Analisi genetica
dei caratteri
quantitativi
19
SOMMARIO
DEL
CAPITOLO
19.1 I caratteri quantitativi presentano
variabilità fenotipica continua
19.2 L’analisi dei caratteri quantitativi è
statistica
19.3 L’ereditabilità misura la componente
genetica della variabilità fenotipica
19.4 I loci dei caratteri quantitativi
corrispondono ai geni che
contribuiscono ai caratteri quantitativi
CONCETTI FONDAMENTALI
Istogramma umano che rappresenta la distribuzione delle
altezze di docenti e studenti dell’Università del Connecticut.
Le donne indossando magliette bianche e gli uomini blu.
Una linea che raffigura la distribuzione continua dell’altezza
adulta è sovraimpressa alla foto.
È
più facile spiegare la connessione tra fenotipi
e genotipi quando la variabilità fenotipica di
un carattere è determinata dalla variabilità di
un singolo gene. Per esempio, gli alleli di un unico
gene determinano se i piselli di Mendel sono lisci
o rugosi. Nelle osservazioni di Mendel non vi sono
altri geni coinvolti e non vi è evidenza di interazioni
tra geni (come l’epistasi) o di interazioni tra gene
e fattori ambientali specifici. La variabilità di altri
geni determina se i baccelli sono rigonfi o grinzosi,
se il fiore è di color porpora o bianco e così via. In
maniera simile, il gruppo sanguigno (A, B, AB oppure
0) è determinato esclusivamente dalla variabilità
ereditata di un singolo gene e l’ambiente nel quale
674
I caratteri quantitativi sono influenzati da
diversi geni e anche dall’ambiente. Essi
sono distribuiti in maniera continua su una
scala fenotipica. Alcuni caratteri quantitativi
sono separati in fenotipi distinti da una
soglia.
Le distribuzioni fenotipiche dei caratteri
quantitativi sono descritte da misure
statistiche che stimano anche i contributi
genetici e ambientali al fenotipo.
Il contributo della variabilità genetica
alla variabilità fenotipica dei caratteri
quantitativi può essere stimato, fornendo
un’indicazione di come i caratteri possano
rispondere alla selezione artificiale.
I geni che influenzano i caratteri quantitativi
sono identificati e mappati utilizzando
incroci genetici e procedure molecolari e
statistiche.
19.1 - I caratteri quantitaivi presentano variabilità fenotipica continua
siete cresciuti non ha alcun effetto su questo risultato.
Tuttavia, in realtà, queste correlazioni dirette tra i
fenotipi e i genotipi non sono comuni. Per esempio,
molti caratteri presentano una variabilità che risulta
da interazioni geniche epistatiche (Capitolo 4). Inoltre,
numerosi caratteri, noti come caratteri poligenici, sono
il risultato dell’influenza di diversi geni, che assortiscono
indipendentemente per produrre un gran numero di
genotipi e fenotipi. L’ereditarietà dei caratteri poligenici
viene detta eredità poligenica. La scoperta che molti
caratteri la cui ereditarietà è determinata da diversi
geni sono influenzati anche da fattori ambientali,
complica ulteriormente le correlazioni tra genotipi e
fenotipi. Pertanto, sia la variabilità genetica che quella
ambientale contribuiscono alla variabilità fenotipica di
alcuni caratteri, che vengono quindi chiamati caratteri
multifattoriali.
Una misura importante dell’influenza di diversi geni e
fattori ambientali sui fenotipi è data dalla valutazione
della variabilità dei caratteri in termini quantitativi
piuttosto che qualitativi. “Semi lisci” rispetto a “semi
rugosi” oppure “sangue del gruppo A” rispetto a
“sangue del gruppo B” sono esempi di differenze
fenotipiche qualitative. I fenotipi qualitativi ricadono
in categorie discrete che corrispondono a genotipi
particolari e che sono spesso chiaramente diversi l’uno
dall’altro. Invece, la variabilità fenotipica quantitativa è
generalmente continua su una scala fenotipica e deve
essere descritta utilizzando delle unità di misura. Per
esempio, si potrebbero utilizzare i chilogrammi per
misurare la variabilità quantitativa del peso dei bovini o
i centimetri per misurare la variabilità quantitativa della
lunghezza delle pannocchie di granturco. I caratteri che
appartengono a quest’ultima categoria vengono detti
caratteri quantitativi. Questo termine si applica anche
ai caratteri che variano lungo un intervallo fenotipico.
Alcuni fenotipi di caratteri quantitativi vengono
misurati in valori come grammi o centimetri, mentre
altri vengono identificati in modalità non numeriche,
come i fenotipi che ricadono in un intervallo di colore
(per esempio, da “bianco” a “nero”).
Lo studio e l’analisi dal punto di vista genetico
dei caratteri quantitativi è l’oggetto di studio
della genetica quantitativa. In questo capitolo
analizzeremo il modo in cui la genetica quantitativa
esamina la variabilità ereditaria dei caratteri poligenici
e multifattoriali. Nel far ciò, illustreremo alcuni dei
modi in cui i genetisti cercano di districare le influenze
genetiche e ambientali sulla variabilità dei caratteri e
descriveremo gli approcci genetici all’interpretazione
degli effetti relativi di questi fattori sui fenotipi dei
caratteri quantitativi.
19.1 I caratteri quantitativi
presentano variabilità fenotipica
continua
Per la maggior parte dei caratteri discussi nei capitoli precedenti, la variabilità fenotipica è controllata
dalla variabilità allelica di singoli geni. I fenotipi di
questi caratteri presentano generalmente variabilità discontinua, cioè manifestano delle differenze
che permettono di assegnare gli organismi a categorie fenotipiche discrete e ben distinte. Gli schemi
della variabilità discontinua permettono di specificare rapporti fenotipici costanti, come un rapporto
di 3:1 nella progenie F2 di organismi F1 autofecondati. Anche nel caso di due geni con interazioni epistatiche che determinano l’espressione fenotipica, i
fenotipi sono discreti e si presentano in rapporti
prevedibili (Sezione 4.3). Invece, i caratteri poligenici e multifattoriali sono controllati da molti geni e
presentano generalmente una variabilità continua,
cioè una variabilità fenotipica distribuita in un intervallo di valori in un continuo ininterrotto.
In questa sezione esploreremo i fattori genetici
che contribuiscono ai caratteri che presentano variabilità continua.
Potenziale genetico
L’altezza adulta umana rappresenta un esempio di
carattere multifattoriale che varia in maniera continua lungo una scala di misura generalmente indicata in centimetri o in pollici. Questa variabilità
continua è illustrata nella fotografia all’inizio del capitolo, in cui circa 150 studenti e docenti dell’Università del Connecticut sono disposti in funzione
dell’altezza.
675
CAPITOLO 19 - Analisi genetica dei caratteri quantitativi
La distribuzione dell’altezza in questo campione,
divisa in incrementi di 1 pollice, varia da 60 pollici
(5 piedi, ovvero 1 metro e 52 centimetri) a 77 pollici (6 piedi e 5 pollici, ovvero 1 metro e 95 centimetri). La lunghezza di ogni fila di individui dietro al
cartello con l’indicazione dell’altezza rappresenta la
frequenza di ogni categoria incrementale.
L’altezza degli adulti è influenzata da un gran numero di geni. Per esempio, uno studio del 2011 effettuato da Matthew Lanktree e numerosi colleghi, servendosi dell’analisi della variabilità genomica umana
e di metodi statistici, ha suggerito che l’altezza adulta
potrebbe essere influenzata da oltre 60 geni. Benché il
numero reale di geni che influenzano l’altezza umana
continui a essere oggetto di studio, le esperienze personali, come pure gli studi di popolazione, suggeriscono che genitori più alti tendono ad avere figli più
alti, mentre genitori più bassi tendono ad avere figli più bassi. Oltre a questa influenza genetica, tuttavia, alcuni fattori ambientali e dello sviluppo possono esercitare un effetto significativo. Se il vostro
corso di genetica riflette il quadro tipico, un’inchiesta tra i vostri compagni rileverà probabilmente che
molti uomini sono più alti dei propri padri e nonni
e che molte donne sono più alte delle proprie madri
e nonne. Queste differenze sono dovute quasi esclusivamente al miglioramento nella salute e nella nutrizione materna e infantile e solo in minima parte a
differenze dell’insieme dei geni che influenzano l’altezza adulta. Studi longitudinali confermano che la
maggior parte della popolazione mondiale sta diventando più alta. Nel corso del XX secolo, l’altezza media delle donne americane è aumentata da circa 157
centimetri nel 1900 a circa 165 centimetri nel 1990.
Un aumento ancora più radicale nell’altezza media
degli adulti può essere notato esaminando le porte
delle case e di altre strutture costruite durante il Medioevo. La maggior parte dei visitatori odierni deve
curvarsi per entrare! Tali osservazioni portano chiaramente alla conclusione che l’altezza degli adulti è
un carattere multifattoriale.
Per comprendere il ruolo della genetica in questo tipo di carattere, dovete immaginare che i genitori trasmettano ai propri figli un “potenziale genetico” per un’altezza adulta massima data; questa
verrà raggiunta se il figlio cresce e si sviluppa in condizioni ideali. Non tutti i figli di una certa coppia di
676
genitori presenterà lo stesso potenziale genetico,
dato che la segregazione e l’assortimento indipendente dei geni che contribuiscono al fenotipo può
produrre diversi genotipi. Questi processi producono la generazione di una prole con diversi genotipi che portano un potenziale genetico per un intervallo di altezze, anche superiori o inferiori a quelle
dei genitori. Mediamente, tuttavia, il potenziale genetico della progenie per l’altezza sarà approssimativamente intermedio rispetto ai potenziali genetici
dei due genitori.
CONCETTI CHIAVE
I fenotipi dei caratteri quantitativi possono essere
misurati su scale quantitative. I caratteri quantitativi prodotti dall’influenza cumulativa di diversi geni
sono poligenici. I caratteri quantitativi che sono influenzati da fattori poligenici, ambientali o dello sviluppo, sono detti multifattoriali.
Effetti dei geni principali e dei geni
additivi
La variabilità fenotipica continua dei caratteri poligenici è il risultato degli effetti di diversi geni che
possono esercitare gradi diversi di influenza. Per
esempio, il gene umano OCA2 presenta diversi alleli
che influenzano fortemente il colore dell’occhio
adulto. Questo è ulteriormente influenzato da altri
geni che agiscono con minor forza rispetto a OCA2.
Un gene come OCA2 è classificato come gene principale, dato che esercita un effetto predominante
sul fenotipo. I geni che esercitano un effetto meno
importante sul fenotipo, ma che contribuiscono
ugualmente a esso, sono classificati come geni modificatori.
D’altro canto, per molti caratteri poligenici la distribuzione fenotipica continua deriva da contributi
incrementali da parte di numerosi geni. Quando i
geni che partecipano ai caratteri poligenici contribuiscono in parti uguali alla variabilità fenotipica totale, essi vengono detti geni additivi. A ogni allele
dei geni additivi può esser attribuito un valore quantitativo che indica il suo contributo a un carattere
poligenico noto come carattere additivo, perché i
fenotipi possono essere predetti sommando i valori
degli alleli. Per alcuni caratteri, tutti i geni additivi
19.1 - I caratteri quantitaivi presentano variabilità fenotipica continua
presentano un effetto approssimativamente uguale.
Nel caso del colore dei fiori, ad esempio, una sola copia di un allele di un gene additivo può contribuire
a un’unità di colore, due alleli contribuiscono a due
unità di colore e così via. Se i geni che controllano
la variabilità ereditaria di un carattere sono additivi,
nessun gene, da solo, esercita un effetto predominante sulla variabilità fenotipica, pertanto vengono
osservate delle differenze incrementali del fenotipo.
Per comprendere il concetto di gene additivo è
necessario pensare ai genotipi e ai fenotipi in maniera
diversa rispetto a quanto abbiamo fatto finora. Poiché
i caratteri controllati dai geni additivi presentano un
fenotipo che è la somma dei valori degli alleli, è possibile che ad alcuni fenotipi corrispondano diversi genotipi. La segregazione e l’assortimento indipendente
degli alleli additivi producono i diversi genotipi, ma
il fenotipo corrispondente a ognuno di essi è basato
sulla somma dei valori degli alleli di tutti i loci che
contribuiscono al fenotipo stesso.
All’inizio del Novecento, in corrispondenza con
la verifica e l’espansione dei principi dell’ereditarietà
di Mendel appena riscoperti, i genetisti iniziarono
a esplorare l’ipotesi che la segregazione degli alleli
di diversi geni giocasse un ruolo nella variabilità fenotipica di particolari caratteri. L’ipotesi, nota come
ipotesi dei geni multipli, propone che gli alleli di
ognuno dei geni che contribuiscono al fenotipo obbediscano ai principi di segregazione e assortimento
indipendente ed esercitino un effetto additivo nella
produzione della variabilità fenotipica.
L’ipotesi dei geni multipli è stata il fondamento
della genetica quantitativa e il genetista delle piante
Hermann Nilsson-Ehle fu uno dei primi a utilizzare questa ipotesi nella sua descrizione, risalente al
1909, del controllo genetico del colore dei chicchi dl
grano. La Figura 19.1 illustra uno dei modelli genetici di Nilsson-Ehle, che descrive la determinazione
del colore del chicco di grano da parte degli alleli additivi di due geni. In questo modello, vengono presi
in considerazione solo gli effetti genetici sul fenotipo. Il modello predice che il colore del chicco occupa uno spettro di colore che va dal rosso intenso
al bianco. I geni A e B presentano due alleli ciascuno.
Gli alleli A1 e B1 sono equivalenti e ognuno di essi
aggiunge una stessa unità di colore al fenotipo. Anche gli alleli A2 e B2 sono equivalenti e nessuno dei
Gameti
Riassunto dei:
Genotipi
Fenotipi
Figura 19.1 Eredità poligenica del colore dei chicchi di
grano controllata da due geni additivi. Ogni allele 1 (sia A1
che B1) aggiunge un’unità di colore, mentre gli alleli 2 (A2 o B2)
non aggiungono unità di colore. Due incroci distinti tra piante
genitrici appartenenti a linee pure producono una generazione
F1 di diibridi con colore rosa del chicco. Sono previste
cinque classi fenotipiche per la progenie F2, con un rapporto
determinato dal numero totale di alleli A1 e B1 nel genotipo.
due aggiunge unità di colore al fenotipo. Secondo il
modello genetico additivo, più alleli “numero 1”(sia
A1 che B1,) contengono il genotipo, più scuro sarà il
colore dei chicchi di grano. Invece, meno alleli numero 1 (oppure più alleli “numero 2”) sono presenti
nel genotipo, più chiaro sarà il colore del chicco. Il
colore rosso scuro è presente quando sono presenti
quattro alleli numero 1 (A1A1B1B1). Viceversa, i chicchi bianchi sono prodotti quando non vi sono copie
di alleli numero 1 nel genotipo (A2A2B2B2).
