LE MALATTIE MUSCOLARI
Le malattie muscolari ricadono in 3 gruppi principali: le distrofie muscolari, le miopatie, le
malattie neurogene.
Le distrofie muscolari sono malattie muscolari ereditarie che di solito provocano una
degenerazione progressiva. Sono classificabili a seconda delle modalità di trasmissione ereditaria,
del quadro clinico dei muscoli interessati e sempre più spesso con tecniche di genetica molecolare
(distrofia tipo Duchenne e Becker).
Le miopatie, sono un gruppo di condizioni con eziologia diversa come:
miopatie infiammatorie, in cui c’è infiammazione primitiva del muscolo molto spesso trattabile
miopatie metaboliche, in cui un problema metabolico primitivo di origine genetica con un impatto
importante sulla funzione muscolare. Esempi: GLICOGENOSI: sono condizioni nelle quali le
cellule accumulano glicogeno per incapacità a metabolizzarlo, dovute a deficienze di enzimi
metabolizzanti tale substrato. Sono geneticamente determinate e trasmesse con modalità di tipo
autosomico recessivo
Le malattie mitocondriali, dovute a mutazioni del DNA mitocondriale, per la loro caratteristica di
compromette la fosforilazione ossidativa, tendono a danneggiare maggiormente gli organi che più
dipendono da questo processo, tra cui i muscoli scheletrici (MIOPATIE MITOCONDRIALI).
Le malattie neurogene sono essenzialmente le sindromi miasteniche, caratterizzate dalla
combinazione di debolezza muscolare e affaticamento. La miastenia grave è la forma che si
incontra più frequentemente. E’ una malattia autoimmune in cui il bersaglio antigenico è il recettore
dell’acetilcolina nella membrana postsinaptica della giunzione neuromuscolare. Le risultanti
anomalie nel numero o nella funzione dei recettori alterano o bloccano la trasmissione dell’impulso
nella giunzione neuromuscolare
DISTROFIE MUSCOLARI
LA DISTROFIA DI DUCHENNE è la più frequente forma di distrofia muscolare; è trasmessa
con eredità recessiva, legata al cromosoma X e pertanto colpisce quasi esclusivamente i maschi.
Infatti, nel maschio che ha solo un cromosoma X, una malattia determinata da un gene posto su tale
cromosoma si produce anche se il gene è recessivo; nella femmina, invece, se il gene è recessivo,
avrà bisogno per determinare la malattia, dello stato omozigote; dovrà cioè essere posto anche
sull’altro cromosoma X. L’incidenza è di circa 1 su 5000 maschi nati. La malattia è dovuta a
delezioni sul gene che codifica la distrofina. Il gene si estende per 2300 kilobasi, è uno dei geni più
1
estesi del genoma ed include 79 esoni. Il solo mRNA ha una lunghezza di 14.000 basi e la proteina
ha 3.685 residui amminoacidici. Proprio per la sua estensione, questo gene presenta un’alta
incidenza di delezioni: il 60% delle mutazioni Duchenne sono delezioni, il 30% mutazioni
puntiformi.
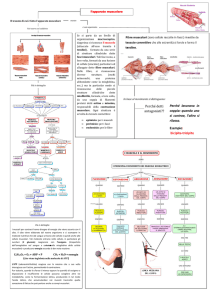
La distrofina è localizzata a livello muscolare, e normalmente ancora la membrana della cellula
muscolare alla matrice extracellulare. E’ pertanto essenziale per il processo di contrazione ed ha un
ruolo meccanico nell’assorbimento degli urti, in modo che essi non danneggino la fibra muscolare. I
soggetti che non esprimono questo gene sviluppano una perdita progressiva della motilità
muscolare che li conduce a perdere la deambulazione entro i primi 10 anni di vita. La malattia
viene diagnostica intorno ai 3 anni, quando il bambino che ha iniziato a camminare regolarmente,
manifesta una sempre maggiore debolezza muscolare, particolarmente evidente all’esordio agli arti
inferiori e che contrasta con l’aspetto ipertrofico del polpaccio (pseudoipertrofia). In realtà, la
biopsia mostra degenerazione e necrosi delle fibre muscolari, che vengono sostituite da tessuto
connettivo e infiltrazione leucocitaria. Il paziente perde via via la funzionalità di tutti i muscoli,
compresi quelli respiratori e la morte che ne consegue è in genere per insufficienza respiratoria.
