti chirali.
Per esempio, (R)- e (S)-BINAL-H.
OR
O Al
O
H
Li+
OR
O Al
O
H
Li+
(S)-BINAL-H
(R)-BINAL-H
O
Per spiegare le stereoselettività osservate è
stato proposto un modello basato su uno
stato di transizione ciclico a sedia.
R
O
S
Al
O
H
R
Li
RL
O
modello dello stato di transizione
esempio:
La disposizione favorita sistema in
posizione equatoriale il sostituente
più grande.
uso di BINAL-H nel primo passaggio di una sintesi di un butanolattone chirale.
OMOM
O
OMOM
CH3
N
NMe2
1. (S)-BINAL-H
SnBu3
2. MOMCl
SnBu3
93% e.e.
O
O
O
82
41
Anche alcuni reagenti riducenti derivati
del borano sono chirali.
Entrambi derivatidell’α-pinene
Cl
BBN
B
O
H
H3C
CH3
CH3
B
R
R
clorodiisopinocanfenilborano
Alpine-borano
modello dello
stato di transizione
L’Alpine-borano è particolarmente efficace per la riduzione asimmetrica di aldeidi, composti βdicarbonilici, chetoni α,β-insaturi e chetoni acetilenici.
OH
O
Alpine-borano
Me
Me
La sua bassa reattività verso altri
chetoni si supera aumentando
l’acidità di Lewis del B, nel
clorodiisopinocanfenilborano.
H
O
Me
OH
Me
(-)-Ipc2BCl
98% e.e.
83
USO DI CATALIZZATORE CHIRALE
H Ph Ph
Reagente CBS.
Viene recuperato al termine
della reazione.
OH
BH3
O
N B
Me
THF
H Ph Ph
O
N+B
H2B
-
Me
O
H
R RL
RS R L
S
H Ph Ph
H Ph Ph
O
N+B Me
O
N+B Me
H2B
O
H
R RL
S
Altre addizioni nucleofile sono state molto
studiate con vari catalizzatori chirali:
H2B
R
S
O
RL
addizione di butillitio alla benzencarbaldeide
H OH
O
H +
Li
cat*
OMe
cat* =
N
Me
N
95% e.e.
OH
cat* =
N
H
90% e.e.
84
42
addizione di dietilzinco alla benzencarbaldeide
H
O
H +
OH
cat*
Zn
N
NMe2
OH
cat* =
cat* =
99% e.e.
H
98% e.e.
OH
85
2. ADDIZIONE CONIUGATA ASIMMETRICA
L’addizione nucleofila a composti carbonilici α,β-insaturi può portare alla generazione di
un nuovo stereocentro in posizione β.
Inoltre, quando l’enolato intermedio viene intrappolato da un elettrofilo, c’è la
possibilità di creare un nuovo centro chirale anche in α.
La configurazione relativa e/o assoluta in queste posizioni si può, in linea di principio,
controllare con le stesse tecniche applicate per l’addizione 1,2- al carbonile.
Nu
-
O
Nu O -
Nu O
E+
E
L’addizione coniugata in tandem con l’alchilazione porta
ad una disposizione trans dei due gruppi che entrano.
O
O
OTHP
O
O
OLi
O-tBu
1. S
S
Ar'
Li
S
S
Ar'
O
2. ArCH2Br
Ar
O
OTHP
t-BuO
O
O
O
Quando nella molecola è presente
un centro stereogenico adiacente il
nucleofilo che si avvicina entra in
trans al gruppo già presente.
86
43
OBn
OBn
1. Me2CuLi
O
O
2. H3O+
O
O
Nell’esempio seguente la configurazione della posizione β è determinata da un solfossido chirale.
O
CH3OS
..
O
O
MeO
1. ZnBr2, ArMgBr
2. Ni-Raney
O
O
(-)-podorizone
MeO
3. LDA, Ar'CO2Et
OMe
O
O
Usando nucleofili ed elettrofili diversi dagli alchili, si possono preparare vari composti carbonilici
α- e β- sostituiti, tra cui α- e β- amminoacidi.
O
O
RR'N
RR'NH
O
1. LiAlH4
O
O
O
2. H2O
NRR'
HO
OH
100% d.e.
