PROGRAMMA CALCOLO
INDICE
UNITA’ 1
Configurazione Elettronica degli Elementi……p.3
UNITA’ 2
Elettrone……..………………………………...p.8
UNITA’ 3
Il Legame Dativo……………………………...p.11
UNITA’ 4
Il Modello Quanto-Meccanico di Atomo….….p.13
UNITA’ 5
La Teoria Atomica di Dalton………...……….p.14
UNITA’ 6
Legame ad Idrogeno………………………….p.15
UNITA’ 7
Legame Covalente………...………………….p.17
UNITA’ 8
Legame Ionico...……………………………...p.19
UNITA’ 9
Modello di Bhor………………………………p.24
UNITA’ 10 Orbitale………...……………………………...p.29
UNITA’ 11 Regola dell’Ottetto..………………………….p.39
UNITA’ 12 Relazione tra volume pressione e temperatura nei
gas………………………………..…………...p.40
UNITA’ 13 connessione tra Caratteri Qalitativi…………...p.45
UNITA’ 14
Distribuzione di Gauss…..………......………p.47
UNITA’ 15
Distribuzione di Bernoulli…...………………p.49
UNITA’ 16
Distribuzione di Poisson.……………………p.51
UNITA’ 17
Legge dei Grandi Numeri…………………...p.53
UNITA’ 18
Teorema Centrale del Limite..………………p.54
UNITA’ 1
CONFIGURAZIONE ELETTRONICA DEGLI
ELEMENTI
In chimica, il termine configurazione elettronica si riferisce alla
disposizione degli elettroni legati; ovvero al loro comportamento attorno
ainuclei di uno o più atomi.
Poiché gli elettroni sono fermioni, essi sono soggetti al principio di
esclusione di Pauli, il quale stabilisce che due fermioni non possono
occupare lo stesso stato quantico contemporaneamente. Questo principio
è fondamentale nel determinare la configurazione degli elettroni negli
atomi: una volta che uno stato viene occupato da un elettrone, l'elettrone
successivo deve occupare uno stato differente.
In un atomo, gli stati stazionari (indipendenti dal tempo) di funzione
d'ondaelettronica
(ovvero
gli
stati
che
sono stati
particolari dell'equazione di Schrödinger HΨ = EΨ dove H è
l'hamiltoniana) vengono detti orbitali, per analogia con la visione classica
dell'elettrone come particella che orbita attorno al nucleo. Per un atomo
multielettronico, con x elettroni, l'espressione corretta della funzione
d'onda deve considerare le coordinate spaziali di tutti gli x elettroni
contemporaneamente. Ciò, in termini matematici, viene espresso dalla
funzione d'onda Ψ = Ψ(n1, n2, n3,...nx). Tuttavia, per gli scopi della
chimica, viene sfruttata una notevole semplificazione utilizzando la
cosiddetta "approssimazione orbitalica": cioè ogni elettrone viene
considerato singolarmente come appartenente ad un atomo idrogenoide e
la carica nucleare Ze, carica che viene utilizzata per calcolare il termine
relativo all'energia potenziale da inserire nell'equazione di Schrödinger,
viene corretta utilizzando la carica nucleare efficace Zeff. Quindi la forma
semplificata della funzione d'onda, utilizzata per descrivere un atomo
polielettronico,
diviene
una
funzione
del
tipo
Ψ
=
Ψ(n1)Ψ(n2)Ψ(n3)...Ψ(nx).
Il quadrato del modulo del valore di Ψ in un punto (ampiezza d'onda
complessa) rappresenta la densità di probabilità di trovare l'elettrone in
quel punto. Gli orbitali di un atomo sono distinti da quattro numeri
quantici: n, l, ml e ms e, per il principio di Pauli, non è possibile che due
elettroni abbiano lo stesso valore per tutti e quattro i numeri.
Numero quantico principale (n)
Il primo numero quantico n, detto numero quantico principale, determina
la distanza media dal nucleo (dimensione dell'orbitale), che aumenta al
crescere di n, e la maggior parte dell'energia dell'elettrone (livello
energetico=periodo). Esso assume tutti i valori interi positivi in ordine
crescente.
Elettroni (e orbitali) che condividono n appartengono allo stesso livello
(detto anche "guscio").
Valore
di n
Lettera
Massimo numero di elettroni nel livello (pari a 2 x
n2)
1
K
2
2
L
8
3
M
18
4
N
32
5
O
50
6
P
72
7
Q
98
...
...
...
Il numero massimo di elettroni nell'n-simo livello di energia è 2n2.
Numero quantico orbitale (l)
Il secondo numero quantico l, detto numero quantico orbitale (o,
impropriamente, numero quantico azimutale o angolare o rotazionale),
corrisponde al momento angolare dello stato. Questi stati prendono la
forma di un'armonica sferica, e sono quindi descritti da polinomi di
Legendre.
I vari stati correlati ai differenti valori di l vengono a volte detti "sottolivelli" o "sotto-gusci", e (principalmente per ragioni storiche) vengono
indicati da lettere, come elencato di seguito:
Valore
di l
Lettera
Massimo numero di elettroni nel sotto-livello (pari a
(2 x l + 1) x 2)
0
s
2
1
p
6
2
d
10
3
f
14
4
g
18
...
...
...
Per ogni valore di n, l assume in ordine crescente tutti i valori interi
compresi tra 0 e n-1 e quindi il guscio n=1 possiede solo il sotto-guscio s,
il guscio n=2 possiede i sotto-gusci s e p, il guscio n=3 possiede i sottogusci s, p e d e così via.
Numero quantico magnetico (ml) e numero quantico di spin (ms)
Ogni sotto-guscio può accogliere 2(2l+1) elettroni. Questo perché per
ogni valore di l il terzo numero quantico ml, detto numero quantico
magnetico, che può essere pensato (in maniera inaccurata) come la
proiezione quantizzata del vettore momento angolare sull'asse z, assume
in ordine crescente tutti i valori interi compresi tra -l e l, e quindi ci sono
2l+1 stati possibili. Ogni stato distinto nlml può essere occupato da due
elettroni con spin opposto (dato dal quarto numero quantico ms,
detto numero quantico di spin, che per ogni valore di ml assume i due
valori -1/2 e +1/2), dando un totale di 2(2l+1) elettroni. Stati con
valori l superiori a quelli mostrati nella tabella sono perfettamente
ammissibili in teoria, ma questi valori sono relativi ad atomi che non sono
ancora stati scoperti.
Ordine di riempimento degli stati quantici e relazione con la struttura
della tavola periodica
Allo stato fondamentale, gli stati quantici di un atomo sono riempiti in
ordine crescente di energia, secondo il principio dell'Aufbau; ovvero, il
primo elettrone va ad occupare lo stato libero con energia più bassa è così
via. Il fatto che lo stato 3d sia più alto, come energia, dello stato 4s, ma
più basso del 4p è il motivo per l'esistenza dei metalli del blocco d.
L'ordine in cui gli stati vengono riempiti è il seguente:
1s
2s
3s
4s
5s
6s
7s
2p
3p
3d 4p
4d 5p
4f 5d 6p
5f 6d 7p
Ciò porta direttamente alla struttura della tavola periodica. Le proprietà
chimiche di un atomo sono largamente determinate dalla disposizione
degli elettroni del guscio più esterno, il guscio di valenza (anche se altri
fattori, come raggio atomico, peso atomico, e l'aumentata accessibilità a
stati elettronici addizionali contribuiscono alla chimica degli elementi,
man mano che le dimensioni degli atomi aumentano).
Progredendo attraverso un gruppo, dall'elemento più leggero a quello più
pesante, i gusci elettronici esterni (quelli che partecipano più facilmente
alle reazioni chimiche) sono tutti nello stesso tipo di orbitale, con forme
simili, ma con un sempre maggiore livello di energia e distanza media dal
nucleo. Ad esempio, i gusci esterni degli elementi del primo gruppo,
introdotto dall'idrogeno, hanno tutti un elettrone nell'orbitale s.
Nell'idrogeno, l'orbitale s è nel più basso stato di energia possibile per
ogni atomo (ed è rappresentato dalla posizione dell'idrogeno nel
primo periodo della tavola periodica). Nel francio, l'elemento più pesante
del gruppo, il guscio esterno si trova nel settimo orbitale, decisamente più
lontano dal nucleo rispetto agli elettroni che riempiono i gusci sottostanti.
Come altro esempio: sia il carbonio che ilpiombo hanno quattro elettroni
nell'orbitale del guscio esterno.
A causa dell'importanza del guscio esterno, le differenti regioni della
tavola periodica sono a volte dette blocchi della tavola periodica,
chiamati secondo il sotto-guscio nel quale risiede l'ultimo elettrone:,
blocco s, blocco p, blocco d, ecc.
Notazioni e semplificazioni
Un esempio della notazione comunemente usata per esprimere la
configurazione elettronica di un atomo, nel caso del silicio, è il seguente:
1s22s2 2p6 3s2 3p2. I numeri sono i numeri dei gusci, n; le lettere si
riferiscono agli stati del momento angolare e i numeri sovrascritti sono i
numeri degli elettroni in quello stato per l'atomo in questione. Una
versione più semplice è quella di elencare il numero di elettroni di ogni
guscio, ad esempio, sempre per il silicio: 2-8-4.
Altra esemplificazione, molto utilizzata nella pratica comune, consiste
nell'evidenziare i gusci più esterni esprimendo i livelli energetici
precedenti tramite abbreviazione che rimanda alla configurazione del gas
nobile immediatamente precedente l'elemento in oggetto. Ad esempio,
considerando sempre il silicio, la configurazione elettronica può essere
espressa nella seguente forma contratta: [Ne] 3s2 3p2, dove [Ne] indica la
configurazione elettronica del neon.
UNITA’ 2
ELETTRONE
L'elettrone è una particella subatomica con carica elettrica negativa di
qe = 1,602 · 10−19 C(carica
elementare),
e
una
massa
di
−31
circa 9,10 · 10 kg (0,511 MeV/c²).
L'elettrone viene comunemente rappresentato dal simbolo e−.
L'antiparticella dell'elettrone è ilpositrone, che si differenzia solo per la
carica elettrica positiva.
Gli atomi consistono di un nucleo (formato da protoni e neutroni)
circondato da elettroni. La massa dell'elettrone è circa 1/1836 di quella di
neutroni e protoni.
