UNIVERSITA’ DEGLI STUDI DI MILANO
Facoltà di Farmacia
Corso di Laurea in Chimica e Tecnologia Farmaceutiche
STUDIO DEL MECCANISMO D’AZIONE
DEL 17β-ESTRADIOLO SULL’ATTIVAZIONE DI NF-κB
Relatore: Chiar.mo Prof. Adriana MAGGI
Correlatore: Dott. Elisabetta VEGETO
Tesi di Laurea di:
Alessandro GUERINI ROCCO
Matr. Nr. 561917
ANNO ACCADEMICO
2003/2004
alla musica degli Dei
INDICE
Pagina
INTRODUZIONE
1
1) Estrogeni e i loro recettori
1
2
5
8
Sintesi
Struttura dei recettori degli estrogeni
Isoforme dei recettori degli estrogeni
2) Meccanismo d’azione degli estrogeni
Via classica ligando-dipendente
a) Coattivatori
b) Corepressori
c) Fosforilazione
Via ligando-indipendente
Via ERE-indipendente
Via non genomica
a) ER e PI3K
b) I recettori di membrana degli estrogeni
3) Infiammazione
Tipi di infiammazione
Cause dell'infiammazione
Le cellule della risposta infiammatoria immediata
a) I granulociti neutrofili
b) I monociti/macrofagi
Mediatori dell'infiammazione
4) Il fattore nucleare κB (NF-κ
κB)
Classificazione dei membri di NF-κB
Attivazione di NF-κB
a) L’inibitore di NF-κB e le chinasi IKK
b) Fosforilazione di NF-κB
c) Acetilazione di NF-κB
Geni regolati da NF-κB
NF-κB e l'infiammazione
i
13
13
14
16
19
20
22
24
25
26
29
29
30
31
32
33
36
44
44
47
49
51
54
55
56
Farmaci che agiscono su NF-κB
NF-κB ed LPS
58
59
5) Estrogeni ed infiammazione del sistema nervoso
62
63
Estrogeni e patologie a componente infiammatoria del sistema
nervoso
a) La sclerosi multipla
b) La leucodistrofia a cellule globoidi
c) L'artrite reumatoide
d) L'ischemia cerebrale
e) La malattia di Alzheimer
Estrogeni ed infiammazione cerebrale sperimentale
Estrogeni ed infiammazione del sistema nervoso in modelli
cellulari
Estrogeni ed NF-κB
63
69
71
73
76
78
79
80
SCOPO DELLA TESI
83
MATERIALI E METODI
84
1) Colture cellulari
84
84
86
86
86
87
88
89
Condizioni di crescita
Terreni per colture cellulari
a) DMEM con rosso fenolo + 10% FBS
b) MEM con rosso fenolo + 10% FBS
c) RPMI 1640 con rosso fenolo + 10% FBS
d) RPMI 1640 + 10% FBS-DCC
e) Strippaggio del siero DCC
2) Preparazione di estratti proteici da lisati cellulari
Procedimento
Soluzioni
a) Lysis buffer per estratti totali di proteine cellulari
b) Lysis buffer per estratti citosolici di proteine cellulari
c) Lysis buffer per estratti nucleari di proteine cellulari
d) TEN buffer
e) PBS 10X
Determinazione delle proteine con il metodo di Bradford
ii
90
90
91
91
91
91
92
92
92
3) Western blot
Procedimento
Immunodetezione
Soluzioni per western blot ed immunodetezione
a) Acrilammide 30%
b) 7,5% separating gel PAGE
c) 4% stacking gel PAGE
d) Running buffer 5X
e) Running buffer 1X
f) Laemmli buffer 3X
g) Blotting buffer
h) TBS buffer
i) Blocking solution
l) Stripping solution
Anticorpi usati nelle western blot
93
93
94
96
96
96
96
96
96
97
97
97
97
97
97
a) Soluzione di blocco
b) Soluzione per anticorpi
c) Soluzione di lavaggio
d) Soluzione di avidin-biotin horseradish peroxidase complex
e) Soluzione di 3,3'-diamminobenzidina
98
98
98
99
99
100
100
100
100
100
101
5) Schema dei trattamenti delle cellule
101
RISULTATI
102
1) Azione del 17β-estradiolo sulla localizzazione citoplasmatica
di p65 in cellule macrofagiche
2) Azione del 17β-estradiolo sulla localizzazione citoplasmatica
di p65 in cellule monocitarie
3) Effetto del 17β-estradiolo sulla degradazione di IκBα
4) Influenza del 17β-estradiolo su IKK
5) Influenza del 17β-estradiolo su MAPK
102
4) Immunocitochimica
Preparazione vetrini
Fissaggio delle cellule
Immunodetezione
Montaggio vetrini
Soluzioni per immunocitochimica per p65
iii
105
107
109
111
6) Influenza del 17β-estradiolo su PI3K
7) Azione del 17β-estradiolo su altri membri di NF-κB
8) Azione del 17β-estradiolo sulla localizzazione citoplasmatica
di p65 in cellule non macrofagiche
113
115
118
DISCUSSIONE
121
BIBLIOGRAFIA
123
iv
INTRODUZIONE
GLI ESTROGENI E I LORO RECETTORI
Gli estrogeni fanno parte della famiglia degli ormoni steroidei, che
comprende anche i glucocorticoidi, i mineralcorticoidi, gli androgeni ed il
progesterone. Gli ormoni steroidei derivano tutti dal colesterolo e
condividono lo stesso nucleo ciclopentano-peridro-fenantrenico. Secondo
la nomenclatura sistematica, gli steroidi con 21 atomi di carbonio sono
denominati pregnani, mentre quelli con 19 e 18 atomi di carbonio sono
denominati androstani ed estrani rispettivamente.
Tutti gli ormoni steroidei esercitano la loro azione attraversando la
membrana plasmatica e legandosi a recettori intracellulari. I complessi
ormone-recettore esercitano la loro azione legando elementi responsivi
all’ormone (Hormone Response Elements, HREs) sul DNA ed
influenzando così la trascrizione dei geni bersaglio.
Gli estrogeni sono sintetizzati nella donna dalle cellule della teca e dalla
granulosa nei follicoli ovarici, dal corpo luteo e dall’unità feto-placentare
durante la gravidanza e nell’uomo dai testicoli. Gli estrogeni naturali sono
il 17β-estradiolo (E2), il maggior prodotto della secrezione ovarica,
l’estrone (12 volte meno potente), prodotto in piccole quantità sia nella
donna che nell’uomo anche dalla corticale del surrene, e l’estriolo (80
volte meno potente), derivato dall’ossidazione degli altri due, che si
verifica principalmente a livello del fegato ed in minor misura in altri
distretti dell’organismo.
Il catabolismo degli estrogeni genera inoltre altre molecole dotate di
1
attività estrogenica, genericamente indicate come catecolestrogeni, per via
dell’anello catecolico che si forma dopo l’ossidazione dell’anello A. I
catecolestrogeni sono generati per idrossilazione aromatica del 17βestradiolo e dell'estrone nella posizione C2 o C4 [1], in vari tessuti [2-7].
Tra i catecolestrogeni un ruolo di primo piano è esercitato dal 4-idrossiestradiolo, considerato un estrogeno a lunga azione, per via dei lunghi
tempi necessari per la sua dissociazione dal recettore. Alcuni studi
indicano come i catecolestrogeni possano avere dei propri recettori
distinti da ERα ed ERβ [8-12]. Gli estrogeni sono necessari per la
maturazione dell’organismo femminile e per le funzioni riproduttive.
Tuttavia hanno una serie di effetti anche sui tessuti non riproduttivi, ad
esempio a livello osseo, cardiovascolare e cerebrale.
Sintesi
La prima reazione nella conversione del colesterolo, che ha 27 atomi di
carbonio, negli ormoni steroidei con 21, 19 e 18 atomi di carbonio,
coinvolge il taglio irreversibile di 6 atomi di carbonio dalla catena laterale
con la formazione di pregnenolone ed isocapraldeide. Questa reazione è
finemente regolata ed è il passaggio limitante nella biosintesi degli
steroidi. L’enzima che catalizza la reazione: “enzima che taglia la catena
laterale legato al citocromo P450” (P450-linked side chain cleaving
enzime, P450scc) o desmolasi, è localizzato sulla membrana mitocondriale
interna. Si tratta di un sistema enzimatico complesso costituito da un
citocromo P450 e dalla ferro/zolfo proteina adrenodossina (una reduttasi
del P450). Dopo il distacco della catena laterale del colesterolo, il
pregnenolone fuoriesce dai mitocondri e si localizza nel reticolo
endoplasmatico dove va incontro a modificazioni sequenziali [13].
2
Estrogeni e progesterone sono i principali prodotti steroidei delle ovaie.
La sintesi degli estrogeni dipende dalla compartecipazione delle cellule
della teca e delle cellule della granulosa. Si parla di modello “due cellule–
due gonadotropine”. Le cellule della teca, stimolate dall’ormone
luteinizzante (LH), producono androstenedione e testosterone, che
diffondono nelle cellule della granulosa, dove sono convertiti
rispettivamente in estrone e in estradiolo sotto l'azione dell'ormone
follicolo stimolante (FSH). L’enzima coinvolto è l’aromatasi, localizzata
a livello del reticolo endoplasmico, che tramite idrossilazione e
deidratazione determina l’aromatizzazione dell’anello A degli androgeni
[14]. L’attività aromatasica è indotta dall’ormone follicolo stimolante
(FSH). La sintesi degli ormoni steroidei nelle gonadi è regolata da un
meccanismo di feed-back negativo, in quanto una volta sintetizzati gli
ormoni steroidei inibiscono la sintesi di quelle stesse sostanze che ne
hanno stimolato la produzione: l'ormone liberante le gonadotropine
(Gonadotropine Releasing Hormone, Gn-RH) secreto dall’ipotalamo ed
FSH e LH secreti dall’ipofisi. Gn-RH è liberato dal nucleo arcuato
dell’ipotalamo in maniera pulsatile ed è quindi pulsatile anche la
secrezione di gonadotropine ipofisarie. Elevati livelli di ormoni sessuali
circolanti esercitano un feed-back negativo sulla sintesi del GnRH [15].
L’ormone luteinizzante induce un aumento rapido e marcato della
capacità del P450scc di convertire il colesterolo in pregnenolone. Inoltre a
lungo termine stimola la sintesi del P450scc e di altri enzimi della
steroidogenesi. L’LH agisce sulle cellule della teca stimolando la sintesi
di androstenedione e testosterone. L’FSH agisce sulle cellule della
granulosa stimolando la trascrizione del gene dell’aromatasi [16].
3
Androstenedione
Testosterone
17-chetoreduttasi
aromatasi
aromatasi
17-chetoreduttasi
Estrone
Estradiolo
Sintesi degli estrogeni dal testosterone e dall'androstenedione
4
Struttura dei recettori degli estrogeni
I recettori degli estrogeni fanno parte della famiglia dei recettori nucleari.
I recettori nucleari si possono dividere in: 1) recettori per gli ormoni
steroidei; 2) recettori per ormoni non steroidei; 3) recettori orfani, i cui
ligandi non sono ancora stati identificati. I recettori per gli ormoni
steroidei hanno un domino A /B più lungo, e formano sempre omodimeri,
sono complessati con le proteine da shock termico (Heat Shock Proteins,
HSPs) e possono interagire con il DNA solo in presenza di uno stimolo. I
recettori per gli ormoni non steroidei formano eterodimeri con RXR, non
sono complessati alle HSP e sono associati al DNA in assenza di uno
stimolo. I recettori dei glucocorticoidi e dei mineralcorticoidi sono
citoplasmatici ed entrano nel nucleo solo in seguito all’attivazione da
parte del ligando. Gli altri recettori, invece, sembrano essere localizzati a
livello nucleare, ma tale fenomeno dipende dalla quantità di recettore
presente in una cellula. Infatti Castoria e collaboratori hanno
recentemente dimostrato che in cellule NIH3T3 il recettore degli
androgeni non va incontro a traslocazione nucleare in seguito al legame
con l'ormone, poiché in queste cellule la quantità di AR espressa è molto
bassa [17].
I membri della famiglia dei recettori nucleari hanno una struttura
conservata che consta di cinque diversi domini [18-20]. 1) La regione
ammino terminale, detta A/B domain, è la meno conservata tra i diversi
membri della famiglia dei recettori nucleari. Contiene un dominio detto
AF-1 (Activation Function 1), che stimola la trascrizione dei geni
bersaglio in modo indipendente dal ligando. 2) Il dominio C o di legame
al DNA (DNA Binding Domain, DBD), è il più conservato e determina la
specificità del recettore rispetto ad una classe di geni: recettori diversi
5
riconoscono infatti diverse sequenze consenso. Nel DBD sono presenti
due strutture digitiformi ad α elica, chiamate “zinc finger”, in cui uno ione
di Zn2+ è coordinato da quattro cisteine. Nella prima si trova il P-box
(Proximal box), che permette di riconoscere una specifica sequenza sul
DNA, mentre nella seconda si trova il D-box (Distal box), che è coinvolto
nella dimerizzazione sul DNA. 3) Il dominio D è una regione “a cardine”
(hinge): collega il dominio C al dominio E ed è sede di legame della
chaperonina hsp 90. Esso può anche contenere sequenze di localizzazione
nucleare (Nuclear Localization Signal, NLS). 4) La regione E, oltre ad
essere il dominio di legame del ligando (Ligand Binding Domain, LBD),
contiene un dominio per la dimerizzazione recettoriale e media
l'interazione con le proteine dello shock termico (HSP). A livello
dell’LBD è localizzato il dominio AF-2 (Activation Function 2),
coinvolto nella trascrizione ligando-dipendente. Infine all'interno del
dominio E è contenuto un NLS. 5) La regione carbossi-terminale F è poco
conservata ed è presente solo in alcuni recettori nucleari, tra cui i recettori
per gli estrogeni.
6
A/B
C
D
E
DBD
F
LBD
AF-2
AF-1
NLS
dimerizzazione
Co-attivatori
Hsp90
Hsp56
DR
C
C
Zn++
C
C
P
D
(C1)
Digitazioni Zn-coordinate
C
C
Zn++
Co-attivatori
C
C
(C2)
Struttura del recettore degli estrogeni
7
Isoforme dei recettori degli estrogeni
Gli estrogeni esercitano le loro numerose azioni biologiche tramite i
recettori degli estrogeni ERα ed ERβ, che si possono considerare come
fattori di trascrizione ligando-dipendenti. La prima prova che l’effetto
degli estrogeni fosse mediato da un recettore è di circa 40 anni fa, grazie
agli studi condotti da Jensen e Jacobson basati sul legame specifico del 17
β-estradiolo nell’utero [21]. Nel 1986 due diversi gruppi di ricerca sono
riusciti a clonare ERα, che si credeva l’unico recettore degli estrogeni.
Nel 1993 Korach e i suoi collaboratori, riuscirono a ottenere topi knockout per ERα. Si ottennero animali vitali di entrambi i sessi, però questi
animali mostravano una residua capacità di legare il 17β-estradiolo a
livello di diversi organi.
Nel 1995 è stato clonato ERβ da una biblioteca di cDNA di prostata di
ratto [22]. Successivamente sono state descritte diverse varianti di
splicing di ERβ. Alcune hanno un dominio N-terminale più esteso, altre
delezioni o inserzioni a livello della regione C-terminale e nell’LBD. Un
cDNA di hERβ, che codifica per una proteina di 530 aminoacidi è stato
clonato nel 1998 [23]. Era più lungo dell’ERβ di ratto per un estensione
di 45 aminoacidi nel dominio N-terminale.
Sono descritte anche diverse varianti di splicing di ERα, ma non si sa se
tutte sono espresse come proteine e se hanno un ruolo biologico o
fisiologico. Esistono anche diversi polimorfismi di ERα e di ERβ. Una
terza isoforma del recettore degli estrogeni, ERγ, è stata clonata, ma per il
momento è stata solo rinvenuta nei teleostei. ERγ si è originato da una
duplicazione genica di ERβ [24].
8
αN
hERα
DNA
LIGANDO
C
595
cardine
AF-2
AF-1
dimerizzazione
hERβ
β
N
DNA
LIGANDO
C
530
Paragone tra la struttura di ERα e di ERβ
da Jonathan G. Moggs and George Orphanides
EMBO reports Vol.2 No.9 September 2001 modificata
9
I DBD di ERα e β mostrano un’omologia di sequenza del 97%. In
particolare il P-box, che conferisce specificità del legame al DNA è
identico per ERα ed ERβ. Tramite il P-box gli ER riconoscono specifiche
sequenze sul DNA, gli elementi responsivi agli estrogeni (Estrogen
Response Elements, EREs). Si tratta di sequenze palindrome, ossia
ripetute in modo invertito e speculare rispetto ad un'asse di simmetria
(inverted repeats). La sequenza consenso è AGGTCX, mentre il numero
di nucleotidi che separa le due sequenze per gli ERE è di n=3 ed influenza
l’efficienza del legame al DNA. La specificità del legame al DNA può
essere cambiata mutando alcuni aminoacidi del P-box. Un mutante di ER,
in cui tre aminoacidi sono stati sostituiti così da trasformare l’ER-P-box
in un GR-like P-box, lega gli elementi responsivi ai glucocorticoidi
invece che l’ERE [25]. La struttura tridimensionale del dominio di legame
al DNA di ERα sia in soluzione che legato a un ERE, è stata risolta nei
primi anni ’90 [26, 27].
Nel secondo “zinc finger” è presente il D-box, un’interfaccia di
dimerizzazione. La dimerizzazione tramite i D-box facilita il legame
cooperativo al DNA e quindi stabilizza l’interazione tra i recettori
nucleari e il DNA. Nel caso di ERα, la dimerizzazione a livello del DBD
aumenta il legame anche a ERE imperfetti aumentando così il numero di
sequenze con cui ERα può interagire [28]. Nella parte C-terminale del
dominio di legame al DNA dei recettori degli estrogeni è presente una
regione ricca di aminoacidi basici richiesta per l’interazione con hsp 90
[29]. In quella regione si trova anche un segnale di localizzazione
nucleare [30].
Gli LBD di ERα ed ERβ mostrano un’omologia di sequenza del 55%,
tuttavia le regioni coinvolte nel legame ad agonisti ed antagonisti e l’AF-2
10
hanno un grado maggiore di omologia. L’analisi delle strutture cristalline
degli LBD di diversi recettori nucleari ha mostrato l'esistenza di una
struttura conservata composta da 12 α eliche, chiamate H1-H12. La
struttura tridimensionale del LBD di ERα legato al 17β-estradiolo, al
dietilstilbestrolo (DES), al raloxifene e al 4-OH tamoxifene è stata
ottenuta dagli studi cristallografici di Brzozowski [31]. Il sito di legame
all’ormone è costituito dalle eliche H3, H6, H8 ed H11 che formano una
tasca idrofobica. Quando l’E2 o il DES, due agonisti di ERα, si legano al
recettore, l’elica H12 cambia disposizione spaziale posizionandosi sopra
la tasca idrofobica, stabilizzando le interazioni fra il recettore ed il
ligando e formando una superficie di aminoacidi importante per il
reclutamento e l’interazione con proteine nucleari dette coattivatori. E’
stata anche risolta la struttura del ligand binding domain di ERβ legato al
raloxifene: la conformazione è simile al complesso ERα/raloxifene. Il
raloxifene ed il tamoxifene hanno una catena laterale ingombrante che si
estende al di fuori dell’LBD, quando sono legati ad ERβ l’elica H12
subisce una modificazione che non comporta lo spostamento sulla tasca di
legame, senza quindi portare ad esporre il sito di legame dei coattivatori.
Questo meccanismo sembra spiegare l’attività antagonista di tamoxifene e
raloxifene su ERβ e similmente su ERα [32]. Comparando il sito di
legame dell’ormone dei recettori degli estrogeni con altri recettori
nucleari, si è visto che questo sito è più largo; ciò può spiegare la capacità
del recettore degli estrogeni di legare molti composti [20]. Altri studi
hanno evidenziato che quando ERβ è legato al fitoestrogeno genisteina
l’elica H12 non assume la conformazione che ha con gli agonisti, forse
perché la genisteina è un agonista parziale [33].
11
B
A
H12
H12
A) Struttura 3D di ERα legato ad un agonista
B) Struttura 3D di ERα legato ad un antagonista
12
MECCANISMO D’AZIONE DEGLI ESTROGENI
Gli effetti biologici dell'estradiolo sono mediati da almeno quattro diverse
vie di segnale: 1) via classica ligando-dipendente; 2) via ligandoindipendente; 3) via ERE-indipendente; 4) via non genomica [34].
Via classica ligando-dipendente
L'azione dei recettori degli estrogeni sui siti ERE è un classico esempio di
azione genomica dei recettori nucleari. In assenza di ligando il recettore
degli estrogeni è sequestrato nei nuclei delle cellule bersaglio,
complessato con le proteine dello shock termico hsp 70, hps 90 e hps 56.
In seguito al legame con l’estrogeno il recettore va incontro ad un cambio
di conformazione, che rende possibile il distacco delle HSP e facilita la
dimerizzazione e il legame del recettore a livello degli elementi
responsivi agli estrogeni [35]. L'ER lega il proprio elemento responsivo
come omodimero ed utilizza entrambe le sue regioni di attivazione AF-1
ed AF-2 per reclutare cofattori. L'interazione di AF-2 con i coattivatori è
estrogeno-dipendente [32, 36]; mentre la regione AF-1 ha un'attività
trascrizionalmente molto debole e generalmente sinergizza con AF-2.
Il complesso ER-estrogeno funge da ponte tra il DNA, con cui interagisce
tramite il DBD, ed i cofattori che formano il complesso. Il complesso
recettore/ligando stabilizza il complesso d’inizio della trascrizione
costituito da diverse proteine con il compito di reclutare la RNA
polimerasi II. Il complesso d’inizio si assembla sul TATA box del
promotore del gene che deve essere trascritto, ed è costituito da vari
fattori, tra cui: TFIID, TFIIB, TFIIA, TFIIF, TFIIE (con attività
13
ATPasica) e TFIIH (dotato di attività elicasica) [37, 38]. L’interazione
può essere diretta o mediata da proteine chiamate coattivatori, alcune di
queste sono molto importanti per gli ER, in quanto sembrano essere
specifiche per questi recettori.
Coattivatori
La maggior parte di ciò che è conosciuto riguardo le interazioni fra ERα e
coattivatori deriva da studi sulla funzione AF-2 [39, 40]. I coattivatori
sono stati suddivisi in famiglie [41]: 1) p160; 2) p300/CBP (CREB
Binding Protein); 3) TRAP/DRIP.
La famiglia di p160 può essere suddivisa in tre sottofamiglie: SRC-1
(Steroid Receptor Coactivator-1), TIF2 (Transcriptional Intermediary
Factor-2) e p/CIP. Tutti gli attivatori di questa famiglia presentano, oltre
alla
sequenza
responsabile
del
riconoscimento
del
complesso
recettore/agonista, una porzione atta all'interazione con i coattivatori della
famiglia di p300/CBP. SRC-1 oltre ai recettori nucleari interagisce anche
con altri fattori di trascrizione tra cui: NF-κB, AP-1, SRF e p53.
I membri della famiglia di p300/CBP sono dotati di attività acetiltransferasica degli istoni. p300/CBP interagisce debolmente con ER, ma il
legame con SRC-1 stabilizza questa interazione. Così si forma a livello
del promotore del gene bersaglio un complesso con attività acetiltransferasica sugli istoni (HAT). Questi ultimi sono acetilati a livello delle
lisine e in questo modo diminuisce la forza del legame fra l'istone ed il
DNA. Ciò facilita la decondensazione locale della cromatina e quindi la
trascrizione [42, 43].
TRAP/DRIP è complesso multiproteico che sembra essere coinvolto nel
reclutamento della RNA polimerasi II.
14
A
E2
E2
B
R
Segnale di
trasduzione?
R
inattivo
R
R
attivo
inattivo
nucleo
attivo
nucleo
R
R
R
R
ERE
ERE
DNA
C
DNA
D
E2
E2
E2
R
R
R
inattivo
citoplasma
attivo
Src
MAPK
nucleo
R
Proteine G
PI3K
K+
Akt
R
nucleo
Non ERE
DNA
A) Via classica ligando-dipendente
B) Via ligando-indipendente
C) Via ERE-indipendente
D) Via non genomica
15
cGMP
cAMP
Ca++
I coattivatori interagiscono con i recettori nucleari per mezzo di tre
sequenze conservate LXXLL (Leucina-X-X-Leucina-Leucina), chiamate
anche NR box [44]. Gli NR box formano α eliche anfipatiche in cui i
residui di leucina formano una superficie idrofobica su una faccia
dell’elica [36, 45, 46]. Lo studio della struttura cristallina di hERα ha
mostrato che gli aminoacidi nelle eliche 3, 4, 5 e 12 formano una
superficie idrofobica che costituisce il principale sito di legame per la
sequenza LXXLL presente nei coattivatori della famiglia di p160 [32].
Corepressori
I corepressori sono proteine in grado di reprimere l’attività trascrizionale
dei recettori nucleari legandosi al loro LBD. Le proteine inibitorie della
trascrizione per essere definite corepressori devono soddisfare quattro
criteri: 1) interagire con il recettore non associato al ligando; 2)
dissociarsi in seguito al legame del recettore con un ligando in grado di
attivarlo; 3) potenziare la repressione del recettore; 4) reprimere la
trascrizione dei geni ai quali sono reclutati [47].
Fra i corepressori i meglio caratterizzati sono NCoR (Nuclear
CoRepressor) e SMRT (Silencing Mediator of Retinoid and Thyroid
hormone receptors), due proteine di circa 270 kDa, che mostrano elevate
omologie strutturali e funzionali. Le regioni che interagiscono con i
recettori nucleari, (Interaction Domain 1 e 2, ID1 e ID2) si trovano nella
regione C-terminale di entrambi i corepressori. Una piccola regione
presente nell’α elica 1 del ligand binding domain dei recettori nucleari,
chiamata CoR-box, è necessaria per l’interazione con SMRT e NCoR.
Una mutazione in questa regione inibisce infatti il legame tra i recettori
nucleari e i due corepressori. La posizione dell’H12 del LBD regola sia
16
Fattori di reclutamento
del complesso di inizio
Fattori di modificazione
degli istoni
Fattori di
rimodellamento della
cromatina
TRAP/DRIP
CBP/p300
P/CAF
p160
ADP+Pi
SWI/SNF
ATP
Recettore
attivato
R
R
HRE
D
TBP
B pol II
Complesso di inizio
della trascrizione
Il recettore degli estrogeni e i coattivatori
17
l’associazione dei corepressori ai recettori nucleari sia il loro rilascio. In
presenza dell’agonista, l’H12 è ripiegata così da formare un “coperchio”
sulla tasca di legame e blocca l’interazione con SMRT e NCoR. Al
contrario questi corepressori si associano agli ER legati agli antagonisti
[48, 49]. In questo caso infatti l’H12 è spostata verso l’N-terminale
dell’LBD; questa conformazione recettoriale non permette il legame dei
coattivatori, mentre favorisce il legame dei corepressori. NCoR e SMRT
hanno all’N-terminale tre domini di repressione, RD1, RD2 e RD3, le cui
sequenze non sono omologhe tra loro. Esistono diversi meccanismi di
repressione. Il più studiato è quello che prevede il reclutamento, da parte
di RD1 e di una regione a valle di RD3, delle deacetilasi degli istoni
HDAC1 e HDAC2. Un altro meccanismo proposto per l'attività dei
corepressori riguarda l’interazione con i fattori basali della trascrizione.
Anche le HDAC3, 4, 5, 6 e il complesso NuRD (Nucleosome Remodeling
and histone Deacetylation) potrebbero essere coinvolti nel meccanismo di
repressione [47].
Montano e collaboratori hanno identificato un nuovo corepressore,
denominato REA (Repressor of Estrogen receptor Activity), che
interagisce sia con ERα che con ERβ (ma non con altri recettori) [50].
Esistono anche corepressori che interagiscono con il dominio AF-2 dei
recettori degli estrogeni in presenza di agonisti. Ad esempio RIP 140
(Receptor Interacting Protein 140) e SHP (Short Heterodimerization
Partner)
mostrano
un’attività
co-regolatoria
negativa
perché
antagonizzano SRC-1 in vivo e competono per il legame ad AF-2 in vitro.
SHP è un particolare recettore nucleare che ha un LBD, ma è privo del
DBD. SHP mostra un’interazione ligando-dipendente con ERα ed ERβ,
che risulta nella repressione della loro attività trascrizionale. Tale
repressione è mediata dall’interazione di SHP con gli ER, tramite due
18
sequenze similari al NR-box. Anche DAX-1 (Dosage-sensitive sexreversal Adrenal hypoplasia congenital X chromosome) ed RTA
(Repressor of Tamoxifene Activity) regolano negativamente l’attività
trascrizionale di ERα ed ERβ. E’ stato ipotizzato che gli ER, utilizzando
SHP e DAX-1 come proteine ponte, possano richiamare proteine
corepressorie ad attività deacetilasica e quindi inibire la trascrizione [51,
52].
Fosforilazione
La fosforilazione degli ER è una modificazione post-traduzionale che
regola l'attività dei recettori nucleari. È stato dimostrato che il recettore
degli estrogeni è fosforilato sia in risposta a dosi fisiologiche di 17βestradiolo sia in assenza dell’ormone da altri stimoli, quali IGF [53].
La Ser 118 è uno degli aminoacidi più conservati per il recettore degli
estrogeni lungo la scala evolutiva. Il suo ruolo nel modulare la
trascrizione è a livello della regione AF-1 e potenzia l’interazione con i
coattivatori [54]. La fosforilazione di Ser 167 aumenta invece la capacità
del recettore di legare il DNA [55, 56].
Gli eventi di fosforilazione che riguardano il recettore degli estrogeni
sono specifici del tipo cellulare in cui sono stati osservati. Nelle cellule
COS-1 sito della fosforilazione ligando-dipendente di hERα è la Ser 118,
mentre nelle MCF-7 è la Ser 167 di hERα ad essere fosforilata, sempre in
presenza di E2, dalla casein chinasi II.
19
Via ligando-indipendente
La fosforilazione degli ER è importante nell’attivazione trascrizionale
indotta da ligando, ma anche in assenza di esso. Infatti, i fattori di
crescita, gli attivatori della protein chinasi A (PKA), i neurotrasmettitori e
le cicline sono in grado di indurre l’attività trascrizionale degli ER
mediante la fosforilazione del recettore [57]. L’N-terminale di ERα
contiene diversi residui di Ser conservati nel dominio AF-1, che sono
bersaglio di fosforilazione. La fosforilazione della Ser 118 di hERα è
indotta da EGF e dipendente dall’attivazione delle MAPK [58]. Anche E2
è in grado di indurre la fosforilazione della Ser 118, ma questo sembra
essere indipendente dalla MAPK, indicando che diverse vie di
trasduzione del segnale possono agire sullo stesso residuo, a seconda della
presenza
o
meno
dell’E2.
La
rilevanza
fisiologica
dell’EGF
nell’attivazione di ERα è dimostrata dal fatto che l’EGF mima gli effetti
dell’estrogeno nell’apparato riproduttivo di femmina di topo e in cellule
epiteliali della ghiandola mammaria [59]. Topi femmina KO per ERα
mancano
di una risposta uterotropica all’EGF, dimostrando il
coinvolgimento di ERα nel mediare l’azione dell’EGF in vivo [60, 61].