RR'NH =
,
N
H
,
N
H
Bu2NH
87
+
CH3O-
O
S
O
..
CH3O
ZnBr2
Zn
S
..
CH3O-
O
RMgCl
O
+
..
H
R = Et
R = Ph
R = allile
H
R
O
O
N
Ms
Me O
N
Ms
Me O
N3
2. NBS
N
O
O
N
CuBr.Me2S
O
R
80% e.e.
92% e.e.
99% e.e.
O 1. MeMgBr
N
Al/Hg
O
+
O
O
O
S
Br
N
Ms
O
N3-
Me O
1. LiOH/H2O
2. H2/Pd-C
OH
N
Ms
NH2
88
44
ADDIZIONE CONIUGATA ASIMMETRICA CON AUSILIARI CHIRALI CARBOIDRATI
L’ addizione 1,4- di composti organici di Al con ossazolidinoni derivati con carboidrati diventa
stereoselettiva
O
O
O
O O
O
O
O
Et2AlCl
N
O
(4 equivalenti)
toluene-esano
-78°C
O
O
O
O O
O
O
O
Et
N
O
O
O
Et
R : S = 94 : 6
O
84%
N-acil galattopiranosido-ossazolidinone
OH
Con l’analogo derivato del glucopiranosio si ha reazione analoga, ma con stereoselettività un po’
minore:
1) Et AlCl
2
O
O
O
N
O
O
O
O
(5 equivalenti)
toluene-esano
O
N
2) NCS
da -40°C a temp. amb.
Et
O
O
Cl
O
Cl O
OH
Et
O
O
O
(2R,3S) : (2R,3R) = 94 : 4
89
64%
ADDIZIONE CONIUGATA ASIMMETRICA CON CATALIZZATORI CHIRALI
La fattibilità dell’addizione coniugata enantioselettiva catalizzata da metalli è relativamente recente
Reattivi di Grignard sono stati addizionati ad enoni usando catalizzatri chirali di rameII
O
R
R''
CuII/L*
R'
+ R''MgX
R
N
Cu
N
H
S
R'
O
O
H
O
N
Cu
ee fino a 87%
Pfaltz, 1994
S
Me
N Me
Cu
ee fino a 76%
Fe
N
PPh2
ee fino a 92%
Sammakia, 1997
van Koten, 1997
legante per il CuII
ee fino a 74%
Lippard, 1988
A partire dal 1997, al posto dei Grignard si è iniziato ad usare reagenti dialchilzinco e
leganti monodentati P-N (fosforammidito)
90
45
O
O
R2Zn, toluene, -30°C
Cu(OTf)3/L*2
n
O
P N
O
R
n
n = 1, R = Me, Et, 98% ee
n = 1, R = iPr, 94% ee
n = 0-2
(S,R,R)
Feringa, 1997
Modifiche del catalizzatore iniziale hanno portato negli anni successivi ad un miglioramento della
selettività
O
H
R N
O
P N
O
NH
P
NH
R' N
H
ee 95%
Leighton
ee 96%
Alexakis
R
PPh2
R'
O
ee 98%
Shi
O
H
N
N
P
N
H
ee 98%
(2001-2004)
91
Hoveyda
Nell’addizione coniugata enantioselettiva di zincoalchili sono stati ottenuti buoni risultati, ma i
reattivi di Grignard hanno dei vantaggi:
sono facilmente disponibili
vengono utilizzati tutti i gruppi del composto organometallico
gli enolati di magnesio che si formano sono più reattivi
realizzare un catalizzatore asimmetrico in grado di evitare
l’addizione 1,2- non catalizzata al carbonile dell’enone
Strategia:
La prima reazione di questo tipo ha usato enoni ciclici come substrati, sali di rame rameoso
e, come leganti chirali, ferrocenildifosfine, disponibili commercialmente
L*
O
O
RMgBr, [Cu], Et2O
L*
Me2N
PPh2
Fe Ph P
2
R
R = Me, Et, Pr, Bu, 90-96% ee, L* = Taniaphos
Me
R = t-Bu, 92% ee, L* = Josiphos
Fe
[Cu] = CuCl, CuBr.SMe2
(2004)
(R,S)
Taniaphos
PR2
R 2P
R = cicloesile
(R,S)
Josiphos
92
46
Successivamente il metodo è stato applicato a composti non ciclici
O
R''
R'
R'
L*
(2005)
R'
X
regioselettività eccellente
ee fino a 99%
L*
Me
Me
Fe
O
R
t-BuOMe o CH2Cl2, -78°C
R’ = alchile, arile; X = OR, SR
(2004)
R''
regioselettività eccellente
ee fino a 99%
L*
RMgBr, CuBr.SMe2 (5% mol)
X
O
R'
t-BuOMe o CH2Cl2, -78°C
R’ = alchile, arile; R’’ = alchile
O
R
RMgBr, CuBr.SMe2 (5% mol)
PPh2
Fe Ph P
2
PR2
R 2P
(R,S)
Josiphos
R = cicloesile
(R,S)
93
prima combinazione catalizzatore-reagente organometallico che funziona con esteri α,β -insaturi
ADDIZIONE DI DERIVATI DI ALLIL BORO
Reagenti allilici del B possono essere utilizzati in una reazione con le aldeidi.
