LEUCEMIE ACUTE
Neoplasie ematologiche caratterizzate da
accumulo, nel midollo e nel sangue periferico,
di cellule indifferenziate (blasti) e carenza di
cellule ematiche mature.
Leucemie acute mieloidi: origine da cellula
staminale pluripotente o, più raramente, da
progenitore granulo-monocitopoietico.
Leucemie acute linfoidi: origine da
progenitore B o T linfocitario, più raramente
da cellula staminale pluripotente
LEUCEMIE ACUTE MIELOIDI
- Più frequenti nell’ età adulta (mediana circa 60
anni). Incidenza globale: circa 4 / 105 / anno,
che aumenta con l’età.
- Primitive o secondarie a malattia
mieloproliferativa
cronica
o
sindrome
mielodisplastica.
- Eziologia sconosciuta in molti casi.
- In alcuni casi secondarie ad esposizione a
mutageni ambientali (radiazioni, benzene, altri
solventi organici, fumo tabacco, pesticidi) o
terapeutici (citostatici).
LEUCEMIE ACUTE LINFOIDI
- Incidenza maggiore in età pediatrica (2-10 anni); più
rare nell’ adulto, ma aumentano dopo i 50 anni.
-Di solito primitive, ad eziologia ignota,
probabilmente diversa secondo le età:
possibile esposizione a carcinogeni ambientali durante
la vita fetale per le forme neonatali (1° anno);
probabili errori nel riarrangiamento geni Ig e recettore
T nelle forme dell’ infanzia – adolescenza;
carcinogeni ambientali per le forme dell’ adulto?
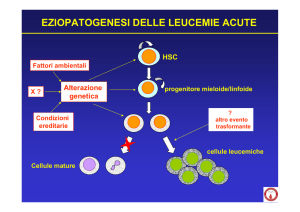
LEUCEMIE ACUTE
Alterazione
genetica
Ulteriore
alterazione
genetica
Cellule staminali
o progenitori
Espansione di
un clone
Ulteriore espansione
Perdita differenziazione
LEUCEMIE ACUTE
Cellula staminale leucemica
Progenitori leucemici
Blasti immaturi ±
rare cellule
differenzianti
LEUCEMIE ACUTE MIELOIDI
ALTERAZIONI GENETICHE (1)
Cromosomiche:
-Traslocazioni bilanciate: t(8;21); inv.16; t(15;17);
nelle LAM; t(11q…) nelle LAM e LAL; t(1;19) nelle
LAL; t(9;22)(Ph1) nella LMC e nelle LAL:
comportano formazione di geni ibridi e produzione
di proteine di fusione che interferiscono con la
trascrizione di geni regolanti differenziazione,
proliferazione, e apoptosi.
LEUCEMIE ACUTE
ALTERAZIONI GENETICHE (2)
Cromosomiche:
-Traslocazioni bilanciate:
t(8;14); t(5;14); t(9;14)… nelle LAL
Traslocano un gene codificante per fattori di
trascrizione che regolano la proliferazione sul
“promoter” dei geni codificanti le catene delle
immunoglobuline (nelle LAL B ) o del recettore
dei T linfociti (nelle LAL T) che sono iperattivi:
deregolazione trascrizionale
LEUCEMIE ACUTE
ALTERAZIONI GENETICHE (3)
Cromosomiche
-Delezioni: -5; 5q-, -7;…. (nelle LAM):
perdita di geni oncosoppressori.
-Trisomie: +8; +21…(nelle LAM) e
-Iperploidie (nelle LAL): amplificazione
genica.
LEUCEMIE ACUTE MIELOIDI
ALTERAZIONI GENETICHE (4)
Mutazioni geniche:
- mutazioni puntiformi o duplicazioni su geni che
codificano per recettori di fattori di crescita o
proteine che trasmettono i segnali proliferativi:
FLT3, Kit, ras,…
Attivazione permanente del segnale proliferativo
- mutazioni puntiformi o delezioni su geni
oncosoppressori: p53, …. (perdita funzionale)
LEUCEMIE ACUTE
SINTOMI E SEGNI DI CARENZA
CELLULE MATURE
Anemia: astenia, dispnea da sforzo, pallore,
tachicardia, edemi declivi.
Granulocitopenia: febbre, infezioni recidivanti.
Piastrinopenia: gengivorragie, epistassi,
petecchie, ecchimosi.
LEUCEMIE ACUTE
SINTOMI E SEGNI DI ESPANSIONE DEL
CLONE NEOPLASTICO:
- dolori o tensione addominale per splenomegalia
e/o epatomegalia;
- dolori ossei per espansione midollare;
- adenopatie superficiali e/o profonde (in LAL);
- cefalea, rachialgie, paralisi nervi cranici per
invasione liquor c.s. (rara, più frequente in LAL).