Altri fattori di crescita coinvolti nell’attivazione di ERα sono l’insulina
[62], i fattori di crescita insulino-simili I e II (IGF-I ed IGF-II) [53], il
fattore di crescita trasformante β (TGF-β) [63] ed il fattore di crescita
trasformante α (TGF-α) [64]. Nella linea cellulare SK-ER3, neuroblasti
umani con espresso stabilmente il recettore per gli estrogeni, l’insulina e i
due fattori di crescita insulino-simili, IGF-I ed IGF-II sono capaci di
indurre differenziamento fenotipico coinvolgendo la via delle MAPK
[53]. In cellule MCF-7 l’attivazione ligando-indipendente di ERα da parte
20
dell’IGF-I è mediata dalla PI3K e da Akt [65]. Akt attiva la funzione AF1 di ERα fosforilando il recettore a livello della serina 167. La mutazione
di questa serina in alanina distrugge l’effetto.
Anche le cicline e le chinasi ciclino-dipendenti (CDK) sono coinvolte
nell’attivazione ligando-indipendente di ERα. Due differenti cicline, A e
D1, sono state identificate come capaci di attivare ER in modo ligandoindipendente. La sovraespressione di ciascuna di queste due proteine
determina un aumento dell’attività di ER in assenza di E2. I meccanismi di
attivazione di ER sono diversi per queste due cicline. L’attivazione di ER
da parte della ciclina D1 non coinvolge la fosforilazione e quindi la
presenza della corrispettiva cdk [66]. La ciclina A invece attiva gli ER
mediante fosforilazione, tramite cdk2 nel dominio AF-1 [67].
L’attivazione di ERα in modo ligando-indipendente non avviene solo
tramite la fosforilazione di residui di serina nel dominio AF-1, ma anche
nel dominio AF-2. In questo processo è coinvolto il cAMP, che attiva la
PKA [68]. Agenti che aumentano il contenuto cellulare di cAMP (la
forskolina, l’acido ocadaico e la tossina colerica) determinano un’attività
trascrizionale di ERα ligando-indipendente; inoltre, queste sostanze
sinergizzano con l’attivazione trascrizionale mediata da E2. Anche la
risposta all’agonista parziale tamoxifene aumenta se c’è un aumento della
concentrazione intracellulare di cAMP. In cellule MCF-7, la trascrizione
dei geni bersaglio di ERα, fra cui il recettore del progesterone (PR), P62,
LIV-1 e catepsina D, può essere stimolata o da un analogo del cAMP
(8Br-cAMP), o da una combinazione di un inibitore della fosfodiesterasi
(IBMX) e della tossina colerica, che aumenta la produzione del cAMP
bloccando la subunità αs delle proteine G. Studi ulteriori hanno
dimostrato che le risposte indotte dal cAMP sono inibite dall’antagonista
21
puro dell’ER, l’ICI 182,780 [69]. Inoltre i domini E/F di ERα sono
sufficienti per l’attivazione da parte della forscolina più IBMX e questo
fenomeno è accompagnato dalla fosforilazione del recettore.
La Tyr 537 nell’ERα umano e la Tyr 541 nel topo localizzate nell’LBD
sono importanti per la regolazione dell’attività di ERα. La struttura
tridimensionale di ERα indica che Tyr 537 si trova nell’ansa che precede
l’elica H12. Tyr 537 viene fosforilata dai membri della famiglia della Tyr
chinasi, quali Src, in assenza di E2. Tuttavia questa fosforilazione non
induce attività trascrizionale, ma regola la capacità di ERα di legare E2
[70].
Anche ERβ va incontro ad attivazione ligando-indipendente, infatti l’EGF
induce la fosforilazione delle Ser 106 e Ser 124 di ERβ tramite
l’attivazione delle MAP chinasi [58]. Questa fosforilazione determina il
reclutamento ligando-indipendente di SRC-1 e il conseguente aumento
dell’attività trascrizionale. Recentemente, è stata studiata l’attivazione
cAMP-dipendente dell’ERβ [69]. In trasfezioni transienti la forskolina più
l’IBM, che aumentano i livelli intracellulari del cAMP, stimolano
l’attività trascrizionale di ERβ. Questo effetto è bloccato da un inibitore
della PKA ed è dipendente dalla presenza di un ERE.
Via ERE-indipendente
I recettori degli estrogeni modulano l'espressione genica agendo anche su
geni che non presentano ERE nei propri promotori. Ad esempio IL-6,
TNFα e MCP-1 non hanno ERE nei loro promotori, ma la loro
trascrizione è inibita dagli estrogeni. In questo caso gli ER non hanno un
effetto trascrizionale diretto, ma si pensa che l'azione degli ER sia
22
mediata dall'interazione con altri fattori di trascrizione.
Infatti parecchi gruppi di ricerca hanno dimostrato che l'E2, agendo
tramite i suoi recettori, è in grado di reprimere l'espressione di IL-6
mediante l'interazione con i fattori di trascrizione della famiglia di NF-κB
[71-76]. L'ER inibisce anche la trascrizione di MCP-1 [77], di TNFα [74]
e di RANTES (Regulated upon Activation Normal T cell Expressed and
Secreted) [78] interferendo con l'azione di NF-κB.
Infatti sia ERα che ERβ possono interagire con il fattore di trascrizione
AP-1 (Activating-Protein-1), costituito da un eterodimero tra Jun e Fos.
Queste due proteine, in seguito all’attivazione della via delle MAPK,
eterodimerizzano per formare il complesso AP-1. A seconda del contesto
cellulare e del gene trascritto l’estrogeno può attivare o sopprimere la
trascrizione genica mediata da AP-1. Infatti l'ER stimola la trascrizione
del gene dell'ovalbumina per interazione con i dimeri Jun/Fos [79] e
potenzia l'attivazione di AP-1 da parte dei fattori di crescita in MCF-7
[80]. In altri contesti l'estrogeno antagonizza l'azione di AP-1, infatti
reprime l'espressione di TNFα [81, 82], di MCP-1 [83] e di molecole di
adesione come ICAM (Intercellular Adhesion Molecule) e VCAM-1
(Vascular Cell Adhesion Molecule type 1) [84, 85].
Un altro esempio di azione indiretta di ER sulla trascrizione è
l’interazione degli ER con il fattore di trascrizione Sp1. In cheratinociti
umani ERβ inibisce la trascrizione di MCP-1 in modo dipendente dalla
presenza di Sp1 [83]. Inoltre, sia ERα che ERβ possono attivare la
trascrizione del gene del recettore dell’acido retinoico (RAR-1), mediante
la formazione di un complesso tra ER e Sp1 sui siti di legame di Sp1
(ricchi di GC) del promotore di RAR-1 [86, 87].
23
Via non genomica
Le prime evidenze di effetti degli estrogeni troppo rapidi per essere
mediati dall'attivazione della trascrizione e della sintesi proteica risalgono
agli anni settanta, quando studi elettrofisiologici dimostrarono che
l'estradiolo è in grado di modulare le concentrazioni intracellulari dello
ione Ca2+ entro pochi secondi dalla stimolazione [88]. Un anno più tardi
Pietras e Szego descrissero una rapida formazione di cAMP in risposta
all'E2, presumibilmente mediata dal legame dell'ormone ad una proteina
recettoriale nella membrana cellulare [89, 90]. Dopo questi studi
pionieristici parecchi gruppi si sono dedicati agli effetti non genomici dei
recettori degli estrogeni [91, 92]. Questi effetti mostrano delle
caratteristiche comuni: 1) sono troppo rapidi per essere compatibili con la
sintesi di RNA e di proteine, dal momento che si verificano entro pochi
minuti dalla somministrazione dell’ormone; 2) avvengono in cellule nelle
quali la sintesi di RNA e proteine è praticamente assente, come gli
spermatozoi; 3) sono riprodotti in presenza di inibitori della trascrizione o
della sintesi proteica; 4) possono essere riprodotti usando ormoni legati
covalentemente a molecole impermeabili alla membrana cellulare [93].
Questi effetti non trascrizionali degli estrogeni comprendono la
regolazione del flusso cellulare di Ca2+ [94-102], la modulazione del
contenuto citoplasmatico di cAMP [103-107], cGMP [108, 109], IP3
[110] e la modulazione di recettori associati alle proteine G [111-116].
Inoltre l'E2 attiva chinasi come la PKA [69, 100, 105], la PKB [117, 118],
la PKC [119, 120], la chinasi Ca2+/calmodulina-dipendente (CAMK)
[121], le MAP chinasi [122-124] e tirosin-chinasi [125]; ed attiva fattori
di trascrizione tra cui CREB [115, 116] ed AP-1 [126].
24
ER e PI3K
Uno degli effetti rapidi degli estrogeni più recentemente descritti è la
modulazione della via della PI3K e di Akt. Le fosfatidilinositolo 3-chinasi
(PI3K) sono delle chinasi importanti per molti processi cellulari. Sulla
base della struttura e della specificità del substrato sono state identificate
tre
classi
di
PI3K:
la
classe
I
fosforila
preferibilmente
il
fosfatidilinositolo-4,5-bifosfato, la classe II il fosfatidilinositolo-4-fosfato
e la classe III il fosfatidilinositolo. La classe I si suddivide a sua volta in
due sottoclassi: IA ed IB. Esistono tre isoforme di PI3K della classe IA:
p110α, p110β e p110δ; queste sono strettamente associate ad una
subunità regolatoria che contiene due dominii SH2 (Src-Homology 2).
Della subunità regolatoria si conoscono tre isoforme: p85α, p85β e p85γ.
Della classe IB è noto un solo membro: PI3Kγ che è associato alla
subunità regolatoria p101; la classe II è formata dalle isoforme:
PI3K-C2α, PI3K-C2β e PI3K-C2γ, mentre la III classe dal solo enzima
Vps34p. Le due classi IA ed IB sono quelle più importanti e studiate.
Le fosfatidilinositolo 3-chinasi sono coinvolte nella sopravvivenza
cellulare,
nel
ciclo
cellulare,
nella
mobilità
cellulare,
nella
degranulazione, nell'immunità, nella regolazione del metabolismo di
glucosio e glicogeno e nella sintesi proteica [127]. Parecchi studi hanno
evidenziato un cross-talk tra le vie di trasduzione del segnale della PI3K e
quelle del recettore degli estrogeni; è stata provata sia un'attivazione di
ER in seguito a fosforilazione da parte di Akt, una chinasi a
serina/treonina attivata dalla PI3K [65, 128-130], sia un'attivazione del
signaling della PI3K in seguito al legame dell'estrogeno al suo recettore.
In cellule MCF-7 è stato dimostrato che l’ingresso in fase S del ciclo
cellulare e l’attività del promotore della ciclina D1 sono mediati
25
dall’attivazione della PI3K e di Akt, indotta dal 17β-estradiolo [131]. La
formazione di un complesso ternario tra ERα, la tirosin chinasi non
recettoriale Src e p85 sembra necessaria per l’attivazione della via della
fosfatidilinositolo 3-chinasi.
Inoltre, a livello della parete vascolare la PI3K e Akt mediano il rapido
rilascio di NO indotto dal 17β-estradiolo tramite l’attivazione della
nitrossido sintasi endoteliale (eNOS) [132]. L’NO attiva la guanilato
ciclasi a livello delle cellule muscolari lisce (SMC) della parete vascolare.
La conseguente produzione di cGMP determina il rilassamento della
muscolatura liscia vasale e inibisce la proliferazione delle cellule
muscolari lisce. È stato dimostrato che la stimolazione di eNOS da parte
di E2 è mediata da una sottopopolazione di ERα, localizzato a livello delle
caveole [133, 134]. Infatti, l’E2, tramite ERα, attiva la fosfatidilinositolo
3-chinasi e Akt, che a loro volta attivano e fosforilano eNOS.
L'attivazione da parte dell'estrogeno di PI3K/Akt è inoltre importante
nella neuroprotezione esercitata dall'ormone [135].
I recettori di membrana degli estrogeni
Gli effetti degli estrogeni che non prevedono la modulazione della
trascrizione suggeriscono l'esistenza di recettori di membrana. Le prime
prove dell’esistenza di una sottopopolazione di recettori degli estrogeni di
membrana risale alla fine degli anni ’70, quando Pietras e Szego
descrissero la presenza di siti di legame per l’estradiolo a livello della
membrana citoplasmatica di cellule endometriali [89]. A tutt'oggi,
nonostante gli effetti non genomici di E2 siano coinvolti in diverse vie di
trasduzione del segnale, non esistono indicazioni chiare circa l'esistenza e
l'identità di recettori di membrana che medino tali effetti. Alcuni gruppi
26
hanno proposto che i recettori di membrana siano gli stessi ERα e ERβ
che traslocano in membrana [112, 136-138], altri che siano nuovi membri
della famiglia degli ER [139-141], altri ancora che siano recettori
accoppiati a proteine G [111, 113, 142], altri, infine, che possano essere
dei recettori tirosino-chinasici simili a quelli dei fattori di crescita [143].
Studi effettuati sulle cellule endoteliali utilizzando estradiolo legato
covalentemente alla BSA, così da impedire l’ingresso dell’ormone nella
cellula, suggeriscono che la rapida attivazione di eNOS indotta da E2 sia
mediata da un ERα di membrana [93]. Infatti cellule endoteliali intatte
legano estradiolo-BSA e sono riconosciute da anticorpi contro ERα,
suggerendo che questo ER di superficie abbia omologia con ERα. L’ICI
182,780 compete con l’E2-BSA nel legame al recettore di superficie.
Inoltre la stimolazione di cellule umane endoteliali con estradiolo-BSA,
così come il 17β-estradiolo, induce una rapida attivazione di eNOS
tramite la via della PI3K.
In cellule EA.hy926 che non esprimono i classici ER, ma una proteina di
46 kDa, riconosciuta da anticorpi contro il dominio C-terminale degli ER,
l’E2 è in grado di indurre la rapida attivazione di eNOS [144, 145].
Siccome una proteina di dimensioni e reattività simili è stata trovata
associata alla membrana plasmatica in cellule MCF-7 [146] si è ipotizzato
che questa proteina sia un recettore di membrana degli estrogeni. Flouriot
e collaboratori hanno dimostrato che questa forma di ER di 46 kDa deriva
da uno splicing alternativo di ERα ed è priva dei primi 143 aminoacidi
(regione AF-1) [147]. È stato successivamente dimostrato che l’E2 attiva
rapidamente c-Src inducendo la formazione di un complesso tra la
proteina di 46 kDa, c-Src e p85 [148]. La formazione di questo complesso
risulta nella successiva attivazione della PI3K, di Akt e di eNOS.
27
Un lavoro del gruppo di Toran-Allerand ha ipotizzato l'esistenza di una
isoforma di ER di membrana diversa da ERα ed ERβ. È stato osservato
che, in espianti neocorticali sia di topo wild-type che knock-out per ERα
(ERαKO), la somministrazione sia del 17α che del 17β-estradiolo
induceva la rapida fosforilazione e l’attivazione delle chinasi regolate da
segnali extracellulari ERK1 ed ERK2. Secondo il lavoro di ToranAllerand questi effetti non sono mediati dai classici ERα ed ERβ, ma da
un recettore associato alla membrana plasmatica, di 62-63 kDa,
immunoreattivo per l’LBD di ERα ma non di ERβ, denominato dagli
autori ER-X [149, 150]. Questo recettore è localizzato a livello di
microdomini simili alle caveole (CLM, omologhi neuronali delle caveole)
nella membrana cellulare di neuroni neocorticali di topi sia wild type che
ERαKO. ER-X è espresso solo nei primi giorni dopo la nascita, ma in
seguito ad attacco ischemico è espresso anche nell’adulto.
Un possibile significato biologico della presenza nella cellula di un pool
di recettori di membrana e di uno di recettori nucleari è la loro
cooperazione. L'E2, tramite il recettore di membrana, può rapidamente
attivare la trascrizione, che viene poi mantenuta attraverso il recettore
nucleare. Il segnale di membrana è importante per le modificazioni posttraduzionali delle proteine, la cui sintesi può essere incrementata dal
recettore nucleare [91]. Attualmente nonostante le prove che ne
suggeriscono l’esistenza il recettore di membrana degli estrogeni non è
stato ancora né isolato, né clonato, né completamente caratterizzato.
28
INFIAMMAZIONE
L’infiammazione, o flogosi, si può definire come un processo dinamico
comprendente l’insieme delle modificazioni reattive che compaiono nelle
strutture vascolari e connettivali di un distretto organico, per arginare e
riparare i danni prodotti da agenti lesivi di diversa natura.
I classici segni clinici dell’infiammazione sono rubor, calor, tumor, dolor
et functio laesa (rossore, calore, gonfiore, dolore e perdita di funzione).
Nonostante l'infiammazione sia un processo localizzato, ai fenomeni
infiammatori locali non rimane estraneo l’organismo nel suo insieme.
Infatti anch’esso risponde agli stimoli flogogeni, sia con modificazioni
neuro-ormonali, sia con l’attivazione del sistema linforeticolare, che
comporta un’esaltazione della fagocitosi ed un aumento della produzione
di anticorpi. Sebbene nella maggior parte dei casi l’infiammazione svolga
un compito difensivo, tendente a soffocare un’azione lesiva o a
circoscriverla ad un territorio limitato, in alcuni casi la reazione difensiva
supera largamente le necessità locali di risposta agli insulti, e produce
essa stessa un danno.
Tipi di infiammazione
Si può distinguere tra infiammazione acuta e cronica.
L’infiammazione acuta è di breve durata: minuti, ore o al massimo pochi
giorni. È caratterizzata da alterazioni vascolari che causano un aumento
del flusso sanguigno, da edema e da migrazione dei leucociti, soprattutto
dei neutrofili provenienti dalla microcircolazione e dal loro accumulo
nella regione del danno.
29
L’infiammazione cronica è un’infiammazione di lunga durata in cui i
processi infiammatori, il danno tissutale ed i tentativi di riparo avvengono
contemporaneamente. È determinata dalla persistenza di uno stimolo
infiammatorio. Le cause possono essere diverse, ad esempio un’infezione
batterica persistente, la prolungata esposizione ad un agente tossico, una
malattia autoimmune, l'invecchiamento sono di cause infiammazione
cronica. È caratterizzata dall'infiltrazione di cellule mononucleate
(macrofagi, linfociti e plasma-cellule), dalla contemporanea presenza di
danno tissutale, angiogenesi e fibrosi.
Cause dell'infiammazione
L’origine di un’infiammazione può essere di ordine fisico (caldo, freddo,
correnti elettriche, ultrasuoni, radiazioni eccitanti ed ionizzanti), chimico
(sostanze irritanti, veleni, tossine microbiche, complesso antigeneanticorpo, prodotti abnormali del metabolismo), biologico (parassiti,
batteri, miceti, protozoi, virus). Le cause fisiche agiscono soprattutto
indirettamente, attraverso la liberazione di sostanze da parte delle cellule
e dei tessuti lesi. Sperimentalmente è facile provocare l’infiammazione
chimica, ad esempio applicando sulla cute sostanze irritanti come l’olio di
croton e la trementina; anche l’aumento in un tessuto della concentrazione
di certi metaboliti endogeni è spesso causa di infiammazione chimica.
Tuttavia le cause biologiche sono tra le più frequenti nell'induzione della
flogosi.
In
essa
l’azione
dei
microrganismi
batterici
dipende
essenzialmente dalla loro capacità moltiplicativa e dall’intervento sui
tessuti di prodotti del loro metabolismo (esotossine), o di costituenti
chimici della parete batterica, che si liberano dopo la morte dei
microrganismi (endotossine). Per fenomeni di glicolisi aerobica ed
30
anaerobica essi provocano nel territorio infiammato la formazione di
acido lattico e CO2, alterando così l’equilibrio acido-basico dei tessuti.
Le cellule della risposta infiammatoria immediata
Nella risposta infiammatoria si ha un'infiltrazione di cellule leucocitarie
nel tessuto interessato dalla flogosi in particolare del lignaggio mieloide.
Nell'infiammazione acuta la popolazione cellulare è prevalentemente
composta dai granulociti neutrofili, mentre più raramente si ritrovano i
granulociti eosinofili, quando entra in gioco una risposta immunitaria di
tipo immediato. Rappresentano una rara eccezione le infiammazioni acute
dominate da essudazione linfocitaria, come capita in alcune infezioni
virali. Nella tipica infiammazione acuta è caratteristica la precoce e
progressiva sostituzione dei granulociti neutrofili con i monociti, cellule
dotate di una capacità di sopravvivenza molto superiore a quella dei
neutrofili che è di soli 3-4 giorni. Con sostanze ad azione irritante
relativamente blanda, come soluzioni di glicogeno, si è visto che
l'accumulo di neutrofili, che inizia immediatamente, raggiunge il valore
massimo dopo 4 ore e quindi declina rapidamente; mentre l'accumulo di
monociti comincia a manifestarsi dopo 4 ore e raggiunge il valore
massimo dopo 18-24 ore. Neutrofili e monociti non migrano
simultaneamente e perciò non reagiscono nello stesso modo agli stimoli
chemiotattici.
In
conclusione,
la
migrazione
dei
monociti
nell'infiammazione acuta prende inizio solamente quando diminuisce
quella dei neutrofili, ma continua per un periodo molto più lungo.
31
I granulociti neutrofili
I granulociti neutrofili sono caratterizzati dal nucleo segmentato e dalla
presenza nel citoplasma di numerosissimi granuli contenenti vari enzimi
proteolitici. Questi, oltre ad essere antimicrobici, possono danneggiare il
tessuto sede di flogosi o comunque esercitare attività flogogena.
I granulociti sono le cellule più frequentemente reperibili negli essudati,
dove svolgono attività fagocitaria verso i batteri o verso materiali estranei,
che possono essere contenuti in vacuoli citoplasmatici di 0,1-1µm.
Sulla membrana cellulare dei neutrofili vi sono dei recettori per le
immunoglobuline e per il complemento, che entrano in gioco nella
immunofagocitosi. Il citoplasma dei neutrofili è ricco di glicogeno. La
produzione di energia nel neutrofilo maturo è affidata più alla glicolisi
(>90%) che alla respirazione. Ciò favorirebbe l'efficienza della cellula nei
tessuti ipossici e negli essudati.
Negli spazi interstiziali i neutrofili sono capaci, con la loro attività di
fagociti, di impedire ai microrganismi patogeni di diffondersi al di fuori
del focolaio dell'infiammazione. Qui però i neutrofili diventano molto
fragili a causa dell'acidità del focolaio infiammatorio o perché lesi da
batteri, che hanno sopraffatto il loro potere fagocitario e microbicida.
Così
in
breve
tempo
vanno
incontro
a
fenomeni
regressivi
(degranulazione, infiltrazione grassa, vacuolizzazione oppure picnosi del
nucleo) ed a fenomeni di autodigestione per l'attivazione delle proteasi
contenute nei lisosomi. I loro frammenti vengono fagocitati e distrutti dai
macrofagi. La sopravvivenza dei granulociti neutrofili nel campo
infiammatorio comporta la secrezione abbondante gli enzimi proteolitici
di cui sono ricchi tali cellule, in modo da fluidificare e digerire il tessuto o
32
le componenti dell'essudato, come la fibrina. Questi processi sono alla
base del fenomeno della suppurazione (infiammazione purulenta).
I monociti/macrofagi
I monociti (fagociti mononucleati del sangue) e i macrofagi (fagociti
mononucleati dei tessuti) hanno una parte centrale nella resistenza alle
infezioni e nell'infiammazione. Inoltre queste cellule assumono
un'importanza fondamentale nell'immunità specifica attraverso una stretta
associazione funzionale con i linfociti, soprattutto quelli della classe T.
I macrofagi derivano dalle cellule staminali emopoietiche del midollo
osseo che, attraverso lo stadio di monoblasti e promonociti, si
differenziano in monociti e come tali entrano in circolo. Da qui, dopo
poco tempo (36-104 ore), i monociti migrano nei tessuti e nelle cavità
sierose in cui si differenziano a macrofagi. Sotto la denominazione di
macrofagi vengono comprese cellule capaci di svolgere un'intensa attività
fagocitaria, anche verso elementi estranei di dimensioni relativamente
grandi, come batteri ed anche cellule intere.
I fagociti mononucleati comprendono: 1) elementi a locazione
endoteliale: le cellule di Kupffer dei sinusoidi del fegato, le cellule che
rivestono i seni linfatici dei linfonodi (cellule litorali) e i seni venosi della
milza e del midollo osseo; 2) cellule reticolari nei tessuti linfatici; 3)
cellule sparse in tutti i connettivi (cellule migranti a riposo di Maximow)
e cellule situate nella tonaca avventizia dei vasi (cellule avventiziali di
Marchand); 4) macrofagi alveolari dei polmoni; 5) cellule della microglia
del sistema nervoso centrale (SNC); 6) macrofagi liberi delle cavità
sierose (macrofagi pleurici e peritoneali); 7) monociti del sangue
(precursori diretti dei macrofagi tissutali).
33
Normalmente i macrofagi dei tessuti non si moltiplicano, se non in certe
circostanze di sovrastimolazione, hanno una lunga durata di vita (100
giorni e più) e sono circa 30 volte più numerosi dei monociti del sangue.
Nell'essudato infiammatorio il numero dei macrofagi comincia ad
aumentare progressivamente quando si va esaurendo l'intervento dei
granulociti neutrofili.
Nell'essudato appena formato i macrofagi non si distinguono praticamente
dai monociti, ma dopo poco tempo cominciano a maturare, cioè cambiano
morfologia ed attitudini funzionali rispetto ai monociti: aumenta il
volume cellulare, il consumo di glucosio, la produzione di lattato,
l'attività fagocitaria, la formazione dei lisosomi, l'attività degli enzimi
idrolitici e la quantità di goccioline lipidiche. Monociti e macrofagi
ricavano energia dalla respirazione e dalla glicolisi.
Una caratteristica importante dei macrofagi è la loro capacità di fagocitare
corpi estranei. La captazione del materiale da fagocitare può avvenire in
due modi: a) attraverso la chemiotassi, con la migrazione dei fagociti
mediante movimenti ameboidi verso le particelle da fagocitare, come
avviene molto attivamente nei focolai infiammatori; b) attraverso il
contatto casuale dei macrofagi con particelle da fagocitare presenti nel
circolo sanguigno e linfatico. L'endocitosi del materiale da fagocitare
avviene attraverso una trasformazione della membrana esterna del
fagocita: vengono emesse delle propaggini digitiformi tentacolari
(pseudopodi), che prima circondano il materiale da fagocitare e poi si
fondono perifericamente formando vescicole (vacuoli citotici o fagosomi)
entro cui viene a trovarsi imprigionata la particella estranea. A questo
punto i granuli (lisosomi) convergono sul fagosoma in formazione, si
fondono con esso, e scaricano il loro contenuto enzimatico nel lume del
34
vacuolo attorno alla particella estranea. Questo processo porta alla
scomparsa dei granuli e si chiama degranulazione.
Le idrolasi acide derivanti dai lisosomi (proteasi, fosfatasi, nucleasi,
glucuronidasi, solfatasi e lipasi) degradano il materiale digeribile
all'interno dei vacuoli fagosomi; inoltre, questi enzimi intervengono nella
digestione di batteri già uccisi precedentemente da vari altri fattori
antimicrobici.
Durante la fagocitosi i macrofagi vanno incontro a un fortissimo aumento
dell'attività metabolica. Il consumo di ossigeno si raddoppia o si triplica,
aumenta la formazione di ossigeno in forma anionica (O2-) e di perossido
di idrogeno (H2O2).
Il significato funzionale dell’aumento dell’attività respiratoria risiede
nella reattività dei prodotti intermedi della riduzione dell'ossigeno che si
formano nei macrofagi, principalmente anione perossido (O2-) e perossido
d'idrogeno (H2O2). Altri prodotti reattivi che si possono formare sono il
radicale idrossilico libero (OH·) ed ossigeno singoletto. Il principale
effetto benefico è di fornire alla cellula un potente sistema microbicida,
che si va ad aggiungere a quello rappresentato dal versamento di enzimi
idrolitici e di altri fattori nel fagosoma. Oltre che per la fagocitosi,
l'esaltazione del metabolismo respiratorio è essenziale per l'attività
citotossica dei macrofagi nei riguardi di vari bersagli cellulari comprese le
cellule tumorali.
Nei fagociti, durante lo scoppio respiratorio, tutto il perossido d'idrogeno
prodotto viene degradato dentro la cellula ad opera di catalasi, perossidasi
e glutatione-perossidasi. Invece una certa quota dell'anione perossido
prodotto viene liberata al di fuori della cellula. Qui si ha di nuovo
formazione di H2O2 ad opera della superossido-dismutasi (SOD) ed
inoltre dalla reazione tra H2O2 e l'anione superossido si genera radicale
35
idrossilico (OH·). Ciò comporta un effetto tossico sul tessuto. Il danno al
tessuto viene poi esaltato dalla liberazione di componenti tossiche da
fagociti morti. L’eccesso di O2- generatosi nel corso dell'attivazione
metabolica è citotossico per lo stesso fagocita che lo produce e finisce per
causare la morte prematura della cellula.
In condizioni normali il radicale O2- viene ugualmente generato come
sottoprodotto tossico del metabolismo ossidativo, mentre può venire
completamente degradato dalla superossido-dismutasi (SOD). Nei
fagociti attivati è stata invece dimostrata una drastica diminuzione di
SOD, cosicché una buona parte di O2- sfugge alla degradazione
enzimatica e si diffonde nell'ambiente extracellulare.
Oltre alle funzioni di fagocitosi, i macrofagi hanno funzione secernente:
producono e secernono una varietà di sostanze biologicamente importanti.
Queste sostanze possono essere raggruppate in tre categorie: 1) enzimi
che agiscono su proteine extracellulari: collagenasi, elastasi, proteasi
lisosomiali, attivatori del plasminogeno; 2) sostanze implicate nei
processi difensivi: componenti del complemento, interferone, lisozima; 3)
fattori che regolano l'attività di altre cellule, citochine e chemochine.
Mediatori dell'infiammazione
Nell’infiammazione è coinvolto un gran numero di mediatori chimici. Nel
plasma sono presenti dei precursori che vengono attivati, in genere in
seguito a scissioni da parte di enzimi proteolitici, per acquisire le loro
proprietà biologiche, come il sistema del complemento. Esistono invece
dei mediatori prodotti dalle cellule, che vengono immagazzinati in genere
all'interno di granuli intracellulari, ed in risposta ad uno stimolo vengono
36
secreti. Le fonti cellulari più comuni sono le piastrine, i neutrofili, i
monociti/macrofagi e le mastcellule.
Tra i più importanti mediatori della risposta infiammatoria abbiamo
l'istamina, la serotonina, il sistema del complemento, la bradichinina, il
sistema della coagulazione, i metaboliti dell'acido arachidonico, il PAF, le
citochine, le chemochine e l'NO.
L'istamina è ampiamente distribuita nei tessuti e si trova
principalmente nelle mastcellule. Viene liberata in risposta a molti
stimoli: danni fisici, reazioni immunitarie, anafilotossine, citochine (IL-1
e IL-8). L'istamina provoca dilatazione delle arteriole ed aumenta la
permeabilità vasale delle venule, però provoca vasocotrizione delle grandi
arterie. Essa rappresenta il mediatore principale della fase immediata
dell'aumentata permeabilità vasale, che si verifica nell'infiammazione
acuta.
