")
PROTEINE E DATABASE (06)
Solo nel corpo umano ci sono più di 100.000 differenti proteine al lavoro. Ma quasi tutti sono costituiti da
appena venti diversi amminoacidi. In poche proteine umane altamente specializzate contengono
selenocisteina, aminoacido molto raro 21^ scoperto nel 1986.
Proteinogenici aminoacidi sono quegli amminoacidi che si trovano nelle proteine e richiedono meccanismo
cellulare codificata nel codice genetico di ogni organismo per la loro produzione isolato. Ci sono 23
proteinogenici amminoacidi, ma solo 21 si trovano negli eucarioti.
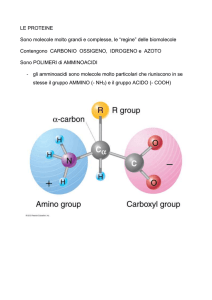
20 sono direttamente codificati dal codice genetico universale, definite come AA standard o canonici), sono
AA alfa poiche catena R è in posizione alfa.
Gli esseri umani possono sintetizzare 11 di questi 20. Gli altri 9 devono essere consumati nella dieta, e così
sono chiamati AA essenziali: sono ISTIDINA, ISOLEUCINA, LEUCINA, lISINA, METIONINA,
FENILALANINA, TREONINA, TRIPTOFANO e VALINA. Selenocisteina viene incorporato nelle proteine
da meccanismi sintetici unici (incorporazione co-traslazionale).
-1986: Selenocisteina è riconosciuto come il 21 ° amminoacido nella sintesi proteica ribosoma-mediata e la
sua incorporazione specifico è diretto dal codone UGA: l'incorporazione è co-traduzionale. È interessante
notare la tripletta di base che codifica questo aminoacido è UGA, normalmente un codone che funziona
come un segnale di STOP in traduzione.
-2002 Pirrolisina è un AA naturale (solitamente indicato come l'amminoacido 22) finora trovato soltanto in
alcune proteine archeali. Pirrolisina è una lisina in un legame ammidico a (4R, 5R)-4-sostituito-pyrroline-5carbossilato. La tripletta di base che codifica questo aminoacido è UAG (codone di stop). Co-traslazionale.
- N-Formilmetionina→ Segnalato anche da alcuni testi come aa naturale nel 1966. Nei batteri, l'inizio della
sintesi proteica viene segnalata dalla formazione di formil-metionil-tRNA ((f-Met)-tRNA). Questa reazione è
dipendente 10-formyltetrahydrofolate, e l'enzima metionil-formil tRNA. Questa reazione non è utilizzato da
eucarioti o Archaea, come la presenza di (f-Met)-tRNA in cellule batteriche non è definito come materiale
intrusivo e rapidamente eliminato.
Polipeptidi→ divisi in corti <50AA =peptidi, lunghi >50AA = proteine
PONTI A DISOLFURO → La formazione di ponti a disolfuro richiede ambienti ossidanti, pertanto di solito
non sono presenti nelle proteine intracellulari, ambiente essenzialmente riducente. Sono invece presenti tra le
proteine extracellulari secrete dalle cellule. Negli eucarioti si formano nel lume del RE primo compartimento
della via secretoria. Possono essere intermolecolari (es. Insulina A e B) oppure intramolecolari, rendendo le
proteine più stabili alla degradazione (es. tossine dei veleni e inibitori proteasi)
Esempio di modificazione post-traduzionali delle proteine
La O-glicosilazione è un processo altamente specifico, che non vede l'aggiunta "seriale" di carboidrati alla
proteina in oggetto, ma sito-specifica. Si svolge completamente nell'apparato del Golgi, dove zuccheri
vengono legati al peptide a livello dell'atomo di ossigeno delle catene laterali di serina o treonina.
Struttura primaria→ FASTA → primo rigo commento preceduto dal segno > in cui appare scritto nome
della proteina e codice del database, 2 riga scrittura degli amminoacidi in fila col codice ad una lettera.
Indica posizione numero dei ponti disolfuro.
Database: luogo in cui sono organizzati in maniera gerarchica i dati classificati per: evoluzione→ sequenza
simile o distanza, filogenesi, raggruppamento in superfamiglie e famiglie proteiche, più profili di
allineamento di sequenze; funzione e struttura.
In ordine storico la madre di tutti i database è National Biomedical Research Foundation (NBRF) che
oggi è Protein Identification Resource (PIR).
