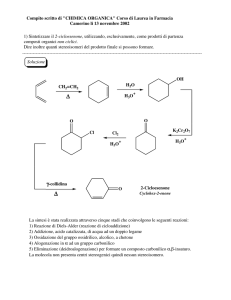
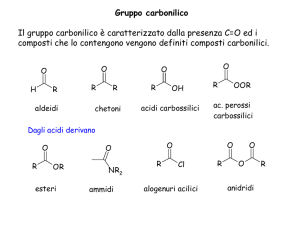
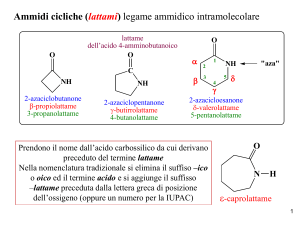
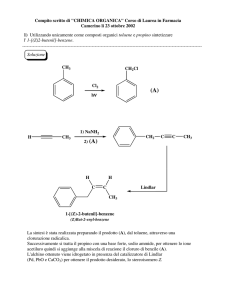
APPUNTI DI CHIMICA GENERALE
La spontaneità delle reazioni chimiche
Si chiamano reazioni spontanee quelle che, nelle condizioni date, formano i prodotti indicati dalla
relativa equazione, senza aiuti provenienti dall’esterno.
Quando si immerge, per esempio, una lamina di zinco, di colore grigio, in una soluzione di solfato
di rame, caratterizzata da una colorazione azzurra, si nota che il metallo si ricopre di un deposito
bruno, mentre l’azzurro della fase liquida diviene sempre meno intenso.
Questo perché si verifica la reazione:
Zn(s) + CuSO4(aq)
Cu(s) + ZnSO4(aq)
Al contrario, l’equazione:
Cu(s) + ZnSO4(aq)
Zn(s) + CuSO4(aq)
è quella di una reazione che non avviene, se non in determinate condizioni operative, ossia con
l’apporto esterno di energia. In altre parole esistono delle reazioni che non si verificano, pur
potendone scrivere e bilanciare la relativa equazione: si tratta di reazioni non spontanee.
La valutazione della spontaneità o non spontaneità di un processo chimico è il fine che si propone la
termodinamica, attraverso lo studio degli scambi di energia, sotto forma di calore e di lavoro, che
accompagnano le trasformazioni chimiche, e dei fattori, come la temperatura, che le possono
influenzare.
•
Entalpia: il contenuto termico delle sostanze
Ogni sostanza, a seconda dei legami chimici che uniscono i suoi atomi e del suo stato fisico, è
caratterizzata da una certa quantità di energia. Nel corso di una reazione chimica, il tipo e il numero
di legami presenti nel sistema cambiano, poiché si rompono quelli che costituiscono le specie
reagenti e se ne formano di nuovi nelle specie prodotte. Inoltre può cambiare anche lo stato fisico
delle sostanze presenti prima e dopo la reazione. Ogni reazione è pertanto accompagnata da una
variazione di energia del sistema, ∆E, che si manifesta sotto forma di calore, Q, e/o di lavoro,L.
Queste tre variabili sono collegate dalla legge:
∆E = Q – L
che esprime il primo principio della termodinamica, noto anche come principio di conservazione
dell’energia:
”in una trasformazione, la somma delle energie del sistema e dell’ambiente è uguale prima e
dopo la trasformazione stessa”.
Esso indica che la variazione di energia ∆E di un sistema è uguale alla somma algebrica del calore
Q e del lavoro prodotto – L.
1
In un sistema chimico il lavoro L diventa significativo soltanto quando si formano sostanze gassose,
perché il gas che si sviluppa compie un lavoro di espansione causato dalla variazione di volume ∆V.
Poiché a pressione costante P, il lavoro del gas è espresso dalla relazione L = P · ∆V, il primo
principio della termodinamica si può scrivere nella forma:
∆E = Q – P · ∆V
Il calore scambiato a pressione costante (Qp) detto calore di reazione, è pertanto:
Qp = ∆E + P · ∆V
Per rappresentare E + P · V è stata introdotta una funzione termodinamica chiamata entalpia, che si
indica con H, il cui valore dipende dall’energia del sistema, dalla pressione e dal volume:
H=E+P·V
Sebbene l’entalpia di una sostanza o di un sistema non possa essere valutata, si può invece misurare
la variazione di entalpia che subisce un sistema in un processo o in una reazione. Nel caso di una
reazione a pressione costante, la variazione di entalpia, ∆H, vale:
∆H = ∆E + P · ∆V
Dalle formule scritte si vede che ∆H = Qp ossia:
la variazione di energia di un sistema, ∆H, in una reazione che avviene a pressione costante,
corrisponde al calore di reazione , Qp, assorbito o sviluppato dal sistema; convenzionalmente
∆H > 0 se il calore è assorbito e ∆H < 0 se il calore è ceduto.
Poiché normalmente le reazioni chimiche avvengono a pressione atmosferica e quindi costante, il
calore Qp che il sistema scambia con l’esterno può essere misurato, consentendo così di calcolare la
variazione di entalpia del sistema. Per rispettare la convenzione adottata per i segni, la relazione tra
calore scambiato dall’ambiente esterno nel corso di una reazione (Qamb) e la variazione di entalpia
del sistema (∆H) è:
Qamb = - ∆H
dove:
∆H = Σ Hprodotti – Σ Hreagenti
Calori di reazione importanti sono il calore o entalpia di formazione di un composto.
L’entalpia di formazione di un composto, ∆Hf, è misurata dal calore scambiato (sviluppato o
assorbito) quando si forma una mole di composto a partire dai suoi elementi.
Dal momento che il valore del ∆H dipende dalla temperatura e dalla pressione alla quale si verifica
una reazione, sia essa di formazione o qualsiasi altra, per rendere confrontabili fra loro i valori dei
∆Hf sono state stabilite certe condizioni sperimentali standard (c.s.) per la loro determinazione.
Si definisce come stato standard di un elemento o di un composto la sua forma molecolare, o di
aggregato, più stabile e più diffusa in natura, alla temperatura T di 298 K (25 °C) e alla pressione P
di 1 atmosfera.
2
Le entalpie determinate in condizioni standard vengono indicate come ∆Hf° nel caso delle entalpie
di formazione di un composto dagli elementi, e come ∆H° se entalpie di reazione. Le entalpie
molari standard degli elementi sono convenzionalmente nulle.
Esempi:
∆Hf° (kJ · mol-1)
reazione
1) 1/2 H2(g) + 1/2 Cl2(g)
HCl(g)
-92,3
2) H2(g) + 1/2 O2(g)
H2O(g)
-242
3) H2(g) + 1/2 O2(g)
H2O(l)
-286
4) 1/2 N2(g) + 1/2 O2(g)
NO(g)
90,4
5) 1/2 N2(g) + O2(g)
NO2(g)
33,1
Le equazioni debbono sempre riportare lo stato delle sostanze in gioco. Come si vede dalle
equazioni 2 e 3 il ∆Hf° è diverso a seconda che venga prodotta acqua liquida o vapore acqueo. Nelle
equazioni, inoltre, si utilizzano coefficienti frazionari poiché ci si riferisce sempre alla formazione
di un’unica mole di composto.
•
Reazioni esotermiche ed endotermiche: gli scambi di calore
Quando in una reazione si passa da reagenti caratterizzati da un alto contenuto di energia a prodotti
con un più basso contenuto, la differenza tra l’energia dello stato iniziale del sistema, quello dei
reagenti, e l’energia dello stato finale rappresentato dai prodotti viene liberata sotto forma di calore
ceduto all’ambiente. Poiché nelle normali condizioni di pressione e temperatura a cui avvengono le
reazioni chimiche, la variazione di energia di un sistema è rappresentata dalla variazione della sua
entalpia (∆H), si può dire che: le reazioni esotermiche, che avvengono cioè con liberazione di
calore nell’ambiente, hanno ∆H < 0.
Esempi:
reazione
Qamb
∆H° (kJ)
1) C3H8(g) + 5 O2(g)
3 CO2(g) + 4 H2O(l)
2506
-2506
2) Mg(s) + 1/2 O2(g)
MgO(s)
602
-602
3) H2(g) + 1/2 O2(g)
H2O(l)
286
-286
4) CH4(g) + 2 O2(g)
CO2(g) + 2 H2O(l)
890
-890
Per una particolare reazione il ∆H° è riferito alle moli di reagenti e di prodotti indicate dai
coefficienti nell’equazione bilanciata.
Anche in questo caso nelle equazioni deve essere sempre specificato lo stato fisico delle sostanze in
gioco.
3
La situazione contraria a quella appena considerata si realizza quando in una reazione reagenti a
basso contenuto energetico formano prodotti ad alta energia: in tal caso è necessario un
assorbimento di calore dall’ambiente.
Pertanto: le reazioni endotermiche, che avvengono cioè con assorbimento di calore nell’ambiente,
hanno ∆H > 0.
Esempi:
reazione
1) C(s) + H2O(g)
CO(g) + H2(g)
Qamb
∆H° (kJ)
-131
131
2) CaCO3(s)
CaO(s) + CO2(g)
-176
176
3) H2O(l)
H2(g) + 1/2 O2(g)
-286
286
-86,6
86,6
4) Cu(s) + H2O(g)
CuO(s) + H2(g)
Come si può notare, la reazione di dissociazione dell’acqua liquida in idrogeno e ossigeno gassosi
richiede la stessa quantità di calore liberata nella sintesi di acqua liquida a partire dagli elementi.
Infatti: la variazione di entalpia di una reazione ha lo stesso valore, con segno cambiato, di quella
della reazione che avviene in senso opposto.
Reazione esotermica ∆H < 0
Reazione endotermica ∆H > 0
H
H
reagenti
prodotti
calore ceduto
calore assorbito
prodotti
reagenti
tempo
•
tempo
La legge di Hess: calcolo teorico del ∆H di una reazione
Quando non è possibile determinare il calore di reazione (∆H) per via sperimentale, si ricorre al
fatto che l’entalpia è una funzione di stato, cioè che le sue variazioni dipendono solo dallo stato del
sistema. Quindi la variazione di entalpia, associata a una data reazione, può essere calcolata
considerando solo lo stato iniziale e quello finale del sistema, indipendentemente dagli stati
intermedi attraverso cui esso passa. Da tale proprietà discende la legge di Hess, secondo la quale:
il ∆H di una reazione, che può essere espressa come somma algebrica di più reazioni, è dato
dalla somma algebrica dei ∆H di queste reazioni.
4
In pratica, quando il ∆H di una reazione non è misurabile, si deve ricercare un gruppo di altre
reazioni di cui sia noto o sia misurabile il ∆H, le quali, opportunamente combinate, diano
un’equazione che parte dagli stessi reagenti e arriva agli stessi prodotti della reazione voluta.
Se si vuole per esempio calcolare il ∆H della reazione:
C(s) + 1/2 O2(g)
CO(g)
e sono noti i valori dei ∆H° delle seguenti reazioni:
1) C(s) + O2(g)
∆H°1 = -393,5 kJ · mol-1
CO2(g)
2) CO(g) + 1/2 O2(g)
∆H°2 = -283,0 kJ · mol-1
CO2(g)
Sottraendo membro a membro le due equazioni si ricava:
C(s) + O2(g) - CO(g) - 1/2 O2(g)
CO2(g) - CO2(g)
che, semplificata, dà
C(s) + 1/2 O2(g)
CO(g)
Ossia la reazione di cui si vuole calcolare il ∆H.
Pertanto, combinando nello stesso modo i relativi ∆H°, quello della reazione in questione è dato da:
∆H = ∆H°1 - ∆H°2
Ovvero in termini numerici:
∆H = -393,5 kJ · mol-1 – (-283,0 kJ · mol-1) = -110,5 kJ · mol-1
Che rappresenta il ∆H°f dell’ossido di carbonio CO.
Più generalmente, la legge di Hess afferma che:
il ∆H di una reazione si ottiene sommando al ∆H°f dei prodotti il ∆H°f dei reagenti, ciascuno
moltiplicato per il proprio coefficiente stechiometrico:
∆H = Σ ∆H°f prodotti – Σ ∆H°f reagenti
Proviamo a calcolare, utilizzando la legge di Hess, il ∆H della reazione:
Cl2(g) + 2 HBr(g)
2 HCl(g) + Br2(l)
Sapendo che i ∆H°f di HCl e di HBr sono rispettivamente –92,0 kJ · mol-1 e –36,4 kJ · mol-1.
Ricordando che i ∆H°f degli elementi presenti nella reazione sono nulli, poiché si fa riferimento al
cloro gassoso e al bromo liquido, cioè al loro stato nelle condizioni standard, si può scrivere:
∆H = 2 · (-92,0) + 0 - [0 + 2 · (-36,4)] = -111,2 kJ
5
•
Entalpia e spontaneità
L’esperienza quotidiana suggerisce che qualsiasi sistema tende spontaneamente ad andare da
situazioni con elevato contenuto di energia potenziale (capacità di compiere lavoro associata alla
posizione di un corpo) a situazioni più stabili, cioè con minore energia potenziale.
Poiché l’energia potenziale di un composto chimico può essere identificata con il suo contenuto di
entalpia, si potrebbe pensare che solo le reazioni esotermiche siano spontanee, dato che procedono
da reagenti ad alto contenuto energetico verso prodotti con basso contenuto, come testimonia il
calore liberato. In realtà si riscontrano molte eccezioni.
La reazione:
Hg(l) + 1/2 O2(g)
HgO(s)
∆H = -90,8 kJ/mol
Pur essendo esotermica, avviene spontaneamente solo a temperature elevate, mentre a temperatura
ambiente non procede.
Al contrario, alcune reazioni o processi endotermici avvengono spontaneamente. L’assorbimento di
calore che si verifica quando alcune sostanze passano in soluzione è sfruttato per la preparazione di
impacchi freddi istantanei (confezioni che contengono acqua e, separatamente, NH4NO3 solido).