La Figura 19.1 mostra un incrocio tra linee pure
di piante rosse e linee pure di piante bianche che
677
CAPITOLO 19 - Analisi genetica dei caratteri quantitativi
presentano diversi fenotipi perché possiedono genotipi diversi.
L’incrocio produce piante F1 diibride (A1A2B1B2)
che presentano un colore rosa intermedio dei chicchi in conseguenza del fatto che portano solo due
alleli numero 1. Incrociando piante della F1 si ottiene una generazione F2 con cinque diversi colori
dei chicchi, ognuno dei quali dipende dal numero
totale di alleli numero 1 nel genotipo. Per questi due
loci, i genotipi possono avere un massimo di quattro alleli numero 1 e un minimo di zero alleli numero 1. I cinque diversi totali che si ottengono per
gli alleli numeri 1 producono i cinque diversi fenotipi nella generazione F2, in percentuali determinate
1
dall’assortimento indipendente. Nella F2, 16
delle
piante porta quattro alleli numero 1 e produce chic4
chi rossi come la pianta parentale, 16
portano tre alleli numero 1 e presentano chicchi di colore rosso
6
chiaro, 16
presentano due alleli numero 1 e chicchi
4
rosa, 16 portano un solo allele numero uno e presen1
tano chicchi di colore rosa chiaro e il rimanente 16
non presenta alleli numero 1 e ha chicchi bianchi
come la pianta parentale.
All’aumentare del numero dei geni additivi che
contribuiscono al carattere, aumenta il numero
delle categorie fenotipiche. La Figura 19.2 illustra
un modello genetico additivo nel quale il colore dei
chicchi di grano è determinato da tre geni. In questo esempio, i geni A, B e C presentano due alleli ciascuno, i cui effetti additivi vengono calcolati nello
stesso modo del sistema a due geni della Figura 19.1.
Le categorie fenotipiche sono determinate dal numero di alleli “1” contenuti nel genotipo. Un incrocio tra piante parentali di linee pure rosso scuro e
linee pure bianche produce una F1 di un colore intermedio (rosa scuro) dovuto al suo fenotipo triibrido (A1A2B1B2C1C2).
L’assortimento indipendente produce una F2 con
sette categorie fenotipiche determinate dai genotipi,
che presentano un massimo di sei alleli 1 e un minimo di zero alleli 1.
CONCETTI CHIAVE
I caratteri quantitativi additivi sono determinati dalla somma dei contributi relativamente uguali di alleli di diversi geni.
678
Variabilità fenotipica continua dovuta a
molteplici geni additivi
Più numerosi sono i fenotipi che si presentano su
una scala di misura limitata, più piccola è la fetta di
distribuzione occupata da ogni categoria e meno
evidente è la demarcazione tra le categorie. La Figura 19.3 mostra cinque istogrammi che illustrano
la distribuzione dei fenotipi della F2 prodotti da numeri diversi di geni additivi, ognuno dei quali presenta due alleli. Come negli esempi precedenti, ogni
allele numero 1 aggiunge un’unità di colore, contrariamente agli alleli numero 2. Le percentuali per
ogni fenotipo sono determinate utilizzando il triangolo di Pascal (Figura 2.15). Notate l’aumento nel
numero di classi fenotipiche via via che il numero
dei geni che contribuiscono al fenotipo aumenta da
uno a cinque. Inoltre, le classi fenotipiche adiacenti
assomigliano sempre più l’una all’altra via via che il
numero di classi aumenta, mescolandosi in una distribuzione fenotipica continua.
Il numero di categorie fenotipiche distinte per
un carattere poligenico prodotto dalla segregazione
di alleli additivi di un dato numero di geni (n) è calcolata come 2n+1.
Per esempio, per tre geni additivi che contribuiscono a un carattere poligenico, n = 3 e il numero di
categorie fenotipiche distinte è 2(3) + 1 = 7.
Nella Tabella 19.1 sono elencate il numero di categorie fenotipiche per diversi numeri di geni che
contribuiscono al fenotipo e viene indicata la frequenza dei fenotipi più estremi in ogni distribuzione. Se sono presenti più di due alleli dei geni che
contribuiscono al fenotipo, il numero di fenotipi
può aumentare.
Segregazione degli alleli nella
produzione dei caratteri quantitativi
Nel 1916, il genetista delle piante Edward East intraprese un esame dettagliato dell’ipotesi dei geni multipli testando la propria abilità a spiegare gli schemi
di variabilità ereditaria che produceva per la lunghezza della corolla (la parte del fiore che produce i
petali) in Nicotiana longiflora. In questa specie di tabacco a fiore lungo, la corolla è rappresentata da una
struttura a forma di tubo, la cui lunghezza può essere misurata e confrontata a quella della corolla di
altre piante.
19.1 - I caratteri quantitaivi presentano variabilità fenotipica continua
Gameti
Gameti
Frequenza della progenie
Numero degli alleli che producono colore
Figura 19.2 Modello additivo a tre geni
Proporzione della progenie
per il colore dei chicchi di grano. Il colore è
determinato dal numero totale di alleli 1 (A1, B1 e
C1) nel genotipo. Gli individui della F2 presentano
sette classi fenotipiche in proporzioni determinate
dall’assortimento indipendente dei tre loci.
679
CAPITOLO 19 - Analisi genetica dei caratteri quantitativi
Tabella 19.1 L’effetto di poligeni sulla
variabilità fenotipica
Frequenza della progenie
Numero degli alleli che producono colore
Proporzione della progenie
(b) Due loci: A1A2B1B2 × A1A2B1B2
Numero degli alleli che producono colore
Frequenza della progenie
East iniziò i suoi esperimenti con linee parentali
pure, una dotata di una corolla corta di circa 40 millimetri di lunghezza e l’altra con una corolla lunga
di circa 90 millimetri (Figura 19.4). Notate che vi è
una leggera variabilità nella lunghezza della corolla
in ogni ceppo parentale, suggerendo che, malgrado
i tentativi di produrre linee pure, le interazioni tra
geni o gli effetti multifattoriali producono un po’ di
variabilità. La progenie F1 di questo incrocio presenta una lunghezza media della corolla di circa 65
millimetri, all’incirca intermedia tra le medie parentali. Questi valori “parentali intermedi” rappresentano un’indicazione del forte controllo genico
della lunghezza della corolla. Ancora una volta, vi è
un certo grado di variabilità attorno al valore medio
della lunghezza della corolla, ma non vi è alcun individuo della F1 che presenta lunghezza della corolla
prossima a quella dei genitori.
East permise alle piante della F1 di autofecondarsi per produrne circa 450 alla F2, tra le quali os-
(a) Un locus: A1A2 × A1A2
Proporzione della progenie
(c) Tre loci: A1A2B1B2C1C2 × A1A2B1B2C1C2
Numero degli alleli che producono colore
Frequenza della progenie
Figura 19.3 Distribuzioni di fenotipi che presentano
geni additivi. I genitori della progenie, in ogni esempio, sono
eterozigoti per tutti i geni. Gli alleli dei geni che contribuiscono
al colore sono indicati come 1. Il numero di categorie fenotipiche
della F2 aumenta all’aumentare del numero di geni additivi.
Proporzione della progenie
(d) Quattro loci: A1A2B1B2C1C2D1D2 × A1A2B1B2C1C2D1D2
3
1
4
2
5
1
16
3
7
1
64
4
9
1
256
11
1
1024
6
13
1
4069
7
15
1
16.384
8
17
1
65.536
9
19
1
262.144
10
21
1
1.048.576
5
680
Proporzione della progenie
(e) Cinque loci: A1A2B1B2C1C2D1D2 E1E2 × A1A2B1B2C1C2D1D2 E1E2
Numero degli alleli che producono colore
Frequenza della progenie
1
Frequenza della progenie
Numero degli alleli che producono colore
Numero
Numero di categorie Frequenza dei
di geni (n) fenotipiche
fenotipi più estremi
Proporzione della progenie
19.1 - I caratteri quantitaivi presentano variabilità fenotipica continua
Percentuale
Percentuale
Percentuale
Genitori
Lunghezza
della corolla
Linee pure
con corolla
corta e lunga
La lunghezza della
corolla è intermedia
tra i genitori, con
varianza dovuta
all’ambiente
La varianza nella
lunghezza della corolla
è genetica e ambientale
Percentuale
Selezione per
diverse lunghezze
Tre generazioni di
selezione per corolla
corta e per corolla
lunga determinano
ceppi che assomigliano
ai genitori
Lunghezza della corolla (mm)
Figura 19.4 Lunghezza della corolla nella pianta di
tabacco. Edward East determinò che la varianza genetica nella
lunghezza della corolla della pianta di tabacco è controllata dagli
alleli di diversi geni (Nicotiana longiflora).
servò una distribuzione più ampia di lunghezza della
corolla rispetto alla F1, benché la lunghezza media
fosse all’incirca la stessa di quella della F1. Nessuna
delle piante della F2 prodotte da East presentava una
lunghezza della corolla pari a quella delle linee parentali pure. Quindi, nel corso di tre ulteriori generazioni a partire dalla F2, East generò in maniera
selettiva le piante in modo da produrre un ceppo a
corolla corta e un ceppo a corolla lunga, riuscendo a
ottenere nuovi insiemi di piante con lunghezza della
corolla all’incirca pari a quella rinvenuta nelle linee
parentali pure originali.
East giunse a due conclusioni generali basate
sulle sue osservazioni. Entrambe le conclusioni sono
coerenti con i modelli di variabilità fenotipica continua dei caratteri quantitativi che abbiamo descritto.
In primo luogo, concluse che la lunghezza della corolla di Nicotiana longiflora, in particolare nella F2,
deriva dalla segregazione degli alleli di geni multipli.
In secondo luogo, East concluse che l’espressione
fenotipica di ogni genotipo è influenzata da fattori
non genetici, cioè interazioni geniche o fattori ambientali che rendono sfumata la connessione tra un
dato genotipo e uno specifico fenotipo. I fattori non
genetici spiegano parzialmente la variabilità attorno
alla lunghezza media della corolla.
L’Analisi genetica 19.1 vi guiderà nell’analisi dei
contributi di poligeni all’altezza della pianta.
Effetti di fattori ambientali sulla
variabilità fenotipica
Districare i fattori genetici e non genetici che determinano la variabilità fenotipica è un compito difficile, ma importante, nel campo della genetica. Negli
umani, per esempio, malattie comuni come le cardiopatie, il cancro e il diabete sono influenzate
dall’ereditarietà, ma anche fattori non ereditari sono
estremamente importanti nello sviluppo della malattia. L’identificazione dei particolari geni e degli
specifici fattori non ereditari che contribuiscono a
queste malattie è lo scopo finale di molte ricerche,
ma per arrivarvi bisogna procedere per piccoli passaggi incrementali che includono la creazione di
modelli per le interazioni tra fattori ereditari e non
ereditari.
La Figura 19.5 presenta un approccio generale
assunto dai modelli di questo tipo. Essa illustra gli
intervalli fenotipici che sarebbero associati ai genotipi A1A1, A1A2 e A2A2 secondo diversi presupposti di
interazioni gene-ambiente. Nella Figura 19.5a, nessuna interazione gene-ambiente è presente e ogni
genotipo corrisponde a un fenotipo distinto. Nella
F2 si osserva una corrispondenza prevedibile tra genotipo e fenotipo, con distribuzione fenotipica discontinua e rapporto fenotipico 1:2:1.
La Figura 19.5b mostra gli intervalli fenotipici
dei genitori e delle generazioni F1 e F2 quando sono
presenti interazioni moderate tra i genotipi e i fattori ambientali. In ogni generazione, ogni genotipo è
681
CAPITOLO 19 - Analisi genetica dei caratteri quantitativi
ANALISI GENETICA 19.1
Il Dott. Ara B. Dopsis, un famoso genetista delle piante, sviluppa diverse linee pure di narcisi. In condizioni ideali di crescita, le piante del ceppo 1 sono le più alte e crescono fino a
un’altezza di 48 centimetri, mentre le piante del ceppo 2 sono le più corte e crescono fino
a 12 centimetri. Il Dott. Dopsis costruisce un modello a tre geni additivi per spiegare l’ereditarietà poligenica dell’altezza della pianta. Presuppone che il ceppo 1 presenti un genotipo A1A1B1B1C1C1 e il ceppo 2 A2A2B2B2C2C2. Nel rispondere alle domande, immaginate che
il genotipo, da solo, determini l’altezza delle piante in condizioni di crescita ideali e che gli
alleli dei tre geni siano additivi.
a. Se queste due linee parentali pure vengono incrociate, quale sarà il genotipo e l’altezza
delle piante della progenie F1?
b. Se viene prodotta una progenie F2, qual è la frequenza attesa delle piante in funzione
dell’altezza in questa generazione?
Strategie per arrivare alla soluzione
Soluzione guidata
Valutazione
1. Identificate l’argomento affrontato dal proble- 1. Questo problema riguarda la valutazione di un modello additima e spiegate la natura della risposta richiesta.
vo a tre geni per l’altezza della pianta, l’applicazione del modello agli incroci tra piante parentali pure di diverse altezze e l’esame della progenie F1 e F2.
2. Identificate le informazioni chiave fornite dal 2. Vengono forniti i genotipi delle piante parentali appartenenti a
problema.
linee pure. Nell’applicare il modello additivo, dobbiamo presumere che il genotipo da solo determini la variabilità nell’altezza
delle piante.
Deduzione
3. Deducete il contributo dato da ogni allele dei 3. L’altezza di 48 cm delle piante del ceppo 1 viene determinata
geni additivi all’altezza nel ceppo1.
da sei alleli di geni additivi. Ogni allele “1” nel genotipo del ceppo 1 dà un contributo di 48 cm/6 = 8 cm all’altezza della pianta.
Suggerimento
Assumete che ogni allele dia un contributo uguale in questo modello genetico additivo.
4. Deducete il contributo dato da ogni allele dei 4. Sei alleli contribuiscono ugualmente all’altezza di 12 cm delle
geni additivi all’altezza nel ceppo 2.
piante del ceppo 2. Ogni allele “2” nel genotipo del ceppo 2 dà
un contributo di 12 cm/6 = 2 cm all’altezza della pianta.
5. Deducete il genotipo dei gameti prodotti da 5. Il ceppo 1 presenta un genotipo A1A1B1B1C1C1 e produce gameti
ogni linea pura.
con il genotipo A1B1C1.
Il ceppo 2 presenta un genotipo A2A2B2B2C2C2 e produce gameti
Suggerimento
con il genotipo A2B2C2.
Le leggi della segregazione e dell’assortimento indipendente si applicano anche ai
geni che controllano i caratteri poligenici.