La serotonina è un altro mediatore vasoattivo con azioni simili a
quelle dell'istamina. È presente nelle piastrine e nelle cellule
enterocromoaffini.
Un certo numero di fenomeni della risposta infiammatoria è
mediato da tre fattori interconnessi e derivati dal plasma: il complemento,
le chinine e il sistema della coagulazione.
Il sistema del complemento consta di 20 componenti proteici
(insieme ai loro prodotti di scissione) presenti in altissime concentrazioni
nel plasma. Questo sistema agisce nelle reazioni immunitarie contro gli
agenti microbici, che culminano con la lisi dei microbi da parte del
complesso di attacco alla membrana (MAC). Durante il processo
infiammatorio vengono prodotti alcuni componenti del complemento che
provocano l'aumento della permeabilità vascolare, la chemiotassi e
37
l'opsonizzazione. I componenti del complemento presenti nel plasma in
forma inattiva vengono numerati da C1 a C9.
I fattori derivati dal complemento influiscono su molti fenomeni
dell'infiammazione acuta: 1) fenomeni vascolari; 2) adesione dei
leucociti, chemiotassi ed attivazione; 3) fagocitosi. Tra i fattori del
complemento, il C3 ed il C5 rappresentano i più importanti mediatori
dell'infiammazione. La loro importanza è anche aumentata dal fatto che
possono essere attivati da una quantità di enzimi proteolitici presenti
nell'essudato infiammatorio, fra i quali enzimi proteolitici liberati dai
neutrofili. Perciò l'effetto chemiotattico del complemento e gli effetti
attivanti il complemento dei neutrofili possono instaurare un ciclo di
migrazione dei neutrofili che si perpetua da solo.
La bradichinina è un potente fattore vasodilatatore che causa anche
contrazione della muscolatura liscia, essa viene rapidamente inattivata
dalla chininasi.
Il sistema della coagulazione è un altro gruppo di proteine
plasmatiche coinvolte nell'infiammazione, infatti durante la conversione
del fibrinogeno in fibrina si formano fibrinopeptidi, che causano aumento
della permeabilità vascolare ed hanno attività chemiotattica sui leucociti.
I sistemi del complemento, della coagulazione e delle chinine vanno
incontro a cross-attivazioni, che aumentano la potenza della risposta
infiammatoria.
Molto importante è anche il ruolo svolto dai vari derivati dell'acido
arachidonico. Tra questi vanno segnalati il trombossano A2 per l'azione
vasocostrittrice, i leucotrieni C4, D4 ed E4 per l'attività vasocostrittrice e
l'aumento della permeabilità vascolare, la PGI2, la PGE1, la PGE2 e la
PGD2 per la vasodilatazione ed il leucotriene B4 e l'HETE per la
chemiotassi.
38
Il PAF è coinvolto nella genesi di molti fenomeni caratteristici
dell'infiammazione, tra cui: la vasodilatazione e l'aumento della
permeabilità vascolare (è 1000 volte più potente dell'istamina), l'adesione
dei leucociti all'endotelio e la chemiotassi, agisce infine sulla
degranulazione [151].
Le citochine sono polipeptidi prodotti da molti tipi di cellule, ma
principalmente da linfociti e macrofagi attivati, che modulano le funzioni
di altri tipi di cellule. Le citochine più importanti come mediatori
dell'infiammazione sono IL-1, IL-6, IFNγ, TNFα, TNFβ e la famiglia
dell'IL-8.
IL-1 e TNFα hanno in comune molte proprietà biologiche e sono prodotti
da macrofagi attivati, TNFβ è prodotto dalle cellule T attivate [152].
La secrezione di questi fattori può essere stimolata da endotossine,
immunocomplessi, tossine, danni fisici e da una varietà di processi
infiammatori. Le citochine possono agire sulla stessa cellula da cui
vengono prodotte (effetto autocrino), su cellule nelle immediate vicinanze
(effetto paracrino) o per via sistemica (effetto endocrino). Le loro azioni
più importanti nell'infiammazione riguardano l'endotelio, i leucociti, i
fibroblasti e l'induzione delle reazioni sistemiche della fase acuta.
L’interleuchina-1 (IL-1) rappresenta uno dei principali effettori della
risposta infiammatoria nel macrofago. Ne sono note due isoforme, IL-1α,
che si trova per la maggior parte associata alla membrana cellulare, ed
IL-1β, che viene invece secreta. Prodotta da cellule delle linee mieloide e
linfoide, essa veicola un segnale immuno-stimolante e pro-infiammatorio
nei confronti di cellule T e B, dei monociti e dei macrofagi [153]. IL-1α e
IL-1β stimolano l'espressione di vari geni associati all'infiammazione e
alle malattie autoimmuni; tra i più importanti la cicloossigenasi 2
39
(COX-2), la fosfolipasi A2 e l'ossido nitrico sintetasi inducibile (iNOS).
IL-1 aumenta anche l'espressione di molecole di adesione come ICAM-1
sulle cellule mesenchimali e VCAM-1 sulle cellule endoteliali. Questa
proprietà promuove l'infiltrazione di cellule dell'infiammazione ed
immunocompetenti nello spazio extravasale [152].
L’interleuchina-6 (IL-6) è considerata una citochina pro-infiammatoria
attiva nella generazione e nella coordinazione della risposta immune. In
particolare, tra i suoi effetti vi sono l’attivazione delle cellule B che
vengono indotte a sintetizzare anticorpi, l’incremento della permeabilità
vascolare e l’induzione delle risposte di fase acuta, ovvero quella serie di
eventi a carico di organi metabolici (fegato) ed esecutivi (cellule
immunitarie) che vanno a supportare l’instaurarsi della difesa
immunitaria. A differenza di IL-1, però, IL-6 può anche veicolare risposte
anti-infiammatorie, inibendo la sintesi di TNFα ed inducendo la sintesi
dei recettori solubili per TNFα ed IL-1, i quali diminuiscono i livelli di
citochine disponibili per l’induzione della risposta infiammatoria [154].
Gli interferoni (IFN) possono essere suddivisi in due gruppi sulla base
delle loro caratteristiche strutturali e dei recettori a cui si legano. Gli IFN
di tipo II comprendono IFNα, IFNβ, IFNω e IFNτ, accomunati dalle
caratteristiche strutturali e recettoriali, mentre la molecola dell’IFNγ (IFN
di tipo I) si differenzia per struttura dalle altre e possiede un recettore
distinto. IFNγ riveste un ruolo di rilievo nella regolazione dell’attività
immunitaria, essendo in grado di indurre l’espressione di molecole di
adesione e del complesso maggiore di istocompatibilità (MHC) di classe
II, di stimolare la risposta umorale e cellulare e l’interazione dei linfociti
con l’endotelio vascolare [155].
40
Il tumor necrosis factor α (TNFα), spesso considerato il prototipo delle
citochine pro-infiammatorie, viene prodotto dai monociti/macrofagi, dalle
cellule dendritiche o dai linfociti ed esercita i suoi effetti su una
vastissima gamma di cellule sulle quali può intervenire per influenzarne
crescita e differenziamento.
TNFα provoca l'aggregazione e l'attivazione dei neutrofili, con aumentata
risposta di queste cellule ad altri mediatori, e la liberazione di enzimi
proteolitici da parte delle cellule mesenchimali, contribuendo al
danneggiamento dei tessuti [153].
IL-8 è secreta da macrofagi attivati ed è un potente chemiotattico ed
attivatore dei neutrofili, mentre ha poca attività sui monociti e sugli
eosinofili. La sua produzione viene indotta principalmente da altre
citochine, come IL-1 e TNFα.
Della stessa famiglia di IL-8 fanno parte MCP-1 (Monocyte
Chemoattractant Protein-1), che è un agente chemiotattico ed attivante per
i monociti, e RANTES (Regulated upon Activation Normal T cell
Expressed and Secreted), fattore chemiotattico per i timociti [156].
Le
chemochine
costituiscono
un
sofisticato
sistema
di
comunicazione usato da tutti i tipi cellulari, comprese le cellule
immunitarie. Le chemochine sono classificate in base alla loro
composizione aminoacidica, in particolare per la presenza di un motivo
conservato di quattro cisteine. La posizione relativa delle prime due
cisteine, separate da un aminoacido non conservato o contigue, permette
la divisione delle chemochine in due sottoclassi: CXC e CC. Inoltre tre
molecole omologhe sono classificate come chemochine: CXC3CL1, che
presenta tre aminoacidi tra le prime cisteine, e XCL1 e XCL2, che non
presentano due delle quattro canoniche cisteine delle chemochine.
41
A tutt'oggi sono conosciute 43 chemochine umane, tuttavia la presenza di
isoforme, polimorfismi e splicing alternativi aumenta notevolmente il
numero di chemochine che agiscono nell'uomo. Esse agiscono legandosi a
specifici recettori accoppiati a proteine G, nell'uomo ne sono stati
riconosciuti 19. Le chemochine influenzano molti aspetti della cellula non
solo la chemotassi e l'adesione, ma anche la proliferazione, la
maturazione, il differenziamento, l'apoptosi e la trasformazione maligna.
Le chemochine sono indispensabili per la risposta infiammatoria in
quanto coordinano la migrazione delle cellule della risposta immune che
avviene sia in seguito all'esposizione ad un agente infettivo sia nel
normale sviluppo del sistema immunitario [157].
L'ossido nitrico è un gas solubile che viene prodotto dall'enzima
NO-sintasi (NOS) che è espresso in tre isoforme denominate nNOS o
NOS-1, costitutivamente espressa a livello prevalentemente neuronale,
iNOS o NOS-2, inducibile, in particolare nelle cellule della serie
leucocitaria, ma anche in talune cellule endoteliali ed epiteliali, negli
epatociti e nei neuroni, ed eNOS o NOS-3, costitutivamente espressa
principalmente a livello delle cellule endoteliali. Bassi livelli di ciascuna
isoforma possono essere espressi anche in tipi cellulari diversi.
La sua emivita in vivo è molto breve, quindi la sua azione è limitata alle
cellule adiacenti al sito in cui viene prodotto.
L'effetto dell'NO deve essere probabilmente ricondotto ai prodotti della
sua interazione con le specie radicaliche dell’ossigeno. Ad esempio la
reazione con l’anione superossido O2-, prodotto dall’attività della
superossido dismutasi, genera il radicale perossinitrito ONOO- a sua volta
instabile, che, in presenza di CO2 come catalizzatore, libera, fra gli altri,
NO2 e CO3- probabilmente i veri responsabili di gran parte degli effetti
tossici inizialmente ascritti ad NO. Questi comprendono effetti di
42
alchilazione del DNA dovuta ad alterazione in senso genotossico di
molecole aggredite da specie radicaliche dell’azoto; oppure alterazione
dell’attività di proteine regolatorie che possono essere nitrate o nitrosate
in corrispondenza di residui importanti per la loro funzione, con un effetto
che può rendersi manifesto a diversi stadi lungo la loro via di segnale,
fino ad influenzare la stessa espressione genica.
Oltre che agire sulla muscolatura liscia vasale, inducendone la
dilatazione, l'NO ha anche altri ruoli nell'infiammazione, fra cui la
riduzione dell'aggregazione e dell'adesività piastriniche.
Inoltre l'NO prodotto dai macrofagi, agendo come radicale libero, è
citotossico sia per i microbi e le cellule tumorali, sia per le cellule sane,
quindi una sua iper-produzione può portare a danno tissutale.
L’induzione della trascrizione di iNOS può essere mediata da una serie di
citochine pro-infiammatorie come IL-1β, TNFα ed IFNγ, che la
modulano attraverso l’attivazione di fattori di trascrizione attivi su
elementi responsivi a STAT1 e ad NF-κB [158].
È stato dimostrato che NO up-regola mediatori pro-infiammatori, come
TNFα, IL-8 e la stessa iNOS. Questi effetti sono mediati dall'attivazione
di NF-κB [159].
43
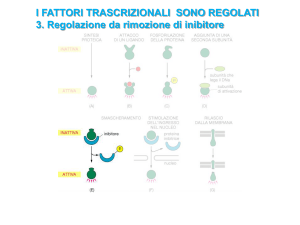
IL FATTORE NUCLEARE κB (NF-κ
κB)
NF-κB è un fattore di trascrizione sequenza-specifico ben conosciuto per
il suo coinvolgimento nell'infiammazione e nella risposta immunitaria
innata. Inoltre è sempre più accertato un suo coinvolgimento nello
sviluppo tumorale [160]. È stato descritto per la prima volta nel 1986
come un fattore nucleare necessario per la trascrizione della catena
leggera κ delle immunoglobuline nei linfociti B, e da qui il nome nuclear
factor-κB [161, 162].
NF-κB è un fattore di trascrizione dimero espresso in modo ubiquitario,
anche se il suo ruolo è più studiato nelle cellule del sistema immunitario.
Nelle cellule B e nelle plasma-cellule, NF-κB è localizzato nel nucleo,
dove lega una regione di dieci paia di basi dell'enhancer intronico kappa e
promuove la trascrizione [163]. Nelle altre cellule è mantenuto
citoplasmatico dal suo inibitore, IκB (Inhibitor of NF-κB) [164-166].
Classificazione dei membri di NF-κB
Le proteine che costituiscono NF-κB appartengono alla famiglia Rel.
Fanno parte della famiglia Rel proteine di Drosophila melanogaster
(Dorsal, Dif e Relish) e di mammifero (p65, RelB, c-Rel, p50, p52)
[167-169]. I vari membri di questa famiglia possono associarsi formando
complessi eterodimerici od omodimerici, eccetto RelB che forma solo
eterodimeri. Il dimero più frequentemente presente è costituito dalla
proteina p65, denominata anche RelA, e dalla proteina p50, chiamata
anche NF-κB1. Ci sono però anche altri dimeri trascrizionalmente attivi
come p50/c-Rel, p65/p65 e p65/c-Rel. Omodimeri di p50 o di p52
44
Struttura 3D dell'eterodimero p65/p50
45
agiscono invece come repressori [170-174]. L’attività costitutiva di
NF-κB nei linfociti B è principalmente attribuibile agli eterodimeri
p50/c-Rel [163, 175]. Tuttavia nella maggior parte delle cellule l’attività
di NF-κB è largamente inducibile e il dimero più diffuso è p50/p65.
Ciascun membro della famiglia Rel contiene all’N-terminale una regione
conservata di 300 aminoacidi, detta RHD (Rel Homology Domain). Il
dominio RHD contiene sequenze coinvolte nel legame al DNA e nella
dimerizzazione. Nell’estremità C-terminale del RHD è presente anche una
sequenza di localizzazione nucleare (Nuclear Localization Signal, NLS).
Il fattore di trascrizione NFAT (Nuclear Factor of Activated T cells) ha
anch’esso un dominio RHD ed è da alcuni considerato membro della
famiglia Rel [176].
Le proteine appartenenti alla famiglia Rel si possono suddividere in due
classi in base alla loro sequenza C-terminale. I membri della prima classe
sono p105/p50, p100/p52 e la proteina di Drosophila Relish. Queste
proteine sono caratterizzate da domini ripetuti di anchirina, presenti anche
nelle proteine della famiglia di IκB. p100, il precursore di p50, e p105, il
precursore di p52, sono esclusivamente citosoliche, perché i domini
ripetuti di anchirina mascherano le loro sequenze di localizzazione
nucleare [177]. Queste due proteine sono poi digerite nel dominio Nterminale a livello di regioni specifiche di 23 aminoacidi ricche di glicina
(Glicine Rich Region, GRR) così da generare p50 e p52. p50 e p52 non
hanno domini di transattivazione e quindi sono trascrizionalmente inattive
[163].
La seconda classe include p65, RelB, c-Rel, Dorsal e Dif. Queste proteine
contengono uno o più domini di transattivazione nella loro regione
C-terminale [178]. In particolare, il dominio di transattivazione di p65
contiene due regioni di transattivazione, una di 30 aminoacidi a livello
46
C-terminale, detta TA1, e una di 90 aminoacidi, adiacente al TA1,
chiamata TA2. Sia nel dominio TA1 che nel dominio TA2 sono presenti siti
di fosforilazione importanti per l’attivazione di p65. Questi domini,
quando legano le sequenze bersaglio, assumono una conformazione ad α
elica.
Attivazione di NF-κB
In assenza di uno stimolo infiammatorio, NF-κB è localizzato nel
citoplasma complessato con una proteina inibitoria chiamata IκB
(Inhibitor of NF-κB), che maschera la sua sequenza di localizzazione
nucleare (NLS). Quando uno stimolo extracellulare attiva la via di NF-κB
si attivano le IKK, chinasi specifiche per IκB, che fosforilano IκBα sulle
serine 32 e 36. Tale fosforilazione promuove la reazione di
poliubiquitinazione sulle lisine 21 e 22 e quindi la rapida degradazione di
IκBα da parte del proteasoma 26S. In assenza di IκB è dunque
smascherata la sequenza di localizzazione nucleare e NF-κB può migrare
nel nucleo dove attiva la trascrizione dei geni bersaglio. Fra i geni
responsivi a NF-κB troviamo anche IκBα. Quando IκBα è stato
risintetizzato, entra nel nucleo, perché ha, all’interno del dominio di
anchirina ripetuto (Ankyrin Repeat Domain, ARD), una sequenza di
localizzazione nucleare che permette il suo ingresso nel nucleo. Qui si
lega ad NF-κB bloccandone l’attività trascrizionale e complessata a NFκB torna nel citoplasma, grazie alla presenza di sequenze di esporto
nucleare (Nuclear Export Sequences, NES) ricche di leucine localizzate
nei
dominii
C-terminale
(aminoacidi
(aminoacidi 45-54) di IκBα [179].
47
265-277)
ed
N-terminale
I membri della famiglia di NF-κB
da Sankar Ghosh, Michael J. May and Elizabeth B. Kopp
Annu. Rev. Immunol. Vol.16 April 1998 modificata
48
Delezioni o mutazioni nelle sequenze NES determinano la localizzazione
nucleare dei complessi NF-κB/IκBα. In più la leptomicina B, inibitore
dell’esportina CRM1, determina un accumulo nel nucleo dei complessi
NF-κB/IκBα inattivi. Esiste quindi un meccanismo di feed-back negativo
che regola l’attività trascrizionale di NF-κB.
È stato quindi proposto un modello dinamico secondo il quale i complessi
NF-κB/IκBα migrano continuamente tra il nucleo e il citoplasma.
Tuttavia la velocità del trasporto dal nucleo al citoplasma è molto
maggiore di quella dal citoplasma al nucleo, in accordo con la prevalente
localizzazione citoplasmatica dei complessi NF-κB/IκBα [180-182]. La
trascrizione di IκBβ invece non è regolata da NF-κB, perciò in cellule in
cui IκBβ è preponderante, l’attivazione di NF-κB è persistente nel tempo.
Gli stimoli in grado di attivare NF-κB possono essere raggi UV, intermedi
reattivi dell’ossigeno, proteine virali, LPS, ma anche diverse citochine
come TNFα ed IL-1.
L'inibitore di NF-κB e le chinasi IKK
Le proteine della famiglia di NF-κB posseggono degli inibitori specifici:
le proteine IκB. Esistono diverse proteine IκB: IκBα , IκBβ, IκBγ, IκBε,
Bcl-3 nei vertebrati superiori e Cactus in Drosophila. Inoltre anche p105 e
p100 hanno regioni simili a IκB. Tutti questi inibitori contengono regioni
di omologia: i motivi ripetuti di anchirina (Ankyrin Repeat Motifs),
necessari per l’interazione proteina-proteina. Ciascun membro della
famiglia IκB differisce per il numero di queste sequenze che conferisce la
specificità per i diversi dimeri NF-κB.
49
Degradazione
nel proteasoma
L'attivazione di NF-κB
da Yumi Yamamoto and Richard B. Gaynor
TRENDS in Biochemical Sciences Vol.29 No.2 February 2004 modificata
50
Sono
anche
presenti
regioni
ricche
di
aminoacidi
coinvolti
nell’interazione con le sequenze di localizzazione nucleare e di legame al
DNA di NF-κB. È presente anche una sequenza C-terminale, detta PEST
(pro-glu/asp-ser-thr), coinvolta nella regolazione della degradazione di
IκB.
La proteina meglio caratterizzata della famiglia è IκBα, una proteina di
circa 37 kDa. Sia IκBα sia IκBβ regolano la maggior parte dei dimeri
p50/p65 e p50/c-Rel. IκBε lega esclusivamente i dimeri p65/p65 e
p65/c-Rel, perciò regola l’espressione di specifici geni come IL-8, il cui
promotore lega preferenzialmente i complessi p65 e c-Rel.
Come detto sopra la fosforilazione di IκB è effettuata dalle IκB chinasi
(IKK). IKK è un complesso proteico e ne fanno parte tre polipeptidi:
IKKα, IKKβ, le subunità catalitiche e IKKγ/NEMO, la subunità inibitoria
[183]. IKKα e IKKβ hanno una struttura molto simile. Entrambe
contengono un dominio protein chinasico all’N-terminale, un motivo a
“cerniera di leucina” (leucine zipper) e un motivo elica-ansa-elica al
C-terminale. IKKγ contiene un motivo a “cerniera di leucina” e il dominio
ad α elica, ma non la regione catalitica. I complessi di IKK nativi trovati
nei mammiferi sembrano essere costituiti da eterodimeri IKKα/IKKβ e da
un numero imprecisato di IKKγ. L’attivazione di IKK dipende dalla
fosforilazione di IKKβ.
Fosforilazione di NF-κB
L’attivazione di NF-κB coinvolge la fosforilazione di p65 da parte di
diverse chinasi. L’LPS, ad esempio, stimola la fosforilazione PKAdipendente della serina 276 nel RHD di p65 e il conseguente reclutamento
51
IL-1
LPS
TNF
MSK 1
PKAc
S 276
1
CKII
IKK
S 529 S 536
RHD
TA2
PKC ζ
TA1
PMA
CaMKiV
Le fosforilazioni di p65
da Linda Vermeulen et al.
Biochemical Pharmacology Vol.64 No.5-6 September 2002 modificata
52
551
di CREB binding protein (p300/CBP) [184]. Ciò stimola l’attività
trascrizionale di p65. Il TNFα induce invece la fosforilazione della Ser
529 nel dominio di transattivazione C-terminale. Questa fosforilazione
aumenta l’attività trascrizionale, ma non influenza la traslocazione
nucleare o la capacità di legare il DNA [185]. È stato poi dimostrato che
la fosforilazione della Ser 529 è effettuata dalla casein chinasi II (CKII).
Inoltre si è visto che l’associazione tra IκBα e p65 inibisce la
fosforilazione di p65 da parte della CKII e che, in seguito alla
degradazione di IκBα, CKII può fosforilare p65 e aumentarne l’attività
trascrizionale [186]. TNFα induce anche la fosforilazione, da parte del
complesso IKK, della Ser 536 nel dominio di transattivazione TA1 di p65
[187].
Recentemente si è scoperto che l’LPS induce anche la fosforilazione della
Ser 536 [188]. IKKβ gioca un ruolo essenziale in questa fosforilazione,
mentre IKKα è solo parzialmente coinvolta. Inoltre la fosforilazione di
p65 sulla Ser 536 aumenta la sua attività trascrizionale [189].
Il PMA (un estere del forbolo) induce la fosforilazione del dominio di
transattivazione TA2 di p65, tra gli aminoacidi 442 e 470 e questa
fosforilazione
aumenta
l’attività
trascrizionale.
Inoltre
una
sovraespressione della Ca2+/Calmodulina chinasi IV determina la
fosforilazione nella regione C-terminale. La PKCζ invece fosforila
l’RHD. Recentemente si è scoperto che p65 è fosforilato a livello della
Ser 276 non solo dalla PKA, ma anche dalla chinasi nucleare MSK-1 in
risposta al trattamento con il TNFα. MSK-1 è a sua volta attivata sia da
ERK che da p38, ed è stata inizialmente identificata come una CREB
chinasi molto potente. La fosforilazione e la conseguente attivazione
trascrizionale di p65 da parte di MSK-1, in risposta al trattamento con il
53
TNFα, è un processo rapido, che raggiunge un massimo 10-15 minuti
dopo l’induzione e ritorna a livelli basali dopo 30 minuti [190].
Il ruolo della fosfatidilinositolo 3-chinasi e di Akt nell’attivazione di NFκB è ancora oggetto di studio. Alcuni lavori hanno evidenziato una
potenziale attività antinfiammatoria della PI3K: l'attivazione della via
della fosfatidilinositolo 3-chinasi porta ad un'inibizione dell'espressione di
geni proinfiammatori [191-195]. Tuttavia altri gruppi hanno evidenziato
come l'uso di inibitori farmacologici della PI3K porti ad una diminuzione
dell'attività di NF-κB, quindi un ruolo proinfiammatorio della cascata
PI3K/Akt [196-199]. Sulla base delle evidenze finora pubblicate si
ipotizza quindi che l'effetto della PI3K su NF-κB dipenda dal tipo
cellulare preso in esame.
Acetilazione di NF-κB
L’acetilazione è un'altra modalità con cui NF-κB può essere regolato. Il
TNFα induce l’acetilazione di p65 impedendo così il legame tra p65 e
IκBα. Responsabile di questa acetilazione in vivo è p300/CBP (CREB
Binding Protein), che possiede attività acetil transferasica degli istoni
(HAT) ed è in grado di acetilare, oltre agli istoni, diversi fattori di
trascrizione. p300/CBP acetila le lisine 218, 221 e 310 di p65.
L’acetilazione della Lys 221 aumenta il legame al DNA e inibisce il
legame ad IκBα di p65. L’acetilazione della Lys 310 è invece necessaria
per la completa attività trascrizionale di p65, ma non ha effetti sul legame
al DNA o ad IκBα. p65 acetilata è successivamente deacetilata per
interazione con la deacetilasi degli istoni HDAC3. Questa deacetilazione
promuove il legame con IκBα e ha come conseguenza il trasporto del
54
complesso nel citoplasma [200, 201].
Un recente lavoro giunge però a conclusioni opposte. Gli autori
sostengono che p65 è acetilato da p300/CBP e PCAF sulle lisine 122 e
123. Inoltre affermano che l’acetilazione impedisce il legame al DNA di
p65 e ne favorisce l’esporto dal nucleo mediato da IκBα [202].
Geni regolati da NF-κB
I geni bersaglio di NF-κB contengono nel loro promotore la sequenza
consenso di legame al DNA di NF-κB ossia GGGRNNYYCC (R=purina,
Y=pirimidina, N=una base qualsiasi), ma sono ammesse variazioni che
conferiscono specificità per i diversi eterodimeri [203, 204].
I principali geni bersaglio di NF-κB sono i geni dell’infiammazione [205].
Possiamo
distinguere
tra
geni
precoci,
la
cui
espressione
è
immediatamente successiva all'ingresso di NF-κB nel nucleo, e geni
tardivi, che vengono espressi in seguito alla seconda fase dell'attivazione
di NF-κB. Tra i più importanti geni precoci abbiamo IκBα, MnSOD e
MIP-2. Tra i geni tardivi, la cui sintesi inizia dopo 90 e 120 minuti
dall'attivazione di NF-κB, vi sono numerose citochine come IL-1, IL-2,
IL-6, IL-8, GM-CSF, INFγ, TNFα ed alcune chemochine come RANTES
e MCP-1 [206]. È stimolata anche la produzione di proteine della fase
acuta.
Queste
includono
la
proteina
amiloide
del
siero
A,
l’angiotensinogeno, componenti del complemento.
Fanno parte dei geni bersaglio anche la COX-2 e iNOS, molto importanti
per le risposte infiammatorie, e proteine di adesione come VCAM-1 ed
ICAM-1. Citochine come IL-1 e TNFα oltre a essere sintetizzate in
risposta a NF-κB sono anche in grado di attivare NF-κB, con un
55
meccanismo di feed-back positivo.
NF-κB e l'infiammazione
L’attivazione di NF-κB è coinvolta nella patogenesi di malattie
infiammatorie croniche, come l'aterosclerosi, l’asma, l’artrite reumatoide
e malattie infiammatorie dell’intestino. In più un'alterata regolazione di
NF-κB è stata riscontrata in malattie come l’Alzheimer, in cui la risposta
infiammatoria è parzialmente coinvolta.
Molti geni proinfiammatori importanti per la patogenesi dell'aterosclerosi
sono regolati da NF-κB, che è presente in forma attivata nella placca
aterosclerotica. Uno dei primi eventi nell'aterogenesi è l'attivazione
dell'endotelio vascolare, che porta al reclutamento di monociti e linfociti
T. Una volta arrivati nella parete vasale, i monociti differenziano a
macrofagi e quindi, una volta fagocitati i lipidi, in cellule schiumose. In
seguito si ha una migrazione di cellule muscolari lisce dalla media
all'intima, con susseguente proliferazione e deposizione di matrice
extracellulare. Ciò porta alla formazione di una placca matura. Gli eventi
acuti si possono originare o da una stenosi del vaso o da una
complicazione trombotica. Alcuni gruppi hanno rilevato la traslocazione
nucleare di NF-κB nell'intima e nella media di lesioni aterosclerotiche, in
cellule muscolari lisce, in macrofagi, cellule endoteliali e linfociti T.
Mentre analisi di pareti vasali sane hanno dimostrato una localizzazione
citoplasmatica di NF-κB [207, 208]. Nel contesto dell'aterosclerosi vi
sono molti stimoli che possono attivare NF-κB, tra cui fattori locali,
come: ingiurie vascolari, LDL modificate, agenti infettivi e citochine
[209].
56
In diversi studi è stato messo in evidenza come l’attivazione dei geni delle
citochine da parte di NF-κB contribuisca alla patogenesi dell’asma, che è
caratterizzato da infiltrazione delle cellule infiammatorie e dalla
deregolazione di citochine e chemochine nel polmone [210].
Anche nell’artrite reumatoide è coinvolta l’attivazione della via di NF-κB.
Nel fluido sinoviale di pazienti colpiti da questa malattia si trovano
elevati livelli di TNFα e di altre citochine [211]. La produzione di TNFα
è indotta da NF-κB, ma TNFα a sua volta stimola l’attivazione di NF-κB,
con un meccanismo di feed-back positivo. Anticorpi contro TNFα o
recettori troncati del TNFα migliorano i sintomi dei pazienti affetti da
artrite reumatoide.
L’attivazione di NF-κB è stata riscontrata in biopsie di campioni
provenienti da pazienti con coliti ulcerative ed il morbo di Crohn [212].
Queste malattie infiammatorie intestinali sono caratterizzate dalla
produzione di citochine proinfiammatorie sia dai macrofagi che dai
linfociti. Il trattamento con antinfiammatori diminuisce i sintomi.
Anormalità nell’attivazione di NF-κB si trovano anche nella malattia di
Alzheimer (AD). Immunoreattività per NF-κB è stata riscontrata nelle
placche precoci, mentre nelle placche mature c’è una riduzione
dell’attività di NF-κB. L’attivazione di NF-κB può quindi essere
coinvolta nell’iniziazione delle placche neuritiche e nell’apoptosi
neuronale
durante
le
fasi
iniziali
dell’Alzheimer
[213].