L’esistenza di reazioni endotermiche spontanee e di reazioni esotermiche che non procedono
spontaneamente indica che lo sviluppo di calore non è una condizione sufficiente a determinare la
spontaneità di una reazione. È necessario tener conto anche di un’altra grandezza: l’entropia.
•
L’entropia: la misura del disordine
Quando si mescola un mazzo di carte è altamente improbabile che esse si dispongano secondo una
sequenza prestabilita. È normale invece trovarle distribuite nel mazzo in modo del tutto casuale.
Analogamente, è altamente probabile che un sistema formato da oggetti diversi si presenti in forma
disordinata; inoltre tale probabilità aumenta con l’aumentare del numero di oggetti presenti.
Gli stati di aggregazione della materia sono contraddistinti dall’ordine-disordine delle particelle. Si
passa dallo stato altamente ordinato, tipico dei solidi, allo stato totalmente disordinato degli
aeriformi.
S
solido
liquido
gas
6
La solubilizzazione di un composto comporta anch’essa un aumento del disordine del sistema
(solvente + soluto) dato che le particelle del soluto, molecole o ioni che siano, passano dallo stato di
ordine e immobilità rappresentato dal reticolo, al movimento e alla loro dispersione tra le molecole
del solvente.
La funzione termodinamica che misura il grado di disordine di un sistema, e quindi la probabilità
della sua esistenza, è chiamata entropia, ed è indicata con S.
L’entropia, che si esprime in J · K-1, venne introdotta da R. Clausius (1850) secondo un approccio
diverso da quello probabilistico proposto successivamente da L. Boltzmann.
La variazione di entropia di una reazione chimica dipende dalle trasformazioni che le molecole
subiscono, oltre che dal grado di disordine relativo allo stato di aggregazione dei prodotti e dei
reagenti. In una reazione chimica un aumento di entropia si verifica quando:
-
reagenti solidi o liquidi formano prodotti gassosi;
il numero delle molecole dei prodotti è maggiore di quello dei reagenti.
S
La variazione di entropia di un sistema in una reazione è indicata con ∆S ed è uguale alla
differenza tra la somma delle entropie dei prodotti e quella delle entropie dei reagenti:
∆S = Σ Sprodotti - Σ Sreagenti
In base a tale definizione si può dire che: nelle reazioni o nei processi in cui l’entropia dei prodotti è
maggiore di quella dei reagenti, si verifica un aumento di disordine del sistema e ∆S > 0, mentre
quando l’entropia dei prodotti è minore di quella dei reagenti lo stato finale del sistema è meno
disordinato di quello iniziale: ∆S < 0.
Esempi:
reazione
∆S (J · K-1)
1) 2 SO2(g) + O2(g)
2 SO3(g)
-189,6
2) 2 H2(g) + O2(g)
2 H2O(g)
-377,4
3) CaCO3(s)
4) 2 NaHCO3(s)
CaO(s) + CO2(g)
160,8
Na2CO3(s) + H2O(g) + CO2(g)
7
334;1
Come si può vedere dai dati riportati, le prime due reazioni presentano ∆S < 0 e quindi una
diminuzione del disordine. Infatti in entrambi i casi si produce una situazione più ordinata perché
dalle tre molecole di reagenti di specie diversa si passa a due molecole uguali di prodotto. Le ultime
due reazioni sono invece caratterizzate da ∆S > 0 perché avvengono con un aumento del disordine.
Infatti da reagenti allo stato solido si passa a più prodotti, alcuni dei quali gassosi. Queste due
reazioni, pertanto, dal punto di vista entropico, dovrebbero essere spontanee. In realtà a temperatura
ambiente non sono spontanee, mentre lo divengono a temperature elevate. Dunque, né il solo fattore
entalpico, né il solo fattore entropico consentono di prevedere se una reazione è spontanea.
È necessario tener conto di entrambi, oltre che della temperatura a cui avviene la reazione.
•
Energia libera
La spontaneità di una reazione o di un processo è misurata dalla variazione di una funzione
termodinamica G, introdotta verso il 1880 da J. W. Gibbs, chiamata, in suo onore, energia libera di
Gibbs. Il ∆G tiene conto contemporaneamente delle tre variabili ∆H, ∆S e T (espressa in K)
secondo la relazione:
∆G = ∆H – T · ∆S
Nei processi spontanei ∆G < 0, nei processi non spontanei ∆G > 0.
Una situazione particolare si verifica quando ∆G = 0.
Il sistema si trova in queste condizioni in uno stato di equilibrio in cui minime variazioni delle
grandezze chimiche o fisiche possono rendere spontanee la formazione dei prodotti oppure la
ricostituzione dei reagenti.
Vediamo ora come influiscono sul segno e sul valore del ∆G i contributi del ∆H e del ∆S di una
reazione, e della T alla quale si opera.
1) Reazione esotermica con aumento di disordine: ∆H < 0 e ∆S > 0
Poiché le temperature assolute sono sempre positive, i segni di ∆H e ∆S assicurano che ∆G < 0 a
qualsiasi temperatura e che quindi la reazione è sempre spontanea. Consideriamo la combustione
del propano:
C3H8(g) + 5 O2(g)
3 CO2(g) + 4 H2O(g)
La reazione è caratterizzata da: ∆H = -2251 kJ · mol-1; ∆S = 98,2 J · K-1 · mol-1.
Nella reazione si libera calore (∆H < 0) ed essa comporta un aumento del disordine (∆S > 0), dato
che il numero di molecole dei prodotti è maggiore di quello dei reattivi. Entrambi i fattori
favoriscono la reazione, per cui a qualsiasi temperatura si avrà ∆G < 0: la reazione sarà sempre
spontanea.
2) Reazione endotermica con diminuzione del disordine: ∆H > 0 e ∆S < 0
È il caso esattamente opposto a quello precedentemente descritto. Una reazione che assorba calore
(∆H > 0) e proceda verso uno stato più ordinato di quello iniziale (∆S < 0) presenterà ∆G > 0 a
qualsiasi temperatura: la reazione non avviene mai spontaneamente.
Consideriamo la reazione opposta a quella di combustione del metano, cioè la produzione di metano
e ossigeno a partire da acqua e anidride carbonica:
CO2(g) + 2 H2O(g)
CH4(g) + 2 O2(g)
8
Per essa si può calcolare: ∆H = 890 kJ · mol-1; ∆S = -243 J · K-1 · mol-1.
Nella reazione si ha assorbimento di calore (∆H > 0) ed essa comporta una diminuzione del
disordine (∆S < 0). Tutti e due i fattori non sono favorevoli alla spontaneità, per cui a qualsiasi
temperatura si avrà ∆G > 0. La reazione non è mai spontanea. Infatti se ciò accadesse, cioè se si
potesse produrre spontaneamente metano partendo da anidride carbonica e acqua, avremmo risolto
il problema delle risorse energetiche!
3) Reazione esotermica con diminuzione del disordine: ∆H < 0 e ∆S < 0
È una possibilità che si verifica frequentemente nelle reazioni chimiche. Poiché il fattore entalpico
(∆H < 0) è favorevole al procedere spontaneo della reazione e quello entropico (∆S < 0) vi si
oppone, acquista importanza il valore della temperatura. Essa moltiplica il fattore entropico
sfavorevole, pertanto alle basse temperature la reazione è spontanea e non lo è alle alte temperature.
Ovviamente il significato di temperatura alta o bassa è relativo a ogni specifico processo in quanto è
collegato ai valori del ∆H e del ∆S. C’è infatti un valore di temperatura: T = ∆H / ∆S al quale il
senso della spontaneità del processo si inverte in funzione del valore dei contributi entalpici ed
entropici. Se si considera, per esempio, la reazione di formazione del triossido di zolfo:
SO2(g) + 1/2 O2(g)
SO3(g)
Essa presenta: ∆H = -98,3 kJ · mol-1; ∆S = -9,48 · 10-2 kJ · K-1 · mol-1.
Pertanto la temperatura alla quale si verifica l’inversione del senso spontaneo della reazione è:
T = -98,3 kJ · mol-1 / -9,48 · 10-2 kJ · K-1 · mol-1 = 1037 K
Al di sotto di questa temperatura ∆G < 0, pertanto la reazione indicata avviene spontaneamente,
mentre al di sopra è spontanea la reazione opposta.
4) Reazione endotermica con aumento di disordine: ∆H > 0 e ∆S > 0
Anche questo è un caso di reazione chimica che si presenta molto spesso. Poiché il fattore entalpico
(∆H > 0) è sfavorevole alla spontaneità della reazione e quello entropico (∆S > 0) la favorisce,
ancora una volta la temperatura è determinante: alle alte temperature la reazione è spontanea,
mentre alle basse temperature non avviene. Anche in questo caso si può determinare la temperatura
alla quale si ha l’inversione del comportamento del sistema con la formula: T = ∆H / ∆S.
Si consideri la reazione:
CaCO3(s)
CaO(s) + CO2(g)
che presenta: ∆H = 179,3 kJ · mol-1; ∆S = 1,608 · 10-1 kJ · K-1 · mol-1.
La temperatura che indica inversione del comportamento è:
T = 179,3 kJ · mol-1 / 1,608 · 10-1 kJ · K-1 · mol-1 = 1115 K
Pertanto solo al di sopra dei 1115 K, ∆G < 0 e la decomposizione del carbonato di calcio è
spontanea, mentre al di sotto di tale temperatura è spontanea la sua formazione a partire da CaO e
CO2.
9
Dissoluzione in acqua
Dissociazione ionica
La dissociazione è il meccanismo di dissoluzione che riguarda i composti ionici (solidi ionici con
reticoli cristallini). Le molecole fortemente polari di H2O disgregano il reticolo, vincendo le forze di
attrazione elettrostatica tra gli ioni di segno opposto che lo formano. Si producono ioni idratati
(solvatazione) che conservano la carica del rispettivo ione.
Es:
Na+Cl-(s)
H2O
Na+(aq) + Cl-(aq)
Ionizzazione
Coinvolge composti formati da molecole polari, l’H2O rompe i legami covalenti polari posti
all’interno di ogni molecola, generando ioni che non esistevano come tali.
Es:
δ+ δHCl(g) + H2O(l)
H3O+(aq) + Cl-(aq)
ione ossonio
Solubilizzazione
Coinvolge i composti molecolari, l’H2O rompe i deboli legami che esistono fra le molecole (legami
intermolecolari), liberando molecole intere, elettricamente neutre, che si disperdono in seno
all’acqua.
H2O
Es:
C12H22O11(s)
C12H22O11(aq) (molecola idratata)
Elettroliti
Sono elettroliti tutte le sostanze che, disciolte in acqua, producono ioni positivi e ioni negativi, sia
mediante ionizzazione, sia mediante dissociazione.
Tutte le soluzioni elettrolitiche sono in grado di condurre la corrente elettrica.
molecole dissociate
Grado di dissociazione (α
α) =
, pertanto sono:
molecole totali
-
elettroliti forti le sostanze completamente dissociate (α
α = 1)
elettroliti deboli le sostanze poco dissociate (α
α < 1)
Sono elettroliti gli acidi, le basi e i sali.
I sali, in presenza di acqua, producono ioni positivi (cationi) e ioni negativi (anioni).
H2O
es:
KBr(s)
K+(aq) + Br-(aq)
(NH4)3PO4(s) + H2O(l)
3 NH4+(aq) + PO43-(aq)
10
Teoria di Arrhenius (1887)
Il chimico danese studiando la dissociazione ionica di sostanze diverse disciolte in acqua e
confrontando la conducibilità elettrica delle soluzioni così ottenute, concluse che:
sono acidi le sostanze che, sciolte in acqua liberano ioni H+ (protoni)
es:
H3O+(aq) + NO3-(aq)
HNO3(s) + H2O(l)
sono basi le sostanze che, sciolte in acqua, liberano ioni ossidrili OHes:
KOH(s)
H2O
K+(aq) + OH-(aq)
La teoria di Arrhenius non spiega il comportamento basico dell’ammoniaca (NH3).
Teoria di Brönsted-Lowry (1923)
Indipendentemente l’uno (danese) dall’altro (inglese), diedero una definizione più ampia di acido e
di base:
-
è un acido una sostanza capace di cedere protoni (H+);
-
è una base una sostanza capace di acquistare protoni (H+).
Pertanto con questa teoria il termine base non indica più soltanto le sostanze che contengono nella
propria formula lo ione idrossido OH-.
coppia coniugata
NH4+ + OHacido
base
Es: NH3 + H2O
base acido
coppia coniugata
coppia coniugata
HCl + H2O
acido base
Cl- + H3O+
base acido
coppia coniugata
Non è possibile definire in assoluto una sostanza come acido o come base, ma questa deve essere
definita solo relativamente ad un’altra sostanza, con la quale reagisce (vedi l’acqua nelle reazioni
sopra rappresentate).
11
Teoria di G. N. Lewis (1926)
Alcune sostanze, come BF3, sciolte in benzene o in tetracloruro di carbonio, fanno cambiare il
colore del tornasole e danno delle reazioni caratteristiche degli acidi ma, non contenendo atomi di
idrogeno, non possono, in base a nessuna delle precedenti teorie, essere considerate come acidi.
La definizione più generale e più recente di acido e di base è stata formulata nel 1926 dallo
statunitense Lewis considerando i legami che queste sostanze potevano formare quando reagivano.
Secondo Lewis, le proprietà acide di una sostanza sono dovute alla sua disponibilità ad accettare
una coppia di elettroni, formando un altro legame, mentre le specie che possiedono una o più coppie
elettroniche non condivise presentano proprietà basiche.
Pertanto:
-
un acido è una specie in grado di accettare una coppia di elettroni;
-
una base è una specie in grado di cedere una coppia di elettroni.
La definizione di base secondo Lewis ripropone, sotto un altro aspetto, quanto era già stato
affermato da Brönsted e Lowry. Infatti le sostanze che accettano protoni hanno una coppia
elettronica disponibile, come l’acqua e l’ammoniaca.
..
..
+
H3O+
H2O : + H
..