Il B nel gruppo uscente è in grado di coordinarsi
con la coppia non impegnata in legame del
carbonile, formando uno stato di transizione ciclico
a sei termini.
O
R
B
O
B
R
H
Il prodotto è un alcossiborano, che viene facilmente idrolizzato all’alcool corrispondente.
La configurazione relativa (sin o anti) del prodotto è determinata dalla configurazione
(E o Z) del doppio legame C-C nell’allile.
H
Et
B
O
Et
anti (racemico)
H
Et
OH
B
O
OH
Et
sin (racemico)
Il termine sin si usa per denotare se i gruppi OH e R sono dalla stessa parte (sopra o sotto il
piano di scrittura) quando la catena di atomi di C è scritta in forma estesa (zigzag). ll termine
94
anti si usa per indicare I gruppi da parti opposte.
47
Se legato al B si ha un gruppo chirale è possibile ottenere un enantiomero singolo dell’isomero sin o
di quello anti.
Anche quando nell’aldeide è presente un centro stereogenico, la configurazione dei gruppi sul B
determina la configurazione assoluta del prodotto.
OH
EtCHO
)2 B
92% e.e.
Et
CHO
BnO
OH
OH
+ BnO
BnO
98
)2 B
:
2
OH
EtCHO
CHO
BnO
92% e.e.
Et
OH
OH
+ BnO
BnO
92
:
8
95
Risultati simili sono stati ottenuti usando altri derivati allilici del B.
OH
EtCHO
B
O
96% e.e.
Et
CHO
OH
OH
O
O
92
O
B
O
8
70% e.e.
CHO
O
:
O
OH
MeCHO
O
O
+
O
OH
OH
O
+
O
92
:
O
O
8
96
48
3. ALCHILAZIONE DI ENOLATI
E’ possibile controllare la regioselettività della formazione di enolato (controllo cinetico e controllo
termodinamico).
Qualsiasi enolato si formi, darà alchilazione con formazione di un prodotto racemico,
a meno che non sia presente un’influenza chirale.
L’alchilazione diastereoselettiva si può ottenere, per esempio, incorporando un
ausiliario chirale.
O
O
O
N
O
Li
N
LDA
O
O
RX
O
O
N
O
R
R = CH2=CHCH2 94% de
R = Et
90% de
O
N
O
Li
O
O
N
LDA
O
O
N
RX
O
O
O
R
R = CH2=CHCH2
R = Et
94% de
97
Una strategia alternativa coinvolge la formazione di un aza-enolato chirale di un’ossazolina,
preparato facendo reagire un acido carbossilico con un amminoalcool. Il gruppo metossimetile
gioca un ruolo cruciale coordinando il catione litio, che è coinvolto nello stadio di strappo del
protone e di alchilazione.
RCH2
N
R
LDA
O
O
N
H
OMe
R'X
Li
O
R
O
H
R
N
O
Me
H2O
Me
H R
R'
CO2H
80-90% ee
R
H
H
-N
R
H
O
N
+
Li
O
N
R'
O
Me
X
Li
O
Me
98
49
Myers ha introdotto le idrazine chirali SAMP e RAMP (I due enantiomeri dell’1-ammino-2-metossimetilpirrolidina) per ottenere l’alchilazione stereoselettiva dei chetoni.