L'elettrone appartiene alla classe delle particelle subatomiche
dette leptoni, che si ritiene siano componenti fondamentali della materia
(ovvero non possono essere scomposte in particelle più piccole).
L'elettrone ha spin semi-intero pari a 1/2, il che implica che è unfermione,
ovvero, rispetta la statistica di Fermi-Dirac e il principio di esclusione di
Pauli, non è quindi possibile avere più elettroni nello stesso stato.
Dal 1914, gli esperimenti dei fisici Ernest Rutherford, Henry
Moseley, James Franck e Gustav Hertzhanno stabilito definitivamente
che l'atomo è composto da un nucleo positivo massivo di cariche positive
circondato da una leggera massa di elettroni. Nel 1913, Il fisico
danese Niels Bohr postula che gli elettroni risiedano in stati di energia
quantizzata, con l'energia determinata dal momento angolare delle orbite
degli elettroni attorno al nucleo. Gli elettroni possono muoversi tra questi
stati, o orbite, in seguito all'assorbimento o all'emissione di un quanto di
energia, un fotone di specifica frequenza. Questa teoria è in grado di
spiegare correttamente le linee di emissione spettrale dell'idrogeno che
questo forma se scaldato o attraversato da corrente elettrica. Ciò
nonostante, il modello di Bohr fallisce nel predire l'intensità delle relative
linee e nello spiegare la struttura dello spettro di atomi più
complessi. I legami chimici tra gli atomi sono spiegati nel 1916
da Gilbert Newton Lewis, come una interazione tra gli elettroni che li
costituiscono. Come è noto che le proprietà chimiche degli elementi si
ripetono ciclicamente in accordo con la legge periodica, nel 1919 il
chimico americano Irving Langmuir suggerisce che questo può essere
spiegato se gli elettroni in un atomo sono strutturati su strati. Gli elettroni
si dispongono in gruppi intorno al nucleo.
Nel 1924, il fisico austriaco Wolfgang Pauli osserva che la struttura a
strati di un atomo può essere spiegata da un set di quattro parametri che
definiscono univocamente lo stato quantico di un elettrone, e un singolo
stato non può essere occupato da più di un singolo elettrone (questa legge
è nota come Principio di esclusione di Pauli). Nonostante ciò, sfuggiva il
significato fisico del quarto parametro che può assumere solo due valori.
Questo fu spiegato dai fisici tedeschiAbraham Goudsmith e George
Uhlenbeck quando suggerirono che un elettrone, oltre al momento
angolare associato alla sua orbita, possa possedere un proprio momento
angolare intrinseco. Questa proprietà è nota come spin, e riesce a spiegare
la misteriosa separazione delle linee spettrali osservate con la
spettrografia ad alta definizione.
L'elettrone, come particella subatomica, appartiene alla classe dei leptoni.
In quanto particella elementare, l'elettrone ha una doppia natura:
corpuscolare ed ondulatoria.
L'antiparticella dell'elettrone è il positrone, di carica uguale in modulo,
ma di segno opposto. Lo scopritore del positrone, Carl David
Anderson propose di cambiare il nome dell'elettrone in negatrone,
lasciando il termineelettrone ad indicare una generica particella di carica
non specificata.
L'elettrone ha una carica elettrica di −1,6021765 × 10−19 C,
una massa pari a 9,11 × 10−31 kg (quest'ultimo valore corrisponde a
1/1836 della massa del protone). Il simbolo comunemente utilizzato per
indicare l'elettrone è e−. La vita media dell'elettrone supera i 4,6 ×
1026 anni.
Secondo i postulati della meccanica quantistica, è possibile rappresentare
l'elettrone per mezzo di una funzione d'onda, dalla quale è possibile
dedurre anche la probabilità di trovare tale particella in un volume
infinitesimo dV. Tuttavia, in base al principio di indeterminazione di
Heisenberg, non è possibile determinare con esattezza laposizione e
la quantità di moto di un elettrone. Il principio afferma infatti che
maggiore è la precisione con la quale si rileva la posizione, minore è la
precisione con la quale è possibile rilevare la quantità di moto, e
viceversa.
Con lo sviluppo degli acceleratori di particelle nella prima metà del
ventesimo secolo, i fisici iniziarono a sondare in profondità nelle
proprietà delle particelle subatomiche. Il primo tentativo riuscito di
accelerare elettroni usando l'induzione magnetica fu fatto nel 1942 da
Donald Kerst. Il suo primo betatrone raggiunse energie di 2.3 MeV
(milioni di elettronvolt), mentre il conseguente raggio beta raggiunse i
300 MeV. Nel 1947, fu scoperta la radiazione di sincrotrone con un
sincrotrone di 70 MeV della General Electric. Questa radiazione era
causata dall'accelerazione di elettroni a velocità prossime a quelle della
luce in un campo magnetico.
Con un gruppo di particelle cariche (beam) di energia di 1.5 GeV, il
primo collider ad alte enegie fu ADONE, che iniziò le operazioni nel
1968. Questa struttura accelerò elettroni e positroni in direzioni opposte,
raddoppiando l'energia effettiva a disposizione, rispetto a collisioni con
un bersaglio statico. Il Large Electron-Positron Collider (LEP) al CERN,
che operò dal 1989 al 2000, raggiunse collisioni di 209 GeV e fece
importanti misure in merito al Modello Standard dell Particelle.
L'LHC, l'ultimo acceleratore del CERN, sostituirà largamente l'uso di
elettroni con l'uso di adroni perché questi sono meno soggetti alla perdita
di energia per radiazione di sincrotrone e quindi è maggiore il rapporto
fra energia acquisita dalla particella e l'energia spesa per ottenerla.
UNITA’ 3
IL LEGAME DATIVO
Nei legami di tipo covalente il doppietto elettronico condiviso dai due
atomi, è costituito da elettroni provenienti l'uno da un atomo e l'altro
dall'altro. I due atomi coinvolti nel legame covalente devono avere
pertanto ciascuno almeno un elettrone spaiato sull'ultimo livello quantico.
Analizzeremo ora il caso in cui la coppia di elettroni comuni è data per
intero da uno solo degli atomi partecipanti al legame, mentre l'altro mette
a disposizione un orbitale vuoto. Questo tipo di legame si chiama
covalente dativo, o semplicemente legame dativo.
Esempi di legame dativo si trovano nella formazione degli ioni ammonio
e idronio. Lo ione H+ è l'atomo di idrogeno privo dell'elettrone e presenta
pertanto l’unico orbitale di cui dispone vuoto. D'altra parte, l'atomo di
azoto della molecola dell'ammoniaca, NH3, presenta nell'ultimo livello
quantico un doppietto elettronico che può essere utilizzato per formare un
legame dativo nel modo illustrato in figura 15.
Il legame dativo può essere anche indicato con una freccia diretta dal
donatore all'accettore del doppietto elettronico. Una volta formatosi il
legame, la carica elettrica si distribuisce sulla struttura complessiva che
quindi si comporta come si trattasse di uno ione formato da un singolo
atomo.
In modo analogo, l'atomo di ossigeno della molecola di acqua, presenta
due doppietti elettronici disponibili per la formazione di altrettanti legami
dativi con lo ione H+. Quando però il primo H+ si unisce alla molecola
d'acqua, lo ione idronio che si forma respinge un secondo H+ che si
avvicina in quanto si tratta ora dell'interazione di due strutture elettriche
dello stesso segno. Lo ione H4O++ infatti non è mai stato osservato
sperimentalmente.
L' atomo di ossigeno può diventare accettore di elettroni pur non
presentando, in condizioni normali, orbitali vuoti. Però, uno dei due
elettroni spaiati che stazionano negli orbitali di tipo p, con un piccolo
apporto di energia, può passare sull'altro orbitale accoppiandosi
all'elettrone già presente in esso e liberando in questo modo un orbitale da
elettroni
Quest'ultima struttura dell'atomo di ossigeno con un orbitale libero
sull'ultimo livello energetico, è meno stabile (si ricordi la regola di Hund)
di quella con gli elettroni distribuiti su tutti gli orbitali. Così facendo,
però, l'atomo di ossigeno si rende disponibile per la formazione di un
composto che prevede legami dativi. Il composto che si forma risulta più
stabile dell'atomo isolato e alla fine il bilancio energetico appare
globalmente positivo.
Due esempi in cui l'atomo di ossigeno funge da accettore di doppietti
elettronici sono la molecola di acido nitrico e quella di acido solforico.
L'azoto presente nella molecola di acido nitroso HNO2 possiede un
doppietto elettronico disponibile per un legame dativo. Se a questo si
aggancia un atomo di ossigeno, si forma l'acido nitrico di formula HNO3.
.
Anche l'acido solforico rappresenta un esempio classico di legame dativo,
anzi, nella sua molecola, si può notare la presenza di due legami di questo
tipo
UNITA’ 4
IL MODELLO QUANTO-MECCANICO
DELL’ATOMO
La teoria di Bohr (detta anche di Bohr-Sommerfeld) lasciava insoluti
numerosi problemi. Nel 1924, il fisico francese L. De Broglie ipotizzò
che, analogamente a quanto si era postulato per i quanti di luce o fotoni
(Einstein, 1905), anche agli elettroni si può attribuire una duplice
natura ondulatoria e corpuscolare. Questa ipotesi, confermata in
seguito sperimentalmente, indica che a una particella di
massa m (espressione della natura corpuscolare) è associabile una
lunghezza d'onda λ (espressione della natura ondulatoria) secondo la
relazione:
dove v è la velocità della particella e h è la costante di Planck. Il fisico
tedesco W. Heisenberg (1927) enunciò il principio di indeterminazione,
secondo cui non è possibile conoscere contemporaneamente velocità e
posizione dell'elettrone. Ciò escludeva la possibilità di attribuire
all'elettrone orbite definite come quelle del modello di Bohr, ammettendo
invece la possibilità di delimitare una regione di spazio intorno al nucleo
dove è massima la probabilità di trovare l'elettrone.
In quegli anni il fisico austriaco E. Schrödinger, approfondendo l'ipotesi
di De Broglie, formulò un'espressione matematica, detta equazione
d'onda di Schrödinger la cui soluzione permette di rappresentare
l'elettrone come una nube di carica negativa la cui densità varia in
funzione della distanza dal nucleo e della direzione presa in esame.