NH3 + H+
NH4+
In queste reazioni, acqua e ammoniaca si comportano come basi, sia secondo la definizione di
Lewis, sia secondo quella di Brönsted e Lowry. La definizione di acido dato da Lewis consente di
classificare come acidi, oltre allo ione H+, anche cationi, detti acidi di Lewis, in grado di formare
legami dativi di coordinazione:
..
+
Ag + 2 NH3
Ag(NH3)2+
o elementi con ottetti incompleti o espandibili:
..
–
+
BF3 + NH3
F3B – NH3
Delle teorie riportate, quella che si presenta più completa, perché è in grado di descrivere il
comportamento di un grande numero di sostanze in ambienti molto diversi, è indubbiamente quella
di Lewis. D’altra parte ciò è comprensibile perché essa punta l’attenzione sul meccanismo di
formazione dei legami, situazione che si verifica in ogni reazione. Se però si limita il campo di
studio al solvente di più largo impiego in grado di scambiare protoni, cioè l’acqua, la teoria di
Brönsted e Lowry è adeguata a spiegare il comportamento di una vastissima gamma di composti.
12
Costante di equilibrio
Quando l’intera massa dei reagenti si è trasformata nei prodotti, la reazione è completa (es: tutte le
combustioni). Tali reazioni si dicono anche irreversibili. La maggior parte delle reazioni invece
non vanno a compimento, sono incomplete, in uno stato di apparente equilibrio (accanto ai prodotti
si trovano reagenti residui), sono reversibili.
Una reazione reversibile può avvenire in entrambe le direzioni:
sintesi
H2(g) + I2(g)
decomposizione
sintesi = reazione diretta;
V = velocità di reazione
2 HI(g)
(1)
decomposizione = reazione inversa
T = tempo
V
reazione diretta
reazione inversa
T
Lo stato di equilibrio del sistema è di tipo dinamico perché a livello microscopico le reazioni
continuano alla stessa velocità.
Legge dell’equilibrio chimico o dell’azione di massa
(Guldberg e Waage, Norvegia 1865)
A una data temperatura costante, il rapporto tra il prodotto delle concentrazioni molari (moli/litro)
dei prodotti della reazione, elevate ciascuna al proprio coefficiente stechiometrico, e il prodotto
delle concentrazioni molari dei reagenti, elevate anch’esse al proprio coefficiente stechiometrico, è
costante.
Es: a 425 °C per la reazione (1)
[HI]2
Keq =
= 54,40
[H2] ⋅ [I2]
•
•
Una costante di equilibrio piccola (Keq < 1) significa che la reazione è spostata a sinistra.
Una costante di equilibrio grande (Keq > 1) significa che la reazione è spostata a destra.
13
Principio di Le Chatelier
Se un sistema all’equilibrio viene perturbato, esso reagisce in un modo da controbilanciare la causa
della perturbazione.
a) Effetto della variazione di concentrazione di una delle specie chimiche del sistema reattivo,
senza variare la temperatura (reazione1):
-
sottraendo HI l’equilibrio si sposta a destra
aggiungendo H2 oppure I2 l’equilibrio si sposta a destra
aggiungendo HI l’equilibrio si sposta a sinistra
non cambia la Keq
b) Effetto della variazione di pressione o di volume nell’ambiente di reazione.
Es:
N2(g) + 3 H2(g)
Keq =
2 NH3(g) ………..1 v. M. + 3 v. M
[NH3]2
2 v.M.
v. M. = volume molare
[N2] ⋅ [H2]3
la reazione avviene spontaneamente con diminuzione di volume:
-
comprimendo l’equilibrio si sposta verso destra
-
decomprimendo l’equilibrio si sposta verso sinistra
non cambia la Keq
Se in una reazione non si ha variazione del volume, un cambiamento della pressione non influisce
sull’equilibrio.
c) Effetto della variazione di temperatura.
endotermica
Es:
N2O4
2 NO2
……… Keq =
esotermica
[NO2]2
[N2O4]
•
reazione endotermica
-
l’aumento della temperatura sposta l’equilibrio a destra
la diminuzione della temperatura sposta l’equilibrio a sinistra
cambia la Keq
•
-
reazione esotermica
l’aumento della temperatura sposta l’equilibrio a sinistra
la diminuzione della temperatura sposta l’equilibrio a destra
14
Prodotto di solubilità (Kps)
L’equilibrio a cui si giunge quando si pone in un solvente una quantità di sostanza maggiore di
quella che vi si può disciogliere è detto equilibrio di solubilità.
L’equilibrio di solubilità del AgCl (sale poco solubile in acqua) viene così rappresentato:
AgCl(s)
Keq =
H2O
Ag+(aq) + Cl-(aq)
[Ag+(aq)] ⋅ [Cl-(aq)]
per cui,
dalla quale si ricava,
[AgCl(s)]
Keq ⋅ [AgCl(s)] = [Ag+(aq)] ⋅ [Cl-(aq)]
Poiché la concentrazione del solido è costante, anche il termine Keq ⋅ [AgCl(s)] sarà costante, esso
viene denominato prodotto di solubilità (Kps), pertanto:
Kps = [Ag+(aq)] ⋅ [Cl-(aq)]
Tutti i conposti ionici solidi hanno un proprio Kps che varia solo con la temperatura. Il prodoto di
solubilità è un numero che rappresenta il prodotto delle concentrazioni degli ioni del composto in
una soluzione satura, elevate ciascuna con il proprio coefficiente stechiometrico.
Esempio: a 25 °C
-
Kps CaSO4 = 2,5 ⋅ 10-5
-
Kps BaSO4 = 1,1 ⋅ 10-10
quindi, il CaSO4 è più solubile del BaSO4.
Esercizi:
1) Il carbonato di stronzio (SrCO3), alla temperatura di 25 °C, ha Kps = 1,6 · 10-9. Determinare le
moli di SrCO3 che si trovano disciolte in un litro di soluzione.
Soluzione:
Kps = [Sr2+] · [CO32-] = 1,6 · 10-9
mol =
1,6 · 10-9
=
16 · 10-10 = 4 · 10-5
15
2) Uno studente cerca di preparare alcune soluzioni con le seguenti concentrazioni ioniche:
[Ca2+] = 10-3 M
[Ag+] = 10-9 M
[Cu2+] = 10-9 M
[Cl-] = 10-3 M
[OH-] = 10-2 M
[SO42-] = 10-4 M
Riferendoti alla tabella, prevedi se si formerà o no un precipitato di:
a) CaSO4
b) Cu(OH)2
c) AgCl
d) Ca(OH)2
a 25 °C
Kps
2,5 · 10-5
1,1 · 10-10
4,8 · 10-9
1,8 · 10-10
1,3 · 10-6
1,6 · 10-19
sostanza
CaSO4
BaSO4
CaCO3
AgCl
Ca(OH)2
Cu(OH)2
Soluzione:
a)
CaSO4 = [Ca2+] · [SO42-] = [10-3] · [10-4] = 10-7 ………..NON si formerà un precipitato
b)
Cu(OH)2 = [Cu2+] · [OH-]2= [10-9] · [10-2]2 = 10-13 ..........SI formerà un precipitato
c)
AgCl = [Ag+] · [Cl-] = [10-9] · [10-3] = 10-12 ……………NON si formerà un precipitato
d)
Ca(OH)2 = [Ca2+] · [OH-]2= [10-3] · [10-2]2 = 10-7 ...........NON si formerà un precipitato
3) Calcolare la solubilità del fosfato d’argento, Ag3PO4, considerando che il prodotto di solubilità
di questo sale in acqua a 25 °C è 8,88 · 10-17.
L’elettrolita forte poco solubile Ag3PO4 si dissocia totalmente in acqua secondo la reazione:
3Ag+ + PO43-
Ag3PO4
In una soluzione satura si instaura l’equilibrio di solubilità:
3Ag+ + PO43-
Ag3PO4 (solido)
Dalla prima reazione si evince che ogni mole di Ag3PO4 si dissocia in 3 moli di ioni Ag+ e una mole
di ioni PO43- e che quindi in una soluzione satura di Ag3PO4 saranno:
[Ag+] = 3s
e
[PO43-] = s
Applicando al secondo equilibrio la legge di azione di massa e sostituendo queste due
concentrazioni, si ha:
Ks = [Ag+]3 [PO43-] = (3s)3 (s) = 27 s4
da cui:
4
s=
4
Ks/27
=
8,88 · 10-17/27
16
= 4,26 · 10-5 M
Prodotto ionico dell’acqua (Kw)
Se si misura la conducibilità elettrica dell’acqua pura con strumenti molto sensibili, si osserva che
essa conduce la corrente elettrica, anche se in minima quantità. In effetti, secondo la teoria di
Brönsted-Lowry, esiste un equilibrio tra le molecole:
H3O+ + OHacido
base
H2O + H2O
base acido
Keq =
[H3O+] · [OH-]
[H2O]
che a 25 °C presenta il seguente valore,
= 3,25 · 10-18
2
La reazione è fortemente spostata a sinistra tanto da poter considerare la concentrazione dell’acqua
costante. Essendo 18 la massa molare dell’acqua, la sua concentrazione molare è:
1000g/L
= 55,5 mol/L
pertanto,
18g/mol
Keq · [H2O]2 = [H3O+] · [OH-] da cui
Kw = Keq · [H2O]2 = 3,25 · 10-18 · (55,5)2 = 1,0 · 10-14 quindi,
Kw = [H3O+] · [OH-] = 1,0 · 10-14 oppure,
Kw = [H+] · [OH-] = 1,0 · 10-14 = prodotto ionico dell’acqua
pertanto, nell’acqua pura:
[H+] = [OH-] = 1,0 · 10-14 = 1,0 · 10-7 mol/L
Il valore del prodotto ionico dell’acqua è costante a temperatura costante e resta tale in tutte le
soluzioni acquose.
Es: se la [H+] è 1,0 · 10-2 mol/L, quale sarà la concentrazione degli ioni OH-?
[1.0 · 10-2] · [OH-] = 1,0 · 10-14
[OH-] =
1,0 · 10-14
1,0 · 10
-2
= 1,0 · 10-12 mol/L
17
Le costanti di acidità e basicità
L’acido acetico in acqua ionizza in questo modo:
CH3COO- + H3O+ per cui,
CH3COOH + H2O
Keq =
[CH3COO-] · [H3O+]
[CH3COOH] · [H2O]
La concentrazione dell’acqua è costante, anche in questo caso può essere incorporata nella costante
di equilibrio:
Keq · [H2O] = Ka costante di dissociazione acida
Ka =
[CH3COO-] · [H3O+]
[CH3COOH]
= 1,8 · 10-5
..........a 25 °C,
l’acido acetico è poco dissociato, la reazione è
spostata a sinistra.
L’acido cloridrico in acqua ionizza in questo modo:
H3O+ + Cl- per cui a 25 °C,
HCl + H2O
Ka =
[H3O+] · [Cl-]
= 1,0 · 107
[HCl]
l’acido cloridrico è molto dissociato, la reazione è
spostata nettamente a destra.
L’ammoniaca in acqua ionizza in questo modo:
NH4+ + OH- per cui,
NH3 + H2O
Keq =
[NH4+] · [OH-]
[NH3] · [H2O]
La concentrazione dell’acqua è costante, anche in questo caso può essere incorporata nella costante
di equilibrio:
Keq · [H2O] = Kb costante di dissociazione basica
Kb =
[NH4+] · [OH-]
[NH3]
= 1,8 · 10-5
........a 25 °C,
la base è poco dissociata, la reazione è spostata
a sinistra.
L’idrossido di sodio in acqua ionizza in questo modo:
NaOH + H2O
OH- + Na (H2O)+ per cui a 25 °C,
18
[OH-] · [Na (H2O)+]
Kb =
[NaOH]
= 5,0 · 100
la base è molto dissociata, la reazione è spostata
destra.
Il prodotto tra la costante acida e la costante basica di una coppia coniugata è uguale al prodotto
ionico dell’acqua:
Ka · Kb = Kw
Esercizi:
Calcola la Ka di NH4+, acido coniugato della base debole NH3, la cui Kb è uguale a 1,8 · 10-5.
Ka · 1,8 · 10-5 = 1,0 · 10-14
Ka =
1,0 · 10-14
1,8 · 10-5
= 5,5 · 10-10
La Ka dell’HCN è 6,2 · 10-10. Calcola la Kb della base CN-.
6,2 · 10-10 · Kb = 1,0 · 10-14
Kb =
1,0 · 10-14
6,2 · 10
-10
= 1,6 · 10-5
Quanto più elevata è la costante di un acido o di una base, tanto maggiore sarà la loro dissociazione:
un acido o una base, sono tanto più forti quanto più elevate sono le loro Ka o Kb.
Per determinare la forza delle basi e degli acidi si può applicare alla costante di equilibrio K la scala
logaritmica definendo: pK = - log K
es:
H2CO3 + H2O
H3O+ + HCO3-
La costante acida di prima ionizzazione dell’acido carbonico è:
Ka = 4,5 · 10-7 pertanto,
pKa = - log 4,5 – log 10-7 = - 0,65 + 7 = 6,35 (acido piuttosto debole)
es:
HNO2 + H2O
H3O+ + NO2-
La costante acida dell’acido nitroso è:
Ka = 5,1 · 10-4 pertanto,
pKa = - log 5,1 – log 10-4 = 0,71 + 4 = 3,29 ( acido medio forte)
Il valore del pKa è tanto più elevato quanto più l’acido è debole (poco dissociato), come si può
rilevare dal confronto tra H2CO3 e HNO2 . Le stesse considerazioni valgono per pKb.
19
Il pH
Premessa
In ogni soluzione acquosa sono sempre presenti ioni H+ e ioni OH- in concentrazioni tali da
verificare il prodotto ionico dell’acqua [H3O+] · [OH-] = 1,0 · 10-14 :
-
quando [H3O+] = [OH-] = 1,0 · 10-7 mol/L la soluzione è neutra;
quando [H3O+] > [OH-] la soluzione è acida;
quando [H3O+] < [OH-] la soluzione è basica.