S-prolina
acido R-glutammico
4 passaggi
OMe
resa 58%
6 passaggi
MeO
N
NH2
N
NH2
SAMP
RAMP
O
O
Strategia:
C
R
resa 35%
H
R
* E
C
Sostituzione elettrofila
(asimmetrica)
scissione
*R N-NH
2
2
N
R
NR*2
C
metallazione
N
H
R
NR*2
- M+
C
elettrofilo
N
NR*2
E
C*
R
equivalente dell’enolato
(chirale)
99
Il gruppo metossimetile gioca un ruolo chiave nel determinare il sito di strappo del protone e
nell’assistere l’avvicinamento dell’alogenuro alchilico.
E X
N
LDA
N
OMe
R'
R''
R
R O. .
.
N
. Li
R .. .
O
R'
R
O
E X
N
N
N
E
R'
R''
OMe
R''
H3C
E X
ECCZCN
La chelazione intramolecolare dell’atomo di Li da parte di OMe (che nel SAMP è sotto il piano
CCNN) porta ad elevata differenziazione diastereofacciale.
esempio
O
+
N
NH2 OMe
SAMP
N
N
LDA
OMe
N
N
Li O Me
H
NR2
H
H
100
50
N
N Li O
Me
Br
Br
O
H3O+
Me
-----Pr
99.5% e.e.
La reazione ha rese elevate ed i prodotti si formano con elevata stereoselettività.
Servono quasi due equivalenti di LDA che, in presenza di LiCl anidro, porta a deprotonazione
cinetica di OH e NH2, generando un O,N-dianione.
Riscaldando la miscela di reazione a 0°C si ha equilibrazione all’enolato più stabile, che
reagisce con l’alogenuro alchilico, formando il prodotto di C-alchilazione.
Quest’ultimo si può idrolizzare in modo efficiente, con poca o nessuna racemizzazione,
semplicemente riscaldando con NaOH acquosa, formando l’α-amminoacido od il suo
derivato N-protetto.
101
Me O
N
OH Me
LDA
NH2
LiCl, -78°C
RX
Me O
0°C
N
OH Me R
N
OLi Me
Un modello operativo per spiegare la
diastereoselettività del passaggio di
alchilazione considera il blocco della
faccia π dell’enolato da parte
dell’alcossido di litio e, forse ancora
più importante, delle molecole di
solvente (THF) associate con il catione
litio.
2. Boc2O
Me OLi
NHLi
N
OLi Me
NH2
O
1. NaOH
NH2
0°C
Me O
NHBoc
HO
R
H
R
resa
e.e.
Et
allile
Bn
97%
91%
88%
>99%
>99%
>99%
OLi
H
OLi
H3C
H3 C
N
H
NH2
RX
102
51
O
O
O
R
N
O
O
Ar
O
N
2.
H3C
R
O
O
N
O
OH
H3C
O
H2C
O
N
SO2
O
N
O
R
1. NaHDMD
O
H2C
1. NaHDMD
N
O
N3
H2C
88%
90%
80%
88%
O
1. LiOH
Ar
+ NH
N
Br
H2C
O
NaN3
O-
3
O
O
Ar
2. NBS
Et
allile
Ph
Bn
2. H2/ Pd-C
O
1. Bu2BOTf
i-Pr2NEt
d.e.
O
Ar
2. ArSO2N3
R
Ar
O
N
O
N3
H 2C
103
4. CONDENSAZIONE ALDOLICA
Nella condensazione aldolica si formano due nuovi centri stereogenici e perciò si producono due
diastereomeri.
O
R'CHO + Me
OH O
base
R'
R
OH O
R
+ R'
R
Me
Me
sin
anti
A differenza dell’addizione di derivati boro allilici (che è irreversibile e perciò sotto controllo
cinetico) la condensazione aldolica è reversibile e si può effettuare sotto controllo cinetico o
sotto controllo termodinamico.
Sotto controllo cinetico, nella maggior parte dei casi, è coinvolto uno stato di transizione ciclico
in cui l’atomo di metallo è coordinato all’O del carbonile dell’altro componente. Il diastereomero
principale dipende dall’enolato coinvolto e si può prevedere usando il cosiddetto modello
Zimmerman-Traxler. Questo assume che lo stato di transizione a sei termini adotti una forma a
sedia e che si applichino i principi generali dell’analisi conformazionale.