Soluzione
6. Determinate il genotipo e l’altezza delle piante Risposta a
della F1.
6. La progenie F1 di queste piante parentali pure avrà il genotipo
A1A2B1B2C1C2. Sulla base del contributo di ogni allele 1 e 2, l’altezza delle piante della F1 prevista è [(3)x(8 cm)] +[(3)x(2 cm)]
= 30 cm.
682
19.1 - I caratteri quantitaivi presentano variabilità fenotipica continua
7. Determinate la frequenza e l’altezza di ogni Risposta b
categoria di piante della F2.
7. La progenie F2 attesa è così composta:
Suggerimento
Numero di alleli
Utilizzate il triangolo di Pascal (Figura 2.15) oppure determinate la probabilità dei genotipi contenenti diversi
numeri di alleli 1 e 2.
Attenzione
Ricordate che alla maggior parte delle categorie appartengono diversi genotipi con il numero corrispondente
di alleli 1 e 2.
Frequenza
Altezza (cm)
1
64
12
5
6
64
18
2
4
15
64
24
3
3
20
64
30
4
2
15
64
36
5
1
6
64
42
6
0
1
64
48
1
0
2
6
1
Per esercitarvi ulteriormente, svolgete i Problemi 8, 9 e 20 on-line all'indirizzo http://hpe.pearson.it/sanders
associato a un intervallo di valori fenotipici e nella F2
si osserva un piccolo grado di sovrapposizione tra gli
intervalli fenotipici di diversi genotipi. Nella Figura
19.5c, si osserva un’interazione sostanziale tra i geni
(a) Nessuna interazione
gene-ambiente
Ogni genotipo corrisponde a
un fenotipo discreto
e l’ambiente. Un ampio intervallo di valori fenotipici
è associato a ogni genotipo e nella F2 si osserva un
significativo grado di sovrapposizione fenotipica tra
i genotipi, tanto che una percentuale importante di
(b) Interazioni geneambiente moderate
Piccole sovrapposizioni tra i
fenotipi nella F2
(c) Interazioni gene-ambiente
sostanziali
Intervalli fenotipici ampi e sovrapposizioni
significative
Figura 19.5 Effetto delle interazioni gene-ambiente. Il fenotipo determinato da un singolo gene con alleli codominanti può
essere modificato dall’azione di fattori ambientali.
683
CAPITOLO 19 - Analisi genetica dei caratteri quantitativi
Distribuzione continua
di suscettibilità genetica
nella popolazione
generale
Soglia di
suscettibilità
genetica
Non affetto
Bassa
Affetto
Media
Suscettibilità genetica
Alta
Figura 19.6 Caratteri soglia. Distribuzione fenotipica continua
e soglia di suscettibilità genetica per un carattere soglia.
eterozigoti presenta fenotipi che si sovrappongono a
quelli degli omozigoti. Le interazioni gene-ambiente
di questo tipo, sono tipiche dei caratteri multifattoriali e possono rendere difficile la determinazione
del genotipo di un organismo semplicemente osservando il suo fenotipo. L’uso di una “scheda di valutazione fenotipica” per predire il risultato dell’ereditarietà poligenica e delle interazioni gene-ambiente
nella determinazione del carattere multifattoriale dell’altezza in una pianta ipotetica è illustrato
nell’Approfondimento sperimentale 19.1.
Caratteri soglia
La maggior parte dei caratteri multifattoriali presenta una distribuzione fenotipica continua, ma alcuni di essi, benché abbiano una distribuzione sottostante ugualmente continua, possono essere
suddivisi in categorie distinte. Tali caratteri vengono chiamati caratteri soglia.
I caratteri soglia vengono riscontrati frequentemente in contesti medici in cui vengono effettuati
dei tentativi, non sempre con successo, di identificare due categorie cliniche “non affetto” (o “normale”) e “affetto” (o “anormale”) e pertanto di distinguere individui che presentano un’anomalia
da quelli che non ne presentano. La grande maggioranza dei membri di una popolazione si trova
dal lato non affetto della soglia e presenta un fenotipo normale. Una piccola percentuale della popolazione, tuttavia, si trova dall’altro lato della soglia
e presenta un fenotipo affetto o anormale. I casi situati al confine tra le due categorie possono essere
difficili da diagnosticare.
684
L’ipotesi genetica che spiega i caratteri soglia
propone che il carattere sia poligenico o multifattoriale e che le categorie fenotipiche sottostanti affette
e non affette rappresentino una distribuzione continua di suscettibilità genetica, termine che indica il
rischio di un organismo di avere un fenotipo affetto
in conseguenza dell’eredità di un particolare genotipo. Ogni membro di una popolazione presenta una
specifica suscettibilità genetica determinata da ereditarietà poligenica. La Figura 19.6 mostra una distribuzione continua di suscettibilità genetica per
una popolazione e la designazione di una soglia che
separa gli individui affetti da quelli non affetti nella
popolazione. La percentuale della popolazione che
si trova a sinistra della soglia di suscettibilità genetica, di gran lunga maggiore, è identificata come non
affetta o normale e il piccolo gruppo a destra della
soglia viene considerato affetto o anormale.
Per esaminare l’applicabilità di questi concetti
alle osservazioni nel mondo reale, a livello della popolazione, vengono utilizzati appositi modelli. In
questi modelli la probabilità di varcare la soglia di
suscettibilità aumenta quando nel genotipo sono
presenti diversi “alleli di suscettibilità”. Per esempio, la Figura 19.7a illustra un ipotetico modello a
tre geni nel quale gli alleli sono designati come 1 oppure 2 a livello di ogni locus e nel quale la suscettibilità genetica aumenta all’aumentare degli alleli numero 1. In questo modello la soglia di suscettibilità è
stata posizionata arbitrariamente in modo che debbano essere presenti cinque alleli 1 per superarla.
Un numero di alleli 1 superiore aumenta la percentuale della progenie che si troverà a destra della soglia di suscettibilità e che presenterà un fenotipo affetto. Questo modello permette di valutare i rischi
derivanti da incroci tra genitori che portano diversi
numeri di alleli di suscettibilità.
La Figura 19.7a illustra l’incrocio 1 tra un genitore con due alleli 1 e un genitore con tre alleli 1. Entrambi i genitori presentano un fenotipo normale,
perché entrambi si trovano sul lato non affetto della
soglia. Tra la progenie di questo incrocio, si prevede
1
che 32
(3%) porterà cinque alleli 1, ma nessuno po1
trà portare sei alleli 1. Pertanto 32
della progenie si
troverà a destra della soglia di suscettibilità e presenterà il fenotipo affetto. La Figura 19.7b presenta
l’Incrocio 2 con genitori diversi che producono un
19.1 - I caratteri quantitaivi presentano variabilità fenotipica continua
Approfondimento sperimentale 19.1
Scheda di valutazione fenotipica: simulazione di un fenotipo quantitativo multifattoriale
In questo Approfondimento sperimentale viene presentata un’attività pratica che illustra un approccio alla creazione
di un modello per un carattere quantitativo multifattoriale.
In questo esempio ipotetico, l’altezza matura di una pianta è sotto il controllo di cinque geni additivi, indicati con le
lettere da A a E. Due alleli per ogni gene danno contributi diversi all’altezza. Ogni allele con il pedice 1 aggiunge 5
centimetri al potenziale genetico e ogni allele con il pedice
2 aggiunge 10 cm. Pertanto, una pianta omozigote per gli
alleli 1 in tutti i loci (A1A1B1B1C1C1D1D1E1E1) ha un potenziale
genetico per un’altezza pari a [(10 alleli)x(5 cm)] = 50 cm,
rispetto alle piante che portano un genotipo composto interamente da alleli 2, che hanno un’altezza potenziale di
[(10 alleli)x(10 cm)] = 100 cm. Le piante che portano genotipi con diversi numeri di alleli 1 e 2 presentano potenziali
diversi per l’altezza, che giacciono a intervalli di 5 cm l’uno
dall’altro in un continuo compreso tra 50 e 100 cm.
A questo punto, poniamoci la seguente domanda: “Quanti alleli 1 e quanti alleli 2 devono essere presenti per avere
un’altezza potenziale di 80 cm?” Ogni genotipo contiene un
totale di 10 alleli, due per ognuno dei cinque loci. Pertanto,
ogni genotipo con sei alleli 2 e quattro alleli 1 produrrà un
potenziale di altezza pari a [(6)x(10) + (4)x(5)] = 80 cm.
Una domanda supplementare potrebbe essere: “Quale
proporzione della progenie di due piante, ognuna con un
potenziale di altezza pari a 75 cm, avrà un potenziale di altezza pari a 80 cm?” Questo problema è più complesso. Le
piante con un potenziale di altezza pari a 75 cm presentano cinque alleli 2 e cinque alleli 1 [(5)(10) + (5)(5) = 75]. I genotipi della progenie che contengono sei alleli 2 e quattro
alleli 1 avranno un potenziale di altezza di 80 cm. Possiamo
utilizzare l’istogramma della Figura 19.3 e per predire la risposta: 210 piante sulle 1024 della progenie (pari al 20,5%)
hanno sei copie di alleli 2 e quattro copie di alleli 1.
Dopo aver esaminato la correlazione tra genotipo e altezza
potenziale in questo modello, valutiamo l’effetto di cinque
fattori ambientali sul raggiungimento dell’altezza:
1. Quantità di acqua
2. Quantità di luce solare
3. Drenaggio del suolo
4. Contenuto in nutrienti del suolo
5. Temperatura
livello più elevato di suscettibilità genetica nella progenie. In questo incrocio, ogni genitore porta tre alleli di suscettibilità, ma nessuno dei due è affetto
perché la soglia di suscettibilità è pari a 5 o più al-
Ogni fattore ambientale può variare da ottimale a scarso.
Supponiamo che l’altezza potenziale completa venga raggiunta quando tutti i fattori sono ottimali. Tuttavia, se uno
o più fattori ambientali sono inferiori all’ottimale, l’altezza
è ridotta. Lo stato di ogni fattore ambientale ha un effetto
sulla crescita. In questo esercizio, supporremo che l’altezza è alterata secondo quanto indicato dalla seguente scala:
Stato del fattore ambientale
Perdita di altezza
Ottimale (O)
0 cm persi
Buono (G)
4 cm persi
Discreto (F)
8 cm persi
Al limite (M)
12 cm persi
Scarso (P)
16 cm persi
Se, per esempio, una condizione è ottimale, due sono buone, una è discreta e una è al limite, la perdita di altezza potenziale è di 28 cm.
La tabella seguente mostra come lo stesso genotipo possa
produrre diversi fenotipi in diverse condizioni ambientali e
come diversi genotipi possano produrre fenotipi simili in
diverse condizioni.
Notate che i primi due genotipi sono identici, ma producono fenotipi diversi a causa delle differenze ambientali. Notate, inoltre, che il terzo genotipo presenta un potenziale di
altezza inferiore, ma, in combinazione con un ambiente ottimale, produce una pianta più alta. Potete provare nuove
combinazioni di genotipi e condizioni di crescita per osservare i diversi risultati.
Genotipo
Stati dei fattori
Potenziale ambientali
di altezza 1 2 3 4 5
Altezza
raggiunta
A1A2B1B2C2C2D1D2E1E2 80 cm
GFOGM
52 cm
A1A2B1B2C2C2D1D2E1E2 80 cm
FMGGF
44 cm
A1A1B1B2C1C2D1D2E1E2 70 cm
OGGGG
54 cm
leli di suscettibilità. Tuttavia, tra la progenie, l’as7
sortimento indipendente predice che 64
(11%) presenterà genotipi che contengono cinque o più alleli
1. Questi individui della progenie si trovano a de-
685
CAPITOLO 19 - Analisi genetica dei caratteri quantitativi
(a) Incrocio 1: A1A2B1B2C2C2 × A1A2B1B2C2C2
(Due alleli di suscettibilità)
(Tre alleli di suscettibilità)
Numero di alleli di suscettibilità
Frequenza della progenie
Non affetto
Affetto
Soglia di
suscettibilità
Proporzione della progenie
(b) Incrocio 2: A1A2B1B2C1C2 × A1A2B1B2C1C2
(Tre alleli di suscettibilità)
(Tre alleli di suscettibilità)
Numero di alleli di suscettibilità
Frequenza della progenie
Non affetto
Affetto
Soglia di
suscettibilità
Proporzione della progenie
Figura 19.7 Modello poligenico per un carattere soglia.
Tutti gli alleli indicati con 1 conferiscono suscettibilità genetica,
tutti gli alleli indicati con 2 non conferiscono suscettibilità; gli
alleli sono additivi. (a) Nell’incrocio 1, la coppia presenta una
1
di generare un bambino affetto. (b) Nell’incrocio
probabilità di 32
7
di generare un
2, la coppia presenta una probabilità di 64
bambino affetto.
L’influenza dei fattori ambientali e dello sviluppo sui fenotipi dei caratteri soglia rappresenta
una componente aggiuntiva importante. Questi fattori sembrano giocare un ruolo fondamentale nel
determinare se i singoli organismi che presentano
una suscettibilità genetica prossima alla soglia finiscano da un lato o dall’altro della soglia stessa. Nel
modello a soglia si ipotizza che gli organismi che
possiedono un’elevata suscettibilità genetica (cioè
che possiedono un genoma con molti alleli di suscettibilità) abbiano il potenziale di sviluppare il fenotipo affetto. Il fenotipo affetto si sviluppa anche in
funzione dell’influenza di altri fattori ereditari, dello
sviluppo o ambientali. Meno spesso, un organismo
può presentare una suscettibilità genetica leggermente inferiore alla soglia, ma l’influenza dei fattori
ambientali può spingere il fenotipo nella categoria
di coloro che sono affetti.
Infine, è importante prendere in considerazione
un altro aspetto relativo alla definizione delle categorie e alla classificazione dei caratteri soglia, in particolare negli esseri umani. Poiché questi caratteri
sono quantitativi e ricadono lungo un continuo, la
determinazione precisa di categorie e fenotipi può
essere inesatta.
Per esempio, è facile classificare la pressione sanguigna di una persona come normale se si trova
all’interno dell’intervallo dei valori normali, o come
anormale se la pressione è molto alta. Tuttavia,
molte persone presentano pressioni elevate “borderline”, che sono difficili da assegnare a una delle
due categorie, pressione normale o alta.
CONCETTI CHIAVE
stra della soglia di suscettibilità e presentano il fenotipo affetto. I genotipi del secondo incrocio presentano un aumento di quasi quattro volte del rischio (il
3% rispetto all’11%) di produrre una prole affetta rispetto al primo incrocio.
Questa differenza è analoga a quella che si può
osservare per un accoppiamento tra individui che
appartengono a una popolazione generale con un
rischio basso di generare un figlio con un carattere
soglia rispetto a un accoppiamento tra genitori che
provengono entrambi da famiglie con precedenti
per questo carattere.