Una concentrazione degli ioni H3O+ espressa mediante le potenze negative di 10 è scomoda da
usare, si preferisce ricorrere a una grandezza, il pH, che è definita come logaritmo negativo della
concentrazione degli ioni H3O+:
pH = - log [H3O+]
pH
0
[H3O+] 1
1
2
3
4
5
6
7
8
9
10
11
12
10-11
10-12
13
14
10-1 10-2 10-3 10-4 10-5 10-6
10-7 10-8
10-9 10-10
[OH-] 10-14 10-13 10-12 10-11 10-10 10-9 10-8
10-7 10-6
10-5
10-4
10-3
10-2
10-1
1
5
4
3
2
1
0
pOH
14
13
12
11
10
9
8
7
6
10-13 10-14
pOH = - log [OH-]
acidità crescente
neutralità
basicità crescente
Pertanto, ne deriva che: pH + pOH = 14
Determinazione del pH, esercizi:
•
di acidi forti monoprotici
-
Calcolare il pH della soluzione ottenuta mescolando 10 mL di HCl 0,1 M con 250 mL di H2O
distillata.
In una soluzione acquosa di un acido forte, avente concentrazione Ca, alla concentrazione
complessiva di ioni H3O+ contribuisce sia la ionizzazione dell’acqua che la ionizzazione completa
dell’acido:
HCl + H2O
H3O+ + ClPoiché gli ioni H3O+ sono in soluzione l’unica specie chimica carica positivamente, mentre sono
presenti due diverse specie cariche negativamente (Cl- e OH-), per l’irrinunciabile bilancio elettrico
deve essere, indicando lo ione idronio in forma semplificata (H+):
[H+] = [OH-] + [Cl-]
Considerando che l’acido è completamente dissociato, risulta:
20
Ca = [Cl-]
[H+] = [OH-] + Ca
ovvero
In genere, se Ca > 10-6 M, la [OH-] può essere trascurata rispetto a Ca, per cui si ha, passando ai
cologaritmi: pH = - log [H+] = -log Ca
La molarità è uguale alla normalità (N). V = volume.
numero equivalenti H+ = N HCl · V HCl = 0,1 equivalenti · 0,010 L = 0,001 equivalenti di H+
Considerando che il volume finale è aumentato, in seguito al mescolamento, sino a 260 mL, si ha:
[H+] = 0,001 equivalenti di H+/0,260 L = 0,00385 moli di H+/L
pH = - log [H+] = - log 0,00385 = 2,4
-
Determina il pH di una soluzione 0,001 M di HCl.
[H3O+] = 10-3 mol/L ……………..pH = -log 10-3 = 3
[OH-] = 10-11 mol/L ……………pOH = -log 10-11= 11
•
di basi forti monoprotiche
-
Determina il pH di una soluzione 0,001 M di KOH.
[OH-] = 10-3 mol/L……………. pOH = -log 10-3 = 3
[H3O+] = 10-11 mol/L ……………..pH = -log 10-11 = 11
•
di acidi deboli monoprotici
[H3O+] =
-
Ka · Ca
dove per Ca s’intende la concentrazione molare dell’acido
Determina il pH di una soluzione 0,85 M di acido acetico (Ka = 1,8 · 10-5).
[H3O+] =
Ka · Ca
=
1,8 · 10-5 · 0,85 =
1,53 · 10-5 =
15,3 · 10-6 = 3,91 · 10-3
pH = - log [H3O+] = - log [3,91 · 10-3] = - log 3,91 – log 10-3 = -0,59 + 3 = 2,41
•
di basi deboli monoprotiche
[OH-] =
-
Kb · Cb
dove per Cb s’intende la concentrazione molare della base
Determina il pH di una soluzione 0,55 M di ammoniaca (Kb = 1,8 · 10-5).
[OH-] =
Kb · Cb
=
1,8 · 10-5 · 0,55 =
0,99 · 10-5 =
9,9 · 10-6
pOH = - log [OH-] = - log [3,15 · 10-3] = - log 3,15 – log 10-3 = -0,5 + 3 = 2,5
pH = 14 – pOH = 14 – 2,5 = 11,5
21
= 3,15 · 10-3
•
di acidi poliprotici e di basi poliidrossiliche
-
Calcolare il pH di una soluzione acquosa di acido solforico 0,1 M, giustificando la scelta
dell’equazione ritenuta più idonea (Ka1 = 102; Ka2 = 1,2 · 10-2).
Dai valori delle Ka si evince che l’acido solforico è un acido forte nei riguardi della prima
ionizzazione, che risulta pertanto completa:
H3O+ + HSO4-
H2SO4 + H2O
mentre può essere considerato debole nei riguardi della seconda ionizzazione, per la quale si può
scrivere:
[H3O+] [SO42-]
HSO4- + H2O
H3O+ + SO42Ka2 =
[HSO4-]
Il successivo schema fornisce un quadro delle concentrazioni delle specie presenti in soluzione
prima e dopo l’aggiunta dell’acido in acqua, trascurando la ionizzazione dell’acqua ed indicando
con x la frazione di ioni HSO4- che ionizza secondo l’equilibrio precedente:
specie:
conc. Iniziale
all’equilibrio
HSO40
Ca – x
H2SO4
Ca
0
SO420
x
H+
0
Ca + x
Sostituendo questi valori nella costante di ionizzazione Ka2, si ottiene:
Ka2 =
[H3O+] [SO42-]
-
[HSO4 ]
(Ca + x) x
=
= 0,012
Ca – x
Trascurando x rispetto a Ca:
[H3O+] = Ca = 0,1 M
pH = - log [H3O+] = - log (0,1) = 1,00
Risolvendo invece l’equazione precedente rispetto ad x, si ha:
x2 + (Ka2 + Ca) x – Ka2 Ca = 0
x=
-(Ka2 + Ca) +
(Ka2 + Ca)2 + 4 Ka2 Ca
-(0,012 + 0,1) +
(0,112)2 + 4 · 0,012 · 0,1
=
2
=
2
= 9,85 · 10-3 M, da cui:
[H3O+] = Ca + x = 0,1 M + 9,85 · 10-3 M = 0,11 M
pH = - log [H3O+] = -log (0,11) = 0,96
Il valore di [H3O+] calcolato operando la precedente semplificazione è circa il 9% inferiore a quello
calcolato con l’equazione di secondo grado, per cui l’approssimazione effettuata non pare del tutto
lecita.
22
Soluzioni tampone
Premessa
Nel sangue umano, il pH è 7,4 e tale valore non deve variare di ± 0,4; una variazione più ampia non
permette la sopravvivenza. La stabilità del pH è dovuta a sistemi che si oppongono alle variazioni
del pH, tali sistemi sono detti tamponi. Nel sangue, il sistema tampone è costituito principalmente
dalla coppia acido carbonico, H2CO3, e ione idrogenocarbonato, HCO3-, cioè dall’associazione di un
acido debole con il suo sale di una base forte.
In generale, una soluzione tampone si basa sulla capacità che gli acidi deboli e le loro basi
coniugate hanno di reagire con una base forte o con un acido forte.
Una soluzione tampone molto comune è quella formata dall’acido acetico, CH3COOH, e da un suo
sale, l’acetato di sodio, CH3COONa.
L’acetato di sodio in acqua si dissocia completamente secondo la reazione:
CH3COO-(aq) + Na+(aq)
CH3COONa(s) + H2O(l)
(1)
L’acido acetico in acqua si ionizza generando il seguente equilibrio:
CH3COO-(aq) + H3O+(aq)
CH3COOH(l) + H2O(l)
[CH3COO-(aq)] · [H3O+(aq)]
Ka =
= 1,8 · 10-5
[CH3COOH(l)]
(2)
l’acido acetico è poco ionizzato, la reazione
è spostata a sinistra.
Per il principio di Le Chatelier, gli ioni CH3COO-, provenienti dalla dissociazione ionica
dell’acetato di sodio, spostano ulteriormente la reazione (2) a sinistra. Perciò, in soluzione ci sarà
una concentrazione elevata di molecole indissociate di CH3COOH e una concentrazione altrettanto
elevata di ioni CH3COO- (quest’ultima dovuta soprattutto alla dissociazione dell’acetato di sodio),
mentre la concentrazione di ioni H3O+, provenienti dall’equilibrio (2) sarà piccola.
Se si aggiungono a questa soluzione piccole quantità di HCl; in quanto acido forte, questo composto
si ionizzerà completamente:
H3O+(aq) + Cl-(aq)
HCl(l) + H2O(l)
[H3O+(aq)] · [Cl-(aq)]
Ka =
per cui,
= 1,0 · 107
l’acido cloridrico è molto ionizzato, la reazione
è spostata nettamente a destra.
[HCl(l)]
Gli ioni H3O+ liberati saranno “catturati” dagli ioni CH3COO-, si formerà altro acido acetico non
ionizzato:
CH3COO-(aq) + H3O+(aq)
CH3COOH(l) + H2O(l)
Pertanto, la quantità di ioni H3O+ nella soluzione varierà pochissimo e il pH rimarrà pressoché
costante.
23
Se si aggiungono invece alla soluzione tampone piccole quantità di NaOH; in quanto base forte,
questo composto si dissocerà completamente:
OH-(aq) + Na+(aq)
NaOH(s) + H2O(l)
[OH-(aq)] · [Na+(aq)]
= 5,0 · 100
Kb =
[NaOH(s)]
per cui,
la base è molto dissociata, la reazione è spostata
a destra.
Gli ioni OH- liberati saranno “catturati” dalle molecole di CH3COOH, si formerà acqua secondo la
reazione:
CH3COOH(l) + OH-(aq)
CH3COO-(aq) + H2O(l)
Anche in questo caso il pH rimarrà pressoché invariato perché la quantità di ioni H3O+ varierà
pochissimo.
Dalla (2) si ricava che:
[CH3COOH]
[H3O ]
+
= Ka
cioè,
[CH3COO-]
la concentrazione molare di H3O+ è proporzionale al rapporto tra la concentrazione molare
dell’acido acetico indissociato e la concentrazione molare del suo ione acetato (apportato anche
dall’acetato di sodio).
Il pH di tale soluzione sarà:
[CH3COOH]
[CH3COOH]
pH = - log Ka - log
ovvero pH = pKa - log
(3)
[CH3COO ]
[CH3COO ]
[acido]
Un sistema è un buon tampone se il rapporto
≅1
[base coniugata]
Il pH di una soluzione tampone non varia al variare della concentrazione della soluzione stessa e
dipende soltanto dal rapporto tra la concentrazione molare dell’acido debole e della sua base
coniugata oppure della base debole e del suo acido coniugato.
/
Nel nostro caso cioè dal rapporto [CH3COOH] [CH3COO-]. Quando il valore di tale rapporto è
uguale all’unità, dalla (3) risulta che pH = pKa .
In definitiva, il pH di soluzioni tampone formate da un acido debole e dal suo sale con una base
forte è dato da:
[acido]
pH = pKa – log
[sale]
……quindi,
pH = pKa + log
[sale]
[acido]
24
Esercizi:
-
Calcolare la variazione di pH conseguente all’aggiunta di 0,001 moli di HCl ad 1 litro di
soluzione 0,01 M in acido acetico e 0,01 M in acetato di sodio.
In una soluzione preparata miscelando in acqua l’acido debole CH3COOH in concentrazione Ca e la
sua base coniugata in forma salina CH3COONa, in concentrazione Cs, si può trascurare senz’altro il
contributo della dissociazione dell’acido alla concentrazione degli ioni CH3COO-, rispetto a quello
della dissociazione completa del sale, per cui si può ragionevolmente considerare:
Cs ≅ [CH3COO-]
Inoltre, poiché la dissociazione di un acido debole è sempre piuttosto limitata, è lecito supporre:
Ca ≅ [CH3COOH]
Sostituendo entrambe queste uguaglianze nella costante di ionizzazione dell’acido, si ottiene:
[CH3COOH]
[H3O ]
+
= Ka
Ca
= Ka
[CH3COO ]
-
Cs
ovvero:
pH = pKa + log
Cs
Ca
Dato che la Ka dell’acido acetico è pari a 1,8 · 10-5, corrispondente ad un pKa di 4,75, prima di
aggiungere HCl il pH della soluzione è dunque:
pH = pKa + log(Cs/Ca) = 4,75 + log (0,01/0,01) = 4,75
Dopo l’aggiunta, essendo HCl un acido forte, tutti i protoni da esso liberati reagiscono con un
equivalente numero di ioni CH3COO- facendo variare il numero di moli di queste specie secondo il
seguente schema:
specie
CH3COOH
CH3COOprima dell’aggiunta
0.01
0,01
dopo l’aggiunta
0,01 + 0,001 = 0,011
0,01 – 0,001 = 0,009
da cui:
pH = pKa + log(Cs/Ca) = 4,75 + log (0,009/0,011) = 4,67
La piccola variazione di pH osservata mostra l’efficace potere tamponante di questa soluzione.
-
Calcolare il pH di una soluzione ottenuta mescolando 40 mL di una soluzione 0,25 M di acido
formico e 10 mL di una soluzione 0,05 M di formiato di potassio. Il pKa dell’acido formico è
3,75.
Determinazione della concentrazione molare delle due sostanze in 50 mL di soluzione:
acido formico:
40 mL · 0,25 M = 50 mL · x M
formiato di potassio:
x M = 40 mL · 0,25 M /50 mL = 0,2 M (Ca)
10 mL · 0,05 M = 50 mL · x M
x M = 10 mL · 0,05 M /50 mL = 0,01 M (Cs)
Determinazione del pH della soluzione tampone:
pH = pKa + log (Cs/Ca) = 3,75 + log (0,01 M/0,2 M) = 3,75 + log 0,05 = 3,75 – 1,3 = 2,45
25
-
A 100 mL di soluzione 0,7 M di HNO2 (pKa = 4,4) vengono aggiunti 30 mL di soluzione 1 M
di NaOH. Calcolare il pH di tale soluzione.