L’immunoreattività per p65 è infatti aumentata in neuroni ed astrociti
nell’immediata vicinanza della placca amiloide in sezioni di cervelli di
pazienti
colpiti
dalla
malattia
di
Alzheimer.
Altri
studi
di
immunoistochimica mostrano che nel cervello di pazienti con l’AD
l’attività di NF-κB è accresciuta e che NF-κB è localizzato
57
costitutivamente nel nucleo [214]. Inoltre il peptide β amiloide (Aβ) può
attivare NF-κB in neuroni in coltura. In più è stato osservato che la
regione “enhancer” in 5’ del gene che codifica βAPP contiene siti di
legame NF-κB [215]. E’ possibile così che l’attivazione di NF-κB nei
neuroni porti all’aumento della produzione di APP.
Farmaci che agiscono su NF-κB
Alcuni farmaci, che sono usati per trattare patologie infiammatorie,
svolgono la loro azione inibendo l'attività di NF-κB. Vi sono tre possibili
meccanismi d'azione: 1) inibizione della fosforilazione e degradazione di
IκBα: è stato dimostrato che alcuni FANS come aspirina, sulfasalazina e
sulindac [216-218], la ciclopentenone prostaglandina 15dPGJ2 [219] e
alcuni composti naturali come il resveratrolo [220] inibiscono l'attività di
IKK, diminuendo la fosforilazione e la degradazione di IκBα; 2)
inibizione dell'attività di NF-κB sulla trascrizione genica: interazioni
dirette tra il recettore dei glucocorticoidi ed NF-κB [221, 222] e il
complesso basale della trascrizione [223] sono state proposte per spiegare
l'effetto inibitorio che i glucocorticoidi hanno sulla trascrizione dei geni
regolati da NF-κB. Anche gli agonisti dei PPAR α e γ inibiscono geni
attivati da NF-κB [224]; 3) induzione dell'espressione di IκBα: un altro
meccanismo d'azione proposto per i glucocorticoidi è l'induzione del gene
di IκBα [225, 226], che esporta NF-κB dal nucleo e lo mantiene inattivo
nel citoplasma [182].
58
NF-κB ed LPS
L’endotossina
batterica
lipopolisaccaride
(LPS)
è
il
principale
componente della membrana esterna della parete dei batteri Gramnegativi. La porzione polisaccaridica dell’LPS è costituita da un nucleo
polisaccaridico e dall’antigene-O. La porzione lipidica è rappresentata dal
lipide A, responsabile della tossicità dell’LPS.
L’LPS è trasportato nel plasma dalla proteina legante l’LPS (LPS Binding
Protein, LBP). È poi riconosciuto dal recettore CD14, espresso nelle
cellule della linea mieloide [227]. CD14 ha la funzione di legare l’LPS e
trasferirlo al complesso del recettore TLR-4 (Toll-Like Receptor-4) e
della proteina accessoria MD-2. I recettori della famiglia TLR sono
proteine transmembrana con un dominio citoplasmatico conservato detto
Toll, presente anche nel recettore dell'IL-1, in grado di trasferire il segnale
all’interno della cellula tramite differenti vie che portano all’attivazione di
fattori di trascrizione. NF-κB è uno dei fattori di trascrizione attivato
dall’LPS.
Il dominio Toll di TLR-4 interagisce con la proteina adattatrice MyD88.
Questa proteina contiene dei domini legati alla morte cellulare (Death
Domains, DD) tramite i quali interagisce con i DD della chinasi IRAK,
nota anche come SIIK (Serine/threonine Innate Immunity Kinase). IRAK
attiva TRAF6 (TNFα Receptor Associated Factor). TRAF6 tramite la
proteina ECSIT (Evolutionarily Conserved Signalling Intermediate in
Toll pathways) attiva MEKK1, che a sua volta attiva IKK mediante
fosforilazione. Inoltre TRAF6 tramite la proteina adattatrice TAB2 attiva
la chinasi TAK1 che fosforila IKK. IKK attivata fosforila IκBα,
provocandone la degradazione. L’LPS stimola anche l’attivazione di tutte
59
le vie delle MAP chinasi: ERK1/ ERK2, JNK e p38 che, a loro volta,
fosforilano e attivano fattori nucleari come Elk-1, Jun, Fos, ATF-1, ATF2 e CREB. Inoltre l'LPS attiva la via di segnale PI3K/Akt.
60
LPS
TLR4
α
IKKα
RAS
α
IkBα
PI3K
MAPKK
ERK1/2
κB
NF-κ
κB
NF-κ
Elk1 SRF
kB-site
SRE
Le vie di traduzione del segnale di LPS
61
ESTROGENI
ED
INFIAMMAZIONE
DEL
SISTEMA NERVOSO
Studi clinici e sperimentali indicano che l'E2 influenza l'attività del
sistema nervoso centrale mediante la modulazione dei processi cognitivi
della postura, del movimento fine, dell'umore e dell'affettività. Inoltre il
17β-estradiolo esercita un'azione protettiva contro la neurodegenerazione
e gli insulti al cervello, effetti che possono spiegare l'azione benefica
dell'ormone sulle capacità cognitive, sulla mobilità e sulla sfera affettiva.
Molte ipotesi sono state avanzate per spiegare il meccanismo dell'azione
neuroprotettiva dell'estrogeno: 1) l'attività trofica degli estrogeni. È stato
osservato che il 17β-estradiolo stimola la crescita dei neuriti, la
differenziazione, la formazione di sinapsi. L'ormone inoltre modula la
sintesi di fattori di crescita come NGF, BDNF, IGF-1, TGFβ ed i relativi
recettori. Durante la maturazione del sistema nervoso centrale l'attività
dell'E2 continua ed assicura che i neuroni mantengano le connessioni
sinaptiche indispensabili per il signaling e la sopravvivenza neurale; 2)
l'E2 può regolare positivamente la sintesi di proteine che proteggono il
neurone dall'apoptosi. Questa azione dell'ormone si esplica, molto
probabilmente,
attraverso
l'attivazione
della
sintesi
di
proteine
antiapoptotiche, come Bcl-2 e BclXL, e l'inibizione dell'espressione di
proteine proapoptotiche come BNIP2 [228]; 3) il 17β-estradiolo può
indurre proliferazione delle cellule staminali per rimpiazzare i neuroni che
sono degenerati; 4) l'ormone può influenzare la risposta infiammatoria
controllando la reattività della microglia e la funzionalità vascolare [92,
229].
62
Estrogeni e patologie a componente infiammatoria del sistema
nervoso
Effetti antinfiammatori degli estrogeni sono stati descritti in malattie
dell’uomo, modelli animali di malattie umane ed in sistemi cellulari.
Numerosi studi mostrano che gli estrogeni hanno un effetto protettivo
contro malattie con una componente infiammatoria ritardandone
l’insorgenza e/o attenuandone i sintomi. Tra le malattie a componente
infiammatoria su cui gli estrogeni hanno azione, ricordiamo: la sclerosi
multipla, la leucodistrofia a cellule globoidi, l’artrite reumatoide,
l’ischemia cerebrale e la malattia di Alzheimer.
La sclerosi multipla
La sclerosi multipla (SM) o sclerosi a placche è una malattia cronica
grave del sistema nervoso centrale, che colpisce prevalentemente soggetti
adulti. È caratterizzata dalla presenza di aree di demielinizzazione,
definite placche, e da infiltrazione di linfociti T e macrofagi a livello di
sistema nervoso centrale. Le placche possono essere ovunque nella
materia bianca del sistema nervoso centrale, ma più frequentemente sono
a livello di midollo spinale, cervelletto e nervi ottici. Gli effetti della
perdita della mielina sulle fibre nervose sono molto gravi: è impedita la
conduzione saltatoria e quindi l’impulso è trasmesso più lentamente lungo
l’assone o addirittura c’è un blocco della conduzione a livello del sito
della lesione [230].
Una specifica causa alla base di questa malattia non è stata ancora
identificata. Sulla base dei risultati delle ricerche scientifiche fino ad ora
condotte, si ritiene che siano coinvolti fattori genetici e ambientali. Gli
63
individui assumono il rischio relativo dell'ambiente in cui trascorrono i
primi quindici anni della loro vita [231]. L’incidenza di SM in chi ha un
parente di primo grado colpito dalla malattia è 20 volte maggiore che
nella popolazione generale. Inoltre, studi su gemelli monozigoti mostrano
che il tasso di concordanza è del 30%. Gemelli eterozigoti mostrano un
tasso di concordanza minore del 5% [232].
La SM sembra essere associata ad alcuni alleli di HLA (Human
Leukocyte Antigen): in particolare l’allele DR2 è associato ad un rischio
relativo di sviluppare la malattia 4 volte superiore nella popolazione
caucasica. Questi risultati suggeriscono che sia fattori genetici che
ambientali, probabilmente virali, sono importanti per lo sviluppo della
malattia. Solitamente l’insorgenza della SM è durante il periodo
riproduttivo. Circa il 70% dei pazienti manifesta i sintomi tra 21 e 40
anni. Solo in rare eccezioni la malattia si manifesta prima dei 10 e dopo i
60 anni.
Così come in altre malattie autoimmunitarie, le femmine sono colpite più
frequentemente dei maschi. Le donne hanno una probabilità di sviluppare
la malattia 2-3 volte maggiore degli uomini.
La sclerosi multipla è spesso caratterizzata da episodi di disfunzioni
neurologiche seguiti da periodi di stabilizzazione e di remissione parziale
o completa dei sintomi. Le ricadute seguono spesso un episodio
d’infezione virale alle vie aeree superiori o al tratto gastrointestinale. In
circa la metà dei casi di SM la malattia progredisce fino a diventare
cronica.
I sintomi clinici della malattia sono interamente attribuibili alla patologia
a livello del SNC. I sintomi più comuni includono insensibilità a livello
delle mani e degli arti inferiori, emiparestesie, visione doppia o a macchie
e altri disturbi visivi che possono culminare nella cecità, problemi
64
cerebellari (mancanza di coordinazione e perdita di equilibrio),
intolleranza al calore, disturbi motori (spasticità, paraplegia, paresi),
incontinenza urinaria, depressione, ansietà.
Non c’è ancora una cura per questa malattia, però ci sono farmaci per
trattarne i sintomi. Interferone β-1β (Betaseron) e interferone β-1α
(Avonex) sono usati con successo per ridurre la frequenza e la severità
delle ricadute. Anche i glucocorticoidi sono utilizzati comunemente nelle
fasi in cui la malattia si aggrava.
La SM è una malattia a base autoimmunitaria. I linfociti T per motivi
ancora sconosciuti vengono sensibilizzati contro la guaina mielinica che
riveste gli assoni dei neuroni del SNC. Una volta attivati i linfociti T
attraversano la barriera ematoencefalica ed entrano nel SNC. I linfociti
sono in grado di attraversare i capillari (diapedesi) in virtù di molecole di
adesione come l’integrina α4, CD4 e VLA-4. Attraversate le pareti dei
capillari, i linfociti T producono metalloproteasi, che permettono di
degradare il collagene di tipo IV della matrice extracellulare. Così i
linfociti T possono raggiungere la materia bianca del SNC. Si pensa che
siano i linfociti Th1 a giocare un ruolo critico nell’inizio e nell’espansione
dei danni al SNC. Questa sottoclasse di linfociti T helper è in grado di
produrre citochine e chemochine.
Alcuni studi hanno dimostrato un aumento di TNFα, MCP-1, MIP-1α,
MIP-1β e RANTES nel SNC di soggetti con sclerosi multipla. Queste
citochine e chemochine richiamano nel SNC cellule mononucleate:
linfociti, macrofagi e plasmacellule e attivano la microglia residente. La
risposta infiammatoria acuta di linfociti, plasmacellule e macrofagi
produce demielinizzazione, in quanto i linfociti T producono citochine
che stimolano i macrofagi e la microglia residente a fagocitare la mielina.
Inoltre gli oligodendrociti stessi, che sono le cellule del SNC che
65
producono la mielina, possono andare incontro ad apoptosi e la gravità
della demielinizzazione è correlata con la quantità di oligodendrociti
eliminati. Nelle fasi precoci della malattia quanti più oligodendrociti sono
preservati a livello della placca più la remielinizzazione rimane possibile.
Se c’è una perdita completa di oligodendrociti la possibilità di una
remielinizzazione diminuisce drasticamente.
Ci sono diverse osservazioni che supportano l’idea che fluttuazioni nei
livelli degli ormoni sessuali siano correlate a cambi nello status della
malattia [233]. Durante la gravidanza, un periodo durante il quale i livelli
di estrogeni sono molto elevati, i sintomi clinici della SM diminuiscono
fino a scomparire [234]. Al contrario, durante il post-partum, in cui i
livelli degli estrogeni sono bassi, i sintomi si aggravano molto [235].
Nelle donne affette da sclerosi multipla peggioramenti della malattia e dei
disturbi neurologici si possono avere nei giorni interessati dal ciclo
mestruale o che lo precedono immediatamente, quindi in momenti
caratterizzati da basse concentrazioni ematiche di estrogeno [236, 237].
Anche l’uso dei contraccettivi orali riduce la disabilità in donne affette da
SM, anche se non sembra diminuire il rischio di malattia [238]. Gli
estrogeni influenzano la produzione di citochine da parte di linfociti Th1
prelevati da donne con SM e messi in coltura [239]. Inoltre durante la
gravidanza diminuiscono i livelli di citochine Th1 proinfiammatorie e
aumentano quelli di citochine Th2 che hanno una funzione inibitoria.
L’Experimental Autoimmune Encephalomyelitis (EAE) detta anche
Experimental Allergic Encephalomyelitis è un modello animale della
sclerosi multipla. Le caratteristiche patogeniche della EAE sono molto
alla simili sclerosi multipla. La EAE è caratterizzata da placche di
demielinizzazione sparse in tutto il SNC che mostrano infiltrazione di
linfociti T, macrofagi e plasmacellule.
66
L’EAE può essere indotta, in ceppi di animali suscettibili, per iniezione di
omogenati di midollo spinale assieme ad adiuvante completo di Freund
(CFA). Anche l’iniezione di componenti purificati della guaina mielinica,
come la proteina basica della mielina (Myelin Basic Protein, MBP), la
proteina della mielina oligodendrogliale (Myelin OligodendroGlial
protein, MOG) e la proteina proteolipidica (ProteoLipid Protein, PLP)
assieme a CFA può indurre la malattia. Topi e ratti sono gli animali più
usati, ma EAE è stata indotta anche in porcellini d’India, conigli, scimmie
rhesus e macachi.
Anche i sintomi sono simili alla sclerosi multipla: le reazioni
infiammatorie a livello del SNC causano una paralisi progressiva che
colpisce prima la coda e le estremità posteriori e poi gli arti anteriori. Alla
fine c’è una completa paralisi ed eventualmente la morte.
E’ stato dimostrato che diversi tipi di cellule sono coinvolte nella
patogenesi della EAE [240]. La malattia è mediata da cellule Th1 CD4+
specifiche contro MBP. A conferma di questo fenomeno è l’esperimento
di induzione di malattia EAE in animali singenici mediante il
trasferimento di linfociti T specifici contro MBP derivati da animali
affetti da EAE. In seguito all’induzione dell’EAE i linfociti specifici
contro la mielina proliferano a livello della milza e dei linfonodi. In
particolare i linfociti T TNFα+ CD4+ sono coinvolti nell’iniziazione e
nella progressione del danno [203, 204, 241]. Sono infatti in grado di
produrre citochine infiammatorie e chemochine che reclutano altre cellule
infiammatorie del sangue.
L’infiltrazione dei linfociti T nel SNC avviene soprattutto nelle fasi
iniziali
della
malattia.
Macrofagi
sono
presenti
nell’infiltrato
infiammatorio di topi e ratti con EAE nel SNC [203, 204, 241], parte di
questi producono TNFα [204]. In topi non immunizzati solo il 20% della
67
microglia produce TNFα, mentre in topi che sviluppano l’EAE circa il
50% della microglia esprime il TNFα [204]. Le cellule dendritiche (DC)
sono cellule in grado di presentare l’antigene, coinvolte nell’induzione
dell’EAE grazie alla loro capacità di attivare i linfociti T specifici contro
MBP. Durante l’EAE le DC sono presenti nell’infiltrato infiammatorio
del SNC e anche a livello di milza e linfonodi [242].
Diversi studi dimostrano che il trattamento con E2 ritarda l’insorgenza
dell’EAE e riduce la gravità dei sintomi in animali immunizzati per
sviluppare l’EAE. Questi effetti si vedono in topi femmina BV8S2 Tg
[203], in topi femmina C57BL/6 wt [204] o KO per citochine Th2 [243] e
in ratti di Lewis [241]. Inoltre il trattamento con E2 riduce l’infiltrazione
di macrofagi e linfociti T nel SNC dell’80% [203, 204] e la percentuale di
linfociti T CD4+ e macrofagi che esprimono TNFα. Inoltre E2 diminuisce
la percentuale di linfociti T CD4+ TNFα+ a livello della milza: E2 quindi
sopprime la generazione sistemica di linfociti T CD4+ [204]. L’effetto
inibitorio sulle cellule TFNα+ della microglia è minore. Il trattamento con
E2 riduce l’infiltrazione di cellule dendritiche nel SNC e la percentuale di
DC a livello della milza e dei linfonodi. E2 riduce la produzione di TNFα
e INFγ ed inibisce la capacità di presentare l’antigene e di attivare
specifici linfociti T contro la mielina delle cellule dendritiche in coltura
[242]. Il trattamento con E2 riduce l’espressione e la produzione di
citochine Th1, chemochine e loro recettori. Questo effetto è stato visto sia
con il metodo dei “ microarray” sia con il metodo “RNAse protection
assay” (RPA). Un recente lavoro mette in evidenza che gli effetti benefici
dell'E2 nell'EAE sono mediati da ERα e non da ERβ [244].
68
La leucodistrofia a cellule globoidi
La leucodistrofia a cellule globoidi (GLD), denominata anche malattia di
Krabbe, è una malattia autosomica recessiva metabolica, che coinvolge la
materia bianca del sistema nervoso centrale e periferico. La causa è un
deficit dell’enzima lisosomiale galattosil-ceramidasi (GALC), dovuto a
una mutazione del gene della galattosil-ceramidasi, localizzato sul
cromosoma 14 nell’uomo. Tale deficit enzimatico porta all'accumulo
dello sfingolipide ceramide galattoside nella sostanza bianca cerebrale,
causando una demielinizzazione. L’insorgenza è prevalentemente nei
primi mesi di vita, colpisce sia i maschi che le femmine con un incidenza
di uno su 40000 neonati.
L’iniziale manifestazione istologica della malattia è la presenza di
materiale positivo alla colorazione con acido periodico di Schiff, sia a
livello extracellulare che all’interno delle cellule microgliali. Queste
cellule accumulano al loro interno i substrati dell’enzima galattosilceramidasi: la galattosil-ceramide e la galattosil-sfingosina (psicosina),
altamente tossica. Così come altre encefalopatie, anche la GLD è
caratterizzata da una demielinizzazione, che coinvolge inizialmente il
sistema nervoso centrale e poi quello periferico. I sintomi sono tremori
degli arti superiori e inferiori, disturbi dell’andatura ed infine paresi
[245].
I modelli animali della GLD sono il topo “twitcher” e il topo
saposina A-/-. Nel 1980 è stata descritta una leucodistrofia autosomica
recessiva del topo “twitcher”, ossia che si muove a scatti, molto simile dal
punto di vista istopatologico e dei sintomi alla malattia umana.
Successivamente è stato dimostrato che il topo “twitcher” è un modello
della patologia umana anche dal punto di vista enzimatico.
69
Il topo saposina A-/- è invece utilizzato come modello per le forme
croniche a insorgenza tardiva della GLD. Questo modello animale è stato
generato introducendo una mutazione nel dominio della saposina A, un
attivatore della glucosil-ceramidasi e della galattosil-ceramidasi. La
mutazione è stata inserita nella prosaposina, una glicoproteina acida che è
precursore di quattro saposine denominate A, B, C e D. Il topo saposina
A-/- sviluppa una lenta e progressiva paralisi alle zampe. Le caratteristiche
biochimiche e patologiche sono qualitativamente identiche, ma più lievi
del topo “twitcher” [246].
È stato osservato che le femmine saposina A-/- che avevano portato a
termine delle gravidanze vivevano più a lungo e mostravano un’evidente
diminuzione dei sintomi neurologici rispetto alle femmine saposina A-/che non avevano avuto gravidanze o rispetto ai maschi. Inoltre, i classici
segni della patologia come l’infiltrazione delle cellule globoidi e la
demielinizzazione erano quasi scomparsi [247].
Nei topi saposina A-/-, così come anche nei topi “twitcher” è stato
riscontrato un aumento dell’espressione di diverse citochine come
MCP-1, TNFα e ciò suggerisce il coinvolgimento di un’infiammazione
secondaria nella patogenesi della GLD. I livelli di queste due citochine
erano molto diminuiti durante la gravidanza.
Per confermare che gli estrogeni fossero responsabili dell’effetto
protettivo della gravidanza, sono stati impiantati nei topi saposina A-/- dei
pellet in grado di rilasciare E2, così da mantenere gli elevati livelli di E2
tipici della gravidanza. I topi trattati con E2 mostravano anch’essi
preservazione della mielina e una netta riduzione dell’infiltrazione delle
cellule globoidi. Questi risultati suggeriscono che la somministrazione di
estrogeni potrebbe essere un’utile terapia supplementare per alcune
leucodistrofie croniche.
70
L’artrite reumatoide
L’artrite reumatoide (AR) è un disordine infiammatorio autoimmunitario
sistemico cronico, che può interessare molteplici organi e tessuti; cute,
vasi sanguigni, cuore, polmoni e muscoli; anche se il bersaglio principale
è costituito dalle articolazioni, dove determina una sinovite proliferativa
non suppurativa, che spesso progredisce fino alla distruzione della
cartilagine articolare ed all'anchilosi.
Le cause dell’AR sono ancora ignote, ma si ritiene che siano coinvolti
fattori genetici ed ambientali. La maggior parte dei soggetti che
sviluppano la malattia (dal 65 all’85%) esprime gli alleli dell’antigene di
istocompatibilità umano (Human Leukocyte Antigen, HLA) DR-4, DR-1
o entrambi. Inoltre è stato riscontrato un alto tasso di concordanza tra i
gemelli monozigoti.
Si pensa che l’iniziatore della malattia sia un agente microbico, come ad
esempio il virus di Epstein-Barr, ma non esistono prove sicure. Circa l'1%
della
popolazione
mondiale
è
affetta
dall'AR.
L’insorgenza
è
prevalentemente tra i 20 e i 40 anni e l’incidenza è da tre a cinque volte
maggiore nelle donne che negli uomini.
Gli autoantigeni coinvolti non sono stati identificati con certezza; tuttavia
autoimmunità contro il collagene di tipo II è stata riscontrata nella
maggior parte dei pazienti affetti da AR. Esistono anche prove che la
glicoproteina 39 della cartilagine sia un autoantigene. Sono coinvolti
principalmente linfociti T CD4+ presenti nelle articolazioni negli stadi
iniziali della malattia. Le cellule CD4+ attivate producono citochine e
attivano a loro volta i linfociti B a produrre anticorpi. Nella maggior parte
dei soggetti affetti dalla malattia sono presenti autoanticorpi, denominati
fattore reumatoide, contro la porzione Fc delle IgG e delle IgM. Si
71
formano così immunocomplessi che si depositano nelle articolazioni
[151]. Nel liquido sinoviale s’infiltrano anche macrofagi che producono
citochine come IL-1 e TNFα. Queste due citochine stimolano la sintesi di
molecole di adesione nei capillari sinoviali, determinando così un
ulteriore accumulo di leucociti nel liquido sinoviale. IL-1 e TNFα
stimolano i condrociti a produrre enzimi degradativi come collagenasi,
stromielinasi ed elastasi determinando così la distruzione della cartilagine
[248]. Si è visto inoltre che i fibroblasti sinoviali e i linfociti T attivati
producono RANKL, stimolando proliferazione e attività degli osteoclasti
determinando così osteoporosi [249].
Numerosi studi clinici mostrano che la AR è sensibile alle fluttuazioni dei
livelli ormonali. Durante la gravidanza, caratterizzata da livelli di
estrogeni molto elevati, i sintomi clinici dell’artrite reumatoide, così come
quelli della sclerosi multipla, diminuiscono fino a scomparire. Questo
fenomeno avviene soprattutto nel terzo trimestre della gravidanza. Al
contrario, durante il post-partum, in cui i livelli degli estrogeni sono bassi,
i sintomi si aggravano molto [250, 251].
Il ruolo degli estrogeni è stato studiato anche in modelli animali.
Sono stati utilizzati topi con artrite indotta da collagene. In questi animali
l’ovariectomia aggrava l’andamento della malattia [252, 253]. Il
trattamento a lungo termine con estrogeni invece diminuisce la severità
dell’artrite e diminuisce la risposta immunitaria contro il collagene di tipo
II. Latham e collaboratori hanno visto che il 17β-estradiolo inibisce lo
sviluppo dell'artrite indotta da collagene, diminuendo la produzione di
IFNγ da parte dei linfociti T ed i livelli di IL-10 e GM-CSF prodotti dalla
cellule dei linfonodi [254].
72
L’ischemia cerebrale
Con il termine ischemia s’intende una locale diminuzione dell’apporto
sanguigno, dovuta ad ostruzione del flusso sanguigno arterioso o a
vasocostrizione [151, 255]. L’ischemia cerebrale può essere dovuta ad
una diminuzione generalizzata del flusso sanguigno secondaria ad eventi
cerebrovascolari, come uno shock o un arresto cardiaco, o ad
un’occlusione delle arterie della circolazione cerebrale. Nel caso di una
diminuzione generalizzata del flusso sanguigno cerebrale, l’ischemia
risultante è globale, mentre un’ostruzione vascolare causa un’ischemia
regionale e spesso un infarto localizzato.
L’infarto cerebrale è la forma più comune di malattie cerebrovascolari e
rende conto del 70-80% di tutti gli eventi cerebrovascolari o ictus. Causa
principale è l’aterosclerosi, che predispone a trombosi vascolare ed a
eventi embolici, risultanti entrambi in un’ischemia localizzata e nel
conseguente infarto cerebrale. Fattori di rischio per l’infarto cerebrale
sono l’ipertensione, il diabete mellito e il fumo che predispongono gli
individui all’aterosclerosi.
Gli infarti si verificano soprattutto nelle aree irrorate dall’arteria cerebrale
media (MCA). Le occlusioni della MCA sono causate da emboli. Infarti
in questa regione sono caratterizzati da emiparesi controlaterale, perdita
della sensibilità nel lato del corpo opposto all’infarto e nel caso che
l’infarto coinvolga l’emisfero cerebrale dominante, l’afasia. Meno
comune è l’occlusione dell’arteria carotide interna, di solito causata da
trombosi. Rami del sistema vertebro-basilare sono spesso colpiti da
aterosclerosi e sono perciò potenziali siti di trombosi e fonti di emboli
ateromatosi.
La risposta infiammatoria gioca un importante ruolo nell’ischemia
73
cerebrale. Studi sull’uomo mostrano che i livelli plasmatici delle
citochine TNFα e IL-6 sono più elevati in pazienti colpiti da ictus che in
soggetti sani controllo [256]. È stato anche dimostrato un aumento
dell’espressione della proteina di adesione ICAM-1 a livello dei vasi
cerebrali. Inoltre un aumento del numero di leucociti, in particolare
monociti/macrofagi e leucociti polimorfonucleati, è stato riscontrato nel
fluido cerebrospinale di pazienti in seguito a ictus.
L’incidenza globale dell’ischemia cerebrale è maggiore nell’uomo che
nella donna in tutto il mondo e aumenta con l’età in entrambi i sessi.
L’ischemia cerebrale è rara nelle donne durante il periodo riproduttivo. In
seguito alla menopausa le differenze d’incidenza tra maschi e femmine si
attenuano notevolmente [257]. Inoltre le donne fino ai 65 anni mostrano
un numero di lesioni aterosclerotiche inferiori agli uomini. Dopo i 65 anni
la frequenza delle lesioni è simile in donne e uomini.
Secondo studi epidemiologici il rischio di ischemia cerebrale e più in
generale di malattie cardiovascolari, è ridotto in donne che sono state
sottoposte alla terapia sostitutiva in seguito alla menopausa. Altri studi
suggerivano che la terapia sostitutiva riducesse la mortalità dovuta
all’ischemia cerebrale [258]. Tuttavia lo studio randomizzato e controllato
HERS ha mostrato un aumento del rischio di eventi cardiovascolari
secondari nel gruppo che aveva seguito la terapia sostitutiva estroprogestinica (HRT) rispetto al gruppo placebo [259]. Inoltre lo studio
WHI ha mostrato che l’HRT aumentava il rischio di ischemia cerebrale
rispetto al placebo [260]. Di conseguenza l'HRT non viene consigliata per
prevenire eventi cardiovascolari cerebrali.
Studi su modelli animali mostrano il ruolo dei processi
infiammatori nell’ischemia cerebrale [256]. In ratti in cui era stata indotta
l’ischemia occludendo l’arteria cerebrale media (MCA), è stato
74
dimostrato un aumento dell’espressione delle citochine IL-1, IL-6, TNFα
e IL-8 e delle chemochine MCP-1 e MIP-1. In topi knock-out per ICAM1, in cui era stata occlusa la MCA, è stato osservato una diminuzione
della dimensione dell’area infartuata.
Gli studi effettuati in modelli animali indicano che il trattamento con
estrogeni protegge il cervello da ischemie globali e focali indotte
sperimentalmente [261-264]. Questi risultati sono stati ottenuti in
differenti specie e con diversi modelli animali. Alcuni studi mostrano che
uno dei meccanismi con cui gli estrogeni esercitano un effetto protettivo
sull’aterosclerosi dipende dalla loro azione sull’endotelio. L’adesione dei
monociti alle cellule endoteliali e l’attraversamento dell’endotelio sono
componenti essenziali della risposta infiammatoria, che accompagna il
processo aterogeno. Uno studio effettuato su conigli nutriti con una dieta
ricca di colesterolo ha mostrato che nei conigli maschi l’adesione
leucocitaria e la migrazione attraverso l’endotelio dei leucociti erano
maggiori che nelle femmine [265]. Inoltre femmine ovariectomizzate
avevano un numero maggiore di leucociti aderenti o sottoendoteliali
rispetto a femmine ovariectomizzate e trattate con E2. Il 17β-estradiolo
inibiva l’adesione leucocitaria diminuendo l’espressione della proteina
VCAM-1.
Il 17β-estradiolo riduce inoltre l’adesione dei leucociti nella circolazione
cerebrale di femmine di ratto sottoposte a ischemia transiente (indotta
tramite occlusione dell’arteria carotide comune destra per 30 minuti) e
riperfusione [266]. E’ stata comparata l’adesione dei leucociti in femmine
di ratto ovariectomizzate, ovariectomizzate trattate con E2 e non
ovariectomizzate. L’adesione dei leucociti è stata misurata prima
dell’ischemia e a differenti tempi dopo la riperfusione. Prima
dell’ischemia l’adesione leucocitaria era molto maggiore nelle femmine
75
ovariectomizzate
rispetto
a
quelle
non
ovariectomizzate
o
ovariectomizzate e trattate con E2. Anche dopo 4 o 6 ore di riperfusione le
ratte ovariectomizzate mostravano percentuali di leucociti aderenti
significativamente maggiori rispetto alle ratte non ovariectomizzate od
ovariectomizzate e trattate con E2.