LEUCEMIE ACUTE
DIAGNOSI
Morfologica: esame al microscopio dello striscio di
sangue periferico e midollare ±
biopsia ossea: blasti > 20% nel midollo o > 10% nel
sangue periferico.
Colorazioni citochimiche per caratterizzare i blasti.
Immunologica: impiego di anticorpi monoclonali che
riconoscono antigeni di differenziazione e linea-specifici
per caratterizzare i blasti.
Citogenetica e molecolare: evidenza di anomalie
cromosomiche e genetiche che caratterizzano sottotipi a
diversa prognosi e terapia.
LEUCEMIE ACUTE MIELOIDI
SOTTOTIPI MORFOLOGICI
M0: mieloblastica indifferenziata, identificabile
solo con anticorpi monoclonali.
M1: mieloblastica senza maturazione: < 10%
cellule granulopietiche maturanti.
M2: mieloblastica con maturazione: cellule
granulopoietiche maturanti > 10%.
M3: promielocitica: cellule leucemiche di aspetto
simil- promielocita; alterazione citogenetica e
molecolare tipica.
LEUCEMIE ACUTE MIELOIDI
SOTTOTIPI MORFOLOGICI
M4: mielo-monocitica: 20 - 80 % cellule
monocitoidi.
M5: monocitica: cellule monocitoidi > 80 %
M6: eritroleucemia: eritroblasti > 50%
M7:
megacarioblastica:
blasti
precursori
megacariocitari, identificabili con anticorpi
monoclonali o con microscopia elettronica.
LEUCEMIE ACUTE
DIAGNOSI
Per terapia, fondamentale distinguere
LAM da LAL e M3 da altre LAM
DIAGNOSI LAM
Colorazioni citochimiche:
POSITIVE le colorazioni per mieloperossidasi e
Sudan Nero (M1 – M4) o esterasi non-specifica
(M4, M5).
Tipizzazione immunologica: espressione
antigeni di linea granulo-monocitopoietica
(mieloperossidasi, CD33, CD13) ± antigeni di
cellule immature (CD34).
DIAGNOSI DI LAL
Colorazioni citochimiche:
NEGATIVE le colorazioni per mieloperossidasi,
Sudan Nero, esterasi non-specifica .
Tipizzazione immunologica: espressione
antigeni precoci di linea B o T linfocitaria (CD19,
CD10, Tdt, CD2) ± antigeni di cellule immature
(CD34); assenza antigeni di linfociti maturi.
SOTTOTIPI IMMUNOLOGICI DI
LAL
LINEA B LINFOCITARIA
Pro – B: CD19 +, CD34 +, CD10 - ;
Common: CD19 +, CD34 +, CD10 + ;
Pre – B: CD19 +, CD10 +, catene µ citopl. + ,
CD34 ± ;
B mature: CD19 +, CD 20 +, Ig +, CD34 -
SOTTOTIPI IMMUNOLOGICI DI
LAL
LINEA T LINFOCITARIA
Pre – T: CD 7 +, CD 34 +, CD3 citoplasmat. +,
CD2 - ;
T : CD7 +, CD3 citoplasm +, CD2 +, CD34 ±
TERAPIA LEUCEMIE ACUTE
TERAPIA CITOSTATICA DI INDUZIONE
aplasia midollare
TERAPIA DI
SUPPORTO
rigenerazione emopoiesi
remissione completa
TERAPIA POST- REMISSIONE
CHEMIOTERAPIA LEUCEMIE ACUTE
INDUZIONE
• LAM
• LAL
Citosina arabinoside
• Vincristina +prednisone +
(ARA-C) +
asparaginasi ±
daunorubicina o
daunorubicina
idarubicina o
mitoxantrone ±
etoposide o 6-tioguanina
PROFILASSI MENINGEA
Metotrexate intra-rachide ±
Radioterapia cranica
TERAPIA DI SUPPORTO
TRASFUSIONI: eritrociti se Hb < 8 g /dl; piastrine se
valore < 10- 20.000 /ul o diatesi emorragica; plasma se
coagulazione intravasc. disseminata.
PROFILASSI ANTI-INFETTIVA: isolamento, cibi
solo cotti, antibiotici e anti-micotici orali.
TERAPIA ANTI-INFETTIVA: indagini
batteriologiche (emocultura) e inizio precoce terapia
antibiotica e.v. ad ampio spettro e a dosi piene in caso di
febbre. Terapia anti-micotica e.v. se persiste febbre dopo
3 giorni di antibiotici.
ANTI-EMETICI, IDRATAZIONE E NUTRIZIONE
PARENTERALE durante e dopo la chemioterapia.