Nelle prime fasi della malattia la fibra muscolare danneggiata rilascia gli enzimi muscolari in
circolo, tra cui la creatin chinasi, la cui misurazione era stata in passato utilizzata per la diagnosi
della malattia, prima della identificazione del gene.
Una forma più lieve di difetto alla distrofina è la distrofia muscolare tipo Becker. I pazienti
Becker perdono la deambulazione più tardivamente verso i 15 anni. La distrofia di Becker è dovuta
anch’essa a delezioni a livello del gene per la distrofina, tuttavia queste, a differenza di quelle del
tipo Duchenne, non causano frameshift. La proteina sintetizzata nella distrofia di Becker è più breve
ma in qualche modo funzionale, sebbene in modo incompleto.
2
MIOPATIE METABOLICHE
GLICOGENOSI: sono condizioni nelle quali le cellule accumulano glicogeno per incapacità a
metabolizzarlo, dovute a deficienze di enzimi metabolizzanti tale substrato. Sono geneticamente
determinate e trasmesse con modalità di tipo autosomico recessivo.
Gli organi più colpiti sono quelli in cui il metabolismo del glicogeno è particolarmente attivo come
il fegato, il muscolo, incluso il miocardio, e il rene. La classificazione delle glicogenosi si basa sulla
natura del deficit enzimatico
Esistono circa 9 forme di glicogenosi. Possono avere interessamento prevalentemente epatico e
renale (tipo I) o più generaliizzato e quindi a carico di miocardio e muscolo scheletrico (tipo II). La
forma V colpisce la fosforilasi muscolare ed è caratterizzata da intolleranza allo sforzo fisico. Il
muscolo scheletrico utilizza, infatti, glicogeno, come substrato energetico per la formazione di
glucosio, che prende poi la via della glicolisi. Se mancano gli enzimi metabolizzanti il glicogeno,
questo si accumula nella cellula, in modo morfologicamente dimostrabile e, vi è una bassa
produzione di energia.
3
4
MIOPATIE MITOCONDRIALI (Eredità patologica matrilare)
Con questo termine si indica la trasmissione dalla madre alla prole dell’informazione
presente nel DNA mitocondriale
IL DNA mitocondriale è soggetto, come quello nucleare, a mutazioni
Le mutazioni a carico del DNA mitocondriale del maschio rimangono confinate in lui e non
trasmesse alla prole
Sono state identificate diverse malattie provocate da mutazioni al DNA mitocondriale. Le
malattie mitocondriali, per la loro caratteristica di compromette la fosforilazione ossidativa,
tendono a danneggiare maggiormente gli organi che più dipendono da questo processo, tra
cui i muscoli scheletrici (MIOPATIE MITOCONDRIALI). La sintomatologia è dovuta in
gran parte ad un difetto energetico per alterata funzione della catena respiratoria. Il processo
di fosforilazione ossidativa richiede l’integrità di almeno 100 proteine; di queste 12 sono
codificate dal genoma mitocondriale, le altre da quello nucleare, vengono sintetizzate nel
citoplasma e da qui traslocate nel mitocondrio dove svolgono la loro funzione.
Le malattie mitocondriali sono molto variabili sul piano clinico, sia per quanto riguarda l'età
di esordio sia per il tipo di evoluzione ed il tessuto coinvolto.
Nell’età adulta, i sintomi più ricorrenti sono l'intolleranza allo sforzo ed il facile
affaticamento.
5
MALATTIE INFIAMMATORIE DELLE ARTICOLAZIONI
•
Le malattie infiammatorie delle articolazioni hanno 4 cause principali: autoimmune (artrite
reumatoide, LES), degenerativa (osteoartrosi), deposizione di cristalli (gotta) e infettive
(artrite tubercolare).
ARTRITE REUMATOIDE
L’artrite reumatoide è una malattia cronica autoimmune che colpisce la membrana sinoviale delle
articolazioni. Tale membrana è una sottile pellicola che riveste la superficie interna di tutte le
articolazioni e che ha come funzione di lubrificare l’articolazione stessa producendo il liquido
sinoviale. Circa l'1% di tutta la popolazione ne è affetto; le donne vengono colpite con rapporto di
4:1 rispetto agli uomini. Può esordire a tutte le età, ma la massima incidenza è fra i 25 e i 50 anni.