La malattia di Alzheimer
La malattia di Alzheimer (AD) è la più comune forma di demenza ed è
responsabile di circa il 50-70% dei casi di demenza. L’insorgenza
dell’AD avviene generalmente in tarda età, ma esistono casi ad
insorgenza precoce collegati a mutazioni del gene del precursore della
proteina amiloide (Amyloid Precursor Protein, APP), della presenilina 1
(PS1) e della presenilina 2 (PS2).
Questa malattia è caratterizzata clinicamente da una progressiva ed
inesorabile alterazione della memoria e delle funzioni cognitive, e
patologicamente dalla presenza di un gran numero di placche neuritiche e
di matasse neurofibrillari. Le placche neuritiche sono grosse lesioni
costituite da depositi di un peptide di 40-42/43 aminoacidi chiamato
peptide β amiloide (Aβ), derivante dall’APP. Le matasse neurofibrillari
sono lesioni intracellulari costituite da filamenti intrecciati della proteina
tau del citoscheletro.
L’APP è una proteina con un singolo dominio transmembrana,
metabolizzata tramite due diverse vie in tutte le cellule. In un caso l’APP
è tagliato nel dominio Aβ, da parte di un enzima denominato α secretasi.
La seconda via comporta un taglio tra gli aminoacidi 671 e 672 dell’APP,
da parte di un enzima denominato β secretasi. Questo frammento è
ulteriormente tagliato dalla γ secretasi. A seconda della posizione del
76
taglio è generato un Aβ di 40 o di 42/43 aminoacidi. Aβ1-42(43) si aggrega
facilmente e forma i depositi di β amiloide [267].
Studi
d’immunoistochimica
post-mortem
hanno
rivelato
che
nell’Alzheimer è presente uno stato d’infiammazione cronica limitato alle
aree del cervello lesionate. Microglia attivata circonda i depositi
extracellulari insolubili delle placche e produce numerosi mediatori
dell’infiammazione. Citochine come IL-1, IL-6 e TNFα e i recettori delle
chemochine CC-R3 e CC-R5, prodotti dalla microglia attivata associata
alle placche, aumentano nell’AD. La microglia attivata secerne anche
proteasi come ad esempio la metalloproteasi 9 (MMP-9). Inoltre l’Aβ
agisce come attivatore del complemento.
L’infiammazione cronica quindi contribuisce ulteriormente al danno
neuronale e alla progressione dell'Alzheimer. Molti studi epidemiologici
ed alcuni studi clinici mostrano infatti che i farmaci antinfiammatori non
steroidei riducono il rischio e rallentano la progressione della malattia
[268].
Diversi studi mostrano come gli estrogeni hanno un effetto protettivo
contro l’AD. Esistono forti prove che la terapia sostitutiva a base di
estrogeni (ERT) riduce il rischio, ritarda l’insorgenza e attenua i sintomi
della malattia di Alzheimer.
Studi effettuati su modelli animali confermano gli effetti protettivi
degli estrogeni. Femmine di porcellino d’India ovariectomizzate
accumulavano maggiori quantità di peptide β-amiloide. Questa situazione
era revertita dalla somministrazione di 17β-estradiolo.
La privazione di estrogeni inoltre aumenta la formazione delle placche in
topi transgenici utilizzati come modello per l’Alzheimer. Le femmine
sviluppano placche amiloidi a circa un anno d’età. Giovani femmine
77
ovariectomizzate mostrano livelli più elevati di Aβ solubile e accumulato
nella placca rispetto alle femmine non ovariectomizzate. Inoltre il
trattamento con E2 reverte questi effetti [269].
Estrogeni ed infiammazione cerebrale sperimentale
È possibile indurre sperimentalmente l'infiammazione nel sistema
nervoso mediante iniezioni di sostanze infiammatorie nel cervello o nel
liquido cefalo-rachidiano.
Uno studio effettuato nel nostro laboratorio ha utilizzato un modello di
infiammazione cerebrale mediante l'iniezione di LPS nel terzo ventricolo
cerebrale ed ha dimostrato che l’E2 ha azione antinfiammatoria nel
cervello [270]. L'iniezione, nel terzo ventricolo cerebrale, di LPS
determina l’attivazione della microglia e il richiamo di monociti dal
sangue verso la zona lesa. La somministrazione dell’E2, a concentrazioni
fisiologiche, prima dell’LPS inibisce fortemente l’attivazione dei
macrofagi mediata dall’LPS in diverse aree del cervello: la regione CA3 e
il giro dentato dell’ippocampo, la corteccia parietale, la corteccia
cingolata, i nuclei amigdaloidi, la corteccia rinale e i nuclei talamici
postero-laterali. L’effetto dell’E2 è dose dipendente e tempo dipendente.
Il 17β-estradiolo esercita il suo effetto antinfiammatorio limitando
l’espressione di mediatori dell’infiammazione come la metalloproteasi 9
(MMP-9) e il recettore della proteina C3 del complemento (C3R).
Utilizzando topi knock-out per ERα e topi knock-out per ERβ si è
dimostrato che l’effetto antinfiammatorio dell’E2 è mediato da ERα. In
topi KO per ERβ infatti l’E2 inibisce l’attivazione della microglia così
come nei topi wild type, mentre nei topi KO per ERα l’E2 non è in grado
78
d’inibire l’attivazione della microglia. Inoltre topi KO per ERα mostrano
in alcune aree del cervello un’attivazione spontanea della microglia che
aumenta con l’età.
Estrogeni ed infiammazione del sistema nervoso in modelli
cellulari
L’azione antinfiammatoria degli estrogeni è stata studiata nelle cellule
della microglia, i macrofagi residenti del SNC [271]. La microglia “a
riposo” (resting) o la microglia a basso grado di attivazione “allertata”
(alerted) contribuiscono alla funzione e alla sopravvivenza neuronale
producendo fattori neurotrofici. Una volta attivata da stimoli infiammatori
la microglia è in grado non solo di attrarre i leucociti grazie alla sintesi di
chemochine, ma anche di produrre un gran numero di citochine, e altri
mediatori dell’infiammazione, come NO, intermedi reattivi dell’ossigeno,
prostaglandine e metalloproteasi della matrice. In una fase iniziale
l’attivazione della microglia è protettiva per il SNC. Tuttavia
un’attivazione eccessiva o prolungata può contribuire a neuropatologie
acute e croniche. Infatti le molecole infiammatorie secrete dalla microglia
contribuiscono al danno tissutale. La microglia attivata produce una serie
di mediatori dell'infiammazione tra cui citochine, chemochine, proteasi,
specie radicaliche dell'ossigeno e prostanoidi.
Per
quanto
riguarda
le
evidenze
sperimentali
circa
il
ruolo
antinfiammatorio dell'E2 sulla microglia, sono stati utilizzati i seguenti
modelli sperimentali: nel nostro laboratorio è stato utilizzato il
lipopolisaccaride che induce una cambiamento morfologico della
microglia, che da resting diventa di tipo ameboide. Inoltre, l’LPS induce
la produzione di citochine, chemochine, NO, prostaglandina E2 (PGE2) e
79
metalloproteasi-9 (MMP-9).
Il pretrattamento con 17β-estradiolo di colture primarie di microglia di
ratto previene l’attivazione morfologica indotta dall’LPS [272, 273].
Inoltre il pretrattamento con E2 riduce la percentuale di cellule iNOS
positive e quindi la produzione di NO. E2 previene anche la produzione di
PGE2 e di MMP-9 indotta da LPS. Questi effetti sono dose dipendenti e
avvengono a concentrazioni fisiologiche di E2 e sono bloccati dall’ICI
182,780. Le cellule di microglia in coltura esprimono sia ERα che ERβ e
questi effetti sono perciò mediati dal recettore degli estrogeni [273, 274].
Un altro modello sperimentale prevede l'impiego della proteina
regolatoria Tat del virus HIV, che è in grado di attivare cellule di
microglia in coltura, aumentando sia l’attività fagocitica, che il rilascio
del superossido e del TNFα tramite le MAPK p42 e p44 [275]. Il
pretrattamento con 17β-estradiolo in concentrazione fisiologica riduce sia
la fagocitosi che il rilascio di TNFα e di superossido indotti da Tat,
interferendo con la fosforilazione e quindi l’attivazione delle MAPK.
Estrogeni ed NF-κB
Numerosi studi indicano un'attività antinfiammatoria dell'estrogeno [270,
272, 273, 275, 276], ma non si ha ancora la certezza sul meccanismo
molecolare di questa azione. Esistono evidenze sperimentali di
un’interazione fra i recettori degli estrogeni e NF-κB. Gli studi fino ad ora
effettuati dimostrano che i recettori degli estrogeni impediscono il legame
di NF-κB ai promotori delle citochine IL-6, MCP-1 e TNFα.
Studi effettuati in cellule Saos2, una linea cellulare derivata da
osteosarcoma umano, e in cellule MCF-7, derivate da tumore al seno
80
umano, hanno mostrato che il 17β-estradiolo contrasta l’induzione della
sintesi dell’IL-6 da parte del TNFα [73]. L’effetto del TNFα sulla
trascrizione dell’IL-6 è mediato da NF-κB, che riconosce specifici siti di
legame sul promotore dell’IL-6. Per comprendere il meccanismo
molecolare d’azione dell’E2 è stato effettuato un EMSA (Electrophoretic
Mobility Shift Assays) su estratti nucleari di HeLa, MCF-7 e Saos2.
Come sonda è stata utilizzata la regione -80/-60, contenente siti di legame
per NF-κB del promotore dell’IL-6, in presenza o in assenza di oligo
competitori. Effettuando l’EMSA, aggiungendo hER tradotti in vitro, si è
visto che ER inibisce il legame al promotore dell’IL-6, soprattutto di cRel e in maniera minore di RelA (p65).
Il 17β-estradiolo diminuisce anche la produzione, indotta da IL-1, della
chemochina MCP-1 (Monocyte Chemoattractant Protein-1), in cellule
MCF-7 [77]. L’effetto è dose dipendente, in un intervallo di
concentrazione di E2 da 10-12 a 10-9 M. Un’azione inibitoria analoga, ma
con potenza inferiore è svolta dagli xenoestrogeni (XE), un gruppo di
composti chimici che legano il recettore degli estrogeni e mimano
l’azione dell’E2. Fanno parte degli xenoestrogeni composti molto lipofili
come il bisfenolo A (BPA) e il nonilfenolo (NP). Nel promotore di
MCP-1 sono presenti due siti di legame di NF-κB: A1 e A2. Mutazioni in
queste regioni determinano la perdita della responsività a IL-1. Tramite
EMSA si è dimostrato che l’aumento di MCP-1 indotto da IL-1 è dovuto
al legame di NF-κB al promotore di MCP-1 e che le subunità di NF-κB
coinvolte sono p50/p65 e p50/c-Rel. Sempre tramite EMSA si è
dimostrato che sia l’E2 che gli xenoestrogeni inibiscono il legame di
p50/p65 (e in modo minore di p50/c-Rel) ai siti di legame per NF-κB sul
promotore di MCP-1, in maniera dose dipendente.
Recentemente è stato dimostrato che E2 diminuisce la trascrizione del
81
gene del TNFα indotta da Tax, una proteina regolatoria del retrovirus
HTLV. Nel promotore del TNFα c’è un elemento responsivo al TNFα
(TNF-RE), che contiene un sito di legame per NFAT e NF-κB [277].
Dopo l’attivazione da parte di Tax, un complesso costituito da p50/p65 e
Jun lega il TNF-RE, attivando la trascrizione. Mediante EMSA effettuata
su estratti nucleari di U2OS, transfettate stabilmente con ERα ed ERβ e
trattate con E2 si è dimostrato che ERα si lega a questo complesso. Questi
dati suggeriscono che ERα reprime la trascrizione del TNFα legando Jun
e p50/p65 tramite interazioni proteina proteina.
82
SCOPO DELLA TESI
Lo scopo di questa tesi è stato lo studio del meccanismo molecolare
dell'azione del 17β-estradiolo sulla risposta infiammatoria nei macrofagi.
Si è cercato di individuare gli intermediari della via di trasduzione del
segnale infiammatorio su cui agisce il 17β-estradiolo per svolgere la sua
azione antinfiammatoria.
83
MATERIALI E METODI
COLTURE CELLULARI
Le
cellule
RAW
264,7
(linea
cellulare
di
macrofagi
murini
immortalizzati), le cellule SK-N-BE (linea cellulare immortalizzata da
neuroblastoma umano da midollo spinale) sono state acquistate dalla
ATCC,
le
cellule
U-937
(linea
cellulare
di
monociti
umani
immortalizzati) sono state gentilmente fornite dal laboratorio dei prof
Simonetta Nicosia e GianEnrico Rovati del Dipartimento di Scienze
Farmacologiche di Milano, le cellule SK-ER3 (clone di SK-N-BE che
esprime stabilmente ERα) sono state generate dal nostro laboratorio
mediante una transfezione stabile delle SK-N-BE [278], le cellule HepG2
(linea cellulare immortalizzata da carcinoma epatico umano) sono state
gentilmente fornite dal laboratorio del prof Cesare Sirtori del
Dipartimento di Scienze Farmacologiche di Milano.
Condizioni di crescita
Le cellule RAW 264,7 sono cresciute in terreno DMEM con rosso fenolo
(Invitrogen) supplementato con 10% FBS, le cellule U-937 e le cellule
SK-N-BE in terreno RPMI 1640 con rosso fenolo + 10% FBS, le cellule
SK-ER3 in terreno RPMI 1640 + 10% FBS-DCC, le cellule HepG2 in
terreno MEM con rosso fenolo + 10% FBS a 37°C in atmosfera di aria
umidificata 95% / CO2 5%. Le cellule RAW 264,7 vengono suddivise due
volte la settimana in piastre Petri da 10 cm2 (Corning) alla densità di
1 x 106 cellule/mL. Le cellule SK-N-BE e le cellule HepG2 vengono
84
suddivise due volte la settimana in piastre Petri da 10 cm2 alla densità di
2 x 105 cellule/mL. Le cellule SK-ER3 vengono divise una volta la
settimana in piastre Petri da 10 cm2 alla densità di 1 x 105 cellule/mL. Le
cellule U-937 vengono suddivise due volte la settimana in flask da 75 cm2
(Corning) alla densità di 1 x 106 cellule/mL. Per gli esperimenti di
Western blot e di immunocitochimica le cellule RAW 264,7 vengono
seminate in DMEM + 10% FBS. Dopo 24 h il medium è rimosso e le
cellule sono incubate in DMEM privo di siero. Dopo 6h le cellule sono
trattate. Per gli esperimenti di Western blot le cellule U-937 sono piastrate
in RPMI 1640 + 10% FBS. Dopo 24 h il medium è rimosso e le cellule
sono incubate in RPMI 1640 privo di siero. Dopo 6h le cellule sono
trattate. Per gli esperimenti di immunocitochimica le cellule SK-N-BE
sono piastrate in RPMI 1640 + 10% FBS. Dopo 24 h il medium è
rimosso e le cellule sono incubate in RPMI 1640 privo di siero. Dopo 6h
le cellule sono trattate. Per gli esperimenti di immunocitochimica le
cellule SK-ER3 sono piastrate in RPMI 1640 + 10% FBS-DCC. Dopo 24
h il medium è rimosso e le cellule sono incubate in RPMI 1640 privo di
siero. Dopo 6h le cellule sono trattate. Per gli esperimenti di
immunocitochimica le cellule HepG2 sono piastrate in MEM + 10% FBS.
Dopo 24 h il medium è rimosso e le cellule sono incubate in MEM privo
di siero. Le cellule sono trattate dopo 6h.
85
Terreni per colture cellulari
DMEM (Dulbecco’s Modified Eagle Medium) con rosso fenolo + 10%
FBS
Polvere di DMEM (Invitrogen)
NaHCO3
Glucosio
Streptomicina-penicillina
Sodio piruvato
FBS
13,38 g/L
1,5 g/L
2,5 g/L
5 mL/L di un mix da 10.000 UI
0,11 g/L
10%
Procedimento
1. Si scioglie la polvere in 900 mL di acqua distillata, mantenendo in
agitazione. Si aggiunge NaHCO3, si lascia sciogliere e quindi si
porta a pH 7,2 con HCl 1N, mantenendo sempre in agitazione.
2. Si porta a volume di 1 L con acqua bidistillata e si prelevano 105
mL di soluzione, che costituiranno il medium incompleto.
Ai restanti 895 mL, si aggiungono gli altri ingredienti.
3. Si sterilizza per filtrazione, utilizzando filtri disposable 0,22 µm
(sotto cappa sterile) e si conserva a 4°C.
MEM con rosso fenolo + 10% FBS
Polvere di MEM (Invitrogen)
NaHCO3
Streptomicina-pennicillina
Sodio piruvato
Mix aminoacidi non essenziali
FBS
9,53 g/L
2,2 g/L
5 mL/L di un mix da 10.000 UI
0,11 g/L
1%
10%
86
Procedimento
1. Sciogliere la polvere in 900 mL di acqua distillata, mantenendo in
agitazione.
2. Aggiungere NaHCO3, lasciar sciogliere e quindi portare il pH a 7.2
con HCl 1N, mantenendo sempre in agitazione.
3. Portare a volume di 1 L con acqua bidistillata.
4. Prelevare 115 mL di soluzione, che costituiranno il medium
incompleto.
5. Ai restanti 885 mL, aggiungere gli altri ingredienti e lasciar agitare.
6. Sterilizzare per filtrazione, utilizzando filtri disposable 0,22 µm
(sotto cappa sterile). Si conserva a 4° C.
RPMI 1640 con rosso fenolo + 10% FBS
Polvere pronta RPMI 1640 con rosso fenolo (Invitrogen)
NaHCO3
2 g/L
Glucosio
2,5 g/L
Sodio piruvato
0,11 g/L
Penicillina-Streptomicina
5 mL/L di un mix da 10.000 UI
FBS
10%
Procedimento
1. Si scioglie la polvere per RPMI rosso in 1 L di acqua distillata,
mantenendo in agitazione. Si aggiunge NaHCO3 e si porta pH a 7,2
con HCl 1 N, sempre in agitazione.
2. Si prelevano 105 mL di soluzione, che costituiranno il medium
incompleto. Ai restanti 895 mL, si aggiungono tutti gli altri
ingredienti, tranne FBS, mantenendo sempre in agitazione.
3. Si filtra con filtro disposable 0,22 µm da 500 mL prima
l’incompleto e poi il medium completo.
87
4. Si aggiungono al medium completo 100 mL di FBS sterile (sotto
cappa). Si conserva a 4°C.
RPMI 1640 + 10% FBS-DCC
Polvere pronta RPMI 1640 (Sigma)
NaHCO3
Glucosio
Sodio piruvato
Mix aminoacidi
Mix vitamine
Pennicillina-Streptomicina
FBS-DCC
2 g/L
2,5 g/L
0,11 g/L
10%
10%
5 mL/L di un mix da 10.000 UI
10%
Procedimento
1. Aggiungere la polvere pronta a 900 mL di acqua distillata, in una
beuta da 1L e sciogliere bene la polvere su piastra agitante. Se il
discioglimento dovesse risultare difficoltoso, aggiungere qualche
goccia di HCl 1 M, facendo attenzione a non abbassare il pH al di
sotto di 6,5.
2. Aggiungere il NaHCO3 e portare il pH a 7,2 con HCl 1 M, sempre
mantenendo in agitazione.
3. Portare a volume di 1 L e prelevare 125 mL della soluzione, che
andranno a costituire il medium incompleto.
4. Ai restanti 875 mL aggiungere tutti gli altri ingredienti.
5. Filtrare prima il medium incompleto e poi il completo utilizzando
filtri disposable 0,22 µm da 500 mL.
6. Si conserva a 4°C.
88
STRIPAGGIO DEL SIERO DCC
24 ore prima disattivare il siero tenendolo in bagnetto per 1 ora a 56°C
lasciare quindi a temperatura ambiente per 30 minuti.
1. Pesare 3 g di carbonio attivo e metterli nella bottiglia da 1 L apposta
per DCC
2. Pesare 0,3 g di dextrano ed aggiungerlo al carbonio
3. Aggiungere 1 L di acqua distillata ed agitare per 30 minuti
4. Suddividere la soluzione in 4 recipienti da super centrifuga,
mantenendola in agitazione
5. Centrifugare per 15 minuti a 2500g a temperatura ambiente
6. Scartare delicatamente il surnatante, in modo tale da non
risospendere il pellet
7. Aggiungere il siero, suddividendolo in modo preciso nei 4
contenitori (500 mL di siero)
8. Agitare per 4 ore nell'incubatore agitante e termostatato a 37°C
9. Centrifugare per 15 minuti a 2500g a temperatura ambiente
10.Filtrare su filtro buchner, utilizzare 3 filtri della Whatman in
sequenza D, A, C
11.Filtrare con filtro disposable 0,45 µm
12.Conservare a -20°C.
89
PREPARAZIONE DI ESTRATTI PROTEICI DA LISATI
CELLULARI
Procedimento
Dopo aver rimosso il medium ed aver lavato con PBS 1X, si prelevano le
cellule dal pozzetto con 1 mL di TEN buffer 1X utilizzando uno
“scraper”, quindi si centrifuga a 13000 rpm per 15 secondi , si aspira il
surnatante in modo da ottenere un pellet di cellule a cui si aggiungeranno
100 µL di lysis.
Per ottenere estratti proteici totali, si fanno 3 cicli di congelamentoscongelamento da -90°C a temperatura ambiente. Quindi si centrifuga a
13000 rpm per 30 minuti per precipitare le membrane, si preleva il
surnatante e si mette in tubi da 1.5mL (Eppendorf) per la quantizzazione
dell’estratto con il metodo di Bradford.
Per ottenere estratti di proteine citoplasmatiche e nucleari separate, si
risospende il pellet di cellule nel lysis citosolico, si lascia in ghiaccio per
15 minuti.
Si aggiunge un quantitativo di NP-40 al 10% pari a circa 1/20 del
quantitativo di lysis citosolico usato, si lascia sul vortex alla massima
velecità per 10 secondi e quindi si centrifuga a 4°C, 7200g per 10 secondi.
Si mette il surnatante, che è l’estratto citosolico in tubi da 1.5mL.
Si risospendere il pellet con lysis nucleare, usando un quantitativo pari a
circa 1/3 del lysis citosolico usato, si lascia in ghiaccio per 15 minuti e
quindi si centrifuga a 4°C, 13500g per 15 minuti. Si trasporta il
surnatante, che è l’estratto nucleare in tubi da 1.5mL.
90
Soluzioni
Lysis buffer per estratti totali di proteine cellulari
Hepes pH 7,9
MgCl2
NaCl
EDTA
Glicerolo (Invitrogen)
Triton (Sigma)
β-mercaptoetanolo (Sigma)
PMSF (Sigma)
Aprotinina (Sigma)
Leupeptina (Sigma)
Acqua
20 mM
5 mM
420 mM
100 nM
20%
0,1%
5 mM
100 nM
10 µg/mL
1 µg/mL
Lysis buffer per estratti citosolici di proteine cellulari
Tris-HCl pH 7.8
MgCl2
KCl
EGTA
Saccarosio
DTT (Sigma)
PMSF
Aprotinina
Leupeptina
Pepstatina A (Sigma)
Acqua
10 mM
5 mM
10 mM
300 nM
300 mM
500 nM
1 mM
1 µg/mL
1 µg/mL
1 µg/mL
Lysis buffer per estratti nucleari di proteine cellulari
Tris-HCl pH 7.8
MgCl2
KCl
EGTA
20 mM
5 mM
320 mM
200 nM
91
Tris-HCl pH 7.8
DTT
Aprotinina
Leupeptina
Pepstatina A
Acqua
20 mM
500 nM
1 µg/mL
1 µg/mL
1 µg/mL
TEN buffer
TRIS pH 8
NaCl
EDTA pH 8
Acqua
40 mM
150 mM
1 mM
PBS 10X
NaCl
KCl
Na2HPO4
K2HPO4
Acqua
80 g/L
2 g/L
11,36 g/L
2 g/L
Determinazione delle proteine con il metodo di Bradford
La determinazione quantitativa delle proteine viene condotta secondo il
metodo colorimetrico di Bradford, basato sull’utilizzo di un reagente in
grado
di
sviluppare
un’intensità
di
colore
proporzionale
alla
concentrazione di proteine presenti in soluzione. Requisito fondamentale
di questo tipo di analisi è la preparazione di una retta di taratura standard,
mediante l’utilizzo di diluizioni successive di una soluzione a
concentrazione nota di proteine.
La retta di taratura è preparata a partire da una soluzione di BSA (Bovine
Serum Albumine) 2 mg/mL (Pierce) da cui si ottengono 8 diluizioni
successive (da 1:4 a 1:30). Queste vengono a loro volta diluite 1:50 in
92
piastre da 96 pozzetti (Costar) in cui sono posti 300 µL di Coomassie
(Pierce) e quindi sottoposte a lettura spettrofotometrica alla lunghezza
d’onda di 595 nm.
Per preparare le diluizioni si utilizza lo stesso mezzo in cui sono disperse
la proteine dei campioni, così come è aggiunto al Coomassie per la lettura
del bianco.
I campioni vengono anch’essi aggiunti al Coomassie in rapporto 1:50. Nel
caso in cui la concentrazione sia tale da dare letture superiori al valore
massimo della retta di taratura, i campioni vengono diluiti e la
concentrazione si ricava per confronto con una nuova retta di taratura,
preparata diluendo la BSA in un mezzo a sua volta diluito dello stesso
fattore.
WESTERN BLOT
Procedimento
1. Si preparano il separating gel al 7,5% e lo stacking gel al 4% di
acrilammide.
2. Si utilizza un apparato per Western Blot (Biorad Mini-Protean II
Cell). Per ogni campione si caricano circa 30 µg di estratto cellulare
sul gel portando tutti i campioni a uno stesso volume con il lysis
buffer utilizzato per l’estrazione delle medesime proteine.
3. A ciascun campione si aggiunge un volume 1:3 di Laemmli buffer
3X. Si denaturano i campioni 5 minuti a 95°C, si centrifuga a 13000
rpm per un minuto, si caricano i campioni sul gel (le proteine
denaturate si legano al SDS, diventano cariche negativamente e
93
migrano attraverso il gel di poliacrilammide in base al loro peso
molecolare). I campioni proteici devono sempre essere tenuti in
ghiaccio; dopo la denaturazione possono essere tenuti a temperatura
ambiente.
4. Nel primo pozzetto deve essere messo il marker per conoscere il
peso molecolare della proteina di interesse, 10µL. Si utilizza il
marker Unstained Precision Protein Standard (Biorad), che deve
essere denaturato come i campioni e caricato. La corsa
elettroforetica è effettuata a 60mA, voltaggio massimo per 2 ore.
5. Per il blotting si usa carta Whatman 3MM e due pezzi di filtro di
nitrocellulosa, membrana Hybond C-extra (Amersham) delle stesse
dimensioni del separating gel.
6. Al termine si colora il filtro per circa 1 minuto con Rosso Ponceau
diluito 1:10 con H2O, per evidenziare l’avvenuto trasferimento e per
localizzare le proteine. (Rosso Ponceau 10X: rosso ponceau (Sigma)
2% w/v, acido tricloroacetico 30% w/v, acido solfosalicilico 30%
w/v). Si sciacqua con H2O distillata e si fa asciugare.
7. Il filtro, decolorato con TBS, può essere conservato a +4°C.
Immunodetezione
1. Si incuba il filtro, mantenendolo in agitazione, in blocking solution
per 1 ora a temperatura ambiente.
2. Si incuba con anticorpo primario overnight a 4°C, sotto agitazione.
3. Si lava in TBST; si effettuano 3-4 lavaggi da 10 minuti a
temperatura ambiente.
4. Si incuba per 1 ora a temperatura ambiente, mantenendo in
agitazione, con anticorpo secondario, diluizione 1:2000, in blocking
94
solution. Come anticorpo secondario, si utilizza un anticorpo
coniugato covalentemente con la perossidasi di rafano (Horseradish
Peroxidase).
5. Si lava in TBST, 3 lavaggi da 10 minuti a temperatura ambiente, si
mette il filtro nuovamente in TBST.
6. In camera oscura si copre il filtro con 2 ml di una miscela di
soluzione A e soluzione B (1 mL di soluzione A + 1mL di soluzione
B) del Kit per la reazione ECL (Enhanced Chemiluminescence,
Amersham). A contatto con la soluzione A e la soluzione B la
perossidasi di rafano coniugata all’anticorpo secondario genera una
reazione chemioluminescente, in grado d’impressionare una lastra
fotografica, permettendo così la detezione della proteina.
7. Si espone la lastra fotografica al filtro per un tempo variabile da
stabilire sperimentalmente a seconda dell’intensità del segnale. Per
sviluppare la lastra, la si mette nel liquido di sviluppo fino a quando
non si vedono comparire le bande. Si sciacqua la lastra e la si mette
nel liquido di fissaggio. Si sciacqua la lastra sotto acqua corrente e
la si lascia asciugare.
8. Si sciacqua il filtro con TBST e si conserva a +4°C. Il filtro può
essere utilizzato per eventuali successive ibridazioni con altri
anticorpi dopo lo “strippaggio” (il filtro deve essere lasciato nella
stripping solution per 30 minuti a 50°C e successivamente
sciacquato con TBS).
95
Soluzioni per western blot ed immunodetezione
Acrilammide 30%
Acrilammide (Biorad)
29,2 g
Bis-acrilammide (Biorad)
0,8 g
Acqua
q. b. a 100 mL
Si deve filtrare non sterilmente e usare guanti e mascherina perché
l’acrilammide e la bisacrilammide sono tossiche.
7,5% separating gel PAGE
Acqua
Acrilammide 30%
Tris 1M pH 8,8
SDS 20%
APS (Biorad ) 10%
Temed (Biorad)
(dosi per 2 gel)
6,8 mL
4,9 mL
8 mL
100 µL
100 µL
10 µL
4% stacking gel PAGE
Acqua
Acrilammide 30%
Tris 1M pH 6,8
SDS 20%
APS 10%
Temed
(dosi per 2 gel)
6,8 mL
1,7 mL
1,25 mL
50 µL
100 µL
10 µL
Running buffer 5X
Tris base (Merck)
Glicina (Biorad)
Acqua
15,2 g/L
65 g/L
Running buffer 1X
Running buffer 5X
SDS
Acqua
20%
1%
96
Laemmli buffer 3X
Tris pH 6,8
Glicerolo
SDS
β-mercaptoetanolo
blu di bromofenolo (Biorad)
150 mM
30%
6%
1,5 mM
0,1%
Blotting buffer
Tris base
Glicina
Metanolo (Merck)
Acqua
3,03 g/L
14,4 g/L
20% v/v
TBS buffer
Tris pH 7,5
NaCl
Acqua
50 mM
150 mM
Blocking solution
TBS buffer
Latte scremato in polvere (Regilait)
TWEEN 20 (Sigma)
5%
0,2%
Stripping solution
Tris-HCI pH 6,7
SDS
β-mercaptoetanolo
Acqua
62,5 mM
2%
100 µM
Anticorpi usati nelle Western blot:
p65 rabbit
β-actina mouse
IκBα rabbit
Fosfo IKK rabbit
Fosfo ERK1/2 mouse
ERK1/2 rabbit
c-Rel rabbit
1:500
1:5000
1:500
1:250
1:1000
1:1000
1:500
97
(Santa Cruz)
(Sigma)
(Santa Cruz)
(Cell Signaling)
(Cell Signaling)
(Cell Signaling)
(Santa Cruz)
p65 rabbit
p50 rabbit
1:500
1:1000
(Santa Cruz)
(Santa Cruz)
IMMUNOCITOCHIMICA
Preparazione vetrini:
Immergere per alcuni giorni i vetrini in HCl 7%. Rimuovere l'HCl dai
vetrini, lavare con acqua bidistillata per 3 volte. Sotto cappa sterile aprire
una petri e mettere nel coperchio dell'alcool e nel fondo acqua per vetrini,
quindi immergere vetrini nell'alcool, poi trasportarli con una pinzetta
nell'acqua. Prendere un vetrino alla volta ed asciugarlo sulla fiamma del
bunsen, infine riporre il vetrino in una piastra da 24 pozzetti sterile.