686
I caratteri soglia sono determinati dall’ereditarietà poligenica e possono inoltre essere influenzati
dai fattori ambientali. Presentano una distribuzione continua della suscettibilità genetica in una popolazione. La maggior parte degli organismi porta
un piccolo numero di alleli di suscettibilità e mostra
il fenotipo non affetto, o normale. Pochi organismi
presentano genotipi con un numero di alleli di suscettibilità sufficiente a collocare l’organismo al di là
della soglia di suscettibilità e a produrre un genotipo anormale (affetto).
19.2 - L'analisi dei caratteri quantitativi è statistica
19.2 L’analisi dei caratteri quantitativi
è statistica
I metodi statistici più spesso applicati, ai giorni nostri, allo studio dei caratteri quantitativi rappresentano una estensione diretta dei contributi dati circa
un secolo fa dal biologo statistico ed evolutivo Sir
Ronald Fisher. Nel 1918, Fisher utilizzò l’analisi statistica per dimostrare che i caratteri quantitativi derivano dalla segregazione di alleli di geni multipli
che presentano un effetto additivo.
Fisher dimostrò inoltre che questi metodi possono rilevare le interazioni tra geni. In più, analizzò
il ruolo delle interazioni gene-ambiente e concluse
che i fattori ambientali contribuiscono alla variabilità continua sfumando le linee di demarcazione
tra le classi fenotipiche. Gli strumenti e gli approcci
qui descritti e di cui Fisher fu pioniere permettono
agli scienziati di identificare le influenze genetiche
sui fenotipi in termini di misura quantitativa piuttosto che di aspetto qualitativo. Nella seguente descrizione e nelle illustrazioni dell’analisi dei caratteri
quantitativi, riesamineremo alcuni concetti della
statistica descritti per l’analisi del Chi-quadrato (Sezione 2.5).
(a) Numero e frequenza delle altezze a intervalli di 3 cm
Altezza (cm)
Numero
Frequenza %
(b) Grafico che mostra la variabilità continua dell’altezza
Il primo passaggio nella quantificazione della variabilità fenotipica di un carattere in una popolazione è
costruire una distribuzione delle frequenze dei valori del carattere stesso su una scala quantitativa.
Una distribuzione delle frequenze mostra la percentuale della popolazione che presenta ogni valore misurato del carattere o che ricade in ogni categoria
definita per il carattere stesso. La Figura 19.8a fornisce un esempio, presentando il numero e la frequenza di ogni categoria designata per l’altezza in
un campione di 1000 maschi in età universitaria. La
Figura 19.8b rappresenta gli stessi dati graficamente, in forma di istogramma, illustrando la distribuzione continua dell’altezza tra questi soggetti.
Gli individui in questo studio sono considerati
un campione casuale. Non sono stati selezionati per
alcun attributo correlato alla loro altezza e pertanto
si presuppone che la distribuzione delle loro altezze
assomigli a quella della popolazione generale di ma-
Numero di individui
Descrizione statistica della variabilità
fenotipica
Altezza in centimetri
Figura 19.8 Altezza adulta dei maschi. La distribuzione
della frequenza dell’altezza di 1000 maschi in età universitaria
viene mostrata sotto forma di (a) tabella (b) istogramma.
687
CAPITOLO 19 - Analisi genetica dei caratteri quantitativi
schi della stessa età. I campioni casuali vengono utilizzati nell’analisi dei caratteri quantitativi per due
ragioni. In primo luogo è spesso impossibile o impraticabile raccogliere i dati relativi a ogni individuo di una popolazione; e, in secondo luogo, i campioni casuali possono essere altrettanto accurati, in
senso statistico, in quanto “campioni” che consistono di popolazioni intere. In maniera analoga, per
la maggior parte degli esami del sangue di routine,
vengono prelevati circa 10 millilitri di sangue, all’incirca due decimi dell’1% del volume di sangue totale di una persona. La quantità presa non è tanto
abbondante da causare problemi fisiologici, ma è
sufficientemente rappresentativa da fornire informazioni attendibili sullo stato di salute di una persona. Dopo la costruzione della distribuzione delle
frequenze, la prima informazione che si ottiene è il
valore medio o media ( x ) della distribuzione. Ricordiamo che questo viene calcolato sommando tutti i
valori del campione e dividendo per il numero totale di individui appartenenti al campione (n; vedere
Sezione 2.5). Per i campioni degli uomini in età universitaria della Figura 19.8, il valore medio dell’altezza è 175,33 cm (circa 68,5 pollici). Le forme delle
distribuzioni di frequenza variano in funzione di diversi fattori, inclusa la dimensione del campione e il
numero delle categorie utilizzate per classificare il
carattere. Pertanto è necessario fornire una descrizione statistica della forma della distribuzione delle
frequenze quando si confrontano i valori del carattere. Per esempio è importante riportare la moda, o
il valore modale, cioè il valore più comune in una
distribuzione. Per i dati relativi all’altezza mostrati
in Figura 19.8, la moda è rappresentata dalla categoria 173-175 cm che contiene 188 valori individuali. Ogni distribuzione possiede inoltre un valore
di mezzo, noto come mediana, o valore mediano.
Nella distribuzione delle altezze, potete pensare
alla mediana come il valore numero 500 (in ordine
crescente) dei 1000 valori della distribuzione. Anche questo valore mediano appartiene alla categoria 173-175 cm. I dati che si ottengono in situazioni
reali sono generalmente asimmetrici, cioè distribuiti in maniera diseguale su un lato o l’altro della media, come illustrato in Figura 19.8. Pertanto, per descrivere la distribuzione delle frequenze, dobbiamo
disporre anche di modalità per misurare (e quindi
688
descrivere) la natura della distribuzione attorno alla
media. Due tipi di misura vengono utilizzati normalmente.
La prima, chiamata varianza (s 2), è una misura
numerica dell’estensione della distribuzione attorno
alla media. Questa misura fornisce un’indicazione
sulla variabilità esistente tra gli individui del campione. Il valore della varianza dipende dalla correlazione tra l’ampiezza della distribuzione e il numero di osservazioni nel campione. Sarà piccolo se
tutte le osservazioni sono vicine alla media, e grande
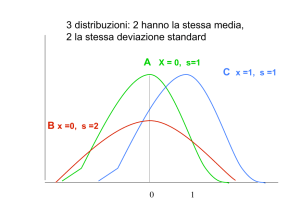
se le osservazioni sono distribuite ampiamente attorno alla media (Figura 19.9). La varianza è determinata sommando il quadrato della differenza tra
ogni singolo valore e la media del campione e dividendo questa somma per il numero di gradi di libertà (df) nel campione. Il numero di gradi di libertà
è pari al numero di variabili indipendenti. Portare
al quadrato le differenze tra i valori individuali e la
media del campione permette di evitare che le differenze negative e positive si annullino le une con le
altre. Per questa ragione la varianza è espressa come
unità al quadrato:
s2 = ∑(xi – x )2/df
Nel caso della variabilità di un fenotipo quantitativo, la varianza viene descritta come varianza fenotipica (V P). Poiché stiamo misurando l’altezza in
centimetri, la varianza sarà espressa in centimetri
quadrati.
La seconda misura che descrive la distribuzione
dei dati è la deviazione standard (s), valore che
esprime la deviazione dalla media nelle stesse unità
della scala utilizzata per misurare il campione.
La deviazione standard (s) viene calcolata come:
s = s2. Nel nostro campione relativo all’altezza di
maschi adulti, VP = s2 = 43,30 cm2 e la deviazione
standard è s = 6,58 cm.
CONCETTI CHIAVE
I caratteri quantitativi sono generalmente misurati
utilizzando un campione casuale. La media è il valore medio del campione e la deviazione dalla media viene misurata come varianza o come deviazione standard.
19.2 - L'analisi dei caratteri quantitativi è statistica
Numero di organismi in ogni categoria fenotipica
Varianza grande con
relativamente pochi
organismi in ogni
categoria
Varianza intermedia
con un numero più
elevato di organismi
in ogni categoria
Varianza piccola con un
numero più elevato di
organismi in un piccolo
numero di categorie
Distribuzione fenotipica
Figura 19.9 Distribuzioni normali. La forma delle curve
che illustrano le distribuzioni normali cambia in funzione della
dimensione del campione e del numero delle classi. La varianza
attorno alla media è, rispettivamente, grande, intermedia e
piccola.
Suddivisione della varianza fenotipica
Un aspetto cruciale dell’analisi della variabilità
dei caratteri quantitativi è l’analisi dei fattori che,
presumibilmente, contribuiscono alla varianza fenotipica (VP, dove P è l’iniziale di phenotype). I fenotipi quantitativi rappresentano il risultato congiunto
di geni, ambiente e interazioni geniche; di conseguenza la varianza fenotipica può essere suddivisa
tra queste influenze. In primo luogo, la varianza fenotipica può essere divisa in due componenti principali: varianza genetica (VG, dove G è l’iniziale di genotype) e varianza ambientale (VE, dove E è l’iniziale
di environment).
Secondo questo assunto, la varianza fenotipica
può essere espressa in termini di varianza genetica
più varianza ambientale: Vp = VG + VE.
In questa espressione, la varianza genetica (VG)
rappresenta quella parte della varianza fenotipica
dovuta alle differenze tra i genotipi. In popolazioni
altamente consanguinee, in cui tutti gli individui
sono omozigoti per gli alleli che controllano un fenotipo quantitativo, VG = 0. Tuttavia, queste popolazioni si ottengono solo per accoppiamenti tra familiari altamente controllati in laboratorio; non si
rinvengono quasi mai in natura a causa della presenza ubiquitaria della variabilità genetica nelle popolazioni naturali. La variazione genetica nelle popolazioni naturali genera individui con genotipi
diversi per i caratteri quantitativi e risulta in una variabilità fenotipica che può essere attribuita direttamente alla variabilità genetica.
La varianza ambientale (VE) è la porzione della
varianza fenotipica dovuta alla variabilità degli ambienti abitati dai singoli membri di una popolazione.
Differenze nell’esposizione al sole, nel contenuto
di acqua e nutrienti del suolo e nell’esposizione ai
parassiti sono esempi di variabilità ambientali che
influenzano la VE nelle piante. Conducendo esperimenti altamente controllati in laboratorio è talvolta possibile controllare tutte le varianti ambientali e produrre una situazione in cui VE = 0. In
natura, tuttavia, tali circostanze non si presentano
praticamente mai. I singoli membri delle popolazioni naturali sperimenteranno quasi certamente
una variabilità delle condizioni ambientali in cui si
imbatteranno. Alcune differenze possono essere sistematiche e prevedibili. Per esempio, i membri di
una popolazione di piante che crescono sotto una
sorgente naturale sperimenteranno delle condizioni
di crescita più umide rispetto alle piante che vivono
sopra la sorgente. Altre variabili ambientali sono
sporadiche o imprevedibili. Per esempio, un’annata secca potrebbe ridurre il flusso di acqua da una
sorgente naturale e incidere più gravemente sulle
piante che vivono sotto la fonte rispetto a quelle che
vivono sopra.
Facciamo un esempio per illustrare la scomposizione di VG e VE come componenti di VP. Supponiamo che vengano generati due diverse linee parentali pure. Ogni ceppo è geneticamente uniforme,
689
CAPITOLO 19 - Analisi genetica dei caratteri quantitativi
con VG = 0; pertanto VP = VE (Figura 19.10a). Le linee pure vengono incrociate per produrre una progenie F1 geneticamente uniforme. Nella F1, VG = 0
perché non vi è variazione genetica tra gli individui
e VP = VE (Figura 19.10b). La produzione di F2 porta
a variabilità genotipica e pertanto alla produzione
di variabilità fenotipica che risulta da una combinazione di varianza genetica e varianza ambientale
(Figura 19.10c). Nella F2, VP = VE + VG. Poiché VE è
stata determinata nei genitori e nella F1, la varianza
genetica può essere calcolata sottraendo la varianza
ambientale dalla varianza fenotipica nella F2. In altre
parole, VG = VP – VE. L’Analisi genetica 19.2 vi permetterà di fare pratica nel determinare la varianza
genetica e ambientale.
Suddivisione della varianza genetica
Ogni differenza allelica che interessa un carattere
quantitativo contribuisce alla varianza genetica in
(a)
Entrambi i ceppi parentali
sono geneticamente
uniformi, pertanto VP = VE
una popolazione, ma non necessariamente tutte le
differenze alleliche contribuiscono nello stesso
modo. In effetti, può essere difficile misurare l’effetto specifico di ogni variante allelica. Tuttavia, la
varianza genetica può essere suddivisa teoricamente
in tre diversi tipi di effetti allelici. La varianza additiva (VA) deriva dagli effetti additivi di tutti gli alleli
che contribuiscono al carattere. La varianza additiva
è il risultato della dominanza incompleta degli alleli
in un locus, che fa sì che gli eterozigoti abbiano un
fenotipo intermedio tra i fenotipi omozigoti. La varianza dominante (VD) è la varianza che deriva dai
rapporti di dominanza tra gli alleli di un eterozigote
che producono un fenotipo non intermedio tra
quelli degli omozigoti (cioè, effetti non additivi degli
alleli dei geni che contribuiscono al fenotipo). Infine, la varianza interattiva (VI) deriva dalle interazioni epistatiche tra gli alleli dei geni che influenzano un fenotipo quantitativo.
Collettivamente queste tre componenti si uniscono per produrre la varianza genetica in un modello riassunto dall’espressione VG = VA + VD + VI.
Utilizzeremo questi valori nella sezione seguente,
per discutere l’ereditabilità.
CONCETTI CHIAVE
(b)
Gli individui della F1 sono
geneticamente uniformi,
pertanto VP = VE
La varianza fenotipica dei caratteri quantitativi consiste nella varianza genetica (VG) e nella varianza ambientale (VE). La varianza genetica può essere suddivisa in componenti separate che tengono
conto della dominanza, dell’additività e delle interazioni epistatiche tra geni.
19.3 L’ereditabilità misura la
componente genetica della variabilità
fenotipica
(c)
La varianza fenotipica
nella generazione F2
risulta dalla varianza
genetica e ambientale
Figura 19.10 Fonti di varianza fenotipica.
690
Uno degli obiettivi della genetica quantitativa è
quello di stimare il grado di influenza della variabilità genetica sulla variabilità fenotipica osservata per
un dato carattere. Si tratta di un compito difficile
quando un carattere è determinato da una combinazione di variabilità genetica, variabilità ambientale e
interazioni gene-ambiente.
Il concetto di ereditabilità è stato sviluppato per
permettere di misurare la percentuale della variabi-
19.3 - L'ereditabilità misura la componente genetica della variabilità fenotipica
ANALISI GENETICA 19.2
Vengono incrociate due linee pure di pomodori, P1 e P2, che producono frutti con pesi medi diversi. Le medie e le varianze delle loro
progenie F1 e F2 sono mostrate nella tabella a destra.
a. Qual è la varianza ambientale (VE) per questo carattere?
b. Qual è la varianza genetica (VG) calcolata a partire dalla F2?