TERAPIA POST-REMISSIONE LAM
CONSOLIDAMENTO
-Uno o due cicli come induzione o con ARA-C ad
alte dosi;
±
-Trapianto di midollo allogenico (se donatore
HLA-compatibile) o autologo se età < 60
TERAPIA POST-REMISSIONE LAL
CONSOLIDAMENTO
Cicli di ARA-C ad alte dosi + mitoxantrone o etoposide o
ciclofosfamide.
MANTENIMENTO
Terapia continua (2-3 anni) con 6-mercaptopurina orale +
metotrexate settimanale ± cicli di vincristina + prednisone
+ daunorubicina o ciclofosfamide.
TRAPIANTO ALLOGENICO
Nei casi a prognosi sfavorevole, se donatore compatibile
RISULTATI TERAPIA LAM
(NON M3)
REMISSIONE COMPLETA
65 - 80 % dei pazienti < 60 anni
10 – 20 % di casi resistenti, 10% mortalità in induzione;
40 – 60 % nei pazienti > 60 anni (se non patologie
associate)
20 – 30 % di casi resistenti e di morti in induzione.
Se NON remissione completa, sopravvivenza 1- 4 mesi.
RISULTATI TERAPIA LAM
( NON M3)
REMISSIONE A LUNGO TERMINE
20 – 60 % se età < 60 (secondo fattori prognostici)
< 10 % se età > 60
SE RECIDIVA: chemioterapia con ARA-C ad alte dosi +
idarubicina o mitoxantrone, poi trapianto, se possibile:
40 - 60 % di seconde remissioni (< 60 anni);
10 -30 % di remissioni a lungo termine; negli altri casi
sopravvivenza < 1 anno dopo la recidiva.
RISULTATI TERAPIA LAL
• BAMBINI
• Remissione completa:
>.90 %
• Remissione a lungo
termine (> 5 anni):
50 – 80 % secondo fattori
prognostici
ADULTI
Remissione completa:
70 – 80 %
Remissione a lungo
termine (> 5 anni):
< 10 – 50 % secondo
fattori prognostici
SE RECIDIVA: ARA-C alte dosi + idarubicina, metotrexate alte
dosi + corticosteroide: 50 - 60 % di seconde remissioni. Poi
trapianto, se possibile.
Remissioni a lungo termine: 20 – 30 % nei bambini, < 10% negli
adulti. Negli altri casi sopravvivenza < 1 anno.
FATTORI PROGNOSTICI LAM
CARIOTIPO: prognosi migliore se t(15;17) (M3),
t(8;21), inv.16;
prognosi pessima se -5, -7, t(3;21), anomalie multiple.
ETA’: prognosi migliore nei giovani, pessima se > 60.
MALATTIA SECONDARIA: prognosi pessima se
LAM secondaria a precedente mal. mieloproliferativa
cron. o sindr. mielodisplastica o precedente esposizione a
citostatici o benzene o radiazioni ionizz. LEUCOCITOSI:
prognosi migliore se GB < 10.000 /ul,
peggiore se > 50.000 /ul.
RISPOSTA AL 1° CICLO: prognosi peggiore se non
remissione con il 1° ciclo.
FATTORI PROGNOSTICI LAL
CARIOTIPO: prognosi pessima se t (9;22) (gene di
fusione BCR/ABL), t (11q;…); migliore se iperdiploide.
ETA’: prognosi migliore tra 2 e 10 anni, poi peggiora
progressivamente.
TIPO IMMUNOLOGICO: prognosi peggiore le pre-T
e pro-B, migliore le “common”.
MASSA NEOPLASTICA: prognosi migliore se GB <
10.000 /ul e non organomegalie, peggiore se GB >
50.000 / ul o grosse adenopatie.
SESSO: prognosi un po’ migliore nelle bambine.
LEUCEMIA ACUTA
PROMIELOCITICA ( M3)
5 % di LAM; più frequente nei giovani adulti.
Presenta, in oltre il 98 % dei casi fusione dei geni PML
e RARα (codifica per recettore nucleare di acido
retinoico), dovuta a traslocazione cromosomica 15;17.
Alla diagnosi è costante un quadro di coagulazione
intravascolare disseminata (C.I.D.), almeno sub-clinica.
Frequente la diatesi emorragica per piastrinopenia e
C.I.D.
Diagnosi sospettata per morfologia, citochimica,
antigeni (spesso CD34-), confermata da dimostrazione
del gene ibrido PML/RARα o da t (15;17).
LEUCEMIA ACUTA
PROMIELOCITICA ( M3)
Le cellule di LAP differenziano in vitro e in vivo
in presenza di concentrazioni farmacologiche di
acido trans-retinoico (ATRA).
L’ aggiunta di ATRA alla chemioterapia ha
radicalmente cambiato la prognosi:
Remissione complete: 90 – 95%
Remissioni a lungo termine: 70 – 80%