Fissaggio cellule:
Aspirare il terreno di coltura, lavare con PBS 1X non sterile, fissare le
cellule per aggiunta di paraformaldeide al 4% per 5 minuti. Aspirare la
paraformaldeide, e lavare per 3 volte con PBS 1X non sterile, mettere
infine PBS + NaN3 conservare a 4°C.
98
Immunodetezione:
1. Togliere il PBS + NaN3, aggiungere la soluzione di blocco per 45
minuti a temperatura ambiente.
2. Incubare con la soluzione contenente l'anticorpo primario overnight a
4°C.
3. Fare 3 lavaggi da 10 minuti, nella soluzione di lavaggio
4. Incubare con la soluzione contenente l'anticorpo secondario per 1 ora a
temperatura ambiente.
5. Fare 3 lavaggi da 10 minuti, nella soluzione di lavaggio
6. Incubare con la soluzione ABC per 1 ora a temperatura ambiente.
7. Lavare due volte con PBS 1X non sterile per 10 minuti.
8. Lavare infine con TRIS HCl 50mM pH 7.5 per 10 minuti.
9. Aggiungere la soluzione con la DAB per circa 5 minuti, fermare la
reazione per aggiunta di PBS 1X non sterile.
10.Lavare con PBS 1X non sterile.
Montaggio vetrini:
Su ogni vetrino mettere una goccia di PBS + glicerolo (1/1), riporre il
vetrino sul porta vetrini, ricoprire con il vetro copri-vetrini, sigillare con
Eukit
99
Soluzioni per immunocitochimica per p65:
Soluzione di blocco
PBS 1X non sterile
BSA (Sigma)
Siero di goat (Gibco)
Triton
3%
10%
0,2%
Soluzione per anticorpi
PBS 1X non sterile
Siero di goat
10%
Anticorpo primario:
p65
1:500
(Santa Cruz)
Anticorpo secondario:
Biotinilato goat anti rabbit IgG 1:200 (Vector Laboratories)
Soluzione di lavaggio
PBS 1X non sterile
BSA
TWEEN (Sigma)
3%
0,1%
Soluzione di avidin-biotin horseradish peroxidase complex
(ABC kit, Vector Laboratories)
10µL di A + 10µL di B per ogni mL di PBS 1X non sterile
agitare su piastra agitante per 30 minuti
100
Soluzione di 3,3’-diaminobenzidina (DAB) (Sigma)
1 pastiglia urea + 1 pastiglia di DAB in 5mL di acqua
vortexare fin quando tutto è ben sciolto
SCHEMA DEI TRATTAMENTI DELLE CELLULE
Negli esperimenti le cellule sono state trattate con i seguenti reagenti:
• 17β-estradiolo (Sigma) 10-9 M 10 minuti seguito dagli stimoli
infiammatori:
• LPS (isotipo 0111:B4, Sigma) 50 µg / mL 30 minuti
• TNFα (Sigma) 20 ng / mL 30 minuti
• IL-1β (Peprotech) 10 ng / mL 30 minuti
• LY 294002 (Sigma) 50 µM 1 ora prima del trattamento con E2
101
RISULTATI
1. Azione del 17β
β-estradiolo sulla localizzazione
citoplasmatica di p65 in cellule macrofagiche
Studi preliminari effettuati dal nostro laboratorio hanno messo in
evidenza che l'E2 è in grado di bloccare il legame di p65 al DNA.
I fattori di trascrizione della famiglia di NF-κB una volta attivati
traslocano dal citoplasma al nucleo. Per questo motivo abbiamo voluto
valutare l'influenza del 17β-estradiolo sulla localizzazione subcellulare di
p65 in cellule dell'infiammazione; ci siamo serviti della linea cellulare
RAW 264,7 in quanto già ben caratterizzata sia nel nostro che in altri
laboratori [274].
La valutazione della localizzazione subcellulare di p65 è stata effettuata
con due tecniche: immunocitochimica e western blot.
Mediante immunocitochimica condotta sulle cellule RAW 264,7
osserviamo che in condizioni di assenza di stimolo la localizzazione di
p65 è citoplasmatica, condizione che non è perturbata dal trattamento con
l'ormone in concentrazione fisiologica.
Il trattamento con LPS, come previsto, porta ad un massivo accumulo
nucleare di p65; il pretrattamento di 10 minuti con il 17β-estradiolo prima
dell'LPS comporta una localizzazione citoplasmatica simile alle cellule
controllo (Figura 1A e B).
Per confermare l'effetto ottenuto tramite immunocitochimica è stata
effettuata una western blot su estratti citoplasmatici e nucleari dalle
cellule RAW 264,7. La tecnica di immunocitochimica permette di
valutare la variazione subcellulare di una proteina, ma non si ha la
102
certezza che l'anticorpo riconosca la proteina in esame; tramite western
blot si ha invece la possibilità di capire se la proteina rilevata
dall'anticorpo ha il medesimo peso molecolare di quella di interesse.
Questo permette di aggiungere informazioni ai dati di immnocitochimica.
I risultati che abbiamo ottenuto per western blot nelle RAW 264,7
confermano quelli precedentemente ottenuti per immunocitochimica.
Infatti il trattamento con LPS porta ad una diminuzione dei livelli di p65
citoplasmatico, mentre il pretrattamento con E2 seguito dall'aggiunta di
LPS al terreno di coltura mostra una quantità di p65 citoplasmatico simile
alle cellule controllo (Figura 1C).
103
B
50
25
50
PS
+L
E
2
β−actina
β−
37
S
50
2
p65
100
LP
75
150
E
100
CT
RL
Densità ottica
β -actina
p65/β
LP
S
E
2+
LP
S
2
CT
R
E L
2
C
LP
S
E
2+
LP
S
CT
R
E L
E
+L
PS
E2+LPS
75
CT
RL
LPS
100
2
E2
E
2
LP
S
C
p65 citoplasm.
(% cell pos /cell tot)
A
Figura 1. Effetto del 17β-estradiolo sulla localizzazione subcellulare di p65
nelle cellule RAW 264,7
A) Immunocitochimica per p65
B) Quantizzazione dell'effetto visto in A) mediante conta cellulare
C) Western blot per p65 su estratti proteici citoplasmatici
104
2. Azione del 17β
β-estradiolo sulla localizzazione
citoplasmatica di p65 in cellule monocitarie
Per valutare se l'effetto osservato nelle RAW 264,7 fosse un meccanismo
conservato nelle cellule mieloidi abbiamo utilizzato le cellule U-937, che
sono monociti umani immortalizzati [279] Utilizzando ancora la tecnica
di western blot osserviamo come nelle U-937 non stimolate p65 sia
prevalentemente citoplasmatico, mentre il trattamento con TNFα
20 ng/mL per 30 minuti porti ad una pressoché totale traslocazione di
p65. Anche in questo caso un breve pretrattamento con l'ormone in
concentrazione fisiologica blocca l'effetto del TNFα (Figura 2A e 2B).
Da questi risultati abbiamo concluso che il 17β-estradiolo è in grado di
influenzare la localizzazione subcellulare di p65 in due tipi di cellule
mieloidi.
105
TN
Fα
E
2+
TN
Fα
75
50
100
50
β−actina
β−
TN
Fα
E
2+
TN
Fα
2
2
CT
RL
E
B
TN
Fα
E
2+
TN
Fα
CT
RL
E
CT
RL
37
E
2
TN
Fα
E
2+
TN
Fα
p65
150
Densità ottica
β -actina
p65/β
2
TN
Fα
E
2+
TN
Fα
CT
RL
E
2
CT
RL
E
A
75
50
50
CT
RL
37
β−actina
β−
100
E
TN 2
Fα
E
2+
TN
Fα
p65
Densità ottica
β -actina
p65/β
150
Figura 2. Effetto del 17β-estradiolo sulla localizzazione subcellulare di p65
nelle cellule U-937
A) Western blot per p65 su estratti di proteine citoplasmatiche
B) Western blot per p65 su estratti di proteine nucleari
106
3. Effetto del 17β
β-estradiolo sulla degradazione di Iκ
κBα
α
L'ipotesi più logica, sulla base delle conoscenze circa la regolazione della
localizzazione subcellulare di p65, che potesse spiegare l'azione di E2
prevedeva che l'estrogeno inibisse la degradazione di IκBα.
Abbiamo così valutato se i livelli di proteina di IκBα fossero aumentati in
seguito al trattamento con E2 ed LPS. Abbiamo effettuato un esperimento
di tempo-dipendenza, analizzando l'azione di LPS dopo 5, 10, 20, 30 e 60
minuti dall'aggiunta di questa endotossina al terreno di coltura. I livelli di
IκBα sono quindi stati valutati mediante western blot sugli estratti
proteici citosolici. Per valutare l'effetto di E2 una serie analoga di
campioni trattati con LPS ai tempi citati sopra è stata previamente trattata
per 10 minuti con l'ormone. Dal grafico di figura 3 vediamo come i livelli
di IκBα inizino a diminuire circa 5 minuti dopo l'aggiunta di LPS e
raggiungano il minimo a 20 minuti. Dopo trenta minuti si osserva un
aumento dei livelli di IκBα. Ricordiamo infatti che IκBα è uno dei geni
precoci indotti da NF-κB. L'aumento di IκBα può essere ricondotto
all'effetto trascrizionale di NF-κB sul promotore di IκBα [206]. Dopo
un'ora di trattamento con LPS i livelli di IκBα sono tornati simili alla
situazione non stimolata.
E' interessante notare che il pretrattamento con 17β-estradiolo non è in
grado di prevenire la diminuzione dei livelli di IκBα, infatti il grafico è
sovrapponibile a quello ottenuto con il solo LPS (Figura 3). Questo
suggerisce che p65 rimane citoplasmatico nonostante si IκBα degradi.
107
S
+L
PS
2
E
2
60'
LP
2
30'
LP
S
E
2+
LP
CT S
R
E L
CT
R
E L
S
+L
PS
2
E
2
LP
S
2+
LP
CT S
RL
E
E
2
E
LP
PS
RL
CT
S
2+
L
E
2
LP
RL
E
CT
20’
10’
5’
75
50
Iκ
κBα
α
37
25
50
β-actina
37
Densità ottica
κBα
α/β
β -actina
Iκ
100
CTRL
E2
LPS
E2+LPS
60
20
5'
10'
20'
30'
60'
Tempo
Figura 3. Effetto del 17β-estradiolo sulla degradazione di IκBα
Western blot per IκBα su estratti totali da RAW 264,7
108
4. Influenza del 17β
β-estradiolo su IKK
La degradazione di IκBα è susseguente alla sua fosforilazione da parte di
IKK, che a sua volta viene attivato mediante fosforilazione [183].
Come ulteriore conferma dei risultati su IκBα tramite western blot
abbiamo valutato la fosforilazione di IKK in seguito ai trattamenti; come
era lecito attendersi i livelli basali di fosforilazione sono quasi nulli,
l'ormone di per sé non influenza questi livelli. L'LPS, come documentato
in letteratura, provoca la fosforilazione di IKK, mentre il pretrattamento
con l'estrogeno non blocca l'attivazione di IKK da LPS, come prevedibile
sulla base dell'esperimento su IκBα (Figura 4).
109
+L
PS
2
E
LP
S
E
2
CT
RL
+L
PS
2
E
LP
S
2
E
CT
RL
100
Fosfo IKK
75
50
β−actina
β−
37
75
50
+L
PS
2
E
LP
S
E
2
25
CT
RL
Densità ottica
β-actina
IKK/β
100
Figura 4. Effetto del 17β-estadiolo su IKK
Western blot per fosfo IKK su estratti totali da RAW 264,7
110
5. Influenza del 17β
β-estradiolo su MAPK
Il legame dell'LPS al suo recettore porta all'attivazione di più cascate di
trasduzione del segnale [227], quindi abbiamo voluto verificare se il 17βestradiolo giocasse un ruolo anche in queste vie.
Abbiamo perciò posto la nostra attenzione sulla via delle MAP chinasi: si
è valutata la fosforilazione di ERK1/2, che poi porta alla loro attivazione.
Come si può vedere dalla figura 5 l'LPS stimola l'attività delle MAPK, ma
l'estrogeno non è in grado di prevenire questa attivazione.
111
+L
PS
2
E
LP
S
2
E
CT
RL
+L
PS
2
E
LP
S
2
E
CT
RL
100
75
50
Fosfo ERK1/2
ERK1/2
37
75
50
+L
PS
2
E
LP
S
E
2
25
CT
RL
Densità ottica
P-ERK1/2/ERK1/2
100
Figura 5. Effetto del 17β-estadiolo sulle MAPK
Western blot per fosfo ERK1/2 su estratti totali da RAW 264,7
112
6. Influenza del 17β
β-estradiolo su PI3K
Un'altra via di segnale attivata da LPS è quella della PI3K, abbiamo
quindi valutato l'azione di E2 anche su questa via di segnale.
Per raggiungere il nostro scopo abbiamo trattato le RAW 264,7 con un
inibitore farmacologico della PI3K: l'LY 294002.
Quindi
abbiamo
fatto
un'immunocitochimica
per
valutare
la
localizzazione subcellulare di p65. Come si vede da figura 6 l'LY 294002
previene l'effetto dell'estrogeno, quindi è necessaria l'attivazione della
PI3K per avere il blocco della traslocazione di p65 mediato dall'ormone.
Da notare che l'inibitore della fosfatidilinositolo 3-chinasi da solo non
influenza la distribuzione subcellulare di p65 (Figura 6).
113
p65 citoplasm.
(% cell pos /cell tot)
100
75
50
25
RL
CT
E2
LP
S
E
PS
+2 L
LY
S
LP
+
LY
2
S
+E
LP
Y
+
L
2
+E
Y
L
Figura 6. Effetto del 17β-estadiolo sulla PI3K
Immunocitochimica per p65 in RAW 264,7
114
7. Azione del 17β
β-estradiolo su altri membri di NF-κ
κB
NF-κB è una famiglia di fattori di trascrizione composta, oltre che da p65,
anche da p50, c-Rel, RelB e p52 [163]. Di conseguenza ci siamo posti la
domanda se l'azione del 17β-estradiolo fosse specifica per p65 o
interessasse altri membri della famiglia di fattori di trascrizione NF-κB.
Per rispondere a questa domanda abbiamo fatto degli esperimenti di
western blot ed immnocitochimica sia in RAW 264,7 che in U-937.
Come risulta dalle figure 7A e 7B anche la traslocazione nucleare di c-Rel
è bloccata dal pretrattamento per 10 minuti con concentrazioni
fisiologiche di estrogeno (Figure 7A e 7B).
Anche p50 in seguito al trattamento con LPS si concentra nel nucleo,
mentre un pretrattamento con 17β-estradiolo riporta la situazione uguale a
quella del controllo (Figura 8).
Da ciò abbiamo concluso che l'ormone femminile è in grado di bloccare
la traslocazione nucleare di tutti i membri della famiglia NF-κB in cellule
infiammatorie, e questo può rendere conto dell'attività antinfiammatoria
dell'estrogeno.
115
α
NF
α
T
F
+
E2
TN
2
L
TR
C
E
α
NF
α
T
F
+
E2
TN
2
A
E
L
TR
C
100
75
c-Rel
50
β−actina
β−
37
75
50
E2
75
50
25
+L
PS
Figura 7. Effetto del 17β-estradiolo su c-Rel
A) Western blot per c-Rel su estratti citoplasmatici da U-937
B) Immunocitochimica per c-Rel in RAW 264,7
C) Quantizzazione dell'effetto visto in B) mediante conta cellulare
116
2
E
C
TR
L
E2+LPS
100
LP
S
C
c-Rel citoplasm.
(% cell pos /cell tot)
LPS
Fα
N
+T
2
B
Fα
TN
E
L
TR
C
2
25
E
Densità ottica
β-actina
c-Rel/β
100
+L
PS
2
E
LP
S
2
E
C
TR
L
+L
PS
2
E
LP
S
2
C
TR
L
E
100
75
50
p50
β-actina
37
Densità ottica
β-actina
p50/β
400
300
200
2
L
TR
C
E
100
S
LP
E
PS
+2 L
Figura 8. Effetto del 17β-estradiolo su p50 in cellule RAW 264,7
Western blot per p50 su estratti nucleari
117
8.
Effetto
del
17β
β-estradiolo
sulla
localizzazione
citoplasmatica di p65 in cellule non macrofagiche
Anche se il suo ruolo principale è nelle cellule del sistema immunitario,
NF-κB è un fattore di trascrizione espresso in modo ubiquitario. Per
questo abbiamo deciso di valutare se l'estrogeno inibisse la traslocazione
di p65 anche in cellule non infiammatorie.
A tale scopo abbiamo condotto un'immunocitochimica su MCF-7,
SK-ER3 ed SK-N-BE. Le cellule MCF-7 sono derivate da un carcinoma
mammario umano positivo per ERα e sono spesso usate per studiare gli
effetti degli estrogeni; le SK-ER3 sono cellule di neuroblastoma
transfettate stabilmente con ERα e sono state generate nel nostro
laboratorio per studiare l'azione del 17β-estradiolo, le SK-N-BE sono il
loro controllo negativo, in quanto non esprimono gli ER.
Ogni tipo cellulare è stato trattato con lo stimolo che produceva
l'attivazione di p65. Infatti abbiamo osservato che LPS non attiva NF-κB
nelle MCF-7, nelle SK-N-BE e nelle SK-ER3, perché queste cellule non
hanno il recettore per l'LPS. Quindi abbiamo usato il TNFα, stimolo che
porta all'attivazione di NF-κB in vari tipi cellulari.
Come si vede dalle figure 9 e 10 in nessuno di questi tipi cellulari un
pretrattamento di 10 minuti con 17β-estradiolo 10-9 M è stato in grado di
prevenire la traslocazione nucleare di p65 (Figura 9 e 10).
L'azione dell'estrogeno è perciò limitata alle cellule che prendono parte
alla risposta infiammatoria.
118
C
E2
TNFα
E2+TNFα
75
50
+T
NF
α
2
E
TN
Fα
2
E
25
CT
RL
p65 citoplasm.
(% cell pos /cell tot)
100
Figura 9. Effetto del 17β-estradiolo su p65 in cellule diverse
Immunocitochimica per p65 in MCF-7
119
A
B
C
E2
C
E2
TNFα
E2+TNFα
TNFα
E2+TNFα
+T
NF
α
2
E
+T
NF
α
2
25
2
E
2
TN
Fα
E
25
50
TN
Fα
50
75
E
75
CT
RL
p65 citoplasm.
(% cell pos /cell tot)
100
CT
RL
p65 citoplasm.
(% cell pos /cell tot)
100
Figura 10. Effetto del 17β-estradiolo su p65 in cellule diverse
A) Immunocitochimica per p65 in SK-N-BE
B) Immunocitochimica per p65 in SK-ER3
120
DISCUSSIONE
Numerosi studi indicano che l'estrogeno blocca la generazione della
risposta infiammatoria nelle cellule macrofagiche [270, 273, 275, 280];
questa proprietà può spiegare il ruolo protettivo degli estrogeni endogeni
ed esogeni in molti modelli animali di patologie a componente
infiammatoria, delle quali l'ormone ritarda l’insorgenza e attenua
i sintomi [243, 281-286]. Il meccanismo molecolare di questa azione deve
essere ancora chiarito.
Lo scopo che ha animato questa tesi è stato quello di determinare quali
eventi molecolari vengano influenzati dal 17β-estradiolo per svolgere la
sua attività inibitoria della via di segnale di LPS.
In particolare ho focalizzato il mio studio sui fattori di trascrizione della
famiglia di NF-κB.
Visto che questi fattori di trascrizione, una volta attivati, migrano nel
nucleo, abbiamo analizzato se l'E2 potesse modificare tale meccanismo di
distribuzione subcellulare di p65.
Un pretrattamento di 10 minuti con il 17β-estradiolo 10-9 M seguito dalla
stimolazione con LPS o TNFα per 30 minuti risulta comunque in una
localizzazione citoplasmatica di p65.
Questa azione dell'ormone non è limitata a p65, ma interessa anche gli
altri membri della famiglia di NF-κB: infatti abbiamo ottenuto gli stessi
risultati quando abbiamo focalizzato la nostra attenzione su p50 e c-Rel.
Sorprendentemente l'E2 non blocca la fosforilazione di IKK, né protegge
IκBα dalla degradazione. Quindi il 17β-estradiolo non agisce sulla via di
IKK/IκBα dell'attivazione di NF-κB né sull'attivazione delle MAP
chinasi.
121
E’ invece coinvolta l'attivazione della PI3K, visto che l'inibitore
farmacologico della PI3K, l'LY 294002 ha bloccato l'effetto del 17βestradiolo. Questo dato è in accordo con i risultati di altri laboratori, che
vedono attività antinfiammatoria della fosfatidilinositolo 3-chinasi [191195],
ed
interazioni
tra
ER
e
la
subunità
regolatoria
della
fosfatidilinositolo 3-chinasi sono state descritte in più sistemi cellulari
[131, 132].
Valutando questi risultati si può ipotizzare che l'azione dell'E2
sull'attivazione di p65 è un evento non genomico, in quanto si esplica in
tempi troppo brevi (dieci minuti) per essere compatibili con una
attivazione della espressione genica.
È importante inoltre notare che il 17β-estradiolo blocca l'attivazione di
p65 in modo indipendente dalla degradazione di IκBα, differenziandosi in
questo dai FANS, che invece agiscono inibendo IKK.
Infine in questa tesi dimostriamo che l'azione inibitoria del 17β-estradiolo
sull'attivazione di NF-κB si esplica solo nelle cellule macrofagiche.
Ulteriori studi sono necessari per meglio caratterizzare gli intermedi che,
oltre alla fosfatidilinositolo 3-chinasi, mediano l'azione dell'estrogeno.
Questi studi potranno servire per individuare dei nuovi bersagli nella via
di attivazione di NF-κB che agiscono a monte della trascrizione di geni
infiammatorii. Lo sviluppo di nuovi farmaci che agiscono in modo simile
all'E2 potrà essere utile per prevenire l'insorgenza di quelle malattie dove
la componente infiammatoria svolge un ruolo cruciale nella loro
patogenesi.
122
BIBLIOGRAFIA
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
Barnea, E.R., N.J. MacLusky, and F. Naftolin, Kinetics of catechol estrogenestrogen receptor dissociation: a possible factor underlying differences in catechol estrogen biological activity. Steroids, 1983. 41(5): p. 643-56.
Fishman, J. and B. Norton, Catechol estrogen formation in the central nervous
system of the rat. Endocrinology, 1975. 96(4): p. 1054-8.
Lane, K.E., M.J. Ricci, and S.M. Ho, Effect of combined testosterone and
estradiol-17 beta treatment on the metabolism of E2 in the prostate and liver
of noble rats. Prostate, 1997. 30(4): p. 256-62.
Bunyagidj, C. and J.A. McLachlan, Catechol estrogen formation in mouse uterus. J Steroid Biochem, 1988. 31(5): p. 795-801.
Paria, B.C., C. Chakraborty, and S.K. Dey, Catechol estrogen formation in the
mouse uterus and its role in implantation. Mol Cell Endocrinol, 1990. 69(1): p.
25-32.
Ball, P. and R. Knuppen, Formation of 2- and 4-hydroxyestrogens by brain,
pituitary, and liver of the human fetus. J Clin Endocrinol Metab, 1978. 47(4):
p. 732-7.
Schutze, N., G. Vollmer, I. Tiemann, M. Geiger, and R. Knuppen, Catecholestrogens are MCF-7 cell estrogen receptor agonists. J Steroid Biochem Mol
Biol, 1993. 46(6): p. 781-9.
Schaeffer, J.M., S. Stevens, R.G. Smith, and A.J. Hsueh, Binding of 2-hydroxyestradiol to rat anterior pituitary cell membranes. J Biol Chem, 1980.
255(20): p. 9838-43.
Vandewalle, B., J.P. Peyrat, J. Bonneterre, and J. Lefebvre, Catecholestrogen
binding sites in breast cancer. J Steroid Biochem, 1985. 23(5A): p. 603-10.
Vandewalle, B., L. Hornez, and J. Lefebvre, Characterization of catecholestrogen membrane binding sites in estrogen receptor positive and negative human breast cancer cell-lines. J Recept Res, 1988. 8(5): p. 699-712.
Etchegoyen, G.S., D.P. Cardinali, A.E. Perez, J. Tamayo, and G. Perez-Palacios, Binding and effects of catecholestrogens on adenylate cyclase activity,
and adrenoceptors, benzodiazepine and GABA receptors in guinea-pig hypothalamic membranes. Eur J Pharmacol, 1986. 129(1-2): p. 1-10.
Philips, B.J., P.J. Ansell, L.G. Newton, N. Harada, S. Honda, V.K. Ganjam,
G.E. Rottinghaus, W.V. Welshons, and D.B. Lubahn, Estrogen receptor-independent catechol estrogen binding activity: protein binding studies in wildtype, Estrogen receptor-alpha KO, and aromatase KO mice tissues. Biochemistry, 2004. 43(21): p. 6698-708.
Miller, W.L., Molecular biology of steroid hormone synthesis. Endocr Rev,
1988. 9: p. 295.
Steinkamph, M.P., C.R. Mendelson, and E. Simpson, Regulation by follicle
stimulating hormone of the synthesis of aromatase cytochrome P-450 in human granulosa cells. Mol Endocrinol, 1987. 1: p. 465.
Matagne, V., M.C. Lebrethon, A. Gerard, and J.P. Bourguignon, In vitro paradigms for the study of GnRH neuron function and estrogen effects. Ann N Y
Acad Sci, 2003. 1007: p. 129-42.
123
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
Simpson, E.R. and S.R. Davis, Minireview: aromatase and the regulation of
estrogen biosynthesis--some new perspectives. Endocrinology, 2001. 142(11):
p. 4589-94.
Castoria, G., M. Lombardi, M.V. Barone, A. Bilancio, M. Di Domenico, D.
Bottero, F. Vitale, A. Migliaccio, and F. Auricchio, Androgen-stimulated DNA
synthesis and cytoskeletal changes in fibroblasts by a nontranscriptional receptor action. J Cell Biol, 2003. 161(3): p. 547-56.
Tsai, M.J. and B.W. O'Malley, Molecular mechanisms of action of steroid/thyroid receptor superfamily members. Annu Rev Biochem, 1994. 63: p. 451-86.
Evans, R.M., The steroid and thyroid hormone receptor superfamily. Science,
1988. 240(4854): p. 889-95.
Petterson, K. and J.A. Gustafsson, Role of estrogen receptor ß in estrogen action. Annu Rev Physiol, 2001. 63: p. 165-92.
Jensen, E.V. and H.I. Jacobson, Basic guide to the mechanism of estrogen action. Recent Prog Horm Res, 1962. 18: p. 387-414.
Kuiper, G.G., E. Enmark, M. Peltro-Huikko, S. Nilsson, and J.A. Gustafsson,
Cloning of a novel receptor expressed in rat prostate and ovary. Proc Natl
Acad Sci USA, 1996. 93: p. 5925-30.
Ogawa, S., S. Inoue, T. Watanabe, A. Orimo, T. Hosoi, and Y. Ouchi, The
complete primary structure of human estrogen receptor ß (hEß) and its heterodimerizazion with ERα in vivo and in vitro. Biochem Biophys Res Commun,
1998. 243: p. 122-6.
Hawkins, M.B., J.W. Thornton, D. Crews, J.K. Skipper, A. Dotte, and P. Thomas, Identification of a third distinct estrogen receptor and reclassification of
estrogen receptors in teleosts. Proc Natl Acad Sci U S A, 2000. 97(20): p.
10751-6.
Green, S. and P. Chambon, Oestradiol induction of a glucocorticoid-responsive gene by a chimaeric receptor. Nature, 1987. 325(6099): p. 75-8.
Schwabe, J.W., D. Neuhaus, and D. Rhodes, Solution structure of the DNAbinding domain of the estrogen receptor. Nature, 1990. 348: p. 458-61.
Schwabe, J.W., L. Chapman, J.T. Finch, and D. Rhodes, The crystal structure
of the estrogen receptor DNA-binding domain bound to DNA: how receptors
discriminate between their response elements. Cell, 1993. 75: p. 567-78.
Kuntz, M.A. and D.J. Shapiro, Dimerizing the estrogen receptor DNA binding
domain enhances binding to estrogen response elements. J Biol Chem, 1997.
272: p. 27949-56.
Chambraud, B., M. Berry, G. Redeuilh, P. Chambon, and E.E. Baulieu, Several regions of human estrogen receptor are involved in the formation of receptor-heat shock protein 90 complexes. J Biol Chem, 1990. 265: p. 20686-91.
Picard, D., V. Kumar, P. Chambon, and K.R. Yamamoto, Signal transduction
by steroid hormones: nuclear localization is differentially regulated in estrogen and glucocorticoid receptors. Cell Regul, 1990. 1: p. 291-99.
Brzozowski, A.M., A.C.W. Pike, Z. Dauter, R.E. Hubbard, T. Bonn, and e. al.,
Molecular basis of agonism and antagonism in the estrogen receptor. Nature,
1997. 389: p. 753-58.
Shiau, A.K., D. Barstand, P.M. Loria, L. Cheng, P.J. Kushner, D.A. Agard,
and G.L. Greene, The structural basis of estrogen receptor/coactivator recognition and the antagonism of interaction by tamoxifene. Cell, 1998. 95: p.
927-37.
124
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
Pike, A.C.W., A.M. Brzozowski, R.E. Hubbard, T. Bonn, A.G. Thorsell, and e.
al., Structure of the ligand-binding domain of oestrogen receptor ß in the presence of a partial agonist and a full antagonist. EMBO J, 1999. 18: p. 460818.
Hall, J.M., J.F. Couse, and K.S. Korach, The multifaceted mechanisms of
estradiol and estrogen receptor signaling. J Biol Chem, 2001. 276(40): p.
36869-72.
McDonnell, D.P. and J.D. Norris, Connection and regulation of the human
estrogen receptor. Science, 2002. 296: p. 1642-44.
Mak, H.Y., S. Hoare, P.M. Henttu, and M.G. Parker, Molecular determinants
of the estrogen receptor-coactivator interface. Mol Cell Biol, 1999. 19: p.
3895-903.
Nikolov, D.B., S.H. Hu, J. Lin, A. Gasch, A. Hoffmann, M. Horikoshi, N.H.