Dei due possibili stati di transizione
che coinvolgono l’enolato Z, è preferito
quello che sistema in posizione
equatoriale il gruppo R’ dell’aldeide,
portando al prodotto sin.
Me
O
Li O
R'
Z
R
H
sin
preferito a
H
Me
O
Li O
R'
R
104
52
O
Applicando la stessa analisi
all’enolato E, si ha la previsione
che verrà preferito l’addotto anti.
E
Li O
R'
Me
H
preferito a
O
Li O
Me
R'
R
H
R
anti
Per apprezzare meglio i risultati seguenti, è necessario fare altre due assunzioni:
1. gli enolati Z sono più stereoselettivi degli enolati E.
2. gli enolati di B sono più stereoselettivi degli enolati di Li.
il legame B-O è più corto del legame Li-O: di conseguenza lo stato di transizione che
coinvolge il B è più “compatto” e gli effetti sterici sono massimi, portando ad una
maggiore stereoselettività.
CHO
OH O
OH O
O
OLi
OLi
LDA
R +
R
R
Z
R = Et
R = i-Pr
R = t-Bu
30
60
>98
:
:
:
E
sin
70
40
<2
64
82
>98
R
R+
Me
Me
anti
:
:
:
36
18
<2
105
O
LDA
CHO
OLi
OLi
OH O
OH O
+
+
Me
Z
LDA
Bu2BOTf
30
E
sin
Me
anti
:
70
64
:
36
98 :
2
98
:
2
Et3N, toluene
Gli enolati degli esteri danno con LDA enolati E con selettività sin/anti scarsa; la selettività può
essere notevolmente migliorata usando enolati di boro.
CHO
OLi
O
LDA
O
Et2O
O
E(Z)
E(Z)
OH O
OH O
O
CH3
sin 49%
+
anti
solo anti
O
CH3
51%
Per enolati che danno lo stesso prodotto principale indipendentemente dalla loro
configurazione, o per reazioni che richiedono acidi di Lewis, si deve sospettare un
controllo termodinamico o uno stato di transizione a catena aperta.
106
53
REAZIONI ALDOLICHE ASIMMETRICHE
Incorporare un ausiliario chirale nell’aldeide o nell’enolato Z porta ad una prevalenza di uno dei due
aldoli sin diastereomerici.
Quando i due componenti contengono un gruppo chirale, influenzeranno entrambi la
stereoselettività della reazione. Questo fenomeno viene descritto come doppia induzione
asimmetrica.
I due gruppi favoriranno la formazione dello stesso isomero (matched pair, "coppia bene
assortita") oppure I loro effetti saranno contrapposti (mismatched pair o “coppia male
assortita”).
Entrambe queste possibilità sono illustrate nello Schema.
OLi
OH O
OTMS
CHO +
Me
Bu t
Me Bu
OH O
OTMS
Bu t
Me
OH O
+
OTMS
OTMS
Me But
t
OLi
CHO +
81 : 19
OH O
OTMS
CHO +
OTMS
Me Me
Me Me
OLi
OH O
+
OTMS
Me Me
87 : 13
OH O
+
OTMS
But
OTMS
Me Me But
45 : 1
107
Nel caso di una matched pair si può ottenere un elevato eccesso diastereomerico.
La strereoselettività preferita dell’enolato chirale si può in parte comprendere considerando le
conformazioni dei due stati di transizione ciclici.
Me
O
Li O
R
H
H
OLi
OTMS
Bu t
+ RCHO
O
Li
R
OTMS
R
Me Bu t
OTMS
But H
Me
O
H
OH O
OH O
H
R
OTMS
OTMS
Me Bu t
But H
Usando enolati di B che hanno elevate selettività diastereofacciali, Evans ha sviluppato reagenti
che possono completamente ribaltare la preferenza stereochimica dell’aldeide.