Viene denominato orbitale atomico la regione di spazio intorno al
nucleo dove è massima la probabilità di trovare l'elettrone. Gli
orbitali nella teoria quanto-meccanica sono descritti per mezzo di numeri
quantici n, l, m, ms di significato analogo a quelli utilizzati nella teoria di
Bohr-Sommerfeld.
UNITA’ 5
LA TEORIA ATOMICA DI DALTON
All'inizio del 1800 lo scienziato inglese John Dalton formulò la
prima teoria atomica della materia scientificamente valida. Dalton si rese
conto che questa ipotesi forniva una perfetta chiave di interpretazione di
tutte le fondamentali leggi della chimica a quei tempi già note (la legge di
conservazione della massa, la legge delle proporzioni definite e la legge
delle
proporzioni
multiple
da
lui
stesso
enunciata).
Il termine atomo (dal greco: indivisibile) fu ripreso dal filosofo greco
Democrito che per primo, nel IV sec. a.C., aveva ipotizzato che la materia
fosse costituita da particelle indivisibili. Quella di Democrito era una
teoria filosofica, non si basava cioè su dati oggettivi e non incontrò
daltronde un grande favore. Nei secoli che seguirono, infatti, le
interpretazioni più seguite furono altre e fu necessario aspettare 2000 anni
perchè queste idee riprendessero piede.
Dal 1800 a oggi la teoria atomica di Dalton ha avuto continue conferme e
da non molti anni, attraverso l'uso di tecniche microscopiche sofisticate, è
stato anche possibile ottenere immagini dirette di alcuni atomi.
I punti principali della teoria atomica di Dalton possono essere così
schematizzati:
•
•
•
•
La materia ha una natura discontinua ed è costituita da particelle
microscopiche non ulteriormnte divisibili (atomi).
Gli atomi di uno stesso elemento sono tutti uguali.
Gli atomi di elementi differenti sono diversi.
Nei composti e nelle reazioni chimiche non possono essere presenti
che numeri interi di atomi (non ha cioè senso parlare di frazioni di
atomi).
Anche la teoria di Dalton tuttavia dovette ben presto essere modificata.
Le nuove scoperte fatte tra la fine del 1800 e l'inizio del 1900
dimostrarono infatti che l'atomo è divisibile e costituito da particelle più
piccole dette subatomiche. Rimane valida la seguente definizione di
atomo:
L'atomo è la più piccola parte di un elemento che
ne mantiene le caratteristiche
UNITA’ 6
LEGAME AD IDROGENO
Il legame a idrogeno o ponte idrogeno è un caso particolare di
interazione fra dipoli. In particolare si tratta di un legame dipolo
permanente - dipolo
permanente in
cui
è
implicato
un atomo di idrogenocoinvolto in un legame covalente con elementi
molto elettronegativi come fluoro, ossigeno o azoto(regola FON), i quali
attraggono a sé gli elettroni di valenza, acquisendo una parziale carica
negativa (δ-) lasciando l'idrogeno con una parziale carica positiva (δ+).
Il legame idrogeno si forma quando la relativamente forte carica positiva
dell'idrogeno viene in contatto con un doppietto elettronico di un gruppo
funzionale di un'altra molecola, il quale lega l'H e viene
definito accettore. Il gruppo dove è legato l'H in maniera covalente viene
detto donatore.
Ad esempio nel radicale idrossile OH e nell'anione idrossido OH- è
presente una parziale carica negativa sull'O e una equivalente positiva
sull'H, e quindi tali composti si polarizzano parzialmente (dipolo
permanente).
Se questo gruppo ne incontra un altro polare (ad esempio un
gruppo carbonile), si crea una interazione elettrostatica.
La forza del legame idrogeno, che è di 20 kJ / mol a temperatura
ambiente nell'acqua pura, dipende dalla permittività elettrica del mezzo;
infatti, essendo un legame elettrostatico per esso vale la legge di
Coulomb. Comunque è nettamente più debole del legame ionico e
del legame covalente, ma è nettamente più forte delle forze di Van der
Waals. Una importante osservazione è che esso è un legame su base
elettrostatica ma altamente direzionale (per es. nell'acqua: ossigeno,
idrogeno, doppietto elettronico, ossigeno debbono essere allineati lungo
lo stesso asse per avere il legame).
Rappresentazione di una α-elica composta da residui di alanina; i
bastoncelli fucsia rappresentano i legami a idrogeno
Il legame idrogeno è presente nell'acqua sia allo stato liquido che allo
stato solido, ed è responsabile della sua relativamente alta temperatura di
ebollizione (se paragonata per esempio all'H2S, che pur avendo peso
molecolaremaggiore è significativamente meno polare). In particolare, se
non esistessero i legami idrogeno, l'acqua bollirebbe a -100 °C.
Una caratteristica peculiare del legame idrogeno è quella di mantenere
le molecole interessate più distanti fra loro rispetto agli altri tipi di
legame: è per questo che il ghiaccio è meno denso dell'acqua (nell'acqua,
infatti, le molecole scorrono l'una sull'altra mentre il ghiaccio assume una
struttura cristallina dovuta proprio ai legami idrogeno)
Il legame idrogeno è presente nelle proteine (principalmente per quanto
riguarda le strutture secondarie: alfa elica ebeta foglietto) e negli acidi
nucleici, è una delle forze che tiene uniti i due filamenti del DNA
UNITA’ 7
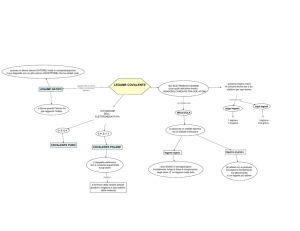
LEGAME COVALENTE
Un legame covalente polare o apolare si viene a instaurare quando
avviene una sovrapposizione degli orbitali atomici di due atomi con una
differenza di elettronegatività inferiore a 1,9. Ciò avviene per una ragione
ben precisa: gli atomi tendono al minor dispendio energetico possibile
ottenibile con la stabilità della loro configurazione elettronica (ad
esempio l'ottetto). Un tipico esempio è fornito dalla combinazione di due
atomi di idrogeno, che porta alla struttura covalente:
H· + ·H --> H:H
Nella molecola finale, H2, i due atomi sono tenuti assieme da una
coppia di elettroni (carichi negativamente) condivisi, i quali attirano
a sé i rispettivi nuclei (carichi positivamente). Un legame covalente è
quindi il risultato di un'interazione elettrostatica che coinvolge i
nuclei. Quando la nube elettronica è distribuita simmetricamente il
legame risulta non polarizzato. In questo caso si parla di legame
covalente omopolare opuro.
Nel caso in cui vi sia un dipolo molecolare permanente, gli elettroni
saranno maggiormente attratti dall'atomo più elettronegativo ed il
legame risulterà quindi polarizzato elettricamente determinando
quindi uno sbilanciamento della nuvola elettronica. In questo caso si
parla di legame covalente eteropolare o più semplicemente polare.
Quando entrambi gli elettroni coinvolti nel legame provengono da
uno solo dei due atomi, mentre l'altro fornisce un orbitale vuoto in
cui allocarli, si parla di legame dativo.
Un legame covalente in cui viene condivisa una sola coppia di
elettroni viene detto legame monovalente (legame semplice), se
vengono
condivise
due
coppie
di
elettroni
viene
detto bivalente (doppio legame) e se le coppie condivise sono tre, si
dice legame
trivalente (triplo
legame).
Esistono
anche
legami tetravalenti, ampiamente studiati in chimica inorganica, e
nel 2005 è stata dimostrata l'esistenza di legami quintupli in molecole
stabili. Il legame monovalente si esprime con un trattino tra i simboli
dei due atomi che vi sono coinvolti, nel caso del legame bivalente il
trattino è doppio e triplo nel caso del legame trivalente e così via.
Esempio
La molecola dell'acqua è un legame covalente polare
tra ossigeno ed idrogeno. Quando i due elementi si fondono in una
molecola d'acqua. L'elettronegatività dell'ossigeno (3,52 ca) prevale
su quella dell'idrogeno (2,11 ca) attirando verso di sè gli elettroni dei
due atomi H. Se pur vero che nel legame a idrogeno la molecola
dell'acqua ha un angolo di legame di 108°3', nel legame covalente la
molecola tende ad essere disposta elettronicamente a 90°. La
molecola dell'acqua diventa cosi un dipolo magnetico. Un altro
esempio è l'acido cloridrico (HCl).
UNITA’ 8
LEGAME IONICO
l legame ionico è un legame chimico di natura elettrostatica che si forma
quando le caratteristiche chimico-fisiche dei due atomi sono nettamente
differenti, e vi è soprattutto una grande differenza di elettronegatività tra i
componenti. Per convenzione si suole riconoscere un legame ionico tra
due atomi quando la differenza di elettronegatività, Δχ è maggiore di 1,9.
Al diminuire di tale differenza cresce il caratterecovalente di un legame.
Nel legame ionico l’attrazione esercitata dal nucleo dell’atomo più
elettronegativo sull’altro atomo, meno elettronegativo, è così forte che la
nuvola di carica elettronica può considerarsi come spostata
completamente sull’elemento più elettronegativo. L’elettrone dell’altro
elemento, meno elettronegativo, viene strappato e un legame ionico è
creato in seguito alla formazione di un catione e un anione. Il legame così
creato è puramente elettrostatico dovuto all’attrazione reciproca (per
la legge di Coulomb) dai due ioni di carica opposta.
A differenza del legame covalente che si produce lungo la direzione
stabilita dagli orbitali di legame, il legame ionico non è direzionale.
L’attrazione tra cariche di segno opposto infatti, non si sviluppa in
un'unica direzione ma agisce con ugual forza, in tutte le direzioni con
simmetria sferica (a pari distanza).
Esempio: il sale da cucina
Uno ione Na+ risulta circondato e attratto da 6 ioni Cl- e viceversa, in una
struttura detta cristallina. Anioni e cationi infatti si dispongono a turno in
uno sconfinato reticolato cubico. Tale disposizione è cristallina perché
macroscopicamente genera un cristallo che riflette la geometria della
struttura ionica.
Anche se la molecola di NaCl esiste allo stato gassoso, allo stato
solido (fatto generale valido per tutti i composti ionici) “non esiste una
molecola ionica”. Quando si indica un composto ionico con una formula,
quindi, non si vuol con essa descrivere una struttura molecolare autonoma
ma soltanto il rapporto numerico esistente nel cristallo fra ioni positivi e
ioni negativi. In pratica parlare di formula molecolare è abusato, mentre
resta valido parlare di formula minima. Allo stesso modo sarebbe più
corretto riferirsi al peso formula piuttosto che al peso molecolare.