Dal punto di vista istopatologico le lesioni reumatoidi sono attribuite alla produzione di anticorpi
anti-immunoglobuline detti fattori reumatoidi (FR). Il fattore reumatoide è un anticorpo per lo più
di tipo IgM diretto contro la regione Fc di self IgG. Gli immunocomplessi così formati precipitano
nel liquido sinoviale, nelle cartilagini e negli endoteli delle arteriole e dei capillari, con capacità di
attrarre fagociti ed innescare una reazione infiammatoria. L’identificazione di FR nel siero non è
patognonomica, in quanto si può trovare anche in soggetti non affetti da A.R. Inoltre una
percentuale rilevante di pazienti affetti da A.R. non presentano F.R. Ciò ha messo in dubbio che il
F.R abbia un ruolo patognomico principale nell’insorgenza della malattia. Varie osservazioni più
recenti tendono ad attribuire un’importanza all’evidenza che i linfociti T helper si rivolgano contro
antigeni posti sulle sierose articolari contribuendo notevolmente al danno articolare. Le cellule T
helper possono produrre citochine flogogene. I macrofagi che vengono attivati e le loro citochine
(p. es., il tumor necrosis factor, il fattore stimolante la formazione di colonie di granulocitimacrofagi) contribuiscono alla migrazione di cellule infiammatorie e al loro deposito nel tessuto
sinoviale. L'aumento delle cellule di rivestimento di derivazione macrofagica è predominante,
insieme a quello dei linfociti e ai cambiamenti vascolari nelle fasi precoci della malattia.
Nelle articolazioni colpite cronicamente la membrana sinoviale, sottile nei casi normali, sviluppa
numerose pieghe villose e si ispessisce per l'ipertrofia e l'iperplasia delle cellule di rivestimento e
per la proliferazione dei linfociti e delle plasmacellule. Le cellule di rivestimento producono una
serie di sostanze diverse, tra le quali la collagenasi e la stromelisina che possono contribuire alla
distruzione della cartilagine; l'interleuchina-1, che stimola la proliferazione dei linfociti e le
prostaglandine. Le cellule infiltranti, disposte inizialmente in sede perivenulare e più tardi a
costituire veri e propri follicoli linfatici provvisti di centro germinativo, sintetizzano interleuchina2, altre citochine, il fattore reumatoide (FR) e altre immunoglobuline. Sono anche presenti depositi
6
di fibrina, fibrosi e necrosi. La membrana sinoviale iperplastica (panno) può erodere la cartilagine,
l'osso subcondrale, la capsula articolare e i legamenti. I leucociti polimorfonucleati (PMN) non sono
cospicui nella membrana sinoviale, ma spesso predominano nel liquido sinoviale. Il nodulo
reumatoide (granuloma), presente in più del 30% dei pazienti, si presenta generalmente a livello
sottocutaneo nei punti sottoposti a maggiore frizione (p. es., gomito). Sono granulomi non specifici
caratterizzati, all'esame anatomopatologico, da una zona necrotica centrale circondata da cellule
mononucleate disposte a palizzata, con il loro maggior asse che si irradia dal centro; il tutto è
circondato da una zona esterna di linfociti e plasmacellule. Si possono trovare segni di vasculite
nella pelle, nei nervi o in numerosi organi viscerali, nei casi gravi di AR, ma questi sono
clinicamente significativi soltanto in alcuni casi. Le lesioni dei vasi possono portare a sofferenza del
polmone, rene, cuore e muscoli scheletrici.
L'OSTEOARTROSI (OA)
L'osteoartrosi (OA), la più comune di tutte le patologie articolari, inizia in modo asintomatico nel 2o
e 3o decennio ed è estremamente diffusa all'età di 70 anni. La quasi totalità dei soggetti intorno ai
40 anni, mostra qualche alterazione patologica delle articolazioni sottoposte al carico, benché una
parte relativamente piccola di essi presenti una sintomatologia. Vengono colpiti entrambi i sessi con
la stessa frequenza, ma l'esordio è più precoce nel maschio. La patogenesi della malattia è
sostanzialmente ignota, ma si ritiene che il condrocita abbia un ruolo di primaria importanza e che
rappresenti sia l’effettore che il bersaglio dei meccanismi patologici che portano alla degenerazione
della cartilagine articolare. Infatti, il processo responsabile della degenerazione del collagene e dei
proteoglicani della matrice cellulare viene messo in atto da enzimi proteolitici (metalloproteasi)
sintetizzati e rilasciati dai condrociti, probabilmente in risposta a citochine infiammatorie come IL-1
e TNF-alfa, prodotti dagli stessi condrociti.