Strategie per arrivare alla soluzione
Ceppo
P1
P2
F1
F2
Peso medio del frutto (g)
6,5
14,2
10,2
9,8
VP
1,6
3,5
2,2
4,0
Soluzione guidata
Valutazione
1. Identificate l’argomento affrontato dal proble- 1. Questo problema riguarda la determinazione della varianza
ma e spiegate la natura della risposta richiesta.
ambientale e della varianza genetica per i dati forniti, relativi a
piante di pomodoro.
2. Identificate le informazioni chiave fornite dal 2. Il peso del frutto e la varianza fenotipica vengono dati per due
problema.
linee parentali pure e per la progenie F1 e F2.
Deduzione
3. Descrivete la correlazione tra VP, VG e VE.
3. VP = VG + VE
4. Identificate i valori di varianza che contribuisco- 4. Le linee parentali pure (P1 e P2) e la progenie F1 sono geneticano a VP in ogni ceppo e generazione.
mente uniformi. Di conseguenza, in questi casi, tutta la varianza
Suggerimento
fenotipica è dovuta alla varianza ambientale e la varianza genePer gli organismi che sono geneticamente
tica non dà contributi. La F2 contiene variabilità genotipica, peridentici VP = VE
tanto sia VG che VE contribuiscono a VP.
Soluzione
5. Determinate VE per questo carattere.
Risposta a
5. Nelle piante geneticamente uniformi di P1, P2 e F1, VG = 0 e in
ogni ceppo VP = VE. La varianza ambientale media in questi ceppi viene calcolata come (1,6 + 3,5 + 2,2)/3 = 2,43 grammi.
6. Determinate VG per questo carattere.
Risposta b
6. VG viene calcolata modificando l’espressione del passaggio 3 in
VG = VP – VE. La varianza genetica per questi dati è VG = 4,0 – 2,43
= 1,57 grammi.
Per esercitarvi ulteriormente, svolgete i Problemi 4, 10, 12 e 14 on-line all'indirizzo http://hpe.pearson.it/sanders
lità fenotipica dovuta alla variabilità genetica. L’ereditabilità è diversa per ogni carattere. La variabilità
fenotipica osservata per un carattere con ereditabilità elevata è ampiamente dovuta alla variabilità genetica e pertanto può essere influenzata fortemente
da programmi di selezione che hanno l’obiettivo di
cambiare la frequenza di un fenotipo in una popolazione. Invece, solo una piccola percentuale della
variabilità fenotipica di un carattere con ereditabi-
lità bassa può essere attribuita alla variabilità genetica e quindi l’espressione del carattere in una popolazione non viene modificata in maniera efficace
da processi di selezione. L’ereditabilità è una misura
importante della potenziale responsività di un carattere alla selezione naturale o artificiale. Questo parametro è di particolare intesse per i biologi evoluzionistici, per i coltivatori e per gli allevatori, che lo
utilizzano per valutare l’impatto potenziale della se-
691
CAPITOLO 19 - Analisi genetica dei caratteri quantitativi
lezione su caratteri di importanza agricola o economica, come descriveremo nel Caso di studio che
conclude questo capitolo.
Due misure di ereditabilità ampiamente utilizzate permettono di valutare le diverse componenti
del contributo dato dalla variabilità genetica alla va2
riabilità fenotipica. L’ereditabilità in senso lato (H )
stima la percentuale della variabilità fenotipica dovuta alla variabilità genetica totale. Questa forma di
2
ereditabilità è definita dall’uguaglianza H = VG/VP.
2
L’ereditabilità in senso stretto (h ) stima la percentuale della variabilità fenotipica dovuta alla variabilità genetica additiva. L’ereditabilità in senso stretto
2
è definita dall’uguaglianza h = VA/VP. Entrambe le
misure di ereditabilità sono espresse come rapporti
che vanno da 0,0 a 1,0. In entrambi i casi, valori più
elevati di ereditabilità indicano un ruolo più importante esercitato dalla variabilità genetica sulla variabilità fenotipica.
L’ereditabilità viene facilmente fraintesa. Una
comprensione errata può portare all’idea sbagliata
che la variabilità genetica dia un contributo più importante alla variabilità fenotipica rispetto a quanto
attualmente supportato dai dati disponibili. Il concetto di ereditabilità è difficile da applicare agli esseri
umani tranne in circostanze particolari (descritte oltre nella discussione degli studi sui gemelli), ma può
essere utilizzato per altri organismi. I seguenti attributi dell’ereditabilità sono fondamentali per comprendere il suo significato:
1. L’ereditabilità è una misura di quanto le differenze genetiche contribuiscano alla variabilità fenotipica di un carattere. In altre parole, l’ereditabilità è elevata quando gran parte della variabilità
fenotipica è prodotta dalla variabilità genetica e
la variabilità ambientale fornisce uno scarso contributo. L’ereditabilità non indica il meccanismo
tramite il quale i geni controllano un carattere,
né dà una misura di quanto un carattere sia prodotto dall’azione dei geni.
2. I valori dell’ereditabilità sono accurati solo per
l’ambiente e la popolazione nel quale sono misurati. I valori dell’ereditabilità misurati in una popolazione non possono essere trasferiti a un’altra popolazione, perché tanto i fattori genetici
quanto quelli ambientali possono differire tra le
popolazioni.
692
3. L’ereditabilità per un dato carattere in una popolazione può cambiare se cambiano i fattori ambientali e modifiche nelle proporzioni dei genotipi in una popolazione possono alterare l’effetto
dei fattori ambientali sulla variabilità fenotipica,
cambiando così l’ereditabilità.
4. Un’elevata ereditabilità non significa che un carattere non sia influenzato dai fattori ambientali.
I caratteri con ereditabilità elevata possono essere molto responsivi a cambiamenti ambientali.
Ereditabilità in senso lato
Abbiamo visto che la varianza genetica (VG) è un valore composito la cui entità deriva dalla somma delle
varianze additiva, di dominanza e interattiva. Purtroppo, la varianza genetica non è sempre facile da
suddividere in queste componenti separate. Per for2
tuna, però, l’ereditabilità in senso lato (H = VG/VP)
può essere utilizzata come una misura generale del
grado di influenza genetica esercitata sulla variabilità fenotipica di un carattere, quando VG non può
essere suddiviso.
In uno studio del 1988 sulla genetica e l’evoluzione del pesce Astyanax fasciatus, Horst Wilkens utilizzò l’analisi dell’ereditabilità in senso lato
per descrivere il contributo genetico all’evoluzione
del tessuto oculare dell’organismo. Alcune popolazioni di questa specie vivono nei corsi d’acqua di caverne sotterranee completamente buie del Messico
orientale e presentano una quantità di tessuto oculare estremamente ridotta rispetto a pesci strettamente imparentati che vivono al di sopra del livello
del suolo. In queste popolazioni, il tessuto oculare
sembra essere stato sottoposto a un rapido cambiamento evolutivo. Gli occhi dei pesci vedenti presentano un diametro di circa 7 cm. In confronto, i pesci Astyanax fasciatus ciechi presentano un tessuto
oculare con un diametro di meno di 2 cm.
Wilkens incrociò Astyanax fasciatus vedenti con
Astyanax fasciatus ciechi, misurò la media e la varianza del tessuto oculare nella F1 e quindi produsse
pesci F2, nei quali prese le stesse misure del tessuto
oculare. Dato che i pesci della F1 sono geneticamente uniformi, la varianza nella quantità del tessuto oculare è dovuta interamente all’ambiente. In
queste generazioni F1, VE era pari a 0,057 cm. Nelle
19.3 - L'ereditabilità misura la componente genetica della variabilità fenotipica
F2, la varianza fenotipica (VP) era pari a 0,563 cm ed
era il risultato sia della varianza genetica che ambientale (VG + VE). L’eredità in senso lato viene calcolata determinando VG e dividendolo per la variabilità fenotipica. In questo caso,
VG = VP -VE = 0,563 – 0,057 = 0,506
2
H = VG/VP = 0,506/0,563 = 0,899
Questo valore di ereditabilità in senso lato, pari a
circa 0,90, significa che circa il 90% della variabilità
fenotipica nelle dimensioni dell’occhio in queste popolazioni di Astyanax fasciatus è dovuta alla variabilità genetica.
Studi sui gemelli
L’ereditabilità può essere quantificata quando è
possibile controllare sia gli accoppiamenti che i fattori ambientali. Tuttavia, quando la variabilità dovuta agli accoppiamenti e quella dovuta all’ambiente
non fanno parte dei parametri sperimentali controllati, l’ereditabilità è molto più difficile (o, a detta di
alcuni, impossibile) da misurare. Questa limitazione
si applica ai tentativi di misurare l’ereditabilità dei
caratteri negli esseri umani. Fortunatamente, gli
studi sulla variabilità fenotipica nei gemelli offrono
nuove prospettive sull’ereditabilità in senso lato dei
caratteri umani.
I gemelli identici, anche noti come gemelli monozigoti (gemelli MZ), sono generati da un solo
evento di fecondazione, seguito dalla divisione
dell’embrione fecondato in due zigoti. I gemelli monozigoti condividono alleli identici. Teoricamente,
l’ereditabilità in senso lato può essere determinata
assumendo che la varianza fenotipica tra di essi sia
completamente attribuibile alla varianza ambientale. Secondo questo assunto, nelle coppie di gemelli
MZ, VP = VE.
I gemelli fraterni, d’altro canto, sono dizigoti
(gemelli DZ), prodotti da due eventi di fecondazione indipendenti che avvengono contemporaneamente. I gemelli dizigoti sono fratelli o sorelle nati
nello stesso momento, ma non sono correlati più
strettamente rispetto a fratelli nati in momenti diversi. Come tutti i fratelli/sorelle pieni i gemelli DZ
hanno una media del 50% di alleli in comune. Per
controllare le differenze tra i sessi, in questi studi
sui gemelli sono stati utilizzati solo gemelli DZ dello
stesso sesso. La varianza fenotipica tra gemelli DZ è
la somma della varianza ambientale più metà della
varianza genetica (il 50% degli alleli non condivisi
dalla coppia media di gemelli DZ): nelle coppie di
gemelli DZ, VP = VE + VG.
Sulla base di queste formule generali per il cal2
colo di H , l’ereditabilità in senso lato dei caratteri
umani può essere stimata tramite metodi che non
discuteremo qui (Tabella 19.2).
Gli studi dei caratteri sui gemelli umani, confrontano generalmente gemelli MZ con gemelli DZ
dello stesso sesso per effettuare delle stime di ereditabilità più accurate. Anche così, gli studi sull’ereditabilità nei gemelli umani sono soggetti a diverse
fonti di errore che portano a valori inaccuratamente
elevati. Di seguito sono elencate le fonti più comuni
di errore:
1. Effetti materni condivisi più forti tra gemelli
identici rispetto a gemelli fraterni. Questi effetti
includono la condivisione delle membrane em-
Tabella 19.2 Alcuni valori di ereditabilità in
senso lato (H2) ottenuti da studi
su gemelli
Carattere
Ereditabilità (H 2), %
Caratteristiche biologiche
Conteggio del totale delle creste
90
delle impronte digitali
Altezza
85
Massima frequenza cardiaca
85
Piede equino
80
Escrezione di amminoacidi
70
Peso
60
Colesterolo totale sierico
60
Pressione sanguigna
60
Indice di massa corporea (IMC)
50
Longevità
29
Caratteri comportamentali
Abilità verbale
65
Indice di socievolezza
65
Indice di temperamento
60
Attitudine alla scrittura
50
Memoria
50
Attitudine alla matematica
30
693
CAPITOLO 19 - Analisi genetica dei caratteri quantitativi
brionali e altri aspetti dell’ambiente uterino che
portano a condizioni di sviluppo maggiormente
simili per i gemelli identici rispetto ai gemelli fraterni.
2. Maggiore somiglianza nel modo di trattare i gemelli identici rispetto ai gemelli fraterni. I genitori, gli altri adulti e i pari tendono a trattare i
gemelli identici in maniera più uguale rispetto a
quanto accade con gemelli fraterni dello stesso
sesso. Ciò conferisce ai gemelli identici un’esperienza sociale e comportamentale simile, mentre
i gemelli fraterni sono trattati più spesso in maniera differente.
3. Maggiore somiglianza nelle interazioni tra fattori
genetici e ambientali nei gemelli identici rispetto
ai gemelli fraterni. I gemelli identici presentano
lo stesso genotipo e sono influenzati in maniera
simile, se non identica, dai fattori ambientali.
D’altro canto, i gemelli fraterni presentano differenze genetiche che possono essere influenzate
in maniera diversa dai fattori ambientali. Ciò può
determinare una varianza maggiore tra i gemelli
fraterni rispetto a quanto avviene per i gemelli
identici.
Date le difficoltà e le potenziali fonti di errore nell’effettuare delle stime di ereditabilità sulla base degli
studi sui gemelli, è probabile che i valori della Tabella 19.2 tendano a essere troppo elevati e non
troppo bassi.
Lo studio dei gemelli identici cresciuti insieme
rispetto a quelli cresciuti in ambienti diversi rappresenta un approccio alternativo per stimare l’influenza dei geni sulla variabilità fenotipica. Tali
studi misurano la concordanza, cioè la percentuale
di coppie di gemelli nei quali entrambi presentano
lo stesso fenotipo per un carattere, rispetto alla discordanza, ovvero la percentuale dei casi in cui
i gemelli presentano fenotipi diversi per un carattere. Le frequenze di concordanza e di discordanza
danno un quadro generale dell’influenza globale dei
geni sui fenotipi. Se la variabilità fenotipica per un
carattere è per il 100% genetica, i gemelli MZ devono essere sempre concordanti nei loro fenotipi,
sia che siano cresciuti insieme che separatamente.
In questo caso, la concordanza sarà del 100%. I gemelli dizigoti condividono una media del 50% dei
694
geni e presenteranno una concordanza di circa il
50% per un carattere la cui variabilità è completamente genetica. Quando la variabilità fenotipica di
un carattere è dovuta interamente a fattori non genetici, d’altro canto, la concordanza tra gemelli MZ
e DZ dovrebbe essere approssimativamente uguale,
ed entrambi i valori dovrebbero essere significativamente inferiori al 100%. Per i caratteri con variabilità fenotipica determinata in maniera significativa
dalla variabilità genetica, la concordanza tra le coppie di gemelli MZ sarebbe sostanzialmente inferiore
al 100%, ma significativamente superiore a quella
dei gemelli DZ. Un certo numero di malattie, malformazioni e altre varianti fenotipiche umane ricadono in quest’ultima categoria. La Tabella 19.3 mostra i valori di concordanza nei gemelli MZ e DZ per
malformazioni comuni e altre anomalie che sono
determinate in buona parte dalla variabilità genetica, ma che sono innescate da fattori ambientali che
giocano un ruolo ancora da chiarire.