Nel nostro caso, cloruro di sodio, la formula NaCl indica che il rapporto
tra le moli di Na+ e Cl- è 1:1 Altro esempio: il cloruro di magnesio,
MgCl2 indica che nel reticolato ionico gli ioni di Mg2+ e Cl- sono presenti
nel rapporto di 1:2.
Numero di coordinazione
In un legame ionico, il numero di ioni che circondano il catione che ha
carica n+ è detto ’’numero di coordinazione’’ dello ione An+ Se si
considerano ioni come sfere rigide, calcolare i numeri di coordinazione
più probabili è possibile grazie al rapporto tra i raggi r+/r-. Nello schema
che segue vi sono i tipi più comuni di coordinazione con la simmetria
spaziale formata dalle particelle, in funzione al suddetto rapporto raggiocatione/raggio-anione. Si osservi che tale rapporto è sempre minore di 1,
essendo il catione di un atomo sempre minore dell’anione corrispondente.
Si deve tenere presente che la geometria trigonale è solo planaere (cioè i
3 anioni e il catione centrale sono complanari).
RAPPORTO r+ / r- NUMERO
COORDINAZIONE
GEOMETRIA
>0,155
3
trigonale
>0,225
4
tetraedrica
>0,414
6
ottaedrica
>0,732
8
cubica
Energie in gioco
Il legame ionico è tipico dei legami tra metalli e non metalli e si realizza
con maggiore probabilità quando un atomo a bassa energia di
ionizzazione si combina con un atomo ad alta affinità elettronica.
Energia di Madelung
Nel reticolo cristallino vi sono forze, quindi energie, di attrazione e
repulsione. Ogni catione attrae a sè ed è attratto dagli anioni. L'energia di
attrazione è negativa ed è calcolata con l'energia di Coulomb che varia
per ogni coppia ione-anione in base a distanza, disposizione geometrica e
numero di coordinazione. Analogamente tra ioni di segno uguale viene a
crearsi una repulsione elettrica. Anche per essa il valore varia a seconda
di distanza, geometria ionica e numero di coordinazione. L'Energia di
Madelung tiene conto di tutte le iterazioni tra ioni. Essa è basata appunto
sull'energia di Coulomb
L'energia di Madelung è quindi una
sommatoria di tutte le energie possibili. Fissato uno ione campione, si
calcolano tutte le energie possibili ad esso legate. Ogni addendo varia
dall'altro per la distanza di legame, che per comodità esprimiamo in
funzione della distanza iniziale r0 e per un fattore moltiplicativo che dice
il numero di ioni coinvolti in quel processo energetico (a parte lo ione
campione stesso. Per capire ecco un esempio che inizia a calcolare
l'energia in un cristallo di salgemma.
Fissato
con R0 la
prima
distanza
calcolata,
le
seguenti
sono
La sommatoria è
negativi(attrazione)
una
e
sequenza di addendi
positivi(repulsione),
alternativamente
di
entità
decrescente.
Se consideriamo solo il numero degli ioni coinvolti e il fattore
motliplicativo che esprime il raggio dell'interazione in funzione del primo
raggio calcolato, R0 si ha una somma che converge a un valore,
detto costante di Madelung, caratteristico proprio per geometria e
coordinazione del cristallo ionico.
Cristalli e solidi ionici
I solidi ionici sono caratterizzati da forti legami di tipo ionico.
Il solido ionico ha una struttura cristallina dalla geometria precisa
che dipende dalle distanze di legame e dal numero di coordinazione. I
nodi reticolari sono occupati da ioni positivi o negativi tra i quali
viene esercitata la forza di Coulomb, si generano così dei legami
ionici direzionali.
I legami covalenti polari si dovrebbero considerare come ibridi con
variabile carattere di legame covalente e ionico, con una tipologia di
legame che può essere più o meno predominante sull'altra. D'altro canto
molti legami ritenuti per semplicità ionici presentano una certa
componente covalente.
UNITA’ 9
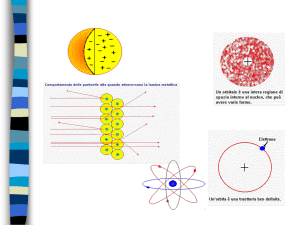
MODELLO DI BHOR
Il modello atomico proposto da Niels Bohr nel 1913 è la più famosa
applicazione della quantizzazione dell'energia, che, insieme all'equazione
di Schrödinger e alle spiegazioni teoriche sulla radiazione di corpo nero,
sull'effetto fotoelettrico e sullo scattering Compton sono la base
dellameccanica quantistica.
Il modello, proposto per l'atomo di idrogeno, ottenne degli eccellenti
risultati,
coincidenti,
entro
il
margine
degli errori,
con
lo spettrosperimentale.
All'inizio del XX secolo lo studio dell'atomo aveva raggiunto un buon
grado di conoscenza. Erano noti, infatti, moltissimi spettri di emissione
diluce proveniente dagli atomi: ovvero delle linee discrete e ben distinte
poste a differenti frequenze.
Una
delle
prime
osservazioni
interessanti
avvenne
nel 1884 quando Johann Balmer, insegnante svizzero, osservò che alcune
righe dello spettro di emissione dell'idrogeno potevano essere calcolate
utilizzando la formula:
Balmer suppose che tale formula fosse, in realtà, un caso particolare di
una legge più generale, che venne trovata da Johannes Rydberg eWalter
Ritz e nota come legge di Rydberg-Ritz:
con n1>n2 ed R la costante di Rydberg.
Con questa legge fu possibile completare lo spettro osservato da Balmer e
si riescono ad ottenere anche le serie di Lyman (n2=1) e Paschen(n2=3).
Furono fatti numerosi tentativi per spiegare teoricamente tali osservazioni
sperimentali, ma il meglio che si riuscì a realizzare fu il modello di
Thomson, lo scopritore dell'elettrone, che suppose che l'atomo fosse un
corpo compatto contenente al suo interno sia la carica positiva, che quella
negativa. Tale modello aveva, però, una pecca: poiché si basava solo
sulla presenza delle forze elettriche, non era in grado di spiegare come
mai il sistema fosse all'equilibrio, né Thomson riuscì mai a determinare
una frequenza tra quelle osservate.
Nel 1911, Hans Geiger e Ernest Marsden, sotto la supervisione di Ernest
Rutherford, realizzarono un esperimento importantissimo per la
comprensione della struttura dell'atomo: bombardando una sottile lamina
d'oro con particelle alfa, notarono che, mentre la maggior parte di esse
subiva deviazioni minime dalla traiettoria iniziale, altre venivano deviate
in misura considerevole, se non addirittura respinte dalla lamina.
Nell'interpretare questo esperimento, Rutherford stabilì che l'atomo fosse
composto da un centro massivo (il nucleo) circondato da cariche
negative. Il modello di atomo proposto da Rutherford soffriva, però, di
una instabilità elettromagnetica e di una instabilità meccanica: poiché
l'elettrone, nel suo moto intorno al nucleo positivo, è sottoposto a
un'accelerazione,
esso
irraggia
energia elettromagnetica della
stessafrequenza del suo moto di rivoluzione, finendo così per cadere sul
nucleo con un moto a spirale. Nel caso di atomi più pesanti, attorno ai
quali ruotino più elettroni, questi ultimi sono soggetti a una repulsione
elettrostatica che rende inoltre meccanicamente instabili le loro orbite,
cosicché, a prescindere dall'irraggiamento, una qualsiasi perturbazione
esterna è sufficiente a scompaginare gli atomi. Fu Niels Bohr a risolvere
le difficoltà del modello di Rutherford, spiegando anche lo spettro di
emissione dell'atomo di idrogeno.
Il modello di Bohr
Bohr, che a quel tempo lavorava con Rutherford, propose un modello
che, applicando all'atomo di Rutherford la quantizzazione dell'energia
introdotta da Planck, riuscì a giustificare lo spettro dell'idrogeno.
Bohr risolse queste difficoltà sulla base di tre postulati:
primo postulato di Bohr: un elettrone può muoversi soltanto su
alcune determinate orbite non-radiative, dette stati stazionari;
Il secondo postulato di Bohr
L'atomo irraggia energia solamente quando, per un qualche motivo, un
elettrone effettua una transizione da uno stato stazionario ad un altro. La
frequenza della radiazione è legata all'energia del livello di partenza e di
quello di arrivo dalla relazione:
dove h è la costante di Planck, mentre Ei ed Ef sono le energie dell'orbita
iniziale e finale (secondo la teoria classica, invece, la frequenza della
radiazione emessa avrebbe dovuto essere uguale a quella del moto
periodico della particella carica). L'energia che l'atomo scambia con il
campo elettromagnetico soddisfa dunque sia il principio
della conservazione dell'energia, sia la relazione tra l'energia e la
frequenza introdotta da Planck. Notiamo, però, che nel suo lavoro Bohr
non chiama in causa i quanti di luce di Einstein, dei quali sarà un deciso
oppositore fino al 1924.
Il terzo postulato di Bohr
Nel modello semplice di Bohr, la carica del nucleo è +Ze, la carica
dell'elettrone è e e l'energia potenziale a distanza r è:
dove k è la costante di Coulomb. L'energia totale di un elettrone che si
muove su un'orbita circolare con velocità v è quindi:
Per ottenere il valore della velocità, e quindi quello dell'energia cinetica,
basta eguagliare la relazione F = ma, dove per l'accelerazione si utilizza
l'espressione per quella centripeta (a = v2/r), con l'attrazione
coulombiana:
e quindi l'energia cinetica risulta essere pari alla metà del valore
assoluto dell'energia potenziale. L'energia totale risulta quindi essere pari
a:
Sostituendo questa nella legge matematica del secondo postulato di Bohr,
si ottiene un'espressione per le frequenze in funzione delle distanze finale
ed iniziale dei livelli interessati dalla transizione:
Questa equazione deve essere consistente con la formula di RydberghRitz, sapendo che ν = c/λ, con c velocità della luce.
I raggi delle orbite stabili, quindi, dovevano essere proporzionali ai
quadrati di numeri interi. Una simile legge di proporzionalità poteva
essere ottenuta ipotizzando che il momento angolare dell'elettrone in
un'orbita stabile fosse pari a:
Questo è il terzo postulato di Bohr, che, in pratica, quantizza il momento
della quantità di moto della particella.