E’, infatti, noto che l’IL-1 è in grado di stimolare la sintesi di metalloproteasi attraverso il
rilascio di NO e di inibire la sintesi di collagene e proteoglicani.
La progressiva riduzione dello spessore cartilagineo e la sua fissurazione hanno importanti
ripercussioni su tutte le strutture articolari; l’osso partecipa con alterazioni di tipo
osteosclerotico reattivo all’alterata meccanica articolare, sviluppa cisti subcondriali e forma
processi osteofitici marginali (crescita di nuovo osso e cartilagine).
7
GOTTA
Le purine sono sostanze importante per l’organismo umano, perché due dei quattro
(cinque) acidi nucleici (adenina e guanina) vengono sintetizzati a partire da questa sostanza.
Se le purine non vengono fornite a sufficienza dall’alimentazione, l’organismo è capace di
fabbricarseli a partire dai tre aminoacidi glicina, asparagina e glutamina.
Gli eccessi di purine (da alimentari e decomposizione di cellule corporee) vengono
trasformati in acido urico. Una parte viene aggiunta dal fegato alla bile ed espulso
nell’intestino, dove una parte specializzata di flora intestinale li usa e decompone. Un’altra
parte viene separata dai tubuli renali ed espulsa nell'urina.
Se accade che ci sia dell’acido urico in eccesso e né l'intestino né i reni riescono più a
espellere tutto, l’acido rimane nell’organismo e, sopra una certa concentrazione, si
cristallizza in diversi organi. Prevalentemente colpisce l'articolazione dell’alluce. Questo si
infiamma e causa dolore. Allora si parla di gotta
E’ caratterizzata dall’aumento della concentrazione ematica di acido urico. La gotta
primaria è una sindrome molto diffusa, caratterizzata da lesioni articolari che dapprima
appaiono acutamente e costituiscono la nota più evidente dell’attacco o accesso gottoso e
poi evolvono in artriti croniche; nei gottosi è inoltre frequente la calcolosi renale, in
rapporto con la continua eliminazione urinaria di forti quantità di acido urico, scarsamente
solubile e quindi facilmente precipitabile in forma di calcoli.
Gli attacchi colpiscono prevalentemente di notte e sono caratterizzati da dolori, dapprima
tenui e poi sempre più forti; dopo questo si ha una progressiva attenuazione del dolore fin
tanto che non scompare del tutto.
Il dolore si associa a tumefazione dell’articolazione colpita. In genere il dolore colpisce una
sola o al massimo due o tre Il dolore si associa a tumefazione dell’articolazione colpita. In
genere il dolore colpisce una sola o al massimo due o tre articolazioni. Le sedi classiche
sono metatarso-falangea dell’alluce, quelle tarso-metatarsiche, quelle tibio-tarsiche, dei
ginocchi o delle mani.
8
L’attacco è in rapporto con la precipitazione di cristalli di acido urico o di urato di sodio dalle
zone periarticolari.
L’inizio dell’attacco gottoso coincide con il passaggio dal sangue ai tessuti di una notevole
quantità di acido urico.
Il depositarsi di acido urico nei tessuti attira i leucociti, che fagocitano i cristalli. Si ritiene che i
cristalli siano lesivi per i granulociti, o per i loro granuli specifici, che liberano all’esterno le
idrolasi acide e le proteine cationiche le quali aumentano la permeabilità vascolare liberando
istamina dalle mastcellule; inoltre le proteasi leucocitarie sono capaci di liberare chinine dai
chininogeni plasmatici. Gli intervalli tra un attacco e l’altro variano da caso a caso, spesso in
rapporto con l’alimentazione. L’introduzione di forti quantità di acidi nucleici e di proteine,
può dar luogo ad un nuovo attacco.
9