CONCETTI CHIAVE
La variabilità in senso lato, che misura il rapporto tra
la variabilità genetica e la variabilità fenotipica di un
carattere quantitativo, viene utilizzata per stimare il
contributo genetico alla variabilità fenotipica. I confronti delle frequenze di concordanza e di discordanza tra i gemelli MZ e DZ danno ugualmente una
misura generale del grado di influenza dei geni sul
fenotipo.
Ereditabilità in senso stretto e selezione
artificiale
2
L’ereditabilità in senso stretto (h = VA/VP) permette
di stimare la proporzione di variabilità fenotipica
dovuta a varianza genetica additiva (VA), cioè alla varianza che risulta dagli alleli di geni additivi. Queste
stime sono particolarmente utili in agricoltura, dove
permettono di prevedere la potenziale responsività
di un carattere, in un animale o in una pianta, alla
selezione imposta attraverso programmi di incroci
selettivi o condizioni di crescita controllate. Valori
elevati di ereditabilità in senso stretto sono correlati
con un grado maggiore di risposta alla selezione rispetto a valori bassi, dato che la varianza genetica
additiva risponde bene alla selezione.
19.3 - L'ereditabilità misura la componente genetica della variabilità fenotipica
Tabella 19.3 Valori di concordanza per
condizioni soglia comuni negli
umani
Carattere
Percentuale di
concordanza
Gemelli MZ Gemelli DZ
Malattia di Alzheimer
60
25
Autismo
70
10
Labbro leporino
40
4
Piede equino
30
2
Dislocazione congenita
dell’anca
35
3
Depressione
70
25
Diabete insulino-dipendente
50
10
Stenosi pilorica
25
3
Disturbo della lettura
70
45
Schizofrenia
60
20
2
La Tabella 19.4 dà esempi di valori h , distribuiti in un ampio spettro di grandezza, per diverse ca2
ratteristiche di piante e animali. Dato che valori h
più elevati corrispondono a una correlazione più
forte con la risposta alla selezione, i biologi predicono che caratteri come il peso corporeo nei bovini,
lo spessore del grasso dorsale nei suini e l’altezza
delle piante di granturco saranno più suscettibili a
modifiche attraverso schemi di selezione artificiale.
D’altra parte, la dimensione della figliata nei suini,
la produzione delle uova nel pollame e il diametro
delle pannocchie nel granturco presentano valori di
2
h bassi e risponderanno meno bene alla selezione.
La stima della risposta potenziale alla selezione
per un carattere inizia con il calcolo di un valore
noto come differenziale di selezione (S), che misura
la differenza tra il valore medio della popolazione
per un carattere e il valore medio del carattere per
gli individui che prendono parte all’accoppiamento.
Supponete, per esempio, che l’obiettivo di un esperimento di selezione artificiale sia quello di aumentare l’altezza delle piante. Scegliere piante con altezza superiore alla media per l’accoppiamento sarà
un modo efficace per aumentare l’altezza della pro2
genie se h è elevato. Se l’altezza media della popolazione è pari a 37,5 cm e l’altezza media delle piante
selezionate per l’accoppiamento è pari a 42 cm, allora S = 42 - 37,5 = 4,5 cm.
La potenziale risposta alla selezione (R) dipende dalla capacità di passare alla progenie la differenza tra il valore medio del carattere degli individui
che hanno preso parte all’accoppiamento e il valore
medio della popolazione. Questa probabilità viene
2
stimata utilizzando la formula R = S(h ). Nel caso
dell’esempio relativo all’altezza della pianta, supponiamo di esaminare l’altezza della pianta di gran2
turco, per la quale h = 0,70 (Tabella 19.4). In questo
caso, R = (4,5 cm)(0,70) = 3,15 cm. In condizioni stabili di crescita, ci si può aspettare che le piante della
progenie abbiano un’altezza pari a quella della media della popolazione più il valore di R, ovvero 37,5 +
3,15 = 40,65 cm. L’ereditabilità in senso stretto può
essere misurata riarrangiando i termini dell’equa2
zione di risposta alla selezione in questo modo: h
2
= R/S. Per l’esempio dell’altezza della pianta, h =
3,15 cm/4,5 cm = 0,70. Il Caso di studio di questo
capitolo sviluppa la questione della risposta alla selezione descrivendo un esperimento di selezione artificiale effettuato oltre 120 anni fa.
Le stime dell’ereditabilità hanno importanti applicazioni pratiche per allevatori, coltivatori e biologi evoluzionisti. Sia che i caratteri siano soggetti
Tabella 19.4 Alcuni valori di ereditabilità in
senso stretto (h2) per animali e
piante
Organismo Carattere
Bovini
Granturco
Cavalli
Suini
Pollame
Ereditabilità (h2)
Peso corporeo
0,65
Produzione di latte
0,40
Altezza della pianta
0,70
Lunghezza della pannocchia
0,55
Diametro della pannocchia
0,14
Velocità in corsa
0,60
Velocità al trotto
0,40
Spessore del grasso dorsale
0,70
Guadagno di peso
0,40
Dimensioni della figliata
0,05
Peso corporeo (8 settimane)
0,50
Produzione delle uova
0,20
695
CAPITOLO 19 - Analisi genetica dei caratteri quantitativi
alla selezione artificiale da parte degli allevatori o
alla selezione naturale, la capacità del valore medio
di un carattere di cambiare in una popolazione dipende dalla sua ereditabilità. Gli allevatori e i biologi
evoluzionisti predicono cambiamenti sostanziali nei
valori medi del carattere (cioè valori più alti di R)
quando l’ereditabilità è elevata, ma cambiamenti
piccoli (o nessun cambiamento) nei valori medi del
carattere quando l’ereditabilità è bassa. In altre parole, i caratteri evolvono quando una parte sostanziale della variabilità fenotipica è dovuta alla variabilità genetica.
La Figura 19.11a presenta tre esempi nei quali
i differenziali di selezione sono gli stessi ma la risposta alla selezione differisce in conseguenza dei
diversi gradi di ereditabilità. Questo confronto illustra il fatto che la risposta alla selezione è prevedi2
bilmente massima quando l’ereditabilità è h = 1,0.
La risposta alla selezione è sostanzialmente inferiore
2
quando l’ereditabilità è h = 0,2 e non vi è risposta
2
alla selezione quando l’ereditabilità è h = 0. La selezione interessa i caratteri quantitativi anche nelle
popolazioni naturali. La Figura 19.11b mostra come
la selezione naturale agisca nel corso di diverse generazioni in tre diversi modi che esercitano effetti
diversi sulle medie e sulle varianze fenotipiche. Nella
modalità nota come selezione direzionale, il valore
fenotipico medio è spostato in una direzione perché
un estremo della distribuzione fenotipica è favorito.
Ciò restringe l’intervallo fenotipico e riduce la varianza fenotipica. Invece, la selezione naturale che
favorisce un fenotipo intermedio rispetto ai fenotipi
estremi, determina una selezione stabilizzante che
riduce la variabilità fenotipica. La selezione distruttiva si presenta quando entrambi i fenotipi estremi
sono favoriti rispetto ai fenotipi intermedi. Il risultato è un aumento nella varianza fenotipica e, potenzialmente, una scissione fenotipica all’interno
della popolazione.
CONCETTI CHIAVE
L’ereditabilità in senso stretto (h2) misura il contributo della varianza genetica additiva alla varianza
fenotipica ed è utilizzata per determinare la potenziale risposta di un tratto quantitativo alla selezione
artificiale o naturale.
696
(a)
Genitore
Prole
Valore fenotipico
Conseguenza:
Stessa media della
popolazione
Leggero cambiamento
nella media della
popolazione
Grosso cambiamento
nella media della
popolazione
La risposta alla
selezione è piccola
La risposta alla
selezione è massima
Conclusione:
La risposta alla
selezione è zero
(b)
Selezione
direzionale
Genitore
Selezione
stabilizzante
Porzione favorita dalla
selezione naturale
Dopo diverse
generazioni
Selezione
distruttiva
Porzione favorita dalla
selezione naturale
Valore fenotipico
Conclusione:
Media e varianza
modificate
Stessa media ma
varianza ridotta
Stessa media ma
varianza aumentata
Figure 19.11 Risposta alla selezione artificiale e naturale.
(a) La risposta alla selezione artificiale dopo una generazione
2
dipende da h . M è il fenotipo medio nella generazione
parentale; MS è il fenotipo medio dei genitori selezionati per la
riproduzione; M’ è il fenotipo medio della prole dopo la selezione;
il differenziale di selezione è S = M – MS. (b) Cambiamenti
attesi nelle medie e nelle varianze fenotipiche dopo diverse
generazioni di selezione naturale.
19.4 - I loci dei caratteri quantitativi corrispondono ai geni che contribuiscono ai caratteri quantitativi
19.4 I loci dei caratteri quantitativi
corrispondono ai geni che
contribuiscono ai caratteri
quantitativi
I geni che contribuiscono alla variabilità di un carattere quantitativo vengono chiamati collettivamente
loci di caratteri quantitativi (QTL, quantitative
trait loci).
Preso singolarmente, un gene che contribuisce
a un carattere quantitativo viene chiamato locus di
un carattere quantitativo. I QTL sono stati inizialmente studiati nelle piante agricole come i pomodori e il granturco, in cui influenzano importanti
attributi, inclusa dolcezza, acidità e colore della
frutta. L’analisi dei QTL si è espansa in maniera importante negli ultimi decenni attraverso l’analisi di
molti caratteri distinti nelle piante e negli animali,
esseri umani inclusi.
In un certo senso, i QTL non sono diversi da altri geni che abbiamo discusso. Per esempio, producono spesso polipeptidi che operano nelle vie metaboliche che producono composti che conferiscono
sapore o colore al frutto. Tuttavia, l’identificazione
dei QTL per analisi sperimentale è diversa dall’identificazione di altri geni che controllano la variabilità
fenotipica, perché i geni che influenzano il carattere
sono molti e la presenza o l’assenza di un particolare
allele non correla bene con fenotipi distinti. Per rilevare e mappare i QTL sono stati sviluppati metodi
statistici specializzati.
Questo processo viene chiamato mappatura dei
QTL e determina l’identificazione delle regioni cromosomiche che presentano una probabilità elevata
di contenere QTL.
Il processo generale di mappatura dei QTL è simile ai metodi utilizzati per determinare l’associazione genetica tra geni. Una regione cromosomica
che contiene probabilmente QTL viene identificata
dalla frequente co-occorrenza di un marcatore genetico specifico come un polimorfismo a singolo
nucleotide (SNP) negli organismi con un particolare fenotipo. Il SNP non è il QTL, ma è geneticamente collegato al QTL. La connessione tra il marcatore genetico e il fenotipo implica che esiste un
QTL in prossimità della posizione genomica che codifica per il marcatore genetico.
Strategie di mappatura dei QTL
La procedura attuale di mappatura dei QTL utilizza
dei marcatori di DNA con posizioni cromosomiche
note per favorire la mappatura e l’identificazione dei
geni. Gli SNP sono particolarmente utili in queste
analisi, insieme ad altri tipi di marcatori del DNA,
come i polimorfismi da lunghezza del frammento di
restrizione (RFLP) e le ripetizioni in tandem a numero variabile (VNTR), nelle quali diversi numeri di
ripetizioni di sequenze nucleotidiche specifiche si
presentano in vari cromosomi.
Negli esperimenti di mappatura dei QTL possono essere utilizzati diversi approcci. Fondamentalmente, tuttavia, la mappatura dei QTL è un processo statistico che ha lo scopo di identificare regioni
dei genomi contenenti marcatori genetici collegati
ai QTL. L’analisi dei QTL può portare all’identificazione della posizione cromosomica potenziale di
un QTL che influenza la variabilità fenotipica di un
carattere quantitativo, ma di per sé non identifica il
QTL vero e proprio. Per l’identificazione dei QTL
sono disponibili altri metodi genetici.
La mappatura dei QTL utilizza i genitori e la progenie prodotta da incroci controllati come la fonte
del DNA necessario all’identificazione di marcatori genetici e come fonte di dati relativi al carattere
quantitativo di interesse. Se, per esempio, un ricercatore vuole identificare i QTL che contribuiscono
a conferire una grossa dimensione ai frutti di pomodoro, incrocerà due linee parentali di pomodori che
differiscono nella dimensione dei frutti. La progenie
F1 di questo incrocio potrebbe quindi essere utilizzata per produrre una progenie F2 oppure, come illustreremo nel seguito, la F1 potrebbe essere utilizzato in un reincrocio con uno dei ceppi parentali.
I marcatori genetici saranno determinati nei ceppi
parentali originali e nella progenie del reincrocio.
Le dimensioni dei pomodori prodotti dalla progenie del reincrocio vengono misurate e i risultati confrontati con i marcatori genetici nelle singole piante.
La Figura 19.12 illustra la struttura di un esperimento di reincrocio progettata per raccogliere dati
relativi ai marcatori genetici e al peso dei pomodori
per l’analisi della mappatura dei QTL. Un ceppo parentale di pomodori che produce frutti di grosse dimensioni, in media 100 grammi (g), contiene dei
marcatori genetici identificati con la lettera L. In ef-
697
CAPITOLO 19 - Analisi genetica dei caratteri quantitativi
Incrocio
parentale:
Frutto
grosso
(100 g)
Frutto
piccolo
(10 g)
Reincrocio
F1:
(60 g)
(100 g)
Progenie del
reincrocio:
e
(da 80 g
a 88 g)
Figura 19.12 Rilevazione e mappatura dei loci dei
caratteri quantitativi (QTL). Piante parentali di pomodoro che
producono frutti grossi (LL) o piccoli (SS) vengono incrociati per
produrre la F1 (LS). La F1 viene quindi reincrociata con il ceppo
a frutto grosso per produrre progenie del reincrocio che può
essere di tipo LL o LS.
fetti, in questa linea esistono molti marcatori collegati ai QTL e, per ogni gene marcatore analizzato,
il ceppo a pomodori grossi avrà due copie del genotipo allelico corrispondente al marcatore del ceppo
grosso, indicate con LL. In maniera simile, un ceppo
che produce pomodori di piccole dimensioni, con
un peso medio di 10 grammi, è caratterizzato dagli
stessi marcatori genetici e ognuno dei loci analizzati
nel genotipo del ceppo piccolo è indicato con SS. La
progenie F1 dell’incrocio grosso X piccolo è eterozigote per ogni locus del marcatore ed è indicata con
LS. Le piante in questo esempio producono pomodori che pesano 60 g. Il reincrocio viene effettuato
con il ceppo a frutto grosso e il genotipo del marcatore sarà LL, se F1 trasmette l’allele del ceppo grosso,
oppure LS, se la F1 trasmette l’allele del ceppo piccolo. La progenie del reincrocio, in questo esempio,
produce pomodori che variano nel peso da 80 g a
88 g. Il peso del pomodoro ottenuto dalle piante del
reincrocio è più grosso rispetto a quello delle piante
della F1 perché le piante del reincrocio sono il risultato di un incrocio tra F1 e il ceppo a pomodoro
grosso.