OBR2O
CHO
N
+
i Pr
O
OH O
OH O
X
Me
+
X
Me
500 : 1
108
54
N
+
O
Me
1 : 500
N
OH O
OH O
OBR2O
+
X'
X' +
Me
Me
CHO
OH O
OH O
OBR2O
CHO
O
36
OBR2O
CHO
N
+
N
:
64
OH O
X
Me
400
O
OBR2O
+
Me Me
OH O
i Pr
CHO
X"
X" +
Me Me
X
+
Me
:
1
OH O
OH O
O
Me
Me
X'
X' +
Me
1 : 660
109
Il decorso stereochimico preferito della reazione che coinvolge l’ausiliario chirale derivato dalla
valina è illustrato nello Schema.
O
Me
O
O
N
Bu2BOTf
O
Me
B
N
R
O
H
RCHO
O
B
O
Me
O
N
i-PrNEt
O
O
OH O
B O
R
N
H
O
R
Me
O
O
N
O
O
Il rapporto sin/anti può essere alterato usando un acido di Lewis, che favorisce la formazione di
uno stato di transizione aperto (non ciclico), dal momento che l’aldeide si coordina all’acido di
Lewis a preferenza dell’enolato di boro.
110
55
O
Me
N
B
O
O
O
Me
Bu2BOTf
N
OH O
O
RCHO
O
R
Me
i-PrNEt
O
N
O
sin
RCHO
Me
O
Et2AlCl
O
R
H
H
B
N
OH O
O
R
O
Me
O
N
O
anti
Et2AlCl
Una situazione in cui la catalisi da acido di Lewis serve sempre è la cosiddetta reazione di
Mukayama, in cui come componente nucleofilo si usa un silil enolato (silil enol etere).
Anche in questo caso si pensa che la selettività anti sia dovuta al fatto che viene favorito uno
stato di transizione aperto.
111
S
O
RCHO
TBSOTf
O
Et3N
N
N
S
O
O
O
O
TiCl4 o
OTBS ZnCl2
S
O
OH
N
O
R
Me
LA
O
S
O
H
H
N
Me
R
OTBS
O
Sono noti altri esempi in cui uno stato di transizione aperto porta prevalentemente al
prodotto sin.
112
56
STRATEGIA ALTERNATIVA
Un approccio alternativo e più breve per ottenere una condensazione aldolica chirale comporta
legare un gruppo chirale al B.
Paterson ha preparato enolati di boro contenenti il gruppo (+)- o (-)-isopinocanfenile.
O
OBR2*
(-)-Ipc2BOTf
2. H2O, MeOH
i-PrNEt
O
1. RCHO
OBR2*
(+)-Ipc2BOTf
1. RCHO
2. H2O, MeOH
i-PrNEt
OH O
R
OH O
R
Masamune ha utilizzato un reagente di dialchilboro con simmetria C2.
CHO
O
OH O
OBR2*
R2*BOTf
SCEt3
SCEt3
99.8% e.e.
E
BR2* =
B
113
Corey ha usato un derivato chirale del B derivato dall’1,2-diammino-1,2-difeniletano.
CHO
O
OBR2*
R2*BOTf
SCEt3
OH O
SCEt3
E
BR2* =
B
O
O
N
N S O
R*2BBr = O S
B
Br
CF3
CF3
CF3
Poiché sono disponibili entrambi gli enantiomeri della diammina, si possono preparare
tutti e quattro gli stereoisomeri del β-idrossiacido.
114
57
CHO
R*2BBr
O
S
S
CH2Cl2
S
OH O
OBR2*
i-PrNEt
97% e.e., 96% d.e.
CHO
R*2BBr
Et3N
O
O
OH O
OBR2*
O
O
toluene
esano
94% e.e., 96% d.e.
In qualche caso reazioni asimmetriche di enolato, compresa la condensazione aldolica, sono
state ottenute usando basi di litio chirali.
MeO
NLi
OH O
OMe
O
1.
resa 61%, 78% e.e.
CHO
2.
MeO
O
O
NLi
OH O
OMe
1.
O
O
CHO
2.
resa 80%, 94% e.e.
115
Una variante recente della condensazione aldolica di Mukayama utilizza
complessi ottenuti con triflato di Zn e leganti chirali
O
(2006)
SiMe3
+
OH O
CHO Zn(OTf) / L
2
solvente
La reazione è stata eseguita in ambiente acquoso
Leganti chirali:
O
O
N
N
N
O
O
N
N
OH
O
O
N
N
N
N
HO
116
58
L
O
% cat (mol)
O
N
OH
temp.