Chua, R.G. Roeder, and S.K. Burley, Crystal strutture of TFIID TATA-box
binding protein. Nature, 1992. 360(6399): p. 40-46.
Nikolov, D.B., H. Chen, E.D. Halay, A.A. Usheva, K. Hisatake, D.K. Lee,
R.G. Roeder, and S.K. Burley, Crystal structure of TFIID-TBP-TATA-element
ternary complex. Nature, 1995. 377(6545): p. 119-128.
Bourguet, W., M. Ruff, P. Chambon, H. Gronemeyer, and D. Moras, Crystal
structure of the ligand-binding domain of the human nuclear receptor RXR-alpha. Nature, 1995. 375(6530): p. 377-82.
Renaud, J.P., N. Rochel, M. Ruff, V. Vivat, P. Chambon, H. Gronemeyer, and
D. Moras, Crystal structure of the RAR-gamma ligand-binding domain bound
to all-trans retinoic acid. Nature, 1995. 378(6558): p. 681-9.
Lee, J.W., Y.C. Lee, S.Y. Na, D.J. Jung, and S.K. Lee, Transcriptional coregulators of the nuclear receptor superfamily: coactivators and corepressors.
Cell Mol Life Sci, 2001. 58(2): p. 289-97.
Kraus, W.L. and J.T. Kadonaga, p300 and estrogen receptor cooperatively activate transcription via differential enhancement of initiation and reinitiation.
Genes Dev, 1996. 12: p. 11540-45.
Hanstein, B., R. Eckner, J. DiRenzo, S. Halachmi, H. Liu, and e. al., p300 is a
component of an estrogen receptor coactivator complex. Proc Natl Acad Sci
USA, 1996. 93: p. 11540-45.
Heery, D.M., E. Kalkhoven, S. Hoare, and M.G. Parker, A signature motif in
transcriptional co-activators mediates binding to nuclear receptors. Nature,
1997. 387(6634): p. 733-6.
Darimont, B.D., R.L. Wagner, J.W. Apriletti, M.R. Stallcup, P.J. Kushner, and
e. al., Structure and specificity of nuclear receptor-coactivator interaction. Genes Dev, 1998. 12: p. 3343-56.
Mc Inerney, E.M., D.W. Rose, S.E. Flynn, S. Westin, T.M. Mullen, and e. al.,
Determination of coactivator LXXLL motif specificity in nuclear receptor
transcriptional activation. Genes Dev, 1998. 12: p. 3357-68.
Hu, X. and M.A. Lazar, Transcriptional repression by nuclear hormone receptors. Trends Endocrinol Metab, 2000. 11: p. 6-10.
Zhang, X., M. Jeyakumar, S. Petukhov, and M.K. Bagchi, A nuclear receptor
corepressor modulates transcriptional activity of antagonist-occupied steroid
hormone receptor. Mol Endocrinol, 1998. 12(4): p. 513-24.
125
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.
59.
60.
61.
62.
Smith, C.L., Z. Nawaz, and B.W. O'Malley, Coactivator and corepressor regulation of the agonist/antagonist activity of the mixed antiestrogen, 4-hydroxytamoxifen. Mol Endocrinol, 1997. 11(6): p. 657-66.
Montano, M.M., K. Ekena, R. Delage-Mourroux, W.R. Chang, P. Martini, and
B.S. Katzenellenbogen, An estrogen receptor-selective coregulator that potentyates the effectiveness of antiestrogens and represses the activity of estrogens.
Proc Natl Acad Sci USA, 1999. 96: p. 6947-52.
Johansson, L., A. Bavner, J.S. Thonsen, M. Fornegardh, J.A. Gustafsson, and
E. Treuter, The orphan nuclear receptor SHP utilizes conserved LXXLL-related motifs for interaction with ligand-activated estrogen receptor. Mol Cell
Biol, 2000. 20: p. 1124-33.
Johansson, L., J.S. Thomsen, A.E. Damdimopoulos, G. Spyrou, J.A. Gustafsson, and E. Treuter, The orphan nuclear receptor SHP inhibits agonist-dependent transcriptional activity of estrogen receptors ER-α and ER-ß. J Biol
Chem, 1999. 274: p. 345-53.
Ma, Z.Q., S. Santagati, C. Patrone, G. Pollio, E. Vegeto, and A. Maggi, Insulin-like grown factors activate estrogen receptor to control the grown and differentiation of the human neuroblastoma cell line SK-ER3. Mol Endocrinol,
1994. 8: p. 910-8.
Endoh, H., K. Maruyama, Y. Masuhiro, Y. Kobayashi, M. Goto, H. Tai, J. Yanagisawa, D. Metzger, S. Hashimoto, and S. Kato, Purification and identification of p68 RNA helicase acting as a transcriptional coactivator specific of the
activation funcion of Human estrogen receptor α. Mol Cell Biol, 1999. 19: p.
5363-72.
Castano, E., D.P. Vorojekina, and A.C. Notides, Phosphorylation of serine167 on the human estrogen receptor is important for estrogen response element binding and transcriptional activation. Biochem J, 1997. 326: p. 149-57.
Denton, R.R., N.J. Koszewski, and A.C. Notides, Estrogen receptor phosphorylation. Hormonal dependence and consequence on specific DNA binding. J
Biol Chem, 1992. 267: p. 7263-68.
Cenni, B. and D. Picard, Ligand-indipendent activaction of steroid receptors:
new roles for old players. Trends Endocrinol Metab, 1999. 10: p. 41-6.
Kato, S., H. Endoh, Y. Masuhiro, T. Kitamoto, S. Uchiyama, and e. al., Activation of the estrogen receptor through phosphorylation by mitogen-activated
protein kinase. Science, 1995. 270: p. 1491-4.
Briand, P., B.K. Lundholt, J. Skouv, and A.E. Lykkesfeldt, Growth response
of breast epithelial cells to estrogen influenced by EGF. Mol Cell Endocrinol,
1999. 153: p. 1-9.
Curtis, S.W., T. Washburn, C. Sewall, R. DiAugustine, J. Lindzey, and e. al.,
Physiological coupling of growth factor and steroid receptor signaling pathways: Estrogen receptor knockout mice lack estrogen-like response to epidermal growth factor. Proc Natl Acad Sci USA, 1996. 93: p. 12626-30.
Ignar-Trowbridge, D.M., K.G. Nelson, M.C. Bilwell, S.W. Curtis, T.F. Washburn, J.A. McLachlan, and K.S. Korach, Coupling of dual signaling pathways:
Epidermal growth factor action involves the estrogen receptor. Proc Natl Acad
Sci USA, 1992. 89: p. 4658-62.
Patrone, C., Z.Q. Ma, G. Pollio, P. Agrati, M.G. Parker, and A. Maggi, Crosscoupling between insulin and estrogen receptor in human neuroblastoma
cells. Mol Endocrinol, 1996. 10(5): p. 499-507.
126
63.
64.
65.
66.
67.
68.
69.
70.
71.
72.
73.
74.
75.
76.
Ignar-Trowbridge, D.M., M. Pimentel, M.G. Parker, J.A. McLachlan, and K.S.
Korach, Peptide growth factor cross-talk with the estrogen receptor requires
the A/B domain and occurs independently of proteine kinase C or estradiol.
Endocrinology, 1996. 137: p. 1735-44.
Ciana, P., S. Ghisletti, P. Mussi, I. Eberini, E. Vegeto, and A. Maggi, Estrogen
receptor alpha, a molecular switch converting transforming growth factor-alpha-mediated proliferation into differentiation in neuroblastoma cells. J Biol
Chem, 2003. 278(34): p. 31737-44.
Campbell, R.A., P. Bhat-Nakshatri, N.M. Patel, D. Costantinidou, S. Ali, and
H. Nakshatri, Phosphatidylinositol 3-kinase/AKT-mediated activation of estrogen receptor α. J Biol Chem, 2001. 276: p. 9817-24.
Zwijsen, R.M., E. Wientjens, R. Klompmaker, J. van der Sman, R. Bernards,
and R.J. Michalides, CDK-indipendent activation of estrogen receptor by ciclin D. Cell, 1997. 88: p. 405-15.
Rogatsky, I., J.M. Trowbridge, and M.J. Garabedian, Potentation of human
estrogen receptor α transcriptional activation through phosphorylation of serines 104 and 106 by cyclin A-CDK2 complex. J Biol Chem, 1999. 274: p.
22296-302.
El Tanani, M.K. and C.D. Green, Two separate mechanisme for ligand-indipendent activation of the estrogen receptor. Mol Endocrinol, 1997. 11: p. 92837.
Coleman, K.M., M. Dutertre, A. El-Gharbawy, B.G. Rowan, N.L. Weigel, and
C.L. Smith, Mechanistic differences in activation of estrogen receptor-α (ER
α) and ERß-dipendent gene expression by AMP signaling pathway(s). J Biol
Chem, 2003.
Tremblay, G.B., A. Tremblay, F. Labrie, and V. Giguere, Ligand-indipendent
activation of the estrogen receptors α and ß by mutation of a conserved tyrosine can be abolished by antiestrogens. Cancer res, 1998. 58: p. 877-81.
Kurebayashi, S., Y. Miyashita, T. Hirose, S. Kasayama, S. Akira, and T. Kishimoto, Characterization of mechanisms of interleukin-6 gene repression by
estrogen receptor. J Steroid Biochem Mol Biol, 1997. 60(1-2): p. 11-7.
Galien, R., H.F. Evans, and T. Garcia, Involvement of CCAAT/enhancer-binding protein and nuclear factor-kappa B binding sites in interleukin-6 promoter inhibition by estrogens. Mol Endocrinol, 1996. 10(6): p. 713-22.
Galien, R. and T. Garcia, Estrogen receptor impairs interleukin-6 expression
by preventing protein binding on the NF-κB site. Nucleic Acids Research,
1997. 25(12).
Deshpande, R., H. Khalili, R.G. Pergolizzi, S.D. Michael, and M.D. Chang,
Estradiol down-regulates LPS-induced cytokine production and NFkB activation in murine macrophages. Am J Reprod Immunol, 1997. 38(1): p. 46-54.
Ray, P., S.K. Ghosh, D.H. Zhang, and A. Ray, Repression of interleukin-6
gene expression by 17 beta-estradiol: inhibition of the DNA-binding activity of
the transcription factors NF-IL6 and NF-kappa B by the estrogen receptor.
FEBS Lett, 1997. 409(1): p. 79-85.
Ray, A., K.E. Prefontaine, and P. Ray, Down-modulation of interleukin-6 gene
expression by 17 beta-estradiol in the absence of high affinity DNA binding by
the estrogen receptor. J Biol Chem, 1994. 269(17): p. 12940-6.
127
77.
78.
79.
80.
81.
82.
83.
84.
85.
86.
87.
88.
89.
90.
91.
92.
93.
Inadera, H., T. Sekiya, T. Yoshimura, and K. Matsushima, Molecular analisis
of the inhibition of monocyte chemoattractant protein-1 gene expression by
estrogen and xenoestrogen in MCF-7. Endocrinology, 2000. 141: p. 50-9.
Kanda, N. and S. Watanabe, 17beta-estradiol inhibits the production of RANTES in human keratinocytes. J Invest Dermatol, 2003. 120(3): p. 420-7.
Gaub, M.P., M. Bellard, I. Scheuer, P. Chambon, and P. Sassone-Corsi, Activation of the ovalbumin gene by the estrogen receptor involves the fos-jun
complex. Cell, 1990. 63(6): p. 1267-76.
Philips, A., D. Chalbos, and H. Rochefort, Estradiol increases and anti-estrogens antagonize the growth factor-induced activator protein-1 activity in
MCF7 breast cancer cells without affecting c-fos and c-jun synthesis. J Biol
Chem, 1993. 268(19): p. 14103-8.
Srivastava, S., M.N. Weitzmann, S. Cenci, F.P. Ross, S. Adler, and R. Pacifici,
Estrogen decreases TNF gene expression by blocking JNK activity and the resulting production of c-Jun and JunD. J Clin Invest, 1999. 104(4): p. 503-13.
An, J., R.C. Ribeiro, P. Webb, J.A. Gustafsson, P.J. Kushner, J.D. Baxter, and
D.C. Leitman, Estradiol repression of tumor necrosis factor-alpha transcription requires estrogen receptor activation function-2 and is enhanced by coactivators. Proc Natl Acad Sci U S A, 1999. 96(26): p. 15161-6.
Kanda, N. and S. Watanabe, 17Beta-estradiol inhibits MCP-1 production in
human keratinocytes. J Invest Dermatol, 2003. 120(6): p. 1058-66.
Galea, E., R. Santizo, D.L. Feinstein, P. Adamsom, J. Greenwood, H.M. Koenig, and D.A. Pelligrino, Estrogen inhibits NF kappa B-dependent inflammation in brain endothelium without interfering with I kappa B degradation. Neuroreport, 2002. 13(11): p. 1469-72.
Caulin-Glaser, T., C.A. Watson, R. Pardi, and J.R. Bender, Effects of 17betaestradiol on cytokine-induced endothelial cell adhesion molecule expression. J
Clin Invest, 1996. 98(1): p. 36-42.
Sun, G., W. Porter, and S. Safe, Estrogen-induced retinoic acid receptor -Sp-1
complex. Mol Endocrinol, 1998. 12: p. 882-90.
Porter, W., B. Saville, D. Hoivik, and S. Safe, Functional synergy between the
transcription factor Sp1 and the estrogen receptor. Mol Endocrinol, 1999. 11:
p. 1569-80.
Kelly, M.J., R.L. Moss, and C.A. Dudley, Differential sensitivity of preopticseptal neurons to microelectrophoresed estrogen during the estrous cycle.
Brain Res, 1976. 114(1): p. 152-7.
Pietras, R.J. and C.M. Szego, Specific binding sites for estrogen at the outer
surfaces of isolated endometrial cells. Nature, 1977. 265: p. 69-72.
Pietras, R.J. and C.M. Szego, Partial purification and characterization of oestrogen receptors in subfractions of hepatocyte plasma membranes. Biochem
J, 1980. 191(3): p. 743-60.
Levin, E.R., Cellular functions of plasma membrane estrogen receptors. Steroids, 2002. 67(6): p. 471-5.
Maggi, A., P. Ciana, S. Belcredito, and E. Vegeto, Estrogens in the nervous
system: mechanisms and nonreproductive functions. Annu Rev Physiol, 2004.
66: p. 291-313.
Simoncini, T., L. Fornari, P. Mannella, G. Varone, A. Caruso, J.K. Liao, and
A.R. Genazzani, Novel non-transcriptional mechanism for estrogen receptor
128
94.
95.
96.
97.
98.
99.
100.
101.
102.
103.
104.
105.
106.
107.
108.
signaling in the cardiovascular system. Interaction of estrogen receptor α with
phosphatidylinositol 3-OH kinase. Steroids, 2002. 67: p. 935-9.
Chaban, V.V., A.J. Lakhter, and P. Micevych, A membrane estrogen receptor
mediates intracellular calcium release in astrocytes. Endocrinology, 2004.
145(8): p. 3788-95.
Tesarik, J. and C. Mendoza, Nongenomic effects of 17 beta-estradiol on maturing human oocytes: relationship to oocyte developmental potential. J Clin Endocrinol Metab, 1995. 80(4): p. 1438-43.
Abraham, I.M., S.K. Han, M.G. Todman, K.S. Korach, and A.E. Herbison,
Estrogen receptor beta mediates rapid estrogen actions on gonadotropin-releasing hormone neurons in vivo. J Neurosci, 2003. 23(13): p. 5771-7.
Mermelstein, P.G., J.B. Becker, and D.J. Surmeier, Estradiol reduces calcium
currents in rat neostriatal neurons via a membrane receptor. J Neurosci, 1996.
16(2): p. 595-604.
Chaban, V.V., E.A. Mayer, H.S. Ennes, and P.E. Micevych, Estradiol inhibits
atp-induced intracellular calcium concentration increase in dorsal root ganglia neurons. Neuroscience, 2003. 118(4): p. 941-8.
Nilsen, J. and R.D. Brinton, Divergent impact of progesterone and medroxyprogesterone acetate (Provera) on nuclear mitogen-activated protein kinase
signaling. Proc Natl Acad Sci U S A, 2003. 100(18): p. 10506-11.
Lagrange, A.H., O.K. Ronnekleiv, and M.J. Kelly, Modulation of G proteincoupled receptors by an estrogen receptor that activates protein kinase A. Mol
Pharmacol, 1997. 51(4): p. 605-12.
Favit, A., L. Fiore, F. Nicoletti, and P.L. Canonico, Estrogen modulates stimulation of inositol phospholipid hydrolysis by norepinephrine in rat brain slices. Brain Res, 1991. 555(1): p. 65-9.
Beyer, C. and H. Raab, Nongenomic effects of oestrogen: embryonic mouse
midbrain neurones respond with a rapid release of calcium from intracellular
stores. Eur J Neurosci, 1998. 10(1): p. 255-62.
Aronica, S.M., W.L. Kraus, and B.S. Katzenellenbogen, Estrogen action via
the cAMP signalin pathway: stimulation of adenylate cyclase and cAMP-regulated transcription. Proc Natl Acad Sci USA, 1994. 91: p. 8517-21.
Minami, T., Y. Oomura, J. Nabekura, and A. Fukuda, 17 beta-estradiol depolarization of hypothalamic neurons is mediated by cyclic AMP. Brain Res,
1990. 519(1-2): p. 301-7.
Watters, J.J. and D.M. Dorsa, Transcriptional effects of estrogen on neuronal
neurotensin gene expression involve cAMP/protein kinase A-dependent signaling mechanisms. J Neurosci, 1998. 18(17): p. 6672-80.
Kelly, M.J. and E.R. Levin, Rapid actions of plasma membrane estrogen receptors. Trends Endocrinol Metab, 2001. 12(4): p. 152-6.
Maus, M., P. Bertrand, S. Drouva, R. Rasolonjanahary, C. Kordon, J. Glowinski, J. Premont, and A. Enjalbert, Differential modulation of D1 and D2 dopamine-sensitive adenylate cyclases by 17 beta-estradiol in cultured striatal neurons and anterior pituitary cells. J Neurochem, 1989. 52(2): p. 410-8.
Palmon, S.C., M.J. Williams, M.T. Littleton-Kearney, R.J. Traystman, D.
Kosk-Kosicka, and P.D. Hurn, Estrogen increases cGMP in selected brain regions and in cerebral microvessels. J Cereb Blood Flow Metab, 1998. 18(11):
p. 1248-52.
129
109.
110.
111.
112.
113.
114.
115.
116.
117.
118.
119.
120.
121.
122.
123.
Russell, K.S., M.P. Haynes, D. Sinha, E. Clerisme, and J.R. Bender, Human
vascular endothelial cells contain membrane binding sites for estradiol, which
mediate rapid intracellular signaling. Proc Natl Acad Sci U S A, 2000.
97(11): p. 5930-5.
Le Mellay, V., B. Grosse, and M. Lieberherr, Phospholipase C ß and membrane action of calcitriol and estradiol. J Biol Chem, 1997. 272: p. 11902-7.
Kelly, M.J. and E.J. Wagner, Estrogen modulation of G-protein-coupled receptors. Trends Endocrinol Metab, 1999. 10: p. 369-74.
Razandi, M., A. Pedram, G.L. Greene, and E.R. Levin, Cell membrane and
nuclear estrogen receptors (ERs) originate from a single transcript: studies of
ERalpha and ERbeta expressed in Chinese hamster ovary cells. Mol Endocrinol, 1999. 13(2): p. 307-19.
Wyckoff, M.H., K.L. Chambliss, C. Mineo, I.S. Yuhanna, M.E. Mendelsohn,
S.M. Mumby, and P.W. Shaul, Plasma membrane estrogen receptors are coupled to endothelial nitric-oxide synthase through Galpha(i). J Biol Chem,
2001. 276(29): p. 27071-6.
Kelly, M.J., J. Qiu, and O.K. Ronnekleiv, Estrogen modulation of G-proteincoupled receptor activation of potassium channels in the central nervous system. Ann N Y Acad Sci, 2003. 1007: p. 6-16.
Gu, G., A.A. Rojo, M.C. Zee, J. Yu, and R.B. Simerly, Hormonal regulation
of CREB phosphorylation in the anteroventral periventricular nucleus. J Neurosci, 1996. 16(9): p. 3035-44.
Honda, K., S. Shimohama, H. Sawada, T. Kihara, T. Nakamizo, H. Shibasaki,
and A. Akaike, Nongenomic antiapoptotic signal transduction by estrogen in
cultured cortical neurons. J Neurosci Res, 2001. 64(5): p. 466-75.
Zhang, L., D.R. Rubinow, G. Xaing, B.S. Li, Y.H. Chang, D. Maric, J.L. Barker, and W. Ma, Estrogen protects against beta-amyloid-induced neurotoxicity
in rat hippocampal neurons by activation of Akt. Neuroreport, 2001. 12(9): p.
1919-23.
Znamensky, V., K.T. Akama, B.S. McEwen, and T.A. Milner, Estrogen levels
regulate the subcellular distribution of phosphorylated Akt in hippocampal
CA1 dendrites. J Neurosci, 2003. 23(6): p. 2340-7.
Colin, I.M. and J.L. Jameson, Estradiol sensitization of rat pituitary cells to
gonadotropin-releasing hormone: involvement of protein kinase C- and calcium-dependent signaling pathways. Endocrinology, 1998. 139(9): p. 3796802.
Ansonoff, M.A. and A.M. Etgen, Estradiol elevates protein kinase C catalytic
activity in the preoptic area of female rats. Endocrinology, 1998. 139(7): p.
3050-6.
Hayashi, T., T. Ishikawa, K. Yamada, M. Kuzuya, M. Naito, H. Hidaka, and
A. Iguchi, Biphasic effect of estrogen on neuronal constitutive nitric oxide
synthase via Ca(2+)-calmodulin dependent mechanism. Biochem Biophys Res
Commun, 1994. 203(2): p. 1013-9.
Watters, J.J., J.S. Campbell, M.J. Cunningham, E.G. Krebs, and D.M. Dorsa,
Rapid membrane effects of steroids in neuroblastoma cells: effects of estrogen
on mitogen activated protein kinase signalling cascade and c-fos immediate
early gene transcription. Endocrinology, 1997. 138: p. 4030-3.
Wade, C.B., S. Robinson, R.A. Shapiro, and D.M. Dorsa, Estrogen receptor
(ER)alpha and ERbeta exhibit unique pharmacologic properties when coupled
130
124.
125.
126.
127.
128.
129.
130.
131.
132.
133.
134.
135.
136.
137.
to activation of the mitogen-activated protein kinase pathway. Endocrinology,
2001. 142(6): p. 2336-42.
Razandi, M., A. Pedram, and E.R. Levin, Estrogen signals to the preservation
of endothelial cell form and function. J Biol Chem, 2000. 275(49): p. 38540-6.
Migliaccio, A., M. Di Domenico, G. Castoria, A. de Falco, P. Bontempo, E.
Nola, and e. al., Tyrosine kinase/p21ras/MAP-kinase pathway activation by
estradiol-receptor complex in MCF-7 cells. EMBO J, 1996. 15: p. 1292-1300.
Jezierski, M.K., A.K. Sturm, M.M. Scarborough, and F. Sohrabji, NGF stimulation increases JNK2 phosphorylation and reduces caspase-3 activity in the
olfactory bulb of estrogen-replaced animals. Endocrinology, 2001. 142(6): p.
2401.
Wymann, M.P., M. Zvelebil, and M. Laffargue, Phosphoinositide 3-kinase signalling--which way to target? Trends Pharmacol Sci, 2003. 24(7): p. 366-76.
Stoica, G.E., T.F. Franke, A. Wellstein, E. Morgan, F. Czubayko, H.J. List, R.
Reiter, M.B. Martin, and A. Stoica, Heregulin-beta1 regulates the estrogen receptor-alpha gene expression and activity via the ErbB2/PI 3-K/Akt pathway.
Oncogene, 2003. 22(14): p. 2073-87.
Stoica, A., M. Saceda, V.L. Doraiswamy, C. Coleman, and M.B. Martin, Regulation of estrogen receptor-alpha gene expression by epidermal growth factor. J Endocrinol, 2000. 165(2): p. 371-8.
Stoica, A., M. Saceda, A. Fakhro, M. Joyner, and M.B. Martin, Role of insulin-like growth factor-I in regulating estrogen receptor-alpha gene expression.
J Cell Biochem, 2000. 76(4): p. 605-14.
Castoria, G., A. Migliaccio, A. Bilancio, M. Di Domenico, A. de Falco, M.
Lombardi, R. Fiorentino, L. Varricchio, M.V. Barone, and F. Auricchio, PI3kinase in concert with Src promotes the S-phase entry of estradiol-stimulated
MCF-7 cells. EMBO J, 2001. 20: p. 6050-9.
Simoncini, T., A.H. Moghadam, D.P. Brazil, K. Ley, W.W. Chin, and J.K.
Liao, Interaction of estrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature, 2000. 407: p. 538-41.
Kim, H.P., J.Y. Lee, J.K. Jeong, S.W. Bae, H.K. Lee, and I. Jo, Nongenomic
stimulation of nitritic oxide release by eatrogen is mediated by estrogen receptor α localized in caveolae. Biochem Biophys Res Commun, 1999. 263(1): p.
257-62.
Chambliss, K.L., I.S. Yuhanna, C. Mineo, P. Liu, Z. German, T.S. Sherman,
M.E. Mendelsohn, R.G. Anderson, and P.W. Shaul, Estrogen receptor alpha
and endothelial nitric oxide synthase are organized into a functional signaling
module in caveolae. Circ Res, 2000. 87(11): p. E44-52.
Honda, K., H. Sawada, T. Kihara, M. Urushitani, T. Nakamizo, A. Akaike, and
S. Shimohama, Phosphatidylinositol 3-kinase mediates neuroprotection by
estrogen in cultured cortical neurons. J Neurosci Res, 2000. 60(3): p. 321-7.
Razandi, M., G. Alton, A. Pedram, S. Ghonshani, P. Webb, and E.R. Levin,
Identification of a structural determinant necessary for the localization and
function of estrogen receptor alpha at the plasma membrane. Mol Cell Biol,
2003. 23(5): p. 1633-46.
Razandi, M., A. Pedram, I. Merchenthaler, G.L. Greene, and E.R. Levin, Plasma Membrane Estrogen Receptors Exist and Functions as Dimers. Mol Endocrinol, 2004.
131
138.
139.
140.
141.
142.
143.
144.
145.
146.
147.
148.
149.
150.
151.
152.
Beyer, C., J. Pawlak, and M. Karolczak, Membrane receptors for oestrogen in
the brain. J Neurochem, 2003. 87(3): p. 545-50.
Das, S.K., J.A. Taylor, K.S. Korach, B.C. Paria, S.K. Dey, and D.B. Lubahn,
Estrogenic responses in estrogen receptor-alpha deficient mice reveal a distinct estrogen signaling pathway. Proc Natl Acad Sci U S A, 1997. 94(24): p.
12786-91.
Gu, Q., K.S. Korach, and R.L. Moss, Rapid action of 17beta-estradiol on kainate-induced currents in hippocampal neurons lacking intracellular estrogen
receptors. Endocrinology, 1999. 140(2): p. 660-6.
Nadal, A., A.B. Ropero, O. Laribi, M. Maillet, E. Fuentes, and B. Soria, Nongenomic actions of estrogens and xenoestrogens by binding at a plasma membrane receptor unrelated to estrogen receptor alpha and estrogen receptor beta. Proc Natl Acad Sci U S A, 2000. 97(21): p. 11603-8.
Filardo, E.J., J.A. Quinn, K.I. Bland, and A.R. Frackelton, Jr., Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor
receptor through release of HB-EGF. Mol Endocrinol, 2000. 14(10): p. 164960.
Anuradha, P., S.M. Khan, N. Karthikeyan, and R.V. Thampan, The nonactivated estrogen receptor (naER) of the goat uterus is a tyrosine kinase. Arch Biochem Biophys, 1994. 309(2): p. 195-204.
Haynes, M.P., L. Li, K.S. Russell, and J.R. Bender, Rapid vascular cell responses to estrogen and membrane receptors. Vascular Farmcology, 2001. 38:
p. 99-108.
Haynes, M.P., D. Sinha, K.S. Russell, M. Collinge, D. Fulton, M. MoralesRuiz, W.C. Sessa, and J.R. Bender, Membrane estrogen receptor engagement
activates endothelial nitric oxide synthase via the PI3-kinase-Akt pathway in
human endothelial cells. Circ Res, 2000. 87(8): p. 677-82.
Marquez, D.C. and R.J. Pietras, Membrane-associated binding sites for estrogen contribute to growth regulation of human breast cells. Oncogene, 2001.
20: p. 5420-30.
Flouriot, G., H. Brand, S. Denger, R. Metivier, M. Kos, G. Reid, V. SonntagBuck, and F. Gannon, Identification of new isoform of human estrogen receptor α (hERα) that is encoded by distinct transcripts and that is able to repress
hER-α activation funcion 1. EMBO J, 2000. 19(17): p. 4688-700.
Haynes, M.P., L. Li, D. Sinha, K.S. Russell, K. Hisamoto, R. Baron, M. Collinge, W.C. Sessa, and J.R. Bender, Src Kinase Mediates Phosphatidylinositol
3-Kinase/Akt-dependent Rapid Endothelial Nitric-oxide Synthase Activation
by Estrogen. J Biol Chem, 2003. 278: p. 2118-23.
Toran-Allerand, C.D., Novel sites and mechanisms of oestrogen action in the
brain. Novartis Found Symp, 2000. 230: p. 56-69; discussion 69-73.
Toran-Allerand, C.D., X. Guan, N.J. MacLusky, T.L. Horvath, S. Diano, M.
Singh, J.E.S. Connolly, I.S. Nethrapalli, and A.A. Tinnkov, ER-X: A novel,
plasma membrane-associated, putative estrogen receptor that is regulated during development and after ischemic brain injury. J Neuroscience, 2002. 22: p.
8391-401.
Robbins, S.L., R.S. Cotran, and V. Kumar, Basic pathology. 2003. 7th edition.
Dinarello, C.A., The IL-1 family and inflammatory diseases. Clin Exp Rheumatol, 2002. 20(5 Suppl 27): p. S1-13.
132
153.
154.
155.
156.
157.
158.
159.
160.
161.
162.
163.
164.
165.
166.
167.
168.
169.
Firestein, G.S., The T cell cometh: interplay between adaptive immunity and
cytokine networks in rheumatoid arthritis. J Clin Invest, 2004. 114(4): p. 4714.
Dijsselbloem, N., W. Vanden Berghe, A. De Naeyer, and G. Haegeman, Soy
isoflavone phyto-pharmaceuticals in interleukin-6 affections. Multi-purpose
nutraceuticals at the crossroad of hormone replacement, anti-cancer and antiinflammatory therapy. Biochem Pharmacol, 2004. 68(6): p. 1171-85.
Kontsek, P., G. Karayianni-Vasconcelos, and E. Kontsekova, The human interferon system: characterization and classification after discovery of novel
members. Acta Virol, 2003. 47(4): p. 201-15.
Sacca, R., C.A. Cuff, and N.H. Ruddle, Mediators of inflammation. Curr Opin
Immunol, 1997. 9(6): p. 851-7.