DATABASE
UNIPROT→ diviso in varie sottosezioni. Uniprot/swissprot è’ un database di sequenze proteiche curato
amano e altamente annotato, ossia vengono incluse le descrizioni, tassonomia, citazioni,, motivi,
classificazioni funzionali e strutturali, le annotazioni specifiche di residui tra cui le variazioni. Spesso sono
gestiti in maniera automatica; uniprot ha uomini che mano a mano controllano le sequenze e le inseriscono
nei database. Uniprot/TREMBL sono proteine tradotte automaticamente dal genoma, sono meno affidabili.
con la stella oro si indica quelli annotati a mano, con la stella argento quelli annotati automaticamente.
Ciascuna entry ha un codice univoco; quando apriamo vediamo prima tutte le annotazioni, poi i riferimenti
testuali e infine la sequenza FASTA con AA divisi in gruppi di 10. In base alla funzione ci dice anche in
quali e quante regioni è divisa. Mostra le modifiche post-traduzionali amminoacidiche. Strumento molto
comune è l’allineamento di sequenze in cui è possibile mettere a confronto più sequenze di diverse proteine
per vedere quali AA hanno in comune; simbologia:
*→ AA sono uguali;
: → AA non sono tutti uguali, ma la sostituzione è conservativo poiche non c’è cambio di funzione,
. → AA non sono tutti uguali e la sostituzione è semiconservativo; poiché sono simili, ma cambia la funzione
→ AA non sono tutti uguali e la sostituzione con conserva niente,
PROSITE→ sia database che strumento di analisi. E’ database di domini proteici, famiglie e siti funzionali.
Si può chiedere informazioni su un certo sito funzionale, cerchiamo sito funzionale e ci appare descrizione,
sezione tecnica che contiene la sequenza AA e referenze. Possiamo anche inserire sequenza FASTA e vedere
se ci sono siti funzionali all’interno. Esistono degli schemi di sequenza che sono molto semplici perciò è
possibile che siano nella nostra sequenza per caso, noi possiamo scegliere se includere anche questi siti
potenziali o no nella ricerca. PROSITE quando gli do una sequenza fa una predizione per similitudine, ma
non è detto che sia corretto. Ci aiuta a predire dove possono avvenire modifiche post-traduzionali.
MYHITS→ molto simile a PROSITE.
INTERPROSCAN → molto simile a PROSITE però effettua la scansione dei domini funzionali
contemporaneamente su più database. Permette di aumentare la probabilità di trovare risultati utili e i risultati
vengono colorati ognuno in modo diverso per differenziare i vari database;
EXPASY→ non è di per se un database , ma è un sito mantenuto dal SIB (istituto svizzero di informatica)
che raccoglie vari database. Vi si trovano sequenze proteiche annotate.
NCBI → più grande database di origine americana in cui possiamo ricercare pubblicazioni su una data
molecola, o fare ricerca su tutti i database per cui a partire dall’inserimento del nome di una molecola in
un'unica schermata mi trovo tutti i risultati che hanno a che fare in qualche modo con quella molecola, divisi
per database; non viene distinto ciò che è stato annotato manualmente o ciò che è stato annotato
automaticamente.
PDB → (protein data bank) non è una banca dati di sequenze, ma di strutture proteiche; possiamo vedere
quindi la struttura tridimensionale (secondaria, terziaria e quaternaria). Faccio una ricerca per parole chiave e
ottengo una lista di risultati, in alcuni casi se siamo fortunati il mio risultato posso trovarlo anche come
molecola del mese, ossia ogni mese viene analizzata una molecola e gli viene fatto un articolo. Possiamo
vedere la strutture tridimensionale delle sequenze presenti su UNIPROT, inoltre vengono evidenziati domini
funzionali; non è detto che ci sia la proteina perché si conosce la struttura di poche, inoltre non è detto che ci
sia la struttura completa. Delle proteine con struttura quaternaria sono presenti tante stecche di sequenze
quante sono le componenti. Inoltre è possibile vedere l’animazione della struttura quaternaria in cui è
possibile evidenziare i vari legami ed è possibile colorare le varie catene polipeptiche con colori diversi. Con
gli altri database si vede una sola subunità poiché ogni gene esprime una sola catena, PDB posso vedere la
struttura composta da tutte le catene.
Spesso troviamo la sigla GO che significa “Gene Ontology → ontologia del gene”,
Un progetto atto a costruire un vocabolario per descrivere geni e prodotti genici attribuibili a ogni
organismo; ossia nominare i geni in maniera univoca in modo che in diverse banche dati appaia lo stesso
nome e sia facilmente rintracciabile, infatti la differenza nel nominare diversamente proteine in luoghi
diversi crea problemi nella creazione dei database e nella ricerca da parte degli studiosi.
")