°C
tempo
h
resa
sin/anti
e.e. (sin)
20
THF-H2O (10%)
0
20
82%
24 (R,R)
20
THF-H2O (10%)
0
96
56%
13 (R,R)
20
THF-H2O (10%)
0
20
73%
90:10
58 (S,S)
20
THF-H2O (50%)
0
72
38%
93: 7
60 (S,S)
N
N
Solvente
HO
O
O
N
N
N
O
O
N
N
N
La configurazione assoluta del prodotto dipende dalla configurazione del
legante usato
117
Il controllo stereochimico della reazione è stato spiegato con il seguente modello:
H2 O
O
N
Me
H
H2O
O
Me3Si
H
N
Zn
H
si
N
O
O
re
Quando l’aldeide si coordina in posizione apicale allo Zn, l’avvicinamento del silil enol
etere dalla faccia re è ostacolato dall’isopropile: l’attacco del nucleofilo avviene in
prevalenza dalla faccia si.
118
59
La condensazione aldolica è uno dei metodi più generali per la formazione
di legami C-C.
☺ Versatile
☺ Efficace
Spesso è usata per costruire i gruppi polioli presenti in molti composti naturali
OH OH OH OH OH OH OH OH OH OH
Me Me Me
Me Me
frammento C9-C27 della (-)-Aflatossina A
Limiti della condensazione aldolica:
Bassa selettività
⇒ Chemioselettività
⇒ Regioselettività
O
O
R'
+
R'
R"
HO
R"
+
R'
HO
O
O
O
O
R"
R"
+
HO
R'
+
R'
HO
R"
119
La versione asimmetrica della reazione aldolica classica richiede l’uso di ausiliari
chirali (per esempio, gli ossazolidinoni di Evans).
Limiti della condensazione aldolica:
Versioni alternative della reazione aldolica (per esempio
la reazione aldolica di Mukayama) richiedono la pregenerazione dell’enolo o dell’enolato mediante quantità
stechiometriche di base o di reagenti sililanti, diminuendo
l’atom economy della reazione.
NEt3
O
Me3SiCl
R'
O
SiMe3
R'
La sfida:
+
O
SiMe3
R'
O
O
H
R"
R'
O
SiMe3
R"
O
OH
R'
trovare o preparare un catalizzatore che possa
effettuare la condensazione aldolica in modo
asimmetrico, senza la formazione preliminare
di un nucleofilo migliore
R"
120
60
La sfida è stata raccolta da Trost, che ha progettato il legante
bis-ProFenolo (bis-ProPhenol)
Ar
OH
Ar
HO
Ar
Ar
OH N
N
B. Trost, J. Am. Chem. Soc., 2000
R
Caratteristiche:
⇒ lega strettamente gli ioni metallici e raggiunge un
livello elevato di riconoscimento molecolare
⇒ mantiene ancora la facilità di scambiare il prodotto,
per raggiungere velocità di turnover ragionevoli
121
Preparazione del legante bis-ProFenolo
OH
Ph
OH
Ph
OH
1. CH2O
Br
Br
2. HBr
CH3
N
H
Ph
OH
Ph
N
HO
Ph
Ph
OH N
CH3
CH3
Applicazione alla reazione aldolica asimmetrica:
R
H
+
resa: 49%
e.e. 68%
resa: 24%
Ar
Ph
Ph
resa: 62%
e.e. 98%
OMe
OH O
OH O
OH O
OH O
OH O
OH O
R
Ar 15% mol Ph2P=S
Ph
Ph
resa: 24%
e.e. 74%
OH O
THF
OH O
OH O
5% mol bis-ProFenolo
10% mol Et2Zn
O
O
Me3Si
resa: 61%
e.e. 93%
resa: 60%
e.e. 98%
O
OH O
Ph
Ph
O
resa: 79%
e.e. 99%
O
OMe
resa: 66%
e.e. 97%
resa: 48%
e.e. 97%
resa: 38%
e.e. 98%
resa: 40%
e.e. 96%
B. Trost, 2000
122
61
R
OH O
5% mol bis-ProFenolo
10% mol Et2Zn
O
O
H
+
Ph
Ph
Ph
OH
OH
OH
resa: 74%
d.r. >49:1
e.e. 96%
resa: 89%
d.r. 13:1
e.e. 93%
OH O
OH O
Ph
Ph
d.r. 30:1
e.e. 92%
Ar
OH
OH O
OH
resa: 83%
R
Ar MS 4, THF, -35°C
OH
OH O
OH
OH O
resa: 65%
resa: 78%
d.r. 35:1
e.e. 94%
d.r. 9:1
e.e. 91%
OH O
OH O
OH O
Ph
O
O
OH
OH
OH
resa: 90%
resa: 97%
d.r. 6:1
e.e. 96%
d.r. 3.4:1
e.e. 95%
resa: 91%
d.r. 5:1
e.e. 87%
123
B. Trost, 2001
Di solito nelle reazioni aldoliche asimmetriche sono stati usati solo nucleofili
semplici. Poco è stato fatto con nucleofili più funzionalizzati
Un donatore importante nella reazione aldolica sarebbe il metil vinil chetone (MVK),
perché sarebbe un “building block” bifunzionale. Però il suo uso è stato limitato dalla
instabilità in ambiente basico, sia del MVK che dei prodotti aldolici.