Raggio di Bohr ed energia fondamentale
A questo punto è abbastanza semplice determinare il raggio dell'orbita,
combinando quest'ultima con la relazione tra energia cinetica e
potenziale:
dove
è il raggio di Bohr del livello fondamentale dell'atomo di idrogeno.
Inoltre, Bohr riuscì a calcolare anche il valore della R costante di
Rydberg:
che utilizzando i valori allora noti per le costanti, è in accordo con il
valore ottenuto dalla spettroscopia.
Infine si possono scrivere tutti i valori possibili dell'energia di un
elettrone in un atomo, scritti in funzione dell'energia fondamentale
dell'atomo di idrogeno:
con
che risulta di circa 13,6 eV. Questo vuol dire che, per estrarre un elettrone
nello stato fondamentale dell'idrogeno, bisogna fornire al sistema
un'energia pari a 13,6 eV. Tenendo conto del fatto che la massa del
nucleo non è infinita (nel caso dell'idrogeno è circa duemila volte la
massa dell'elettrone) e che quindi il nucleo stesso ruota intorno al centro
di massa dell'atomo, si introduce una lieve dipendenza della costante di
Rydberg dalla massa del nucleo, migliorando così l'accordo con i dati
sperimentali.
UNITA’ 10
ORBITALE
In meccanica quantistica ed in chimica quantistica è necessario
generalizzare il concetto classico di orbita per renderlo compatibile
col principio di indeterminazione di Heisenberg. Infatti la meccanica
quantistica prevede che non sia possibile associare contemporaneamente
ad una particella una posizione ed una quantità di moto ben definita. Il
concetto di orbita di un elettrone è sostituito da quello di orbitale, ossia la
parte dello spazio entro la quale è massima la probabilità di trovare una
particella. In questo contesto non ha senso studiare la traiettoria seguita
da un corpo ma se ne studiano gli autostati. Formalmente un orbitale è
definito
come
laproiezione della funzione
d'onda sulla base della
posizione.
Questa nomenclatura è stata introdotta dopo il modello atomico proposto
da Niels Bohr e l'esperimento di Rutherford.
L'emissione di una radiazione durante la rotazione degli elettroni intorno
al nucleo portava alla conseguenza teorica per la quale l'elettrone avrebbe
dovuto perdere gradualmente energia fino a collassare sul nucleo con un
movimento a spirale, fenomeno che in realtà non si osserva
sperimentalmente. Inizialmente si postulò l'esistenza di un'infinità
discreta, di un numero finito di orbite possibile, senza che vi fosse un
modello fisico, in grado di giustificare questo assunto. Bohr fornì una
spiegazione in base al dualismo onda-particella: due onde in fase si
sommano, mentre due onde in opposizione di fase si annullano.
I movimenti di elettroni lungo orbite fuori fase, cresta d'onda contro
ventre, sarebbero distrutti dal fenomeno dell'interferenza. Per cui,
possono avere luogo solo movimenti a lunghezza d'onda in fase, che
definiscono gli orbitali, e, per essere in fase, sono multipli interi di un
valore base, lacostante di Planck.
In chimica si distingue, in generale, tra orbitale atomico ed orbitale
molecolare ma in fisica il concetto di orbitale viene usato per descrivere
un qualsiasi insieme di autostati di un sistema.
Solitamente in chimica, per favorirne la visualizzazione, un orbitale
atomico viene approssimato con quella regione di spazio attorno al
nucleoatomico in cui la probabilità di trovare un elettrone è massima
(massima densità di probabilità) ed è delimitata da una superficie sulla
quale il modulo dell'ampiezza della funzione d'onda è costante
(generalmente normalizzata a uno).
La forma di un orbitale s e di uno dei tre orbitali p. Al centro degli assi si
trova il nucleo. L'asse z è perpendicolare al piano di lettura
In altre parole, una regione di spazio attorno ad un nucleo atomico in cui
la probabilità di trovarvi un elettrone è massima (di solito superiore ad un
limite convenzionalmente fissato nel 90%) è usata per rappresentare
graficamente unorbitale atomico di quell'elettrone.
Visivamente, tale orbitale può essere meglio rappresentato mediante una
nuvola la cui intensità del colore è proporzionale alla densità di
probabilità di trovare l'elettrone in quel punto e con forme tali dal
comprendere il 90% della probabilità elettronica. Quest'ultima, in ogni
punto dello spazio attorno al nucleo, è pari al quadrato delmodulo della
funzione d'onda dell'elettrone nel punto stesso.
Considerando il campo coulombiano di simmetria sferica, moltiplicando
il quadrato della funzione d'onda ψ2 per il volume elementare dτ, uguale
in questo caso a 4πr2dr, è possibile calcolare la probabilità che ha un
elettrone di trovarsi in uno spazio sferico definito dallo spessore dr della
sfera di raggio r. In particolare, usando la forma Pdr, risulta P = 4πr2ψ2 e
questo valore di P viene definito funzione di distribuzione radiale.
Il numero e l'estensione degli orbitali atomici è deducibile dalla soluzione
dell'equazione di Schrödinger per un elettrone confinato nella buca
delpotenziale elettrico generato dal nucleo ed è correlato ai numeri
quantici che identificano il livello energetico in cui si trova l'elettrone
stesso.
Il numero quantico principale n, che può assumere valori interi non
inferiori a 1, definisce il livello
dell'energia (autovalore dell'equazione diSchrödinger), l'estensione
dell'orbitale ed il numero totale di nodi, considerando come nodo
anche una superficie sferica a distanza infinita dal nucleo;
Il numero quantico azimutale (o numero quantico angolare) l, che
può assumere valori interi positivi compresi tra 0 ed n-1, a cui è legato
il numero di nodi non sferici e, indirettamente, la simmetria
dell'orbitale;
Il numero quantico magnetico ml, che può assumere valori interi
compresi tra +l e -l, a cui sono legati il tipo di nodo - planare o conico
- la sua orientazione nello spazio e la molteplicità degli orbitali.
In base al principio di esclusione di Pauli, ogni orbitale può contenere al
massimo due elettroni, dato che essi sono fermioni. Gli orbitali vengono
riempiti partendo da quelli ad energia minima (stato fondamentale) e
riempiendo, via via, quelli ad energia superiore; se sono presenti degli
orbitali degeneri (ovvero più autostati per un unico autovalore, come ad
esempio i tre orbitali p) gli elettroni si distribuiscono preferenzialmente in
modo da occuparne il maggior numero.
La disposizione degli elettroni negli orbitali atomici costituisce
la configurazione elettronica di un atomo, dalla quale dipendono la
reattività, lavalenza e la geometria delle molecole che questi va a
comporre.
s (l=0) p (l=1)
n=1
d (l=2)
m=0
m=0
m=±1
s
pz
px
py
f (l=3)
m=0
m=±1
dz2
dxz
m=±2
dyz
dxy
m=0 m=±1
dx2-y2
fz3
fxz2
fyz2
n=2
n=3
n=4
n=5
n=6
n=7
...
...
...
...
...
...
...
...
...
...
...
...
...
...
...
...
...
...
...
...
...
...
Esempi
idrogeno: 1 elettrone nell'orbitale 1s -> 1s1
con un elettrone spaiato, è in grado di formare un legame semplice
con gli altri atomi
elio: 2 elettroni nell'orbitale 1s -> 1s2
non ha elettroni spaiati, non è in grado di formare legami con gli altri
atomi
azoto: 2 elettroni nell'orbitale 1s, 2 nel 2s, 3 nel 2p -> 1s2 2s2 2p3
con tre elettroni spaiati - uno in ogni orbitale 2p - è in grado di formare
tre legami (ammoniaca: NH3)
ossigeno: 2 elettroni nell'orbitale 1s, 2 nel 2s, 4 nel 2p -> 1s2 2s2 2p4
con due elettroni spaiati - un orbitale 2p ne alloca due, gli altri due uno
ciascuno - è in grado di formare due legami (acqua: H2O).
Il modello, però, costruito così semplicemente, non è perfettamente
compatibile con i dati sperimentali. Se, ad esempio, l'azoto lega tre atomi
a sé tramite i suoi orbitali p, allora l'ammoniaca dovrebbe avere i suoi
legami a 90° di distanza l'uno dall'altro. Sappiamo, dai dati sperimentali,
che non è così; l'angolo formato da due legami N-H è di circa 107°.
Il carbonio ha la seguente struttura: 1s2 2s2 2p2 - due elettroni spaiati negli
orbitali p; però l'unico composto del carbonio in cui questi scambia due
legami è l'ossido di carbonio, C=O: in tutti gli altri suoi composti il
carbonio forma con gli atomi vicini quattro legami.
Orbitali atomici ibridi
Gli orbitali atomici convenzionali vengono ottenuti risolvendo
l'equazione di Schrödinger per sistemi idrogenoidi (ovvero un nucleo
carico positivamente attorno al quale orbita un unico elettrone). Questi
formano una base completa per descrivere tutti gli stati del sistema.
Tuttavia, quando ci sono due o più elettroni che interagiscono fra di loro,
questi orbitali non sono più autostati del sistema. Invece che definire un
nuovo insieme di orbitali, per ogni possibile numero di elettroni attorno al
nucleo, si preferisce, solitamente, descrivere tutti i sistemi
comecombinazione lineare degli orbitali, ottenuti per atomi idrogenoidi.
In chimica queste combinazioni vengono solitamente chiamate orbitali
ibridi.
due
orbitali
ibridi sp allineati
lungo
l'asse
s + p → dell'orbitale p originario che puntano in direzioni opposte,
quindi con un angolo di 180° fra loro
tre orbitali ibridi sp2 che giacciono sul piano formato dai due
s + 2 p → orbitali p di partenza e puntano ai tre vertici di un triangolo
equilatero, quindi con un angolo di 120° fra loro
quattro orbitali ibridi sp3 che puntano ai quattro vertici di un
s+3p→
tetraedro, quindi con un angolo di 109,5° fra loro
Forma degli orbitali sp2 del carbonio.
Forma degli orbitali sp3 del carbonio.