Facendo reagire le due soluzioni, avviene la seguente reazione di neutralizzazione:
NO2- + Na+
HNO2 + NaOH
+ H2O
La situazione del numero di moli di ciascuna specie presente prima e dopo il mescolamento nella
soluzione è la seguente:
specie
prima
dopo
HNO2
n acido=MV=(0,7) (0,1)= 0,07
n acido=0,07 – 0,03= 0,04 (Ca)
NO2n sale = 0
n sale=0 + 0,03= 0,03 (Cs)
OHn=MV=(1) (0,03)= 0,03
n=0,03 – 0,0 = 0
da cui:
pH = pKa + log (Cs/Ca) = 4,4 + log (0,03/0,04) = 4,4 + log 0,75 = 4,4 – 0,12 = 4,28
Composti anfoteri
Esistono alcuni composti che, in soluzione acquosa, si dimostrano capaci di comportarsi sia da acidi
(neutralizzando una base), sia da basi (neutralizzando un acido). Questi composti vengono detti
anfoteri.
Esempio:
se si fa reagire
Al2(SO4)3 + 6 KOH
2 Al (OH)3 + 3 K2SO4
precipitato bianco
…..aggiungendo un eccesso di KOH,
Al (OH)3 + OHacido
Al (OH)-4
…..aggiungendo invece un acido (H+)
Al (OH)3
base
+ 3 H3O+
Al3+ + 6 H2O
…………..pertanto, Al (OH)3 può neutralizzare sia le basi, sia gli acidi.
26
Idrolisi
L’interazione fra sali e acqua viene detta idrolisi.
•
Idrolisi di sali provenienti da acidi forti e basi deboli: idrolisi acida, pH < 7.
Esempio: cloruro d’ammonio
NH4Cl(s) + H2O(l)
NH4+(aq) + Cl-(aq)
Lo ione NH4+ reagisce con l’acqua secondo la reazione d’equilibrio
NH4+ + H2O
NH3 + H3O+ la cui costante è Ka = 5,68 ⋅ 10-10
Per quanto riguarda invece la reazione di equilibrio
Cl- + H2O
HCl + OH- la costante di basicità è Kb = 1⋅ 10-21
Ka > Kb pertanto la soluzione di cloruro d’ammonio risulta acida.
•
Idrolisi di sali provenienti da acidi deboli e basi forti: idrolisi basica, pH > 7
Es: acetato di sodio
CH3COONa(s) + H2O
CH3COO-(aq) + Na+(aq)
Lo ione CH3COO- reagisce con l’acqua:
CH3COO- + H2O
CH3COOH + OH- la cui costante di basicità Kb = 5,68 ⋅ 10-10
Per quanto riguarda la reazione di equilibrio
Na+ + 2 H2O
NaOH + H3O+ la costante di acidità è Ka = 2 ⋅ 10-15
Kb > di Ka pertanto la soluzione di acetato di sodio sarà basica.
•
Nelle soluzioni di sali da acido forte/base forte (es: NaCl) oppure da acido debole/base debole
non si verificano equilibri capaci di alterare l’equilibrio di ionizzazione dell’acqua: le soluzioni
hanno pH neutro.
Neutralizzazione
Secondo la teoria di Arrhenius, si può definire neutralizzazione la reazione tra un acido e una base,
nella quale gli ioni H3O+ dell’acido reagiscono con gli ioni OH- della base dando H2O:
H3O+ + OH2 H2O
Quando un acido e una base reagiscono in quantità equivalenti*, si possono verificare situazioni
differenti:
• se un acido e una base sono di uguale forza, la soluzione sarà neutra;
• se l’acido è forte e la base debole, la soluzione sarà acida;
• se l’acido è debole e la base è forte, la soluzione sarà basica.
27
*Peso equivalente o grammo equivalente o massa equivalente (meq)
1) di un sale dissociato in ioni è data dal rapporto tra la massa molare (M) del sale e il numero di
cariche unitarie positive che esso genera dissociandosi (valenza ionica):
2 Na+ + SO42-
Na2SO4
meq Na2SO4 = M/2 = 142,05/2 = 71,02 g
2) di un acido è data dal rapporto tra la massa molare (M) dell’acido e il numero di ioni H+ che la
sua molecola libera, dissociandosi in soluzione:
HCl
H+ + Cl-
meq HCl = M/1 = 36,46 g
H2SO4
2 H+ + SO42-
meq H2SO4 = M/2 = 98,08/2 = 49,04 g
3) di un idrossido è pari al rapporto tra la sua massa molare (M) e il numero di ioni OH- che esso
libera, dissociandosi in soluzione:
Ca(OH)2
Ca2+ + 2 OH-
meq Ca(OH)2 = M/2 = 74,01/2 = 37,005 g
Normalità o soluzione normale
La normalità (N) di una soluzione, è il numero di equivalenti contenuti in un litro di soluzione:
- soluzione N di H2SO4 = 49,04 g in un litro;
- soluzione N di HCl = 36,46 g in un litro;
- soluzione N di NaOH = 40 g in un litro;
- soluzione 2N di NaOH = 80 g in un litro.
Esercizio:
Calcolare il pH della soluzione ottenuta unendo 150 mL di HCl 0,1 M con 200 mL di KOH 0,2 M.
a)
b)
HCl + KOH
H+ + OH-
K+ + Cl- + H+ + OHH2O
Per entrambe le sostanze la molarità è uguale alla normalità (N). V = volume.
numero equivalenti H+ = N HCl · V HCl = 0,1 equivalenti · 0,15 L = 0,015 equivalenti di H+
numero equivalenti OH- = N KOH · V KOH = 0,2 equivalenti · 0,2 L = 0,04 equivalenti di OHLo ione H+ essendo in difetto, viene completamente consumato nella reazione di neutralizzazione
con OH- e resta in soluzione solo l’eccesso di OH-. Considerando che il volume finale è aumentato,
in seguito al mescolamento, sino a 350 mL, si ha:
concentrazione base (Cb) = (0,04 – 0,015) equivalenti di OH-/0,350 L = 0,0714 moli di OH-/L
pH = pKw – pOH = 14 + log Cb = 14 + log (0,0714) = 14 – 1,15 = 12,85
28
Le reazioni di ossidoriduzione
Nella moderna nomenclatura , al posto della valenza, si preferisce utilizzare il numero di
ossidazione (n.o.) per indicare la carica che ogni atomo in una molecola o in uno ione poliatomico
assumerebbe se gli elettroni di legame fossero assegnati all’atomo più elettronegativo.
Esistono sei regole per assegnare il numero di ossidazione:
1. Allo stato elementare, tutti gli atomi hanno numero di ossidazione zero.
Es: Zn H2
N2
P4
Cu
O2
Fe
S8
2. La somma dei numeri di ossidazione di tutti gli atomi di un composto deve essere uguale a zero
(per le molecole neutre), uguale alla carica complessiva dello ione (nel caso di ioni poliatomici).
+1 -2
+1 +6 -2
Es: H2O
+6 –2
SO42-
H2SO4
+5 -2
NO3-
3. Gli ioni monoatomici hanno n.o. uguale alla loro carica.
-1
Es:
+1
Cl-
+2
Na+
+3
Ca2+
Al3+
+2
Fe2+
+3
Fe3+
4. In un legame covalente, si attribuiscono formalmente i due elettroni di legame all’atomo più
elettronegativo.
+1 -1
Es: HCl
+1 -1
HF
-3 +1
NH3
5. In tutti i composti (tranne gli idruri metallici) l’H ha sempre n.o. +1.
+1 -1
Es: NaH
+2 -1
CaH2
6. In tutti i composti (tranne i perossidi e OF2) l’ossigeno ha sempre numero di ossidazione –2.
+1 -1
Es: H2O2
+2 -1
OF2
Nelle reazioni di ossidazione il numero di ossidazione aumenta; nelle reazioni di riduzione il
numero di ossidazione diminuisce. La reazione di ossidazione avviene quasi sempre con la perdita
di uno o più elettroni, mentre la reazione di riduzione si verifica con l’acquisto di uno o più
elettroni.
ossidazione
-5, -4, -3, -2, -1, 0, +1, +2, +3, +4, +5, +6, +7
riduzione
es:
0
0
2 Mg + O2
-
due atomi di Mg perdono 2 e- ciascuno e si ossidano:
due atomi di O acquistano 2 e- ciascuno e si riducono:
+3 -2
+2 -2
Fe2O3 + 3 CO
-
+2 -2
2 MgO
0
- 4 e+ 4 e-
+4 -2
2 Fe + 3 CO2
3 atomi di C perdono 2 e- ciascuno e si ossidano:
2 atomi di Fe acquistano 3 e- ciascuno e si riducono:
29
- 6 e+ 6 e-
Bilanciamento delle reazioni redox
-
Scrivi la reazione bilanciata tra acido triossonitrico (V), HNO3, e solfuro di idrogeno, che
produce S, monossido di azoto e H2O.
a) reazione non bilanciata:
+1+5-2
+1 -2
+2 -2
HNO3 + H2S
0
+1 -2
NO + S + H2O
Gli elementi che variano il numero di ossidazione sono:
+5
+2
-2
0
N + 3 e- N, riducendosi (red) e S - 2 e- S, ossidandosi (ox).
b) poiché il numero di elettroni acquistati e perduti deve essere uguale….
HNO3
+ H2S
+5
NO + S + H2O
+2
X2 N + 3 e-
N
+5
red ………………………che diviene: 2 N + 6 e-
-2
X3 S -2 e-
0
S
-2
ox
…………….che diviene: 3 S
-6 e-
+2
2N
0
3S
c) riporta i coefficienti così calcolati nella reazione data.
2 HNO3
+ 3 H2S
2 NO + 3 S
+ H2O
d) completa il bilanciamento per l’H2O.
2 HNO3 + 3 H2S
2 NO + 3 S + 4 H2O
Elettrochimica
Poniamo in becher separati una lamina di zinco, in:
a) soluzione di HCl
si nota che lo zinco passa in soluzione e si sviluppano bollicine di gas e calore…
Zn
- 2 e2 H+ + 2 ereazione complessiva: Zn + 2 H+
Zn2+ (ossidazione)
H2
(riduzione)
Zn2+ +
H2
b) soluzione di CuSO4
si nota un deposito di rame metallico sullo zinco, mentre questo si consuma passando in
soluzione; la soluzione originariamente azzurra si scolora e si osserva un lieve sviluppo di
calore.
30
Zn
- 2 e2+
Cu + 2 e-
Zn2+ (ox)
Cu
(red)
reazione complessiva: Zn + Cu2+
Zn2+ + Cu
In questi due casi lo zinco ha dimostrato di possedere una maggiore capacità sia dell’idrogeno, sia
del rame di perdere elettroni, cioè di ossidarsi.
Questi esempi evidenziano che:
- le rezioni redox sono spontanee, infatti sono sempre accompagnate da uno sviluppo più o meno
evidente di calore.
- alcuni ioni sono più abili di altri a catturare elettroni (tendenza alla riduzione).
Celle elettrochimiche: le pile
Lo spostamento di elettroni nelle reazioni di ossido-riduzione, è utilizzato per ottenere energia
elettrica. Abbiamo visto che, se si immerge una lamina di zinco in una soluzione contenente ioni
Cu2+, si verifica un trasferimento diretto di elettroni dallo zinco metallico agli ioni Cu2+, attraverso
una reazione che si può considerare come la somma di due semireazioni, una di riduzione e una di
ossidazione. Facendo svolgere le due semireazioni in due recipienti distinti, ma opportunamente
collegati, gli elettroni liberati nel recipiente in cui avviene l’ossidazione, tenderanno a trasferirsi nel
recipiente ove avviene la riduzione, generando un flusso di elettroni, cioè una corrente elettrica. I
due sistemi metallo-soluzione, che corrispondono alle due semireazioni, sono chiamati semipile; il
dispositivo complessivo è definito cella elettrochimica o pila.
La pila Daniell
Si basa sulla reazione tra zinco metallico e ioni Cu2+. Una semipila è formata da una soluzione M di
ZnSO4 in cui è immersa una lamina di Zn, l’altra semipila è formata da una soluzione M di CuSO4
in cui è immersa una lamina di rame. Le due lamine metalliche (elettrodi) sono collegate da un filo
conduttore.
Galvanometro (amperometro)
e-
e-
ZINCO –
ANODO
+
2 e-
2 e-
ponte salino di KCl (aq) che chiude il circuito
+
Cu2+
Zn2+
Zn2+
Zn
ZnSO4 M
RAME
CATODO
Cu2+
Cu
CuSO4 M
In tutte le pile l’elettrodo su cui avviene la reazione di ossidazione viene denominato anodo ed è
considerato l’elettrodo negativo, in quanto fornisce elettroni al circuito esterno; l’elettrodo su cui
avviene la reazione di riduzione viene denominato catodo ed è considerato l’elettrodo positivo in
quanto attira gli elettroni del circuito esterno.
31
I potenziali standard di riduzione
L’intensità del flusso di elettroni, è tanto maggiore quanto più forte è la tendenza di una delle due
semipile a catturare elettroni rispetto all’altra. Purtroppo, il valore assoluto di tale tendenza, definita
potenziale di una singola pila, non è misurabile, mentre con un particolare strumento, detto
potenziometro, è possibile misurare il valore relativo, cioè la differenza dei potenziali (d.d.p.) di due
semipile. Come semipila di confronto, chiamata elettrodo di riferimento, è stato scelto l’elettrodo
di idrogeno, al cui potenziale è stato assegnato per convenzione il valore zero. La semireazione è:
2 H+
+ 2 e-
H2
Il confronto di vari elettrodi con quello dell’idrogeno, viene effettuato in condizioni standard (es: 25
°C, [ ] M), si determina così per ogni semireazione, il potenziale di riduzione standard (E°).
L’elemento che ha potenziale maggiore si riduce; l’elemento che ha potenziale minore si ossida.
Determinazione del potenziale di riduzione: equazione di Nernst
A 25 °C è:
E’ = E + 60 log [ox]/[R]
E’ = potenziale di riduzione
E = potenziale di riduzione standard
[ox] = concentrazione dell’ossidante
[R] = concentrazione del riducente
L’equazione di Nernst consente di calcolare il potenziale di riduzione E’ di una coppia redox in
funzione del rapporto fra le concentrazioni dell’ossidante ox e del riducente R, essendo noto E.