Le uniche reazioni aldoliche enantioselettive riportate con il MVK sono quelle che
usano il complesso di Zn del legante di Trost, Bis-ProFenolo
R
OH O
5% mol bis-ProFenolo
20% mol Et2Zn
O
O
H
+
OH O
MS 4, toluene o THF
OH O
OH O
R
OH O
OH O
OH O
OSiMe2tBu
resa: 56%
e.e. 91%
resa: 56%
resa: 74%
resa: 37%
resa: 59%
e.e. 91%
e.e. 86%
e.e. 85%
d.e. > 99%
O
resa: 46%
e.e. 87%
OH O
OH O
OH O
tBuMe2Si
O
O
resa: 66%
resa: 33%
e.e. 92%
e.e. 44%
resa: 49%
e.e. 98%
OH O
tBuMe2Si
O
resa: 50%
e.e. 91%
124
B. Trost, 2005
62
O
O
R
5% mol bis-ProFenolo
10% mol Et2Zn
+
H
EtO OEt
MS 4, THF
R'
EtO OEt
EtO OEt
TES
resa: 76%
e.e. 99%
e.e. >98%
e.e. >95%
e.e. >98%
OH O
OH O
TES
TES
resa: 84%
resa: 79%
OH O
EtO
EtO OEt
R'
OH O
TBuMe2Si O
EtO OEt
TES
EtO OEt
TES
resa: 75%
O
EtO OEt
OH O
OH O
OH O
OH O
R
EtO OEt
EtO OEt
resa: 68%
resa: 61%
resa: 61%
e.e. 37%
e.e. 80%
e.e. 87%
B. Trost, 2004
La reazione è stata applicata alla sintesi della Fostriecina, un estere citotossico isolato
da Streptomyces pulveraceus.
5% mol bis-ProFenolo
10% mol Et2Zn
O
O
+
H
EtO OEt
MS 4, THF
BDMS
B. Trost, 2005
OH O
EtO OEt
BDMS
resa: 67%
e.e. 99%
O
O
H3C
O
OH
OH OH
O
ONa P
O HO O
OH
OH
125
OH
H3C
12 passaggi dal piruvato di etile
OH
Sviluppi recenti:
H
R
+
OH
CH3
resa: 84%
resa: 91%
e.e. 81%
e.e. 92%
e.e. 68%
OH
OMe
OH
OMe
MeO
NO2
resa: 95%
SiMe3
MeO
OH
O2N
NO2
OMe
R'
OH
OH
OH
OH
OMe
R
toluene
R'
3 equiv
1 equiv
OH
OH
10% mol bis-ProFenolo
3 equiv Me2Zn
O
resa: 78%
resa: 89%
resa: 87%
e.e. 83%
e.e. 75%
e.e. 92%
OH
OH
OH
O
SiMe3
SiMe3
OMe
OMe
OMe
resa: 74%
resa: 79%
resa: 81%
resa: 86%
resa: 95%
resa: 87%
e.e. 97%
e.e. 84%
e.e. 84%
e.e. 82%
e.e. 99%
e.e. 85%
OEt
O
B. Trost, 2006
… Il successo è confermato dal fatto che siano disponibili commercialmente … 126
63
127
64