L'ibridazione porta ad avere un gruppo di orbitali degeneri in cui gli
elettroni andranno a distribuirsi occupandone il più possibile; prendiamo
l'esempio del carbonio, la cui configurazione elettronicastabile è:
1s2 2s2 2p2
E diventa, in ibridazione sp3:
1s2 2(sp3)4
In questa configurazione ibrida, il carbonio presenta quattro elettroni
spaiati, ognuno in un orbitale sp3, configurazione che spiega i quattro
legami formati dal carbonio nei suoi composti e la geometria tetraedrica
delle molecole in cui compare (vedi alcani).
Invece, in ibridazione sp2,
(vedi alcheni):
solo
due
orbitali p vengono
ibridati
1s2 2(sp2)3 2p1
Analogamente, in ibridazione sp, solo un orbitale p viene ibridato
(vedi alchini):
1s2 2(sp)2 2p2
Similmente all'ibridazione sp3 del carbonio, la configurazione elettronica
dell'azoto cambia in questo modo:
1s2 2s2 2p3 → 1s2 2(sp3)5
Allocare cinque elettroni in quattro orbitali sp3 significa avere un orbitale
completo di due elettroni e tre orbitali contenenti un elettrone spaiato.
Questo spiega non solo i tre legami che l'azoto forma nei suoi composti,
ma anche l'angolo di 107° tra due legami - l'orbitale che ospita i due
elettroni tende a comprimere gli altri tre, distorcendo la regolare
geometria del tetraedro.
I due elettroni allocati nell'orbitale non coinvolto nel legame possono
essere però impiegati per formare un legame dativo, tale comportamento
è alla base del comportamento basico dell'ammoniaca e delle ammine.
Ultimo esempio è l'ossigeno, la cui configurazione elettronica cambia in
questo modo:
1s2 2s2 2p4 → 1s2 2(sp3)6
Allocare sei elettroni in quattro orbitali sp3 significa avere due orbitali
completi di due elettroni ciascuno e due orbitali contenenti un elettrone
spaiato. Questo spiega i due legami che l'ossigeno forma nei suoi
composti ed anche l'angolo di 105° tra i due legami, tipico
della molecolad'acqua - i due orbitali completi non impegnati nei legami
tendono a comprimere gli altri due, distorcendo la regolare geometria
del tetraedro in misura ancora maggiore a quanto visto nell'esempio
precedente.
L'ibridazione è un processo che richiede energia, dato che gli orbitali p si
trovano ad un livello energetico leggermente superiore a quello dei
corrispondenti orbitali s, tuttavia questa energia è ampiamente
compensata dalla maggiore stabilità dei legami che l'atomo ibridato è in
grado di formare.
Le ibridazioni tra orbitali s e p non sono le uniche esistenti. Gli elementi
di transizione possono formare ibridi più complessi (es. d2sp3), tipici
dei composti di coordinazione.
Orbitali molecolari
Un orbitale molecolare è un orbitale esteso a due o più atomi uniti da
un legame covalente. Si può visualizzarlo come il prodotto della fusione
per sovrapposizione di due orbitali atomici.
Quando la sovrapposizione avviene lungo la congiungente i due nuclei,
l'orbitale molecolare prende il nome di σ (sigma); quando la
sovrapposizione avviene perpendicolarmente all'asse che unisce i due
nuclei, ovvero sopra e sotto i medesimi, l'orbitale molecolare prende il
nome di π (pi greco).
Rappresentazione grafica dell'orbitale molecolare σ del legame C-C
dell'etano, per sovrapposizione di orbitali sp3. Le proporzioni sono state
alterate per evidenziarlo. I lobi minori dei due orbitali ibridi sono stati
omessi
Rappresentazione grafica dell'orbitale molecolare π del legame C=C
dell'etene, per sovrapposizione degli orbitali p non coinvolti
nell'ibridazione sp2. Le proporzioni sono state alterate per evidenziarlo.
Il legame C-C rappresentato da una retta è un legame σ analogo al
precedente.
Una funzione d'onda che vada a descrivere il moto di un elettrone attorno
a più nuclei in presenza di altri elettroni risulta estremamente complessa,
una possibilità di trattare gli orbitali molecolari è l'approssimarli facendo
ricorso ad una combinazione lineare degli orbitali atomicida cui essi
derivano per sovrapposizione (metodo LCAO, da linear combination of
atomic orbitals).
Secondo il metodo LCAO, la sovrapposizione di due orbitali atomici
produce due orbitali molecolari, uno a bassa energia, detto legante, che
corrisponde alla somma delle funzioni d'onda dei due orbitali; uno ad alta
energia, detto antilegante, che corrisponde alla sottrazione delle funzioni
d'onda dei due orbitali. Gli orbitali non leganti invece non risultano il
frutto di alcuna sovrapposizione (i non leganti puri), sono ininfluenti
riguardo alla stabilità energetica della struttura molecolare ma influiscono
sulla reattività chimica.
La sovrapposizione di n orbitali atomici in legami delocalizzati, come nel
caso
dei composti
aromatici o
dei dieni
coniugati,
produce
altrettanti norbitali molecolari a energie diverse.
Applichiamo a titolo di esempio il metodo LCAO per ottenere gli orbitali
molecolari di una struttura relativamente semplice, quale quella
delfluoruro di idrogeno (simbolo chimico HF). Innanzi tutto occorre
avere presente le configurazioni elettroniche degli atomi che
compongono la molecola:
per l'idrogeno (H): 1s1
per il fluoro (F): 1s2 2s2 2p2x 2p2y 2p1z
Adesso analizziamo le combinazioni lineari possibili, ovvero le
combinazioni di due diversi orbitali aventi energia comparabile e
medesima orientazione spaziale (da notare come risulti utile esprimere le
simmetrie lungo i tre assi cartesiani indicandole a pedice degli orbitali
orientabili): risulta sovrapponibile solamente l'orbitale 1s dell'idrogeno
con gli orbitali 2s e 2px (con x asse internucleare di legame) del fluoro.
Ciò significa determinare, tramite l'equazione di Schrödinger, i valori
fisicamente accettabili della seguente funzione d'onda:
ψ = c1φ1s(H) + c2φ2s(F) + c3φ2px(F)
valori che identificano tre orbitali molecolari di tipo σ. Inoltre, gli orbitali
2py e 2pz del fluoro restano inalterati in quanto manifestano simmetria π e
non esistono orbitali dell'idrogeno che possano combinarsi con questi;
essi origineranno un orbitale molecolare non legante π. In definitiva otto
elettroni totali (1 di H + 7 di F) assumono per la molecola HF la
configurazione 1σ2 2σ2 1π4, con 1σ orbitale molecolare legante, 2σ
orbitale essenzialmente non legante e l'orbitale 1π non legante puro.
Esiste anche un orbitale molecolare 3σ vuoto e di tipo antilegante.
Dividendo per due il risultato della differenza tra gli elettroni contenuti
negli orbitali leganti e quelli contenuti negli antileganti, si ottiene l'ordine
di legame che in questo caso vale uno.
UNITA’ 11 REGOLA DELL’OTTETTO
La regola dell'ottetto è una regola empirica introdotta da Gilbert Newton
Lewis per spiegare in modo approssimato la formazione di legami
chimici tra gli atomi, usabile a rigore solo per gli atomi dei gruppi
principali (quelli con numerazione romana) della tavola periodica.Ciò
spiega la condizione particolare di stabilità di un atomo, ossia quando
questo possiede il livello elettronico esterno completo, ma considerato il
fatto che il primo livello può contenere al massimo due elettroni sarebbe
meglio parlare di "regola dell'ottetto-duetto".
Tutti gli elementi tendono ad avere una configurazione elettronica stabile
(s2 p6), ovvero a divenire non reattivi o comunque poco reattivi.
Gli
elementi
dei
primi gruppi
della
tavola
periodica perdono elettroni attraverso
un
processo
denominato ionizzazione assumendo in tal modo la struttura elettronica
del gas nobile che li precede; gli elementi del VI e VII gruppo tendono
invece ad acquistare elettroni liberando energia dettaaffinità elettronica e
raggiungendo la struttura elettronica del gas nobile che segue.
Un'importante eccezione è costituita da idrogeno ed elio che, possedendo
solamente un orbitale s, raggiungono una configurazione completa con
due elettroni. Un'altra eccezione è rappresentata dai metalli di transizione,
nel cui guscio di valenza possono essere ospitati fino a 18 elettroni e si
dice che hanno ottetto espanso. Gli elementi a partire dal terzo periodo,
analogamente ai metalli di transizione, possono sfruttare gli orbitali d
espandendo anche loro l'ottetto (ad esempio PCl5 e SCl6).
Dalla regola dell'ottetto segue che i gas nobili non formano legami, anche
se in condizioni particolari è stato possibile ottenere composti di gas
nobili (soprattutto di Xeno), in particolare ossidi, fluoruri e clatrati.
UNITA’ 12
RELAZIONE TRA VOLUME PRESSIONE E
TEMPERATURA NEI GAS
Un gas è un aeriforme caratterizzato da una temperatura critica inferiore
alla temperatura ambiente; gli aeriformi per cui ciò non avviene si
trovano nello stato di vapore.
In pratica, un gas può anche essere definito come un aeriforme non
condensabile a temperatura ambiente.
Inoltre, per estensione, tutti gli aeriformi che si trovano ad una
temperatura superiore a quella critica vengono detti gas: un esempio è
dato dal vapore d'acqua, caratterizzato da una temperatura critica
superiore a quella ambiente (374 °C), viene definito come "gas d'acqua"
solo quando viene portato a superare questa temperatura (temperatura
critica).
Il gas, come tutti gli aeriformi, rappresenta lo stato della materia in cui le
forze interatomiche e intermolecolari tra le singole particelle di una
sostanza sono così piccole che non c'è più un'effettivacoesione tra di esse.
Gli atomi o le molecole del gas sono liberi di muoversi assumendo
ciascuno una certa velocità: le particelle atomiche o molecolari del gas
quindi interagiscono urtandosi continuamente l'un l'altra. Per questo un
gas non ha un volume definito ma tende ad occupare tutto lo spazio a sua
disposizione, e assume la forma del contenitore che lo contiene,
riempiendolo completamente. Un altro vincolo che può limitare il volume
di un gas è un campo gravitazionale, come nel caso dell'atmosfera
terrestre.
Nel linguaggio corrente si dice comunque che una data sostanza "è un
gas" quando la sua temperatura di ebollizione è molto al di sotto della
temperatura ambiente, cioè quando si trova normalmente allo stato di gas
sulla Terra. Per esempio è normale dire che "il metano è un gas mentre il
ferro non lo è", sebbene il metano possa benissimo trovarsi allo stato
liquido (raffreddato al di sotto di -161 °C) e il ferro allo stato gassoso
(riscaldato oltre i 2750 °C).