Forza elettromotrice di una pila o potenziale di una pila
E° semireazione di riduzione + E° semireazione di ossidazione
es:
riduzione: 2 Ag+ + 2 eossidazione:
Cu - 2 e-
Ag
E° = + 0,80 V
Cu2+
E° = - 0,34 V
pertanto il potenziale standard della pila è:
E° pila = 0,80 + (-0,34) = 0.46 V
…..la f. e. m. si misura in volt (V).
32
Elettrolisi
Le pile sono dispositivi in cui si produce energia elettrica mediante reazioni redox spontanee. Le
celle elettrolitiche sono invece dispositivi in cui si svolge il processo inverso, cioè dispositivi in cui
si spende energia elettrica per far avvenire reazioni redox non spontanee (elettrolisi).
Una cella elettrolitica è costituita da un recipiente contenente l’elettrolita allo stato fuso, o in
soluzione, in cui sono immersi due elettrodi. Questi vengono collegati ad un generatore di corrente
continua. Nella cella elettrolitica l’elettrodo collegato al polo negativo del generatore viene
denominato catodo, quello collegato al polo positivo viene denominato anodo.
Quando i due elettrodi, collegati ai poli del generatore, vengono immersi nella soluzione, in questa
si verifica una doppia migrazione degli ioni presenti nell’elettrolita. Gli ioni positivi, i cationi,
vengono attratti dall’elettrodo negativo, il catodo, dove catturano elettroni e si riducono,
contemporaneamente, gli ioni negativi, gli anioni, vengono attratti dall’elettrodo positivo, l’anodo,
dove cedono elettroni e si ossidano.
eanodo
e+
-
catodo
Se si confrontano gli schemi di funzionamento di una pila e di una cella elettrolitica, si nota che in
entrambi i casi, l’anodo è l’elettrodo in cui avviene la reazione di ossidazione e il catodo è
l’elettrodo in cui avviene la reazione di riduzione. Invece, il segno dei due elettrodi è opposto.
Infatti l’anodo è negativo nella pila, positivo nella cella elettrolitica; il catodo è positivo nella pila,
negativo nella cella elettrolitica.
Alcuni processi elettrolitici
- elettrolisi dello ioduro di potassio:
se si fonde lo KI, si rompono i legami tra gli ioni che costituiscono il solido, K+ e I-, che sono liberi
di muoversi. Se si immergono nella massa fusa due elettrodi di grafite e li si collega ai poli di una
batteria da 10 V, si ha l’elettrolisi del sale. Al catodo avviene la reazione di riduzione K+ + eK
mentre all’anodo avviene la reazione di ossidazione 2 I- - 2 eI2 …. reazione globale
2 K+ + 2 I-
2 K + I2 .
In definitiva, si ottengono potassio al catodo e iodio all’anodo.
-
elettrodeposizione: cromatura, zincatura, argentatura, doratura.
33
Le leggi quantitative dell’elettrolisi
• Prima legge di Faraday
La massa delle sostanze che si trasformano agli elettrodi è direttamente proporzionale alle quantità
di carica elettrica che fluisce nella cella elettrolitica.
• Seconda legge di Faraday
In una cella elettrolitica, al passaggio di 96500 C (coulomb) di carica elettrica, ad entrambi gli
elettrodi, si scarica una quantità di sostanza pari alla sua massa equivalente.
1 F (faraday) = 96500 C
La seconda legge si può interpretare così: ogni semireazione redox che avviene in una cella
elettrolitica, coinvolge un numero di elettroni diverso nei diversi casi.
Per esempio nell’elettrolisi di KCl, CuCl2, FeCl3 allo stato fuso, il numero di elettroni richiesti per
la scarica dei cationi cambia.
KCl
K+
CuCl2
FeCl3
+ Cl-
K+ + 1 e-
K
Cu2+ + 2 Cl-
Cu2+ + 2 e-
Cu
Fe3+ + 3 Cl-
Fe3+ + 3 e-
Fe
al catodo:
La stessa quantità di carica (1 F) necessaria perché al catodo si depositi una mole di K, consentirà il
deposito di ½ mole di Cu, e di 1/3 di mole di Fe.
M
Massa equivalente =
numero di e- scambiati
……..1 F scarica una massa equivalente.
34
APPUNTI DI CHIMICA ORGANICA
Premessa
La chimica dei composti del carbonio viene chiamata chimica organica e le relative sostanze
vengono chiamate composti organici.
Ancora all’inizio del 1800 si riteneva che le sostanze proprie degli organismi viventi non potessero
essere sintetizzate in laboratorio perché, per la loro formazione, si pensava fosse necessaria una
misteriosa “forza vitale” che solo gli organismi possedevano.
La corrente di pensiero che sosteneva questa concezione si chiamava vitalismo e uno dei suoi
massimi rappresentanti, il chimico svedese Jöns Jacob Berzelius, coniò nel 1807 il termine
“composti organici” per i composti del mondo vivente, mentre definì “composti inorganici “ quelli
del mondo minerale, considerato privo di forza vitale.
Il superamento della teoria del vitalismo e la nascita della chimica organica moderna risale al 1828.
In quell’anno, il chimico tedesco Friedrich Wöhler, allievo di Berzelius, sintetizzò in laboratorio,
riscaldando una soluzione di cianato d’ammonio, l’urea, una sostanza sintetizzata dal fegato negli
organismi ureotelici, che comprendono anche l’uomo, ed eliminata con l’urina.
NH4+ OCN-
O=C
NH2
NH2
Dopo la scoperta di Wöhler, i chimici sintetizzarono, in laboratorio, altri composti costituenti degli
organismi viventi. Tutti questi composti avevano come caratteristica comune la presenza del
carbonio nella loro composizione elementare. Pertanto, il termine “chimica organica” si riferisce
alla chimica dei composti del carbonio.
Oggi, la chimica organica comprende sia i composti naturali del carbonio che fanno parte del
mondo vivente, sia sostanze completamente nuove, sintetizzate in laboratorio, tanto che, si
conoscono circa 5 milioni di sostanze pure che contengono carbonio.
Ibridazione del carbonio: teoria di Pauling
Struttura elettronica del C:
6
C = 1s2 2s2 2p2
Con quattro e-, il guscio di valenza del C è pieno a metà. Gli atomi di C non hanno una forte
tendenza ne a perdere tutti i loro e- di valenza e diventare C4+ ne una forte tendenza ad acquistare 4
e- e diventare C4-, preferiscono formare 4 legami covalenti condividendo con altri atomi gli enecessari per raggiungere la stabilità rappresentata dall’ottetto di Lewis.
35
Esempi:
1) legami semplici C — C e legami sostanzialmente omopolari C — H
H H
|
|
H — C — C — H etano
|
|
H H
2) legami semplici polarizzati
δCl
δ|δ+ δCl — C — Cl tetracloruro di carbonio
|δCl
3) legami covalenti multipli
δ- δ+ δO=C=O
anidride carbonica o diossido di carbonio
δ+ δH—C≡N
acido cianidrico
Ibridazione sp3 del C (tetraedrica)
2p2
●
●
●
●
●
●
●
●
● sp3
2s2
● ●
●
un e- 2s è promosso
su un orbitale 2p
l’orbitale 2s e i 3 orbitali 2p
si mescolano per formare 4
orbitali ibridi sp3 equivalenti
1s2
● ●
● ●
● ●
Energia
Esempio: metano CH4
H
|
C
H
H
H
C tetraedrico = 4 orbitali ibridi orientati ai vertici di un tetraedro con angoli di 109,5°.
Nel metano ci sono 4 legami semplici C — H (sp3 — s) di tipo σ.
L’ibridazione sp3 del C è tipica degli alcani.
36
Ibridazione sp2 del C (trigonale)
2p2
●
●
●
●
● p
●
●
●
● sp2
2s2
● ●
●
un e- 2s è promosso
su un orbitale 2p
l’orbitale 2s e i 2 orbitali 2p
si mescolano per formare 3
orbitali ibridi sp2 equivalenti,
rimane un orbitale p
1s2
● ●
● ●
● ●
Energia
Esempio: etilene o etene C2H4
H
H
C=C
H
H
C trigonale = 3 orbitali ibridi orientati ai vertici di un triangolo equilatero con angoli di 120° e sullo
stesso piano.
Nell’etene ci sono 4 legami semplici C — H (sp2 — s) di tipo σ e un doppio legame C = C (sp2 —
sp2 e p — p) σ + π. La molecola dell’etene è planare.
L’ibridazione sp2 del C è tipica degli alcheni.
Ibridazione sp del C (lineare)
2p2
●
●
●
●
●
●
●
● p
● sp
2s2
● ●
●
un e- 2s è promosso
su un orbitale 2p
l’orbitale 2s e i 1 orbitale 2p
si mescolano per formare 2
orbitali ibridi sp equivalenti,
rimangono 2 orbitali p
1s2
● ●
● ●
● ●
Energia
Esempio: acetilene o etino C2H2
H—C≡C—H
C lineare = 2 orbitali ibridi sulla stessa linea con angolo di 180°.
Nell’etino ci sono 2 legami semplici C — H (sp — s) di tipo σ e un triplo legame C ≡ C (sp2 — sp2,
p — p e p — p) σ + 2π. La molecola dell’etino è lineare.
L’ibridazione sp del C è tipica degli alchini.
37
Isomeria
Sono isomeri i composti con stessa formula molecolare o grezza, ma diversa formula di struttura:
-
isomeria conformazionale negli alcani e nei cicloalcani
Gli isomeri conformazionali differiscono a seguito di una libera rotazione attorno al legame
semplice carbonio-carbonio. In conseguenza di questa rotazione, sono possibili un numero infinito
di strutture chiamate conformeri o rotameri:
H
H
HH
H
H
H
H
H
H
HH
H
H
H
HH
H
H
H
H
H
H
H
Nella molecola dell'etano è possibile immaginare due casi limite di sistemazione di un gruppo
metilico rispetto all'altro gruppo metilico. Queste due conformazioni vengono indicate con il nome
di conformazione eclissata e conformazione sfalsata. Per una questione di ingombro sterico e a
causa della repulsione delle nuvole elettroniche dei legami C-H, la conformazione sfalsata è più
stabile della conformazione eclissata.
Nell'analisi conformazionale del cicloesano è possibile individuare il conformero più stabile detto a
sedia che presenta un anello privo di tensioni torsionali in quanto tutti i legami sono sfalsati.
Il conformero a barca è in rapido equilibrio con la conformazione a sedia perchè è meno stabile in
quanto presenta nell’anello due legami eclissati.
38
-
isomeria configurazionale o cis – trans nei cicloalcani
I cicloalcani presentano un tipo di isomeria detta configurazionale o cis – trans. Con il termine
configurazione di un atomo si intende la particolare posizione spaziale degli atomi o dei gruppi
atomici che lo circondano.
Isomeri cis – trans dell’ 1,2 – dibromociclopentano: il prefisso cis indica che i due sostituenti ( i
due atomi di bromo) sono dalla stessa parte rispetto al piano dell’anello carbonioso; il prefisso trans
indica che i due sostituenti sono da parti opposte rispetto al piano dell’anello carbonioso.
Isomeria cis – trans dell’ 1,2 – dimetilciclopentano:
-
isomeria di struttura negli alcani
Quando un idrocarburo ha un numero di atomi di carbonio uguale o maggiore di quattro si verifica
il fenomeno dell’isomeria, cioè alla stessa formula molecolare corrispondono due o più composti
che hanno struttura diversa. Isomeri di struttura del butano, C4H10:
CH3
|
CH3 – CH – CH3
CH3 – CH2 – CH2 – CH3
n – butano
Isomeri di struttura del pentano, C5H12:
CH3 – CH2 – CH2 – CH2 – CH3
2 – metilpropano o isobutano
CH3
|
CH3 – CH – CH2 – CH3
n – pentano
2 – metilbutano
CH3
|
CH3 – CH – CH3
|
CH3
2,2 – dimetilpropano
- isomeria di posizione:
| |
|
|
C – C – C – C – OH
| |
|
|
1 – butanolo
39
| |
| |
C–C–C–C
| |
| |
OH
2 – butanolo
|
C
|
| |
C–C–C
|
| |
OH
2 – metil – 2 – propanolo
o alcool terz – butilico
| |
|
|
C – C – C – C – OH
| |
|
|
- isomeria di catena:
1- butanolo
| | |
|
C – C – C – C – OH
| |
|
|
1- butanolo
- isomeria di gruppo
funzionale:
-
| |
| |
C–C–O–C–C
| |
| |
etere dietilico
stereoisomeria geometrica cis – trans negli alcheni
L’isomeria cis – trans negli alcheni dipende dalla rigidità del doppio legame che impedisce la
rotazione degli atomi di carbonio coinvolti, i quali inoltre, devono legare entrambi due atomi o due
gruppi diversi. Gli isomeri cis e trans sono costituiti da molecole che hanno gli stessi tipi di legami
tra atomi, ma diversa disposizione nello spazio. L’isomero cis ha i gruppi simili dalla stessa parte,
mentre l’isomero trans ha i gruppi dalla parte opposta rispetto all’asse del doppio legame.
I due isomeri, cis e trans, hanno un diverso punto di fusione (i trans fondono sempre a temperatura
più alta del cis) e di ebollizione, ma soprattutto una diversa polarità. Queste differenze consentono
di separare e identificare l’isomero cis dal trans. L’isomero trans, essendo maggiormente
simmetrico, è meno polare del cis o del tutto apolare. Isomeri cis e trans dell’acido butendioico:
HOOC
COOH
H
C= C
H
COOH
C=C
H
acido maleico (tossico)
cis – butendioico
HOOC
H
acido fumarico (intermedio del ciclo di Krebs)
trans – butendioico
L’acido cis butendioico o acido maleico fonde a 130 °C; è di sapore sgradevole e tossico; molto
solubile in acqua è più forte dell’acido fumarico. Non è mai stato isolato da prodotti naturali: è però
facilmente preparato per idrolisi della sua anidride (a sua volta ottenuta per ossidazione del
benzene). È impiegato nell’industria tessile come colorante, nell’industria delle materie plastiche e
in molte sintesi organiche.