La Tabella 19.5 mostra i dati relativi al peso dei
pomodori per 10 piante del reincrocio (1-10) e i dati
relativi al marcatore genetico per due geni, marca-
698
tore A (MA) e marcatore B (MB), che non sono associati l’uno con l’altro e sono localizzati in parti
diverse del genoma. In un esperimento reale di reincrocio di QTL possono essere esaminate diverse
centinaia di piante del reincrocio e ogni pianta può
essere genotipizzata per dozzine di marcatori genetici che, idealmente, sono distribuiti a una distanza,
l’uno dall’altro, di 5-10 centimorgan (cM) nel genoma. Questo numero elevato di marcatori genetici e la loro stretta prossimità massimizzano la possibilità di identificare la posizione dei QTL rilevati
dall’analisi.
Nella Tabella 19.5, il peso medio dei pomodori
derivanti da piante del reincrocio è di 84 grammi.
Viene effettuato il confronto del peso medio dei pomodori per le piante LL rispetto alle piante LS per
ogni marcatore. Non vi è praticamente alcuna differenza di peso per MA (LL = 83,8 g rispetto a LS =
84,2 g), ma per MB, le piante LL producono pomodori che sono più pesanti di 4 grammi rispetto alla
media dei pomodori derivanti dalle piante LS (LL =
86,0 g rispetto a LS = 82,0 g). Questi dati suggeriscono la presenza di un QTL che influenza il peso
Tabella 19.5 Analisi dei QTL per il peso dei
pomodori nella progenie di un
reincrocio
Pianta del
reincrocio
Peso medio
del frutto (g)
Marcatori
MA
MB
1
86
LS
LL
2
82
LL
LS
3
85
LL
LL
4
88
LL
LL
5
81
LS
LS
6
83
LS
LS
7
84
LL
LL
8
80
LL
LS
9
84
LS
LS
10
87
LS
LL
Peso medio totale
84
Peso medio LL
83,8
86,0
Peso medio LS
84,2
82,0
19.4 - I loci dei caratteri quantitativi corrispondono ai geni che contribuiscono ai caratteri quantitativi
dei pomodori in prossimità di MB. Viceversa, non vi
è evidenza di QTL localizzati in prossimità di MA.
Utilizzando un gran numero di marcatori genetici collocati a intervalli regolari di qualche cM sul
cromosoma, l’analisi di mappatura dei QTL può potenzialmente rilevare la posizione di qualsiasi QTL
che influenzi un fenotipo dovuto a un carattere
quantitativo. Generalmente, in un genoma vengono
identificati un gran numero di QTL.
Andrew Paterson e colleghi pubblicarono, nel
1988, uno studio che mappava nel genoma del pomodoro 15 QTL che influenzavano il peso del
frutto, l’acidità del frutto e la quantità di solidi solubili nel frutto. Ogni carattere presenta una sua importanza dal punto di vista agricolo e messi insieme
essi determinano la qualità e la produzione di salsa
di pomodoro dal frutto. Lo studio di Paterson utilizzava 70 marcatori di DNA disposti a intervalli medi
di 20 cM lungo il genoma del pomodoro. Collettivamente, questi marcatori coprivano circa il 95% dei
12 cromosomi che costituiscono il genoma del pomodoro.
Le piante parentali erano due specie correlate
e infertili: un pomodoro domestico (Lycopersicon
esculentum) e un pomodoro selvatico a frutto verde
dell’America meridionale (Lycopersicon chmielewskii). Gli ibridi F1 furono reincrociati a L. esculentum, producendo 237 piante del reincrocio destinate
all’analisi. Tutte le piante del reincrocio furono fatte
crescere in condizioni identiche in modo da minimizzare l’influenza dei fattori ambientali sui caratteri di interesse. I singoli frutti ottenuti dalle piante
del reincrocio furono testate per il peso (grammi),
il contenuto di solidi solubili (percentuale) e l’acidità (pH) del frutto. Sono stati così identificati e localizzati sei geni che influenzavano il peso del frutto,
cinque che influenzavano l’acidità e quattro che influenzavano il contenuto di solidi solubili, in regioni
specifiche di nove cromosomi del genoma del pomodoro (Figura 19.13).
Identificazione dei geni QTL
Dato che la mappatura dei QTL identifica la posizione dei geni che influenzano i caratteri quantitativi, ma non i geni stessi, per identificare questi ultimi è necessaria un’ulteriore analisi genetica. Per
acquisire le informazioni che permettono di identi-
Cromosoma
Peso del frutto
Acidità
Solidi solubili
Figura 19.13 Mappatura dei QTL nel pomodoro domestico
(Solanum lycopersicon). Vengono mappati numerosi QTL che
influenzano il peso, l’acidità e la percentuale di solidi solubili
del frutto dei pomodori. Le designazioni dei marcatori di DNA
vengono date sotto ogni cromosoma e la distanza (in cM) tra i
marcatori è indicata da un numero al di sopra dei cromosomi.
ficare il gene, i ricercatori utilizzano le linee quasi
isogeniche (NIL, near isogenic lines), chiamate anche linee di introgressione (IL, introgression lines).
Queste linee sono derivate dalla progenie del reincrocio prodotta come descritto sopra. Diversi individui della progenie del reincrocio sono auto-fecon-
699
CAPITOLO 19 - Analisi genetica dei caratteri quantitativi
dati per molte generazioni per formare linee
altamente consanguinee. I ceppi risultanti sono
quasi isogenici, cioè sono geneticamente identici
per quasi tutti i geni. Le linee differiscono l’una
dall’altra, tuttavia, perché portano diversi crossingover che hanno introdotto alleli diversi in prossimità della posizione di un QTL. Le differenze introdotte sono chiamate introgressioni, dando pertanto
il proprio nome a queste linee.
La Figura 19.14a illustrata sei linee di introgressione (da IL1 a IL6) derivate da un incrocio tra due
linee parentali originali, una specie domestica e una
specie selvatica. I colori dei cromosomi illustrano
i crossing-over che producono delle differenze tra
le linee di introgressione. Le posizioni del crossingover sono identificate dall’analisi dei marcatori genetici e ogni linea di introgressione è caratterizzata
per un fenotipo del carattere. Nella figura, le barrette a destra di ogni linea indicano le differenze
percentuali tra il fenotipo della specie IL e quello
della specie parentale domestica. Due potenziali regioni QTL, QTL-A e QTL-B, contengono variabilità dei segmenti del crossing-over. La maggiore differenza positiva di percentuale rispetto al fenotipo
(a)
Specie
domestica
Specie
selvatica
Produzione di linee
di introgressione
Differenza del carattere
(%) rispetto alla specie
domestica
Figura 19.14 Analisi
dei QTL in linee di
introgressione.
(a) Sei linee di introgressione
(da IL1 a IL6) prodotte
per accoppiamento tra
specie domestiche e specie
selvatiche presentano diversi
pattern di ricombinazione
in una regione contenente
due QTL. La differenza di
espressione del carattere tra la
specie domestica e ogni IL è
data in percentuale.
(b) L’analisi di Brix 9-2-5 in
13 linee di introgressione
identifica i SNP che alterano
l’attività dell’invertasi della
parete cellulare. Il SNP in
posizione 2878 esercita
un’influenza sostanziale sulla
funzione della invertasi della
parete cellulare.
(b)
Attività dell’invertasi
della parete cellulare (%)
Linea di introgressione
700
Effetto
fenotipico
19.4 - I loci dei caratteri quantitativi corrispondono ai geni che contribuiscono ai caratteri quantitativi
della specie domestica si osserva per IL2 e IL3, che
portano cromosomi che dopo il crossing-over contengono il DNA della specie selvatica in prossimità
di QTL-A e il DNA della specie addomesticata in
prossimità di QTL-B.
Per identificare i geni che determinano variabilità dei QTL, devono essere identificati e analizzati
i ”geni candidati”, ovvero quelli che sono potenzialmente responsabili della variabilità osservata. I geni
presenti nelle regioni QTL-A e QTL-B sono localizzati esaminando le sequenze di DNA e tra le linee di
introgressione vengono identificate le varianti di sequenza dei geni candidati. Vengono quindi studiate
le differenze di sequenza rilevate per determinare se
esse correlano con la variabilità fenotipica.
La Figura 19.14b illustra i risultati dell’analisi
sperimentale delle linee di introgressione del pomodoro effettuata da Eyal Fridman e colleghi nel 2004
allo scopo di identificare i geni che contribuiscono al
valore Brix nel pomodoro. Il valore Brix del frutto si
riferisce al contenuto dei solidi solubili totali, di cui
zuccheri e acidi sono i costituenti principali. Fridman e colleghi crearono un gran numero di IL da
un incrocio iniziale tra la specie di pomodoro domestico (Solanum lycopersicum) e un parente selvatico
(Solanum pennellii).
Fu quindi studiato il valore Brix delle specie parentali e di ognuna delle IL e un QTL per cui era
stato ottenuto un valore Brix elevato, Brix 9-2-5,
fu studiato intensivamente. Il sequenziamento del
DNA dei 484 nucleotidi (posizioni da 2799 a 3283)
di Brix 9-2-5 ha rivelato le cinque varianti SNP mostrate nella figura. Il QTL Brix 9-2-5 corrisponde a
un segmento del gene LIN5 del pomodoro che produce l’enzima invertasi della parete cellulare (CW
invertase). Nella figura, vengono mostrate le posizioni dei SNP in 13 IL che portano una ricombinazione a livello di o in prossimità di Brix 9-2-5. La
barretta a destra di ogni IL indica la sua differenza
percentuale nell’attività della invertasi della parete
cellulare rispetto a S. lycopersicum. I risultati mostrano che, quando è presente la sequenza di S. pennellii, l’attività della invertasi della parete cellulare è
significativamente superiore rispetto a S. lycopersicum. La SNP in posizione 2878 (evidenziata da un
riquadro) era fortemente correlata con l’aumento
dell’attività della invertasi della parete cellulare.
L’analisi della sequenza del DNA e della proteina
ha rivelato che questo SNP produce una differenza
nella sequenza amminoacidica che altera l’attività
dell’invertasi della parete cellulare.
CONCETTI CHIAVE
I loci dei caratteri quantitativi (QTL) vengono mappati in posizioni cromosomiche specifiche tramite
l’associazione genetica tra i QTL e i marcatori variabili di DNA.
Studi di associazione genomica
La disponibilità sempre più diffusa di informazioni
relative al sequenziamento del genoma ha aperto
una nuova strada all’identificazione dei QTL in un
gran numero di specie, inclusa quella umana. Noto
come studio di associazione genomica (GWAS), il
metodo cerca di collegare la presenza di una variante di sequenza di un marcatore di DNA a un
QTL che influenza un fenotipo specifico. La correlazione tra un marcatore genetico e il fenotipo è per
“associazione,” cioè gli organismi che portano una
variante particolare avranno una maggiore probabilità di presentare un certo fenotipo rispetto ad altri
che portano una variante diversa. La valutazione
dell’associazione è quantitativa, cioè esprime la percentuale degli organismi con un marcatore genetico
che presenta anche un certo fenotipo rispetto alla
percentuale che presenta il fenotipo ma non il marcatore genetico.
Un vantaggio del GWAS rispetto agli approcci di
mappatura dei QTL è che questo permette di scansionare l’intero genoma per i QTL tramite un esame
statistico delle varianti del marcatore associate alla
variabilità fenotipica. I risultati statistici positivi
che indicano un’associazione identificano delle regioni cromosomiche che possono essere ispezionate più da vicino per verificare la presenza di geni
che influenzano il carattere. Un secondo vantaggio
del GWAS è che possono essere analizzati organismi appartenenti a popolazioni ad accoppiamento
casuale. Invece di richiedere incroci controllati e
la formazione di linee di introgressione, come descritto in precedenza, lo GWAS utilizza i “casi”,
ovvero organismi che presentano un particolare
701
CAPITOLO 19 - Analisi genetica dei caratteri quantitativi
fenotipo, e li confronta ai “controlli” che non lo presentano, per valutare l’associazione tra i marcatori
del QTL e quel fenotipo.
Lo GWAS si basa sulla tendenza degli alleli di
marcatori genetici strettamente associati a mostrare
linkage disequilibrium (vedete la discussione nella
Sezione 5.6 per un ripasso). Combinazioni specifiche di alleli in linkage disequilibrium avvengono
con frequenze significativamente superiori rispetto
a quanto ci si aspetterebbe se queste avvenissero a
caso. Il linkage disequilibrium avviene perché la ricombinazione non ha rimescolato gli alleli in combinazioni casuali. Gruppi di alleli in linkage disequilibrium formano aplotipi lungo segmenti di
cromosomi (Sezione 5.6). Se un gruppo di SNP
strettamente associati formano un aplotipo, l’identificazione di un SNP particolare per un marcatore
significa che altri SNP che fanno parte dello stesso
aplotipo si trovano probabilmente in posizioni vicine. La presenza di SNP negli aplotipi può essere
correlata con la presenza (affetto) o assenza (non affetto) di un fenotipo particolare, come una malattia influenzata geneticamente. L’esame statistico
dell’associazione tra un SNP e un fenotipo malato è
simile a un test del chi quadro (Sezione 2.5). Come
per l’analisi del Chi-quadro, la significatività del risultato è basata sui valori di P. In questo esame statistico l’ipotesi nulla è che l’occorrenza di un certo
SNP e di un particolare fenotipo è determinata dal
caso. Un valore di P inferiore a 0,05 indica che l’associazione non è il risultato del caso e l’ipotesi nulla
viene respinta.
L’analisi statistica GWAS non prova che un QTL
nella o vicino alla posizione del SNP influenzi il fenotipo. Piuttosto, fornisce una prova statistica che
suggerisce che un QTL possa essere situato in prossimità. Ulteriori analisi molecolari possono identificare i geni candidati e collegare una specifica variabilità allelica direttamente alla produzione di un
certo fenotipo. Lo GWAS è stato applicato all’analisi di numerose malattie e disturbi umani influenzati dai QTL. Gli studi GWAS hanno permesso di
scoprire numerosi geni o posizioni di SNP in cui potrebbero essere presenti dei QTL.