Un gas può essere approssimato ad un gas ideale quando si trova ad una
temperatura "molto maggiore" della sua temperatura critica, ossia che T >
> Tcr e convenzionalmente si intende che i due termini devono differire di
almeno un ordine di grandezza). Ciò equivale a chiedere
che
.
La temperatura critica si colloca sul punto di massimo della curva del
liquido-vapore (a forma di campana). All'interno della campana, il fluido
cambia di fase, mentre al di sopra resta allo stato gassoso qualunque sia la
pressione cui è sottoposto. Imponendo che T > > Tcr[, la curva del
liquido-vapore può non essere rappresentata nel diagramma di
Andrews (diagramma pressione-volume), non è visibile se si adotta una
scala normale.
Etimologia e storia del termine gas
Il termine "gas" fu coniato da un chimico fiammingo belga Jean Baptiste
van Helmont nel 1630. Sembra derivi, come spiegò Leo Meyer, dalla
trascrizione della sua pronuncia della parola greca Χαος (caos) che lui
fece diventare geist; ma Weigand e Scheler interprerarono l'origine
etimologica dal tedesco gascht (fermentazione): quindi sarebbe, secondo
loro, inizialmente usata dal chimico van Helmont per indicare la
fermentazione vinosa. Comunque, tralasciando l'etimologia, sappiamo
per certo che il chimico di Bruxelles van Helmont all'età di 63 anni fu il
primo a postulare l'esistenza di sostanze distinte nell'aria che così chiamò
nei suoi saggi pubblicati dal figlio Mercurio van Helmont. Pochi anni
dopo l'irlandese chimico Robert Boyle enunciò che l'aria era costituita da
atomi e da vuoto e solo dopo 140 anni le affermazioni di Boyle e di van
Helmont si dimostreranno vere.
I gas perfetti
In fisica e in termodinamica si usa generalmente l'approssimazione detta
dei gas perfetti: il gas cioè viene considerato costituito da atomi
puntiformi, che si muovono liberi da forze di attrazione o repulsione fra
loro e le pareti del contenitore: questa approssimazione conduce a
formulare la legge nota come equazione di stato dei gas perfetti, che
descrive, in condizioni di equilibrio termodinamico, la relazione fra
pressione, volume e temperatura del gas:
dove P è la pressione, V il volume occupato dal gas, n il numero
di moli del gas, R la costante universale dei gas perfetti e T è
la temperatura. Per esempio, una mole di gas perfetto occupa 22,4 litri a
temperatura di 0 °C e pressione di 1 atmosfera.
Da questa legge ne discendono poi altre due:
La legge di Boyle
Per una certa massa di gas a temperatura costante, il prodotto del volume
del gas V per la sua pressione P è costante.
Cioè per una certa massa di gas a temperatura costante, le pressioni sono
inversamente proporzionali ai volumi. La figura geometrica che ha per
equazione l'espressione è una iperbole equilatera. La legge di Boyle è una
legge limite vale cioè con buona approssimazione ma non in modo
assoluto per tutti i gas. Un gas perfetto o gas ideale che segua
perfettamente la legge di Boyle non esiste. Le deviazioni dal
comportamento dei gas reali sono assai piccole per un gas che si trovi a
bassa pressione e ad una temperatura lontana da quella di liquefazione.
La trasformazione isoterma è quindi una variazione del volume e della
pressione mantenendo costante la temperatura.
La prima legge di Gay Lussac
Un gas perfetto che alla temperatura di 0 °C occupa un volume V° e che
viene riscaldato mantenendo costante la pressione occupa alla
temperatura t un volume Vt espresso dalla legge
in cui V0 è il volume occupato dal gas a 0 °C e α0 è pari a 1/273,15. La
temperatura è espressa in gradi Celsius. La trasformazione isobara è una
variazione del volume e della temperatura a pressione costante. In un
diagramma pressione-volume è rappresentata da un segmento parallelo
all'asse dei volumi. Quindi la variazione di volume che subisce un gas per
la variazione di temperatura di ogni grado centigrado ammonta a 1/273
del volume che il gas occupa a 0 centigradi.
La seconda legge di Gay Lussac
La relazione che intercorre tra pressione-volume e quella tra temperatura
e volume, permette di ricavare la relazione tra la pressione di un gas e la
temperatura quando si operi a volume costante. Un gas perfetto che alla
temperatura di 0 °C ha una pressione p° e che viene scaldato mantenendo
costante il volume si trova, alla temperatura t,a una pressione pt espressa
dalla legge:
La trasformazione isocora è una variazione della pressione e della
temperatura che avviene mantenendo costante il volume.
Oltre alle leggi summenzionate, per i gas perfetti vale anche la legge di
Avogadro: a pari condizioni di temperatura e pressione, se due gas
occupano lo stesso volume allora hanno lo stesso numero di molecole.
I gas reali
Un tentativo di produrre un'equazione che descriva il comportamento dei
gas in modo più realistico è rappresentato dall'equazione dei gas reali.
Le correzioni apportate all'equazione dei gas perfetti sono due: si tiene
conto del volume proprio delle molecole, che non sono quindi più
considerate puntiformi, e si considerano le interazioni tra molecole che
venivano
trascurate
nel
caso
dei
gas
perfetti.
La prima correzione ha l'effetto di rendere non indefinitamente
comprimibile il gas; il suo riscontro empirico è la liquefazione cui vanno
soggetti i gas reali se compressi (e raffreddati) a sufficienza.
L'altra correzione fa sì che i gas reali non si espandano infinitamente ma
arrivino ad un punto in cui non possono occupare più volume (questo
perché tra gli atomi si stabilisce una forza molto piccola, dovuta alla
variazione casuale delle cariche elettrostatiche nelle singole molecole,
chiamata Forza
di
van
der
Waals).
Per questo la legge dei gas perfetti non fornisce risultati accurati nel caso
di gas reali, soprattutto in condizioni di bassa temperatura e/o alta
pressione, mentre diventa più precisa in caso di gas rarefatti, ad alta
temperatura e a bassa pressione, cioè quando forze intermolecolari e
volume
molecolare
diventano
trascurabili.
L'equazione dei gas reali si può ricostruire tenendo quindi conto del fatto
che il volume a disposizione del gas sarà (V - nb), dove b è il volume
occupato da una mole di particelle e n è il numero di moli di gas
considerate, e la pressione sarà invece corretta di un fattore a/V2 che tiene
conto delle forze di attrazione fra atomi. Dunque l'equazione, detta
anche equazione di Van der Waals, risulta:
Questa equazione non è valida in ogni caso, ma solo in particolari
condizioni, ma è molto importante in quanto si può identificare all'interno
di essa un significato fisico. Un'equazione che invece ci da un'esatta
visione dello stato del gas reale è l'equazione del viriale (di cui si parla
più specificamente alla parola Equazione di stato). Troviamo dei
coefficienti che hanno solo significato matematico e che si trovano
tabulati per ogni sostanza gassosa quali a,b,c,....
UNITA’ 13
CONNESSIONE TRA CARATTERI QUALITATIVI
La teoria della connessione studia la dipendenza tra le mutabili, cioè la
dipendenza tra caratteri statistici qualitativi.
Due caratteri sono indipendenti se non esiste alcuna relazione tra essi.
Se due caratteri qualitativi non sono dipendenti allora sono connessi.
Le tabelle, a doppia entrata, in cui vengono rappresentate le frequenze di
due caratteri vengono dette tabelle di connessione.
L'indipendenza tra due caratteri si ottiene quando tutte le frequenze
congiunte soddisfano la relazione
In una tabella di connessione si chiama contingenza cij di una casella la
differenza tra la frequenza osservata nij e quella teorica in caso di
indipendenza Nij :
cij = nij - Ni
Una contingenza positiva indica che quella particolare associazione fra
modalità dei due caratteri si è presentata più spesso che in caso di
indipendenza.
L'indice di contingenza chi-quadro di Person permette di ottenere una
misura della connessione tra i caratteri:
Questo indice permette di verificare la dipendenza o l'indipendenza di
due caratteri statistici:
l'ipotesi nulla(H0) di indipendenza al livello di significatività
H0 :i caratteri sono indipendenti
contro l'ipotesi alternativa
H1 :i caratteri sono dipendenti.
UNITA’ 14
DISTRIBUZIONE DI GAUSS
La gaussiana (curva di Gauss) è un concetto matematico abbastanza
avanzato, ma che ha notevoli implicazioni con il mondo reale. Molte
persone ritengono la matematica arida e finiscono per odiarla ("non sono
portato per i numeri"). Questa posizione può essere senz'altro giustificata
da un insegnamento troppo nozionistico della materia, insegnamento che
fa danni notevoli perché si riscontra che chi ha scarso spirito matematico
ben difficilmente comprende a fondo la realtà. Per spirito matematico non
s'intende la conoscenza delle scienze matematiche, ma la comprensione
(a volte intuitiva) di ciò che della matematica ha un'applicazione
concreta, anzi concretissima.
È vero che molte nozioni sono assolutamente inutili per chi non le userà
poi nella sua professione. Pensiamo alla trigonometria, utilissima a un
ingegnere, ma inutile a una commessa, a un giornalista ecc. Che
importanza "pratica" (cioè per la comprensione del mondo) ha sapere che
sen2a+cos2a=1? Nessuna. La stessa cosa invece non può dirsi per altri
concetti: la curva di Gauss (da Karl Friedrich Gauss, grande
matematico tedesco) ne è un esempio.
Anzi, questo articolo sarà propedeutico a molti altri di alimentazione o di
sport che spiegheranno concetti semplicissimi ma fondamentali.
Armatevi quindi di buona volontà e provate a seguirmi in questa
esposizione divulgativa della curva gaussiana.
La distribuzione
Quando dobbiamo giudicare un evento possiamo descriverlo con la
distribuzione dei suoi possibili valori. Se lancio una moneta il
valore testa ha probabilità 0,5 e idem ne ha il valore croce. Avremo una
distribuzione a due soli valori, ognuno dei quali ha probabilità 0,5. La
somma dei valori possibili dà l'unità (cioè la certezza, o esce testa o esce
croce: non si considera la possibilità che la moneta resti in piedi!).