L’acido trans butendioico o acido fumarico sublima a 200 °C, fonde a 287 °C ed è un composto
intermedio del ciclo di Krebs o ciclo dell’acido citrico, pertanto, è essenziale nella respirazione
cellulare. Industrialmente viene ottenuto per isomerizzazione dell’acido maleico oppure per
fermentazione ossidativa o fumarica dei carboidrati. Trova impiego nell’industria alimentare come
sostituto dell’acido tartarico o dell’acido citrico e come antiossidante.
40
Isomeria ottica o enantiomeria
Osserva le tue due mani accostate. Apparentemente ti sembrano uguali, in realtà esse godono di una
proprietà particolarissima. Quando provi a sovrapporle a palmi entrambi volti verso il basso o
entrambi volti verso l’alto, le dita non coincidono. Prova ora a porre davanti a uno specchio la tua
mano destra, col palmo rivolto allo specchio: quest’ultimo riflette esattamente l’immagine del
palmo della tua mano sinistra. Ne puoi dedurre che ogni mano non è sovrapponibile alla sua
immagine speculare. Tutte le strutture che godono di questa proprietà si dicono chirali. In chimica
sono chirali le molecole che presentano un carbonio chinale*. Un carbonio è chirale quando è legato
a quattro atomi o gruppi atomici diversi.
La presenza in una molecola di un carbonio chirale le conferisce la proprietà del tutto particolare di
esistere in due isomeri diversi pur avendo la stessa formula di struttura. I due isomeri differiscono
per la disposizione nello spazio degli atomi o dei gruppi atomici legati all’atomo di carbonio chirale
e risultano uno l’immagine speculare dell’altro.
Si definiscono enantiomeri o antipodi ottici gli isomeri a immagine speculare, tali che le loro
strutture spaziali non sono comunque sovrapponibili.
X
X
|
|
C*
C*
H
Y
Y
H
Z
Z
H
O
H
C
H
C*
O
C
OH
CH2OH
D (+) gliceraldeide
HO
C*
H
CH2OH
L (-) gliceraldeide
Gli enantiomeri sono sostanze otticamente attive perché hanno comportamento opposto nei
confronti della luce polarizzata, vengono chiamati pertanto isomeri ottici o antipodi ottici.
L’isomero D (+) della gliceraldeide fa ruotare il piano di vibrazione della luce polarizzata verso
destra. Esso ha potere rotatorio destrogiro, indicato col segno (+) o con la lettera D.
L’isomero L (-), invece, fa ruotare il piano di vibrazione della luce polarizzata dello stesso angolo,
ma verso sinistra. Esso ha quindi potere rotatorio levogiro, indicato col segno (-) o con la lettera L.
Una miscela in parti uguali di due enantiomeri è otticamente inattiva e si definisce racemo.
Gli enantiomeri hanno le stesse proprietà chimiche, salvo che nei confronti dei reagenti otticamente
attivi, rispetto ai quali manifestano velocità di reazione diverse.
41
I composti organici
Idrocarburi
•
Alcani
Gli alcani sono idrocarburi alifatici saturi con formula generale CnH2n+2, ciascun atomo di
carbonio ha quattro orbitali ibridati sp3.
Il primo della serie omologa è il metano, CH4.
Formula di struttura e modellino molecolare: ibridazione del C sp3, forma tetraedrica, angoli di
legame di 109,5°, quattro legami semplici di tipo σ.
Modo di rappresentare gli idrocarburi
Esempio: propano
C3H8 formula bruta o molecolare
H H H
| |
|
H–C–C–C–H
| |
|
H H H
CH3 – CH2 – CH3
CH3CH2CH3
formula di struttura
formula razionale
formula compatta
formula minima internazionale (gli atomi di H sono sottintesi, gli atoni di C sono
rappresentati dalle estremità di ogni singolo segmento)
modello a spazio pieno
modello a sfere e bastoncini
42
Nomenclatura degli alcani
Per i primi quattro della serie si usano i nomi tradizionali, mentre dall’alcano con cinque atomi di
carbonio in poi si ricorre ai nomi sistematici attribuiti dalla IUPAC, secondo cui: il nome di un
alcano si ricava dal prefisso greco che indica il numero di atomi di carbonio presenti
nell’idrocarburo, seguito dal suffisso –ano.
CH4 metano
CH3 – CH2 – CH3 propano
CH3 – (CH2)3 – CH3 pentano
CH3 – (CH2)5 – CH3 eptano
CH3 – (CH2)7 – CH3 nonano
CH3 – (CH2)9 – CH3 undecano
CH3 – (CH2)11 – CH3 tridecano
CH3 – (CH2)13 – CH3 pentadecano
CH3 – (CH2)15 – CH3 eptadecano
CH3 – (CH2)17 – CH3 nonadecano
CH3 – CH3 etano
CH3 – (CH2)2 – CH3
CH3 – (CH2)4 – CH3
CH3 – (CH2)6 – CH3
CH3 – (CH2)8 – CH3
CH3 – (CH2)10 – CH3
CH3 – (CH2)12 – CH3
CH3 – (CH2)14 – CH3
CH3 – (CH2)16 – CH3
CH3 – (CH2)18 – CH3
butano
esano
ottano
decano
dodecano
tetradecano
esadecano o cetano
ottadecano
eicosano
Per gli isomeri di struttura più semplici si ricorre al prefisso n- (normal) e iso- per indicare
rispettivamente catene lineari o ramificate.
CH3
|
CH3 – CH – CH3
CH3 – CH2 – CH2 – CH3
n – butano
isobutano (2-metilpropano)
L’uso di questi prefissi nel caso di idrocarburi con cinque o più atomi di carbonio non è però
sufficiente per indicare tutte le loro possibili disposizioni, perciò la IUPAC ha fissato le regole per
l’attribuzione del nome a un qualsiasi alcano, indipendentemente dalla sua complessità. I nomi e le
formule dei più comuni radicali alchilici sono:
metile – CH3
etile – CH2 – CH3
n-propile – CH2 – CH2 – CH3
sec-propile (isopropile)
– CH – CH3
|
CH3
sec-butile CH3 – CH – CH2 – CH3
|
CH3
|
CH3 – C – CH3
|
n-butile – CH2 – CH2 – CH2 – CH3
isobutile – CH2 – CH – CH3
|
CH3
isopentile – CH2 – CH – CH2 – CH3
(isoamile)
|
CH3
I radicali alchilici prendono il nome dall’alcano corrispondente sostituendo al suffisso –ano la
desinenza –ile.
terz-butile
Per esempio, per determinare il nome IUPAC di un composto si procede nel modo seguente:
1. Si individua la catena principale, cioè quella costituita dal maggior numero di atomi di carbonio
disposti in sequenza (può anche essere spezzata).
43
2. Si individuano i radicali alchilici legati alla catena principale.
3. Si numerano gli atomi di carbonio della catena principale in modo da attribuire a quelli cui sono
legati i radicali alchilici i numeri più bassi possibile.
catena principale
CH2 – CH3
|
CH3 – CH2 – CH2 – CH2 – CH – CH – CH2 – CH3
8
7
6
5
4
3|
2
1
CH3
radicali alchilici
4. Il nome del composto considerato si ottiene indicando il numero della posizione e il nome dei
radicali alchilici e ponendo alla fine il nome dell’alcano della catena principale.
5. I radicali alchilici sono indicati per ordine alfabetico, se nella catena sono presenti più radicali
alchilici uguali se ne indica il numero con un prefisso (di-, tri-, tetra-, penta- ecc.). il nome del
composto considerato è 4-etil-3-metilottano.
6. Nei composti in cui sono legati alla catena principale atomi o gruppi di atomi diversi dai radicali
alchilici, per esempio alogeni, si ricorre alle stesse regole di numerazione
CH3
|
CH3 – C – CH2 – CH2 – CH3
|
Cl
2-cloro-2-metilpentano
•
Cicloalcani
I cicloalcani sono idrocarburi alifatici saturi in forma ciclica con formula generale CnH2n, ciascun
atomo di carbonio ha quattro orbitali ibridati sp3. I loro nomi si ottengono facendo precedere dal
prefisso ciclo- il nome dell’alcano a catena aperta corrispondente.
Modellino molecolare del ciclopentano, C5H10: forma a struttura quasi planare, angoli di legame di
108°, legami semplici di tipo σ.
44
•
Alcheni
Gli alcheni sono idrocarburi alifatici insaturi con formula generale CnH2n quando è presente un
solo doppio legame. Ciascun atomo di carbonio coinvolto nel doppio legame è ibridato sp2.
Il primo della serie omologa è l’etene o etilene, C2H4.
Modellino molecolare: ibridazione del C sp2, forma trigonale-planare, angoli di legame di 120°,
doppio legame C = C (σ + π) e quattro legami semplici C – H di tipo σ.
I nomi degli alcheni derivano da quelli degli alcani corrispondenti per sostituzione del suffisso –ano
con il suffisso –ene. Nel nome deve essere inoltre indicata la posizione nella catena del primo
carbonio che forma il doppio legame assegnando a questa il numero più basso possibile:
propene o propilene CH2 = CH – CH3
Radicali: dall’etene CH2=CH2 si ricava l’etenile CH2=CH― chiamato comunemente vinile;
dal propene si ricava il 2-propenile CH2=CH―CH2― chiamato comunemente allile.
Isomeri del butene: CH2 = CH – CH2 – CH3
1 – butene
CH3 – CH = CH – CH3
2-butene
Il 2-butene, inoltre, può presentarsi in due forme stereoisomere a seconda di come sono disposti i
gruppi sostituenti rispetto al doppio legame:
cis-2-butene
trans-2-butene
Gli stereoisomeri di alcheni sostituiti asimmetricamente sul doppio legame vengono in generale
indicati con i prefissi E e Z, che fanno riferimento alle seguenti regole di priorità:
- più alto è il numero atomico dell’atomo legato al carbonio del doppio legame, più alta è la sua
priorità (Br > Cl > O > N > C > H);
- se due atomi connessi al carbonio del doppio legame hanno la stessa priorità, si prosegue lungo
la catena dei gruppi sostituenti fino al punto in cui la priorità dei due gruppi differisce;
- quando nei gruppi sostituenti sono presenti legami multipli, i legami doppi vengono scritti come
due legami singoli e i tripli come tre legami singoli per ciascuno dei due atomi coinvolti.
Se i gruppi a più alta priorità si trovano dalla stessa parte rispetto al doppio legame, il nome del
composto è preceduto dalla lettera Z, in caso contrario si antepone la lettera E.
45
1
H
3
4
2
1
Cl
C=C
H3C
4
H3C
CH3
2
3
C=C
H
(E)-2-cloro-2-butene
Cl
CH3
(Z)-2-cloro-2-butene
1
H3C
5
4
H3C – H2C
3
2
C=C
CH3
H3C
5
H
(E)-3-metil-2-pentene
H3C – H2C
3
2
C=C
4
H
1
CH3
(Z)-3-metil-2-pentene
Considerando il composto di sinistra, vediamo che nel carbonio a destra del doppio legame il
gruppo con maggior priorità è il –Cl, mentre nel carbonio a sinistra è il CH3: poiché si trovano in
posizione opposta al doppio legame la denominazione sarà E. Nel caso di destra, invece, i due
gruppi a priorità maggiore (–Cl e –CH3) si trovano dalla stessa parte rispetto al doppio legame, per
cui la denominazione sarà Z.
Gli isomeri cis o (Z) presentano interazioni molecolari più intense rispetto ai corrispondenti isomeri
trans o (E) e hanno pertanto punti di ebollizione più alti.
I dieni sono un gruppo di idrocarburi insaturi caratterizzati dalla presenza nella loro molecola di
due doppi legami. I loro nomi si ottengono da quello dell’alcano corrispondente, premettendo i
numeri che indicano le posizioni dei doppi legami e utilizzando il suffisso –diene.
CH2 = CH – CH = CH – CH3
1,3-pentadiene
1,3-ciclopentadiene
Gli alcheni contenenti più di due doppi legami vengono generalmente chiamati polieni.
CH2 = CH – CH = CH – CH = CH – CH3
1,3,5-eptatriene
•
Alchini
Gli alchini sono idrocarburi alifatici insaturi con formula generale CnH2n - 2 quando è presente un
solo triplo legame. Ciascun atomo di carbonio coinvolto nel doppio legame è ibridato sp.
Il primo della serie omologa è l’etino o acetilene, C2H2.
Modellino molecolare dell’etino o acetilene: ibridazione del C sp, forma lineare , angoli di legame
di 180°, triplo legame C ≡ C (σ + 2π) e due legami semplici C – H di tipo σ.
Secondo le regole IUPAC i nomi degli alchini derivano da quelli degli alcani corrispondenti per
sostituzione del suffisso –ano con il suffisso –ino.
46
CH ≡ C – CH3
propino
CH ≡ C – CH2 – CH3
1-butino
CH3 – C ≡ C – CH3
2-butino
CH3 – C ≡ C – CH2 – CH3
2-pentino
cicloottino
•
Aromatici
Il principale esponente degli idrocarburi aromatici è il benzene, C6H6.
Nel 1865 il chimico tedesco August Kekulè propose per il benzene una formula di struttura ciclica a
sei atomi di carbonio uniti mediante legami singoli e doppi alternati, che si avvicendano
continuamente l’uno con l’altro, secondo l’equilibrio:
Benzene: forme limite di risonanza di Kekulè
La struttura del benzene viene attualmente spiegata in termini di orbitali, ammettendo per ciascun
carbonio un’ibridazione di tipo sp2 speciale, dove gli orbitali 2p non ibridi si sovrappongono sopra e
sotto il piano dell’anello carbonioso tanto da formare una caratteristica nube elettronica a doppia
ciambella (delocalizzazione).