Uno studio del 2001 effettuato da Yasunori
Ogura e colleghi utilizzava lo GWAS per identificare
diverse regioni cromosomiche associate alla morbo
702
di Crohn (MC), una malattia infiammatoria cronica
dell’intestino che colpisce la popolazione umana
con una prevalenza di 150-200 casi ogni 100.000 individui. La gravità del MC è altamente variabile, da
relativamente lieve a potenzialmente fatale. I clinici
descrivono la gravità del MC utilizzando una scala
che registra la natura quantitativa del carattere, rendendo il MC una malattia candidata per l’analisi
dei QTL. L’eziologia del MC è ignota, ma un’ipotesi predominante propone che questa malattia risulti da una risposta infiammatoria ai batteri e ad altri microrganismi della microflora intestinale. Il MC
si raggruppa in famiglie e la suscettibilità alla malattia è ereditaria, ma influenzata da molteplici geni.
Nello studio di Ogura e colleghi, l’evidenza statistica
più importante di associazione tra un marcatore genetico e un gene di suscettibilità viene dalla regione
cromosomica 16q12. Un gene identificato inizialmente come NOD2 e successivamente rinominato
CARD15 (dominio di reclutamento e attivazione
delle caspasi, caspase activation and recruitment domain, membro 15), influenza probabilmente la suscettibilità alla MC.
CARD15 codifica per 12 esoni che dirigono la
produzione di una proteina costituita da 1040 amminoacidi. Ogura e colleghi sequenziarono gli esoni
e gli introni di CARD15 in 12 pazienti con MC provenienti da diverse famiglie che presentavano diversi
casi di MC. Effettuarono inoltre lo stesso sequenziamento genico in quattro individui sani di controllo.
Lo studio ha permesso di identificare una stessa inserzione del paio di basi C-G a livello del nucleotide
3020 dell’esone 11 in tre dei 12 pazienti con MC.
L’inserzione, indicata con 3020insC, induce una
mutazione frameshift che genera un codone di stop
prematuro, rendendo la proteina mutante più corta
(1007 amminoacidi). Ogura e colleghi svilupparono
un test di PCR (Reazione a catena della polimerasi)
per la mutazione 3020insC ed esaminarono 101 pazienti i cui genitori erano eterozigoti per l’allele selvatico e l’allele 3020insC. Dei 101 pazienti con MC,
68 erano omozigoti per 3020insC (Figura 19.15a).
L’analisi biochimica mostra che la proteina mutante
prodotta da questo gene possiede solo una piccola
parte dell’attività della proteina selvatica. Questa diminuita capacità riduce la sensibilità del sistema immunitario all’invasore microbico e, tramite un mec-
19.4 - I loci dei caratteri quantitativi corrispondono ai geni che contribuiscono ai caratteri quantitativi
(a)
(b) SNP significativamente associati
con morbo di Crohn
Cromosoma
Gene
Controlli
Allele selvatico
Allele 3020insC
Omozigote per
l’allele mutante
Selvatico Marcatore di
peso molecolare
Portatori eterozigoti
Figura 19.15 Rilevamento di 3020insC in CARD15 in una famiglia con morbo di Crohn. (a) Le elettroforesi su gel dei prodotti
della PCR relative a quattro membri di una famiglia sono presentate nelle piste da 1 a 4. Nella corsia 5 è presente un controllo di
tipo selvatico e nella corsia 6 un marcatore di peso molecolare. (b) Sette QTL che influenzano l’espressione della morbo di Crohn,
identificati da studi GWAS.
canismo che deve ancora essere chiarito, determina
ill MC.
Le mutazioni di CARD15 non sono l’unica causa
di MC; numerosi pazienti con MC non portano
3020insC o altre mutazioni note del gene. Una ricerca pubblicata nel 2007 dal Wellcome Trust Case
Control Consortium, un gruppo composto da dozzine di gruppi di ricerca che lavorano in collaborazione, ha confermato il ruolo fondamentale di
CARD15 nella suscettibilità al MC e ha identificato sei geni aggiuntivi che influenzano, anch’essi,
la suscettibilità a questa malattia (Figura 19.15b). Lo
stesso studio collaborativo ha esaminato con un approccio GWAS altre malattie autoimmuni, incluso
il diabete di tipo 1 (insulino-dipendente) e l’artrite
reumatoide. Le analisi GWAS dell’artrite reumatoide e del diabete di tipo I hanno identificato diversi
geni che influenzano la suscettibilità. In maniera interessante, tuttavia, le analisi hanno identificato an-
che due geni di suscettibilità, uno sul cromosoma 1
e l’altro sul cromosoma 6, che sono condivisi da entrambe le malattie. Questa scoperta fornisce un’importante informazione clinica, suggerendo che parte
dei meccanismi coinvolti nello sviluppo di queste
malattie sono simili. Questa informazione potrebbe
rivelarsi utile nella loro diagnosi e nel loro trattamento.
CONCETTI CHIAVE
Gli studi di associazione genomica (GWAS) permettono di localizzare i QTL identificando un linkage disequilibrium tra alcuni marcatori di DNA e un QTL
in popolazioni ad accoppiamento casuale. Un’analisi statistica simile all’analisi del Chi-quadrato permette di valutare l’associazione tra marcatori genetici e QTL.
703
CAPITOLO 19 - Analisi genetica dei caratteri quantitativi
Caso di studio
Selezione artificiale per il contenuto di olio e proteine nel granturco
704
(a) Medie relative al contenuto in olio
% di olio
Illinois high oil
Reverse high oil
Reverse low oil
Illinois low oil
Generazione
(a) Medie relative al contenuto proteico
Illinois high protein
% di proteine
Un esperimento di selezione artificiale in corso ormai da
125 anni fornisce un chiaro esempio di responsività alla selezione dei caratteri quantitativi. L’esperimento, che consiste nella selezione artificiale per cambiamenti del contenuto in olio e in proteine del granturco (Zea mays), fu
iniziato nel 1886 da Cyril Hopkins, allora professore di agronomia all’Università dell’Illinois. Da allora, non ha smesso di
funzionare.
L’esperimento di Hopkins presenta due parti distinte,
una focalizzata sulla selezione di ceppi separati di granturco che presentano contenuto elevato e basso di olio
nei chicchi e l’altra che si focalizza sull’aumento e la diminuzione del contenuto proteico dei chicchi di granturco.
L’esperimento sul contenuto in olio iniziò con un singolo
ceppo di granturco che conteneva all’incirca il 5% di olio.
Oltre 100 generazioni di selezione hanno permesso di produrre ceppi di granturco che differiscono radicalmente nel
contenuto in olio. Ai due estremi troviamo un ceppo indicato con Illinois high oil (IHO) che contiene oggi più del 20%
di olio nei chicchi e un altro ceppo, indicato come Illinois
low oil (ILO), che praticamente non ne contiene (Figura
19.16a). Il cambiamento del contenuto in olio dei chicchi
è stato continuo, a causa della selezione artificiale applicata
a ogni generazione.
Alla generazione 50, a qualche pianta di ogni ceppo è
stata applicata un’inversione dei criteri di selezione artificiale. Alcune piante del ceppo IHO sono state reindirizzate
a un esperimento che aveva lo scopo di invertire la direzione del cambiamento, abbassando il contenuto in olio.
Quando iniziò l’esperimento inverso, il contenuto in olio
del ceppo IHO era all’incirca pari al 12%; ma nel corso delle
50 generazioni successive, il contenuto in olio del ceppo reverse high oil (RHO) fu ridotto al 5%. Inoltre, alla generazione
50, alcune piante del ceppo ILO furono reindirizzate a un
esperimento volto a invertire la direzione della modifica del
loro contenuto in olio. Il ceppo ILO era al 2% di olio al momento dell’inizio del protocollo sperimentale, ma il contenuto del ceppo reverse low oil (RLO) raggiungeva il 5% alla
generazione 100. I risultati di questo esperimento ancora in
atto indicano che è possibile ottenere cambiamenti continui del contenuto in olio applicando in maniera coerente
dei criteri di selezione che favoriscono un contenuto basso
o elevato di olio nei chicchi. Gli esperimenti inversi indicano che la diversità genetica viene mantenuta da molti
dei geni coinvolti nella produzione di olio, anche dopo numerose generazioni di selezione intensa.
L’esperimento sul granturco dell’Illinois ha inoltre selezionato i chicchi per il loro contenuto proteico in un esperimento che ha seguito in maniera quasi identica il protocollo e i risultati dell’esperimento sul contenuto in olio
dei chicchi. La Figura 19.16b traccia la divergenza di due
Reverse high
protein
Illinois low protein
Reverse low
protein
Generazione
Figura 19.16 Risultati della selezione a lungo termine
nel granturco. (a) Selezione per il livello di olio nei chicchi.
(b) Selezione per il contenuto proteico dei chicchi di
granturco.
ceppi di piante, Illinois high protein (IHP) e Illinois low protein (ILP), che sono state prodotte dagli stessi fondatori
dell’esperimento sull’olio. Come nel caso dell’esperimento
sull’olio, alla generazione 50 fu iniziato un esperimento inverso per valutare la possibilità di invertire l’aumento proteico nel ceppo IHP e la perdita proteica nel ceppo ILP. I
ceppi inversi, reverse high protein (RHP) e reverse low protein
(RLP), hanno manifestato l’inversione di alcune delle modifiche proteiche maturate dopo le prime 50 generazioni.
Nel 2004, Cathy Laurie e colleghi intrapresero un’analisi dei QTL delle piante del progetto Illinois sulla selezione
dell’olio nel granturco per cercare di identificare i QTL associati alla produzione dell’olio. Utilizzando l’approccio
Riassunto
GWAS, identificarono oltre 50 QTL potenziali che influenzano il contenuto in olio e che rendevano conto di circa la
metà della variabilità genetica totale nella produzione di
olio. Nessun QTL determinava un cambiamento superiore
all’1% nel contenuto in olio, suggerendo che un gran numero di geni, ognuno con un’influenza molto piccola sulla
produzione dell’olio, sono responsabili del contenuto in
olio dei chicchi. L’analisi di Laurie conferma che un gran
numero di QTL, agendo in maniera additiva, sono responsabili del contenuto in olio del granturco. La risposta alla
selezione regolare e sostenuta osservata nell’esperimento
di Hopkins è l’effetto della selezione su molti loci, ognuno
dei quali esercita un’influenza molto piccola sul contenuto
in olio.
RIASSUNTO
19.1 I caratteri quantitativi presentano
variabilità fenotipica continua
I caratteri quantitativi fenotipici sono poligenici e sono
descritti da scale di misura a cui possono essere assegnati
dei valori su base quantitativa.
I fenotipi dei caratteri multifattoriali risultano dall’ereditarietà poligenica e dall’influenza dei fattori ambientali.
Molti caratteri quantitativi presentano una distribuzione fenotipica continua. Quelli influenzati da un numero maggiore di geni avranno una maggiore probabilità di mostrare una variabilità continua. Per i caratteri
soglia, invece, è probabile che si osservi una variabilità
discontinua del fenotipo.
I caratteri soglia sono dovuti ad alleli additivi e presentano una soglia di suscettibilità che separa una categoria
fenotipica (non affetta) da un’altra (affetta). La soglia di
suscettibilità viene oltrepassata quando un numero sufficiente di alleli additivi si accumula nel genotipo.
19.2 L’analisi dei caratteri quantitativi è
statistica
I caratteri quantitativi vengono analizzati utilizzando
metodi statistici che valutano la media, la mediana, la
moda e la varianza della distribuzione fenotipica di un
carattere quantitativo.
La distribuzione di frequenza per l’intervallo fenotipico
viene descritta dalla varianza o dalla deviazione standard
dei valori del campione. Nel caso dei fenotipi di un carattere quantitativo, la varianza fenotipica (VP) rappresenta
una misura utile della distribuzione del campione.
La varianza fenotipica di un carattere è data dalla somma
della varianza genetica (VG) e della varianza ambientale
(VE).
La varianza genetica si suddivide in varianza additiva
(VA), varianza di dominanza (VD) e varianza interattiva
(VI), dovuta alle interazioni epistatiche tra i geni che determinano il fenotipo.
19.3 L’ereditabilità misura la componente
genetica della variabilità fenotipica
L’ereditabilità è una misura dell’entità del contributo
della varianza genetica alla varianza fenotipica totale.
L’ereditabilità in senso lato (H2) misura il rapporto tra
la varianza genetica e la varianza fenotipica (VG/VP). Un
modo per applicare l’analisi dell’ereditabilità in senso
lato agli umani è rappresentato dagli studi sui gemelli,
che fornisce una stima generale dell’ereditabilità.
L’ereditabilità in senso stretto (h2) misura il rapporto tra
la varianza genetica additiva e la varianza fenotipica (VA/
VP).
L’ereditabilità in senso stretto viene utilizzata per predire la risposta alla selezione (R) di un carattere alla selezione artificiale.
19.4 I loci dei caratteri quantitativi
corrispondono ai geni che contribuiscono
ai caratteri quantitativi
La mappatura dei QTL viene utilizzata per determinare
la posizione di potenziali QTL nel genoma utilizzando
metodi estremamente simili a quelli usati nella mappatura per ricombinazione.
La mappatura dei QTL richiede incroci controllati e
analisi dei cromosomi ricombinanti.
I geni specifici che influenzano i fenotipi dei caratteri
quantitativi vengono identificati e la loro variabilità caratterizzata attraverso l’analisi del locus del QTL candidato.
Gli studi di associazione genomica (GWAS) scansionano l’intero genoma degli organismi in popolazioni ad
accoppiamento casuale per ottenere prove statistiche
della presenza di QTL.
705
CAPITOLO 19 - Analisi genetica dei caratteri quantitativi
PAROLE CHIAVE
carattere multifattoriale
675
gene principale
676
soglia di suscettibilità genetica
carattere poligenico
675
genetica quantitativa
675
carattere quantitativo
675
geni additivi (caratteri additivi)
676
studio di associazione genomica
(GWAS)
684
701
suscettibilità genetica
684
carattere soglia
684
ipotesi dei geni multipli
677
concordanza
694
deviazione standard (s)
688
linea di introgressione (IL) (linea
quasi isogenica)
699
differenziale di selezione (S)
695
locus di carattere quantitativo
(QTL)
697
discordanza
694
varianza additiva (VA)
690
mappatura dei QTL
697
distribuzione delle frequenze
687
varianza ambientale (VE)
689
mediana (valore mediano)
688
ereditabilità
690
varianza dominante (VD)
690
moda (valore modale)
688
varianza fenotipica (VP)
688
2
ereditabilità in senso lato (H )
variabilità continua
675
variabilità discontinua
675
varianza (s2)
688
692
risposta alla selezione (R)
695
varianza genetica (VG)
689
ereditabilità in senso stretto (h )
692
selezione direzionale
696
varianza interattiva (VI)
690
eredità poligenica
675
selezione distruttiva
696
gene modificatore
676
selezione stabilizzante
696
2
Problemi proposti e risposte all'indirizzo http://hpe.pearson.it/sanders
706