Rot, A. and U.H. von Andrian, Chemokines in innate and adaptive host defense: basic chemokinese grammar for immune cells. Annu Rev Immunol, 2004.
22: p. 891-928.
Lin, H.L. and S. Murphy, Regulation of astrocyte nitric oxide synthase type II
expression by ATP and glutamate involves loss of transcription factor binding
to DNA. J Neurochem, 1997. 69(2): p. 612-6.
Muhl, H. and J. Pfeilschifter, Endothelial nitric oxide synthase: a determinant
of TNFalpha production by human monocytes/macrophages. Biochem Biophys Res Commun, 2003. 310(3): p. 677-80.
Greten, F.R. and M. Karin, The IKK/NF-kappaB activation pathway-a target
for prevention and treatment of cancer. Cancer Lett, 2004. 206(2): p. 193-9.
Sen, R. and D. Baltimore, Inducibility of kappa immunoglobulin enhancerbinding protein Nf-kappa B by a posttranslational mechanism. Cell, 1986.
47(6): p. 921-8.
Sen, R. and D. Baltimore, Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell, 1986. 46(5): p. 705-16.
Ghosh, S., M.J. May, and E. Kopp, NF-κB and Rel proteins: Evolutionarily
conserved mediators of immune responses. Annu Rev Immunol, 1998. 16: p.
225-60.
Baeuerle, P.A. and D. Baltimore, I kappa B: a specific inhibitor of the NFkappa B transcription factor. Science, 1988. 242(4878): p. 540-6.
Baeuerle, P.A. and D. Baltimore, Activation of DNA-binding activity in an
apparently cytoplasmic precursor of the NF-kappa B transcription factor.
Cell, 1988. 53(2): p. 211-7.
Baeuerle, P.A., M. Lenardo, J.W. Pierce, and D. Baltimore, Phorbol-ester-induced activation of the NF-kappa B transcription factor involves dissociation
of an apparently cytoplasmic NF-kappa B/inhibitor complex. Cold Spring
Harb Symp Quant Biol, 1988. 53 Pt 2: p. 789-98.
Verma, I.M., J.K. Stevenson, E.M. Schwarz, D. Van Antwerp, and S. Miyamoto, Rel/NF-kappa B/I kappa B family: intimate tales of association and dissociation. Genes Dev, 1995. 9(22): p. 2723-35.
Grilli, M., J.J. Chiu, and M.J. Lenardo, NF-kappa B and Rel: participants in a
multiform transcriptional regulatory system. Int Rev Cytol, 1993. 143: p. 1-62.
Kopp, E.B. and S. Ghosh, NF-kappa B and rel proteins in innate immunity.
Adv Immunol, 1995. 58: p. 1-27.
133
170.
171.
172.
173.
174.
175.
176.
177.
178.
179.
180.
181.
182.
183.
184.
Hansen, S.K., L. Guerrini, and F. Blasi, Differential DNA sequence specificity
and regulation of HIV-1 enhancer activity by cRel-RelA transcription factor. J
Biol Chem, 1994. 269(35): p. 22230-7.
Hansen, S.K., P.A. Baeuerle, and F. Blasi, Purification, reconstitution, and I
kappa B association of the c-Rel-p65 (RelA) complex, a strong activator of
transcription. Mol Cell Biol, 1994. 14(4): p. 2593-603.
Brown, A.M., M.W. Linhoff, B. Stein, K.L. Wright, A.S. Baldwin, Jr., P.V.
Basta, and J.P. Ting, Function of NF-kappa B/Rel binding sites in the major
histocompatibility complex class II invariant chain promoter is dependent on
cell-specific binding of different NF-kappa B/Rel subunits. Mol Cell Biol,
1994. 14(5): p. 2926-35.
Kang, S.M., A.C. Tran, M. Grilli, and M.J. Lenardo, NF-kappa B subunit regulation in nontransformed CD4+ T lymphocytes. Science, 1992. 256(5062):
p. 1452-6.
Plaksin, D., P.A. Baeuerle, and L. Eisenbach, KBF1 (p50 NF-kappa B homodimer) acts as a repressor of H-2Kb gene expression in metastatic tumor cells.
J Exp Med, 1993. 177(6): p. 1651-62.
McKay, L.I. and J.A. Cidlowski, Molecular Control of Immune/Inflammatory
Responses: Interaction between Nuclear Factor-κB and Steroid Receptor-Signaling Pathways. Endocr Rev, 1999. 20: p. 435-59.
Nolan, G.P., NF-AT-AP-1 and Rel-bZIP: hybrid vigor and binding under the
influence. Cell, 1994. 77(6): p. 795-8.
Henkel, T., U. Zabel, K. van Zee, J.M. Muller, E. Fanning, and P.A. Baeuerle,
Intramolecular masking of the nuclear location signal and dimerization domain in the precursor for the p50 NF-kappa B subunit. Cell, 1992. 68(6): p.
1121-33.
Gilmore, T.D., The Rel/NF-κB signal transduction pathway: introduction. Oncogene, 1999. 18: p. 6842-44.
Prigent, M., I. Barlat, H. Langen, and C. Dargemont, IkappaBalpha and IkappaBalpha /NF-kappa B complexes are retained in the cytoplasm through interaction with a novel partner, RasGAP SH3-binding protein 2. J Biol Chem,
2000. 275(46): p. 36441-9.
Johnson, C., D. Van Antwerp, and T.J. Hope, An N-terminal nuclear export signal is required for the nucleocytoplasmic shuttling of IκBa. EMBO J, 1999.
18: p. 6682-93.
Tam, W.F., L.H. Lee, L. Davis, and R. Sen, Cytoplasmic sequestration of Rel
proteins by IκBα requires CRM1-dependent nuclear export. Mol Cell Biol,
2000. 20: p. 2269-84.
Huang, T.T., N. Kudo, M. Yoshida, and S. Miyamoto, A nuclear export signal
in the N-terminal regulatory domain of IκBα controls cytoplasmic localization
of inactive NF-κB/IκBα complexes. Proc Natl Acad Sci USA, 2000. 97: p.
1014-9.
Karin, M., The beginning of the end: IκB Kinase (IKK) and NF-κB Activation.
J Biol Chem, 1999. 274: p. 27339-42.
Zhong, H., R.E. Voll, and S. Ghosh, Phosphorylation of NF-κB p65 by PKA
stimulates transcriptional activity by promoting a novel bivalent interaction
with the coactivator CBP/p300. Mol Cell, 1998. 1: p. 661.
134
185.
186.
187.
188.
189.
190.
191.
192.
193.
194.
195.
196.
197.
198.
199.
Wang, D. and A.S. Baldwin, Activation of nuclear factor-κB-dependent transcription by tumor necrosis factor –α mediates through phosphorylation of
RelA/p65 on serine 529. J Biol Chem, 1998. 273: p. 29441.
Wang, D., S.D. Westerheide, J.L. Hanson, and A.S. Baldwin, Tumor Necrosis
factor α-induced phosphorylation of RelA/p65 on Ser529 is controlled by casein
kinase II. J Biol Chem, 2000. 275: p. 32592-7.
Sakurai, H., H. Chiba, H. Miyoshi, and W. Sugita Toriumi, IκB kinases phosphorylate NF-κB p65 subunit on serine 536 in the transactivation domain. J
Biol Chem, 1999. 274: p. 30353-6.
Guerini Rocco, A., Unpublished data. 2003.
Yang, F., E. Tang, K. Guan, and C.Y. Wang, IKKß plays an essential role in
the phosphorylation of RelA/p65 on serine 536 induced by lipopolysaccharide. Journal of Immunology, 2003. 170: p. 5630-5.
Vermeulen, L., G. De Wilde, S. Notebaert, W. Vanden Berghe, and G. Haegeman, Regulation of the transcriptional activity of the nuclear factor-κB p65
subunit. Biochemical Pharmacology, 2002. 64: p. 963-70.
Guha, M. and N. Mackman, The phosphatidylinositol 3-kinase-Akt pathway limits lipopolysaccharide activation of signaling pathways and expression of inflammatory mediators in human monocytic cells. J Biol Chem, 2002. 277(35):
p. 32124-32.
Diaz-Guerra, M.J., A. Castrillo, P. Martin-Sanz, and L. Bosca, Negative regulation by phosphatidylinositol 3-kinase of inducible nitric oxide synthase expression in macrophages. J Immunol, 1999. 162(10): p. 6184-90.
Bowling, W.M., M.W. Flye, Y.Y. Qiu, and M.P. Callery, Inhibition of phosphatidylinositol-3'-kinase prevents induction of endotoxin tolerance in vitro. J
Surg Res, 1996. 63(1): p. 287-92.
Park, Y.C., C.H. Lee, H.S. Kang, H.T. Chung, and H.D. Kim, Wortmannin, a
specific inhibitor of phosphatidylinositol-3-kinase, enhances LPS-induced NO
production from murine peritoneal macrophages. Biochem Biophys Res Commun, 1997. 240(3): p. 692-6.
Tengku-Muhammad, T.S., T.R. Hughes, A. Cryer, and D.P. Ramji, Involvement of both the tyrosine kinase and the phosphatidylinositol-3' kinase signal
transduction pathways in the regulation of lipoprotein lipase expression in
J774.2 macrophages by cytokines and lipopolysaccharide. Cytokine, 1999.
11(7): p. 463-8.
Arbibe, L., J.P. Mira, N. Teusch, L. Kline, M. Guha, N. Mackman, P.J. Godowski, R.J. Ulevitch, and U.G. Knaus, Toll-like receptor 2-mediated NF-kappa
B activation requires a Rac1-dependent pathway. Nat Immunol, 2000. 1(6): p.
533-40.
Jones, B.W., T.K. Means, K.A. Heldwein, M.A. Keen, P.J. Hill, J.T. Belisle,
and M.J. Fenton, Different Toll-like receptor agonists induce distinct macrophage responses. J Leukoc Biol, 2001. 69(6): p. 1036-44.
Thomas, K.W., M.M. Monick, J.M. Staber, T. Yarovinsky, A.B. Carter, and
G.W. Hunninghake, Respiratory syncytial virus inhibits apoptosis and induces
NF-kappa B activity through a phosphatidylinositol 3-kinase-dependent pathway. J Biol Chem, 2002. 277(1): p. 492-501.
Madrid, L.V., M.W. Mayo, J.Y. Reuther, and A.S. Baldwin, Jr., Akt stimulates
the transactivation potential of the RelA/p65 Subunit of NF-kappa B through
135
200.
201.
202.
203.
204.
205.
206.
207.
208.
209.
210.
211.
212.
213.
214.
215.
216.
utilization of the Ikappa B kinase and activation of the mitogen-activated protein kinase p38. J Biol Chem, 2001. 276(22): p. 18934-40.
Chen, L., W. Fischle, E. Verdin, and W.C. Greene, Duration of nuclear NF-κ
B action regulated by reversible acetylation. Science, 2001. 293: p. 1653-7.
Chen, L., Y. Mu, and W.C. Greene, Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-κB. EMBO J, 2002. 21: p. 6539-48.
Kiernan, R., V. Brest, R. NG, M.P. Courdat, S. Messaoudi, C. Sardet, D.Y. Jin,
S. Emiliani, and M. Benkirane, Post-activation turn-off NF-κB-dependent
transcription is regutated by acetylation of p65. J Biol Chem, 2003. 278: p.
2758-66.
Matejuk, A., K. Adlard, M. Silverman, A.A. Vandenbark, and H. Offner, 17ßestradiol inhibits cytokine, chemochine and chemochine receptor mRNA expression in the central nervous system of female mice with experimental autoimmune encephalomyelitis. Journal of Neuroscience Research, 2001. 65: p.
529-42.
Ito, A., A.C. Buenafede, A. Matejuk, A. Zamora, M. Silverman, J. Dwyer,
A.A. Vandenbark, and H. Offner, Estrogen inhibit systemic T cell expression
of TNF-α and recruitment of TNF-a +T cell and macrophages into the CNS of
mice developing. Clinical Immunology, 2002. 102: p. 275-82.
Pahl, H.L., Activactors and target genes of Rel/NF-κB transcription factors.
Oncogene, 1999. 18: p. 6853-66.
Saccani, S., S. Pantano, and G. Natoli, Two waves of nuclear factor kappaB
recruitment to target promoters. J Exp Med, 2001. 193(12): p. 1351-9.
Brand, K., S. Page, A.K. Walli, D. Neumeier, and P.A. Baeuerle, Role of nuclear factor-kappa B in atherogenesis. Exp Physiol, 1997. 82(2): p. 297-304.
Bourcier, T., G. Sukhova, and P. Libby, The nuclear factor kappa-B signaling
pathway participates in dysregulation of vascular smooth muscle cells in vitro
and in human atherosclerosis. J Biol Chem, 1997. 272(25): p. 15817-24.
Monaco, C. and E. Paleolog, Nuclear factor kappaB: a potential therapeutic
target in atherosclerosis and thrombosis. Cardiovasc Res, 2004. 61(4): p. 67182.
Bochner, B.S., B.J. Undem, and L.M. Linchtenstein, Immunological aspects of
allergic asthma. Annu Rev Immunol, 1994. 12: p. 295-335.
Feldmann, M., F.M. Brennan, and R.N. Maini, Role of cytochines in rheumatoid arthritis. Annu Rev Immunol, 1996. 14: p. 397-440.
Podolsky, D.K., Inflammatory bowel disease. N Engl J Med, 2000. 325: p.
928-37.
Yamamoto, Y. and R.B. Gaynor, Therapeutic potential of inhibition of the NFκB pathway in treatment of inflammation and cancer. J Clin Invest, 2001. 107:
p. 135-42.
Perkins, N.D., The rel/NF-κB family: friend and foe. TIBS, 2000. 25: p. 43440.
Mattson, M.P. and S. Camandola, NF-κB in neuronal plasticity and neurodegenerative disorders. J Clin Invest, 2001. 107: p. 247-54.
Wahl, C., S. Liptay, G. Adler, and R.M. Schmid, Sulfasalazine: a potent and
specific inhibitor of nuclear factor kappa B. J Clin Invest, 1998. 101(5): p.
1163-74.
136
217.
218.
219.
220.
221.
222.
223.
224.
225.
226.
227.
228.
229.
230.
231.
232.
233.
Yin, M.J., Y. Yamamoto, and R.B. Gaynor, The anti-inflammatory agents
aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature,
1998. 396(6706): p. 77-80.
Yamamoto, Y., M.J. Yin, K.M. Lin, and R.B. Gaynor, Sulindac inhibits activation of the NF-kappaB pathway. J Biol Chem, 1999. 274(38): p. 27307-14.
Rossi, A., P. Kapahi, G. Natoli, T. Takahashi, Y. Chen, M. Karin, and M.G.
Santoro, Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IkappaB kinase. Nature, 2000. 403(6765): p. 103-8.
Holmes-McNary, M. and A.S. Baldwin, Jr., Chemopreventive properties of
trans-resveratrol are associated with inhibition of activation of the IkappaB
kinase. Cancer Res, 2000. 60(13): p. 3477-83.
De Bosscher, K., M.L. Schmitz, W. Vanden Berghe, S. Plaisance, W. Fiers,
and G. Haegeman, Glucocorticoid-mediated repression of nuclear factor-kappaB-dependent transcription involves direct interference with transactivation.
Proc Natl Acad Sci U S A, 1997. 94(25): p. 13504-9.
Scheinman, R.I., A. Guadalberto, C.M. Jewell, J.A. Cidlowski, and A.S. Baldwin, Characterization of mechanisms involved in transrepression of NF-κB by
activated glucocorticoid receptors. Mol Cell Biol, 1995. 15: p. 943-53.
Nissen, R.M. and K.R. Yamamoto, The glucocorticoid receptor inhibits NFkappaB by interfering with serine-2 phosphorylation of the RNA polymerase II
carboxy-terminal domain. Genes Dev, 2000. 14(18): p. 2314-29.
Ricote, M., A.C. Li, T.M. Willson, C.J. Kelly, and C.K. Glass, The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature, 1998. 391(6662): p. 79-82.
Auphan, N., J. DiDonato, C. Rosette, A. Hemberg, and M. Karin, Immunosuppression by glucocorticoids: inhibition of NFκB activity through induction of I
κB synthesis. Science, 1995. 270: p. 286-90.
Scheinman, R.I., P.C. Cogswell, A.K. Lofquist, and A.S. Baldwin, Role of
transcriptional activation of IκBa in mediation of immunosuppression by glucocorticoids. Science, 1995. 270: p. 283-6.
Guha, M. and N. Mackman, LPS induction of gene expression in human monocytes. Cellular Signalling, 2001. 13: p. 85-94.
Belcredito, S., E. Vegeto, A. Brusadelli, S. Ghisletti, P. Mussi, P. Ciana, and
A. Maggi, Estrogen neuroprotection: the involvement of the Bcl-2 binding
protein BNIP2. Brain Res Brain Res Rev, 2001. 37(1-3): p. 335-42.
Vegeto, E., P. Ciana, and A. Maggi, Estrogen and inflammation: hormone generous action spreads to the brain. Mol Psychiatry, 2002. 7(3): p. 236-8.
Steinman, L., Multiple Scerosis: a coordinated immunogical attack against
myelin in the central nervous system. Cell, 1996. 85: p. 299-302.
Sadovnick, A.D. and G.C. Ebers, Epidemiology of multiple sclerosis: a critical
overview. Can J Neurol Sci, 1993. 20(1): p. 17-29.
Ebers, G.C., A.D. Sadovnick, N.J. Rische, and a.t.C.C.S. Group, A genetic basis for familiar aggregation in MS. Nature, 1995. 377: p. 150-1.
Gregoire, A.J., R. Kumar, B. Everitt, A.F. Henderson, and J.W. Studd, Transdermal estrogen for treatment of severe postnatal depression. Lancet, 1996.
347: p. 930-3.
137
234.
235.
236.
237.
238.
239.
240.
241.
242.
243.
244.
245.
246.
247.
248.
249.
250.
Thompson, D.S., L.M. Nelson, A. Burns, J.S. Burks, and G.M. Franklin, The
effects of pregnancy in multiple sclerosis: a retrospective study. Neurology,
1986. 36(8): p. 1097-9.
Damek, D.M. and E.A. Shuster, Pregnancy and multiple sclerosis. Mayo Clin
Proc, 1997. 72(10): p. 977-89.
Pozzilli, C., P. Falaschi, C. Mainero, A. Martocchia, R. D'Urso, A. Proietti, M.
Frontoni, S. Bastianello, and M. Filippi, MRI in multiple sclerosis during the
menstrual cycle: relationship with sex hormone patterns. Neurology, 1999.
53(3): p. 622-4.
Zorgdrager, A. and J. De Keyser, Menstrually related worsening of symptoms
in multiple sclerosis. J Neurol Sci, 1997. 149(1): p. 95-7.
Villard-Mackintosh, L. and M.P. Vessey, Oral contraceptives and reproductive factors in multiple sclerosis incidence. Contraception, 1993. 47(2): p. 1618.
Jansson, L. and R. Holmdahl, Estrogen-mediated immunosuppression in autoimmune diseases. Inflamm Res, 1998. 47: p. 290-301.
Brocke, S., A. Gaur, C. Piercy, A. Gautam, K. Gijbels, C.G. Fathman, and L.
Steinman, Induction of relapsing paralysis in experimental encephalomyelitis
by bacterial superantigen. Nature, 1993. 365: p. 642-4.
Hoffman, G.E., W.W. Le, A.Z. Murphy, and C.L. Koski, Divergent effects of
ovarian steroids on neuronal survival during experimental allergic encephalitis in Lewis rats. Experimental Neurology, 2001. 171: p. 272-84.
Liu, H.Y., A.C. Buenafede, A. Matejuk, A. Ito, A. Zamora, J. Dwyer, A.A.
Vandenbark, and H. Offner, Estrogen inhibition of EAE involves effects on
Dendritic cell funcion. Journal of Neuroscience Research, 2002. 70: p. 238-48.
Ito, A., B.F.J. Bebo, A. Matejuk, A. Zamora, M. Silverman, A. Fyfe-Johnson,
and H. Offner, Estrogen treatment down-regulates TNF-α production and reduces the severity of experimental autoimmune encephalomyelitis in cytokine
knockout mice. Journal of Immunology, 2001. 167: p. 529-42.
Polanczyk, M., A. Zamora, S. Subramanian, A. Matejuk, D.L. Hess, E.P. Blankenhorn, C. Teuscher, A.A. Vandenbark, and H. Offner, The protective effect
of 17beta-estradiol on experimental autoimmune encephalomyelitis is mediated through estrogen receptor-alpha. Am J Pathol, 2003. 163(4): p. 1599-605.
OMIM, Online Mendelian Inheritance in Man.
Matsuda, J., M.T. Vanier, Y. Saito, J. Tohyama, K. Suzuki, and S. Kunihiko,
A mutation in the saposin A domain of the sphingolipid activator protein (prosaposin) gene results in a late-onset, chronic form of globoid cell leukodystrophy in the mouse. Human Molecular Genetics, 2001. 10: p. 1191-9.
Matsuda, J., M.T. Vanier, Y. Saito, K. Suzuki, and S. Kunihiko, Dramatic
phenotypic improvement during pregnancy in a genetic leukodystrophy: estrogen appears to be a critical factor. Human Molecular Genetics, 2001. 10: p.
2709-15.
Muller-Ladner, U., Molecular and cellular interaction in rheumatoid synovium. Curr Opin Rheumatol, 1996. 8: p. 210.
Rodan, G.A. and T.J. Martin, Therapeutic approaches to bone diseases. Science, 2003. 289(5484): p. 1508-21.
Silman, A. and P. Brennan, Timing of pregnancyin relation to the onset of
rheumatoid arthritis. Arthritis Rheum, 1992. 35: p. 152-5.
138
251.
252.
253.
254.
255.
256.
257.
258.
259.
260.
261.
262.
263.
264.
265.
266.
Wluka, A.E., F.M. Cicuttini, and T.D. Spector, Menopause, oestrogens and
arthritis. Maturitas, 2000. 35: p. 183-99.
Holmdahl, R., L. Jansson, and M. Anderson, Female sex hormones suppres
development of collagen induced arthritis in mice. Arthritis Rheum, 1986. 29:
p. 1501-9.
Holmdahl, R., L. Jansson, B. Meyerson, and L. Khareskog, Oestrogen induced
suppression of collagen arthritis: longterm oestradiol treatment of DBA/1
mice reduces severity and incidence of arthritis and decreases the anti-type II
collagen immune response. Clin Exp Immunol, 1987. 70: p. 372-8.
Latham, K.A., A. Zamora, H. Drought, S. Subramanian, A. Matejuk, H. Offner, and E.F. Rosloniec, Estradiol treatment redirects the isotype of the autoantibody response and prevents the development of autoimmune arthritis. J
Immunol, 2003. 171(11): p. 5820-7.
Rubin, E.M. and J.L. Faber, Pathology. J.B. Lippincot Company, 1994. 2th
edition.
Pantoni, L., C. Sarti, and D. Inzitri, Cytokines and Cell adhesion molecules in
cerebral ischemia. Arterioscler Thromb Vasc Biol, 1998. 18: p. 503-13.
Hurn, P.D. and L.M. Brass, Estrogen and stroke: a balanced analysis. Stroke,
2003. 34: p. 338-41.
Hurn, P.D. and I.M. Macrae, Estrogen as a neuroprotectant in stroke. J Cereb
Blood Flow Metab, 2000. 20: p. 631-52.
Hulley, S., D. Grady, T. Bush, C. Furberg, D. Herrington, B. Riggs, and E.
Vittinghoff, Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women. JAMA, 1998.
280: p. 605-13.
investigators, W.G.f.t.W.s.H.I., Risks and benefits of estrogen plus progestin
in healthy postmenopausal women: principal results from the Women's Health
Initiative randomized controlled trial. JAMA, 2002. 288: p. 321-33.
Dubal, D.B., M.L. Kashon, L.C. Pettigrew, J.M. Ren, S.P. Finklestein, S.W.
Rau, and P.M. Wise, Estradiol protects against ischemic injury. J Cereb Blood
Flow Metab, 1998. 18(11): p. 1253-8.
He, Z., Y.J. He, A.L. Day, and J.W. Simpkins, Proestrus levels of estradiol
during transient global cerebral ischemia improves the histological outcome
of the hippocampal CA1 region: perfusion-dependent and-independent mechanisms. J Neurol Sci, 2002. 193(2): p. 79-87.
Rusa, R., N.J. Alkayed, B.J. Crain, R.J. Traystman, A.S. Kimes, E.D. London,
J.A. Klaus, and P.D. Hurn, 17beta-estradiol reduces stroke injury in estrogendeficient female animals. Stroke, 1999. 30(8): p. 1665-70.
Simpkins, J.W., G. Rajakumar, Y.Q. Zhang, C.E. Simpkins, D. Greenwald,
C.J. Yu, N. Bodor, and A.L. Day, Estrogens may reduce mortality and ischemic damage caused by middle cerebral artery occlusion in the female rat. J
Neurosurg, 1997. 87(5): p. 724-30.
Santizo, R., S. Anderson, S. Ye, H. Koenig, and D. Pelligrino, Effects of estrogen on leukocyte adhesion after transient forebrain ischemia. Stroke, 2000.
31: p. 2231-5.
McCarthy, J.B., A.L. Barker-Gibb, S.E. Alves, and T.A. Milner, TrKA Immunoreactive Astrocytes in Dendritic Fields of the Hippocampal Formation
Across Estrous. GLIA, 2002. 38: p. 36-44.
139
267.
268.
269.
270.
271.
272.
273.
274.
275.
276.
277.
278.
279.
280.
281.
282.
Hardy, J., Amyloid, the presenilins and Alzheimer's disease. Trends Neurosci,
1997. 20: p. 154-9.
McGeer, P. and E. McGeer, Local neuroinflammation and the progression of
Alzheimer's disease. Journal of NeuroVirology, 2002. 8: p. 529-38.
Gandy, S., Estrogen and Neurodegeneration. Neurochemical Reserch, 2003.
28: p. 1003-8.
Vegeto, E., S. Belcredito, S. Etteri, S. Ghisletti, A. Brusadelli, C. Meda, A.
Krust, S. Dupont, P. Ciana, P. Chambon, and A. Maggi, Estrogen receptor-alpha mediates the brain antiinflammatory activity of estradiol. Proc Natl Acad
Sci USA, 2003. 100: p. 9614-9.
Hanisch, U.K., Microglia as source and target of cytochines. GLIA, 2002. 40:
p. 140-55.
Vegeto, E., G. Pollio, P. Ciana, and A. Maggi, Estrogen blocks inducible nitric
oxide syntase accumulation in LPS-activated microglia cells. Experimental gerontology, 2000. 35: p. 1309-16.
Vegeto, E., C. Bonincontro, G. Pollio, A. Sala, S. Viappiani, F. Nardi, A. Brusadelli, B. Viviani, P. Ciana, and A. Maggi, Estrogen prevents the lipopolysaccharide-induced inflammatory response in microglia. J Neuroscience, 2001.
21: p. 1809-18.
Vegeto, E., S. Ghisletti, C. Meda, S. Etteri, S. Belcredito, and A. Maggi, Regulation of the lipopolysaccharide signal transduction pathway by 17beta-estradiol in macrophage cells. J Steroid Biochem Mol Biol, 2004. 91(1-2): p. 5966.
Bruce-Keller, A., S.W. Barger, N.I. Moss, J.T. Pham, J.N. Keller, and A. Nath,
Pro-inflammatory and pro-oxidant properties of the HIV protein Tat in a microglial cell line: attenuation by 17ß-estradiol. Journal of Neurochemistry,
2001. 78: p. 1515-24.
Zancan, V., S. Santagati, C. Bolego, E. Vegeto, A. Maggi, and L. Puglisi, 17ßestradiol decreases nitric oxide sinthase II sinthesis in vascular smooth muscle
cells. Endocrinology, 1999. 140: p. 2004-9.
Tzagarakis-Foster, C., R. Geleziunas, A. Lomri, J. An, and D.C. Leitman,
Estradiol repress human T-cell leukemia virus type 1 Tax activation of tumor
necrosis factor-α gene transcription. J Biol Chem, 2002. 277: p. 44772-7.
Agrati, P., M. Garnier, C. Patrone, G. Pollio, S. Santagati, E. Vegeto, and A.
Maggi, SK-ER3 neuroblastoma cells as a model for the study of estrogen influence on neural cells. Brain Res Bull, 1997. 44(4): p. 519-23.
Vegeto, E., G. Pollio, C. Pellicciari, and A. Maggi, Estrogen and progesterone
induction of survival of monoblastoid cells undergoing TNF-alpha-induced
apoptosis. Faseb J, 1999. 13(8): p. 793-803.
Hayashi, T., K. Yamada, T. Esaki, E. Muto, G. Chaudhuri, and A. Iguchi, Physiological concentrations of 17beta-estradiol inhibit the synthesis of nitric oxide synthase in macrophages via a receptor-mediated system. J Cardiovasc
Pharmacol, 1998. 31(2): p. 292-8.
Jansson, L., T. Olsson, and R. Holmdahl, Estrogen induces a potent suppression of experimental autoimmune encephalomyelitis and collagen-induced arthritis in mice. J Neuroimmunol, 1994. 53(2): p. 203-7.
Dubal, D.B., H. Zhu, J. Yu, S.W. Rau, P.J. Shughrue, I. Merchenthaler, M.S.
Kindy, and P.M. Wise, Estrogen receptor alpha, not beta, is a critical link in
140
283.
284.
285.
286.
estradiol-mediated protection against brain injury. Proc Natl Acad Sci U S A,
2001. 98(4): p. 1952-7.
Ashcroft, G.S., S.J. Mills, K. Lei, L. Gibbons, M.J. Jeong, M. Taniguchi, M.
Burow, M.A. Horan, S.M. Wahl, and T. Nakayama, Estrogen modulates cutaneous wound healing by downregulating macrophage migration inhibitory
factor. J Clin Invest, 2003. 111(9): p. 1309-18.
Cuzzocrea, S., S. Santagati, L. Sautebin, E. Mazzon, G. Calabro, I. Serraino,
A.P. Caputi, and A. Maggi, 17beta-estradiol antiinflammatory activity in carrageenan-induced pleurisy. Endocrinology, 2000. 141(4): p. 1455-63.
Miyamoto, N., M. Mandai, I. Suzuma, K. Suzuma, K. Kobayashi, and Y. Honda, Estrogen protects against cellular infiltration by reducing the expressions
of E-selectin and IL-6 in endotoxin-induced uveitis. J Immunol, 1999. 163(1):
p. 374-9.
Hodgin, J.B. and N. Maeda, Minireview: estrogen and mouse models of atherosclerosis. Endocrinology, 2002. 143(12): p. 4495-501.
141
Ringraziamenti
Ringrazio la Professoressa Adriana Maggi, per avermi dato la possibilità di svolgere
questa tesi presso il suo laboratorio,
e la Dottoressa Elisabetta Vegeto per avermi seguito da vicino ed aver coordinato in
modo egregio il mio lavoro.
Inoltre voglio ringraziare tutti i componenti del laboratorio Maggi, delle persone
molto preparate e disponibili; in particolare le “cellule” per avermi sopportato per
questi 18 mesi.
Ringrazio i miei genitori per tutto il loro affetto ed il loro supporto, che sono stati
molto importanti durante i miei studii.