Se analizziamo la distribuzione di un campione di persone che seguono
un certo programma televisivo per decadi di età, magari otteniamo un
grafico di questo tipo:
Le cose si complicano quando ho molti valori possibili, addirittura
infiniti.
Supponiamo per esempio di effettuare tante misurazioni di una stessa
grandezza con uno strumento; avremo risultati differenti, dovuti
all'inevitabile imprecisione del nostro strumento e del nostro operato, che
sono detti errori accidentali. Se rappresentiamo le misure ottenute su un
grafico, se il numero di misurazioni è molto grande, al limite infinito, la
curva
che
otterremo
è
proprio
la
curva
di
Gauss.
Si tratta di una curva dalla classica forma a campana che ha un massimo
attorno alla media dei valori misurati e può essere più o meno stretta a
seconda della dispersione dei valori attorno alla media; la dispersione si
misura con la deviazione standard: praticamente una delle proprietà della
gaussiana è che il 68% delle misurazioni differisce dalla media meno
della deviazione standard e che il 95% meno di due deviazioni standard:
quindi maggiore è la deviazione standard, più la gaussiana è "aperta" e
più c'è la possibilità che la media (il punto più alto) non sia
rappresentativo di tanti casi.
Anche nel caso della curva di Gauss l'area sottesa dalla curva vale 1
perché la somma delle probabilità di tutti i valori dà 1, cioè la certezza.
Un esempio reale
La distribuzione di Gauss è spesso detta normale. L'aggettivo è
significativo perché indica che moltissimi fenomeni possono essere
descritti da una curva gaussiana o Gauss-like (cioè simile).
Se è vero che la gaussiana vale per una popolazione infinita di
misurazioni e per eventi del tutto casuali, è altresì vero che curve a
campana (Gauss-like) possono descrivere facilmente molti fenomeni; per
detti
fenomeni
anche
i
concetti
di mediae
di deviazione
standard continuano a essere validi, anche se spesso solo il primo può
essere
definito
con
una
notevole
precisione.
Supponiamo di considerare l'altezza degli italiani maschi. Analizziamo
un campione di 1.000 soggetti. Probabilmente otterremmo una curva a
campana, centrata attorno a una media, del tipo 174 cm di media con una
"deviazione standard" di circa 20 cm, cioè il 95% dei soggetti analizzati
sarebbe compreso fra 154 cm e 194 cm.
UNITA’ 15
DISTRIBUZIONI DI BERNOULLI
La variabile casuale bernoulliana, dal nome dello scienziato
svizzero Jakob Bernoulli (1654-1705), è la più semplice di tutte
le variabili casuali. È una variabile dicotomica, dunque con due sole
possibili realizzazioni (0 e 1), cui sono associate le rispettive probabilità p
e 1-p.
A volte il termine variabile casuale bernoulliana è usato riferendosi
alle variabili casuali binomiali.
La distribuzione di Bernoulli
La distribuzione di Bernoulli è la distribuzione di probabilità discreta
associata ad una variabile casuale bernoulliana, che assume valore 1 con
probabilità di successo e valore 0 con probabilità di
insuccesso
. Sia quindi X una variabile casuale bernoulliana, si
ha che:
La funzione di probabilità f di questa distribuzione è:
Il valore atteso di una variabile casuale di Bernoulli X è:
,
mentre la sua varianza è:
L'indice di skewness e la curtosi sono rispettivamente:
;
.
La curtosi tende ad infinito per valori molto bassi o alti di p, ma
per
la distribuzione di Bernoulli presenta il valore della curtosi
più basso di qualsiasi altra distribuzione: -2.
La funzione generatrice dei momenti è:
Distribuzioni collegate
Se
sono variabili casuali Bernoulli indipendenti e
identicamente distribuite con probabilità di successo pari a p, allora X =
X1 + X2 +...+ Xn, è una variabile casuale binomiale B(n;p).
UNITA’ 16
DISTRIBUZIONI DI POISSON
Nell'ambito della teoria delle variabili casuali con distribuzione
composta di Poisson si intende la somma di un numero casuale
poissoniano di variabili casuale identiche e indipendenti. In particolare si
pone
dove N è una variabile casuale poissoniana con valore atteso λ, e
sono variabili casuali
indipendenti da N.
indipendenti
identicamente
distribuite
e
Allora la somma
è una distribuzione di Poisson composta (dove se N = 0, allora Y è 0.)
Se le n variabili casuali sono identicamente distribuite come un arbitraria
variabile casuale X, con valore atteso µ, secondo momento m2 e terzo
momento m3 si ottengono i seguenti parametri
valore atteso = λµ
varianza = λm2
coefficiente di assimetria = λm3
Alcune composte di Poisson
Se
sono
distribuite
come
la variabile
casuale
logaritmica allora la composta di Poisson è una variabile casuale
binomiale negativa.
UNITA’ 17
LEGGE DEI GRANDI NUMERI
La legge dei grandi numeri è fondamentale nella teoria delle variabili
casuali. Quella che segue è una sua formulazione in termini semplici ed
intuitivi.
Essa afferma che, se E è un evento e p è la sua probabilità di successo,
cioè la probabilità del verificarsi di E in una prova, allora la frequenza
relativa dei successi in n prove indipendenti converge in probabilità a p,
quando n tende a infinito,
dove "converge in probabilità" è un concetto che non definiamo in senso
accurato, ma si può intendere in un senso intuitivo (se il numero di prove
effettuate è sufficientemente grande, la frequenza relativa dei successi
nelle n prove si avvicinerà sempre più alla probabilità di successo nella
singola prova, via via che n cresce).
Questo teorema, formulato da Jakob Bernoulli (1654-1705), fornisce una
possibile giustificazione della legge empirica del caso, secondo la quale
la frequenza relativa di un evento tende a stabilizzarsi all'aumentare del
numero delle prove.
La legge dei grandi numeri stabilisce il comportamento asintotico della
frequenza relativa e non dice nulla sulla possibilità di successo di una
singola prova condizionata a quelle precedenti (che resta sempre p);
quindi, questa legge non dice che l'osservazione di - per esempio - 10
teste aumenta la probabilità che venga croce all'undicesima prova. Questo
fraintendimento è l'errore più comune nel quale incorrono i giocatori
d'azzardo, che scommettono sull'evento che non si verifica da più tempo,
convinti che, per questo stesso fatto, esso si debba verificare.
UNITA’ 18
TEOREMA CENTRALE DEL LIMITE
La famiglia della distribuzioni di probabilità normali ha
un'importante proprietà che la rende utilizzabile in un ampio raggio di
applicazioni: questa proprietà è il teorema centrale limite e riguarda la
distribuzione della somma o della media aritmetica di un campione scelto
a caso di osservazioni. Siccome media aritmetica e somma dei dati
differiscono tra loro per una costante moltiplicativa, d'ora in poi quello
che si dirà riferito alla media aritmetica varrà sostanzialmente anche per
la somma, fatti salvi alcuni aggiustamenti di costanti moltiplicative.
La formula che segue esprime quella che d'ora in avanti definiremo
"media campionaria".
Capita spesso di trovarsi di fronte a somme o medie artimetiche: ad
es., le vendite mensili di un'azienda consistono nella somma delle vendite
dei singoli rappresentanti.
Il teorema centrale limite afferma che, in condizioni abbastanza
generali, somme e medie di misurazioni casuali ricavate da
una popolazione tendono a possedere approssimativamente una
distribuzione a forma di campana, nel senso che si spiegherà in seguito.
La rilevanza di questo concetto è forse meglio comprensibile se ci si
avvale di un esempio. Quest'esempio è un'applicazione del metodo di
montecarlo, un metodo di campionamento simulato, nel quale si simula la
situazione nella quale si vuole calcolare la probabilità di un certo evento.
Si consideri una popolazione di lanci di dadi, generata lanciando un
dado un'infinitamente grande numero di volte, con distribuzione di
probabilità data dalla seguente immagine:
Si estragga un campione di n = 5 misurazioni dalla popolazione
lanciando cinque volte un dado e si prenda nota delle cinque
osservazioni, come indicato nella seguente tabella:
Si noti che i numeri osservati nel primo campione erano y = (3, 5, 1,
3, 2). Si calcoli la somma delle cinque misurazioni e la media
campionaria, . Per scopi sperimentali, si ripeta la procedura di
campionatura un centinaio di volte, o, preferibilmente, un numero
maggiore di volte. Si costruisca ora un istogramma della frequenza
per per i cento campioni e si osservi la distribuzione risultante nella
prima immagine.
Compare un risultato interessante: benché i valori di y nella
popolazione (Y = {1, 2, 3, 4, 5, 6}) siano equiprobabili e perciò
posseggano una distribuzione di probabilità che è perfettamente
orizzontale, la distribuzione delle medie campionarie scelta dalla
popolazione possiede una distribuzione con un addensamento al centro e
una densità minore sulle code a sinistra e a destra. Inoltre, si osserva che,
se si ripete l'esperimento delineato in precedenza per un campione più
grande (es.: n = 10), si noterà che la distribuzione delle medie
campionarie tende ad avvicinarsi sempre più alla forma di una campana,
via via che n cresce.
Pertanto, il teorema centrale limite, che si riferisce a "qualunque"
popolazione da cui si estraggano dei campioni, dice che:
Si traggono campioni casuali di n osservazioni da una popolazione con
media µ e scarto quadratico medio finito (o deviazione standard) σ.
Allora,
quando n è
grande,
la
media
campionaria sarà
approssimativamente distribuita normalmente, con media uguale a µ e
scarto quadratico medio σ/n 1/2. L'approssimazione diventerà sempre più
accurata via via che n cresce.
Il teorema centrale limite è importante innanzitutto perché spiega il
motivo per cui alcune misurazioni tendono a possedere
approssimativamente una distribuzione normale. Si può immaginare
l'altezza umana come composta da un numero di elementi - ognuno dei
quali casuale - associati con variabili come l'altezza della madre e del
padre, l'ambiente, la dieta, ecc. Se ognuno di questi elementi tende ad
aggiungersi agli altri per fornire la misurazione dell'altezza, allora
l'altezza è la "somma" di un numero di variabili casuali e il teorema
centrale limite può essere applicato e fornire una distribuzione delle
altezze che è approssimativamente normale.
In secondo luogo, l'altro e più importante contributo del teorema
centrale limite si esplica nell'inferenza statistica, perché molte procedure
statistiche di verifica di ipotesi fanno uso di questo teorema.