La forma esagonale della molecola e la delocalizzazione dei 6 elettroni 2p del benzene è confermata
dai dati sperimentali che evidenziano la presenza di 6 legami C–C identici, di lunghezza intermedia
(1,40 Å) fra quella di un legame singolo (1,54 Å) e quella di un doppio legame (1,33 Å), e l’assenza
di tre doppi legami C=C.
47
Pertanto, la rappresentazione del benzene e del radicale fenile, −− C6H5, che tiene conto della
delocalizzazione dei 6 elettroni 2p è la seguente:
Nomenclatura degli aromatici
I derivati del benzene sono gli idrocarburi aromatici o areni. Questi composti hanno nomi
comuni, accettati molto spesso dalla IUPAC, in alternativa, per i benzeni monosostituiti viene
indicato il nome del gruppo sostituente seguito da –benzene:
Toluene
o metilbenzene
stirene
o vinilbenzene
clorobenzene
fenolo
nitrobenzene
Per i derivati disostituiti è necessario precisare le posizioni reciproche dei gruppi legati all’anello,
indicando con un numero la posizione degli atomi di carbonio, o ricorrendo ai prefissi orto- (o-),
meta- (m-) e para- (p-). Se sono presenti più di due sostituenti si ricorre alla numerazione.
o-xilene
(1,2-dimetilbenzene)
m-xilene
(1,3-dimetilbenzene)
1-metil-3-nitrobenzene
(m-metilnitrobenzene)
p-xilene
(1,4-dimetilbenzene)
1-cloro-4-nitrobenzene
(p-cloronitrobenzene)
48
Gli areni costituiti da anelli che hanno un lato in comune sono detti idrocarburi aromatici
policiclici, hanno nomi comuni accettati dalla IUPAC.
naftalene o naftalina
antracene
fenantrene
pirene
benzo(a)pirene o 3,4-benzopirene
Il 3,4-benzopirene è una sostanza cancerogena presente nel fumo di sigaretta.
Gli areni, come tutti gli idrocarburi, sono insolubili in acqua e solubili in solventi apolari, come il
tetracloruro di carbonio. Il benzene viene utilizzato nella preparazione dei coloranti, resine
sintetiche, materie plastiche, prodotti farmaceutici, detersivi e insetticidi. È molto tossico e
l’esposizione a dosi elevate dei suoi vapori può provocare anemia, leucemia e danni genetici. Per
questo motivo nell’uso come solvente è sostituito dal toluene e dagli xileni, la cui tossicità è
inferiore. Il benzene è presente anche nelle benzine e costituisce, pertanto, un pericoloso inquinante
atmosferico. Un derivato del benzene è l’acido acetilsalicilico o aspirina (estere dell’acido
salicilico con acido acetico, di formula C9H8O4).
Il toluene è utilizzato per la produzione degli esplosivi come il trinitrotoluene (TNT). Gli xileni
sono utilizzati per la produzione di polimeri e di fibre sintetiche.
I composti aromatici attualmente non comprendono solo idrocarburi o anelli a 6 atomi, ma anche
anelli a 5 o 7 atomi, di cui possono far parte atomi diversi dal carbonio.
furano
pirrolo
49
piridina
Gruppi funzionali
Un gruppo funzionale è un gruppo di atomi che conferisce alla molecola un comportamento
chimico caratteristico. Composti caratterizzati dalla presenza nelle loro molecole di uno stesso
gruppo funzionale formano una classe.
•
Alogenuri alchilici
Gli alogenuri alchilici sono idrocarburi in cui uno o più atomi di idrogeno sono sostituiti da
altrettanti atomi di alogeni, che ne rappresentano il gruppo funzionale.
CH3Cl
clorometano
(cloruro di metile)
CH2BrCH2CH3
CH3CHBrCH3
1-bromopropano
(bromuro di propile)
2-bromopropano
(bromuro di isopropile)
CH2=CHCl
cloruro di vinile
I CFC o clorofluorocarburi, commercialmente noti come Freon, sono derivanti dal metano e
dall'etano per sostituzione degli atomi di idrogeno con atomi di cloro, fluoro, bromo. Sono usati
come propellenti nelle bombolette spray e come sostanze criogene per impianti frigoriferi; dal 1987
ne è stato limitato l’uso (in particolare quelli contenenti cloro) a causa della loro interazione con
l’ozono atmosferico.
•
Alcoli
Gli alcoli sono derivati dagli idrocarburi per sostituzione di un atomo di idrogeno con il gruppo
ossidrile –OH. L’atomo di carbonio su cui è legato il gruppo ossidrile deve essere ibridato sp3.
La nomenclatura IUPAC degli alcoli si ottiene sostituendo la lettera finale dell’idrocarburo
corrispondente con il suffisso –olo e indicando la posizione numerica più bassa possibile del
carbonio al quale è legato il gruppo ossidrilico.
CH3OH
metanolo
(alcol metilico)
CH3CH2OH
etanolo
(alcol etilico)
CH3CH2CH2OH
1-propanolo
(alcol n-propilico)
CH3CHOHCH3
2-propanolo
(alcol isopropilico)
cicloesanolo
Gli alcoli possono essere primari, secondari o terziari a seconda del carbonio (primario,
secondario o terziario) a cui è legato l’ossidrile (con R si intende un radicale alifatico o aromatico).
R
R
|
|
R – CH2 – OH
R – CH – OH
R – C – OH
|
R
alcol primario
CH3CH2CH2CH2OH
1-butanolo
(alcol n-butilico)
alcol secondario
alcol terziario
CH3CH2CHCH3
|
OH
2-butanolo
(alcol sec-butilico)
50
CH3
|
CH3C – OH
|
CH3
2-metil-2-propanolo
(alcol ter-butilico)
Gli alcoli possono essere monovalenti (etanolo), bivalenti (glicol etilenico), trivalenti (glicerolo)
ecc., a seconda del numero di ossidrili presenti nella catena idrocarburica.
CH2 - OH
|
CH2 – CH2
CH – OH
CH3CH2 – OH
|
|
|
OH OH
CH2 – OH
etanolo
1,2-etandiolo
(glicol etilenico)
1,2,3-propantriolo
(glicerina o glicerolo)
Alcoli più comuni
Il metanolo è un liquido incolore, velenoso e miscibile con l’acqua; viene utilizzato come additivo
per le benzine, per la sintesi della formaldeide e del dimetiltereftalato, composto base per ottenere il
polimero PET (polietilentereftalato).
L’etanolo è un liquido incolore molto volatile e infiammabile; viene prodotto industrialmente per
idratazione dell’etilene oppure per fermentazione alcolica degli zuccheri attuata da lieviti o funghi
unicellulari. Soltanto quest’ultimo può essere avviato al consumo alimentare (vino, birra e distillati
alcolici). Viene impiegato anche come solvente, combustibile e carburante.
Il glicol etilenico è un diolo che si scioglie in acqua e di questa ne determina l’abbassamento
crioscopico; per questa sua proprietà viene impiegato come antigelo nei circuiti di raffreddamento
delle automobili. Viene impiegato anche per la sintesi di polimeri.
La glicerina o glicerolo è un triolo che si ottiene a partire dal propene oppure nel processo di
saponificazione dei trigliceridi. Viene utilizzata nell’industria dei cosmetici e per preparare
esplosivi (nitroglicerina o trinitroglicerina).
•
Fenoli
I fenoli sono composti che contengono un gruppo –OH legato direttamente a un anello aromatico.
Sono liquidi incolori o solidi a basso punto di fusione, si scuriscono all’aria perché si ossidano
facilmente.
fenolo
2-metilfenolo
(o-cresolo)
1,4-benzendiolo
(idrochinone)
Il fenolo viene prodotto per ossidazione del toluene. In natura i fenoli sono molto diffusi nelle
piante. L’odore della vaniglia e dei chiodi di garofano è dovuto ai composti fenolici vanilina e
eugenolo. Alcuni fenoli trovano applicazione nell’industria alimentare come antiossidanti. Il
butilidrossianisolo (BHA o E 320) viene impiegato a tale scopo per le patatine fritte confezionate e
nelle gomme da masticare.
51
vanilina
eugenolo
butilidrossianisolo (BHA)
I composti analoghi agli alcoli che al posto del gruppo ossidrilico – OH hanno il gruppo solfidrilico
– SH (detto anche gruppo tiolico o mercaptanico) si chiamano tioalcoli o tioli o mercaptani.
Caratteristica comune a quasi tutti i mercaptani è di possedere un intenso odore sgradevole. In
particolare l'etantiolo viene aggiunto, in piccole quantità, al gas naturale per rilevarne la presenza in
caso di perdite accidentali. Sono sintetizzati trattando alogenuri alchilici con ioni solfidrato HS- in
eccesso: R-X + HS- → R-SH + X-. Si formano anche nei vini per reazione tra acido solfidrico e
alcoli originando mercaptani con classici odori di cipolla e aglio.
Il metantiolo è il più semplice dei mercaptani
Il legame O-H degli alcoli è più forte del legame S-H dei tioli, pertanto, i tioli presentano una
acidità maggiore rispetto ai corrispondenti alcoli. Così l'etantiolo (pKa=10,5) è più acido del
corrispondente etanolo (pKa=18).
•
Eteri
La molecola di un etere è formata da un atomo di ossigeno legato a due radicali alchilici o arilici o
misti. Si possono pertanto considerare derivati dall’acqua per sostituzione dei due idrogeni con
radicali carboniosi: R – O – R.
Il nome IUPAC degli eteri viene ottenuto assegnando alla catena idrocarburica più lunga il nome
del corrispondente alcano o idrocarburo aromatico e indicando, assieme all’ossigeno, quella più
corta come sostituente alcossidico. La nomenclatura tradizionale indica in ordine alfabetico i gruppi
idrocarburici, premettendoli alla parola etere.
CH3 – O – CH3
metossimetano
(dimetil etere)
CH3CH2 – O – CH3
metossietano
(etil metil etere)
52
CH3CH2 – O – CH2CH3
etossietano
(dietil etere)
metossibenzene
(fenil metil etere o anisolo)
ossirano
(ossido di etilene)
etere terz-butilmetilico
(MTBE)
L'anisolo è un composto aromatico che presenta un metossido (CH3O-) unito all'anello aromatico
del benzene. Possiede il tipico odore e sapore dell'anice, ed è contenuto in essa.
L'ossido di etilene o ossirano è il più semplice composto eterociclico contenente ossigeno, e più
specificamente è il più semplice degli epossidi (eteri ciclici in cui l'ossigeno è uno degli atomi di un
anello a tre termini). L’ossirano viene utilizzato come disinfettante per strumenti chirurgici e come
materia prima per la produzione di resine epossidiche.
L’etere terz-butilmetilico è un composto organico di sintesi derivante dal metanolo e dal 2-metil-2propanolo o alcol t-butilico. Viene impiegato come additivo per la benzina per aumentarne il
numero di ottano, in sostituzione del piombo tetraetile e del benzene.
Gli eteri hanno bassi punti di ebollizione, sono fortemente infiammabili ma poco reattivi, vengono
impiegati come solventi. Il dietil etere veniva usato un tempo come anestetico, attualmente, come
solvente per oli e grassi.
I composti analoghi agli eteri che al posto dell’atomo di ossigeno hanno un atomo di zolfo si
chiamano tioeteri o solfuri organici: R – S – R.
Molti tioeteri hanno un cattivo odore come molti altri composti organici contenenti zolfo.
CH3CH2 – S – CH2CH3
dietiltioetere o dietilsolfuro
•
Aldeidi e chetoni
|
Il gruppo funzionale delle aldeidi e dei chetoni è il carbonile, – C = O.
Come gli alcheni, i composti carbonilici sono trigonali-planari intorno al doppio legame e hanno
angoli di 120° (ibridazione sp2 del C).
Nelle aldeidi il gruppo carbonilico è legato da una parte sempre all’idrogeno e dall’altra a un
radicale: R – CHO, fa eccezione l’aldeide formica o metanale, H – CHO.
Il nome IUPAC delle aldeidi si ottiene sostituendo il suffisso –o dell’alcano da cui esse derivano
con il suffisso –ale. I nomi tradizionali delle aldeidi derivano invece dagli acidi carbossilici
corrispondenti.
metanale
(aldeide formica o formaldeide)
etanale
(aldeide acetica)
propanale
(aldeide propionica)
benzaldeide
La formaldeide in soluzione acquosa al 37% è commercialmente nota con il nome di formalina.
L' aldeide acetica a temperatura ambiente è un liquido incolore volatile e infiammabile dall'odore
pungente e irritante. È una sostanza tossica, probabilmente cancerogena. Tracce di acetaldeide sono
contenute anche nel fumo di tabacco. È facilmente ossidabile ad acido acetico.
53
L’aldeide propionica è un isomero strutturale dell'acetone. A temperatura ambiente è un liquido
incolore, con un odore fruttato leggermente irritante.
La benzaldeide è il composto più semplice della classe delle aldeidi aromatiche nonché quello più
sfruttato a livello industriale. In natura si trova nelle mandorle amare e nei semi di albicocca e di
pesche. Benché la benzaldeide si trovi già in natura, si preferisce produrla industrialmente per via
sintetica partendo dal toluene. A temperatura ambiente si presenta come un liquido incolore o giallo
pallido volatile con un caratteristico odore di mandorle amare. Sebbene la benzaldeide possa essere
impiegata come solvente, il suo principale utilizzo è quello di precursore per la sintesi di diversi
composti organici, dai farmaci agli additivi per le plastiche. È anche un importante intermedio per
la preparazione di profumi e fragranze come anche nella sintesi di coloranti anilinici.
Nei chetoni il gruppo carbonilico è legato a due radicali alchilici o arilici o misti: R – CO – R.
Il nome IUPAC dei chetoni si ottiene da quello dell’alcano corrispondente alla catena più lunga, per
sostituzione del suffisso –o dell’alcano con il suffisso –one, e indicando con il numero più basso
possibile la posizione del carbonile. Nella nomenclatura tradizionale si elencano in ordine alfabetico
i nomi dei radicali presenti, facendoli seguire dalla parola chetone.
2-propanone
(dimetil chetone o acetone)
2-butanone
(etil metil chetone)
cicloesanone
acetofenone
(fenil metil chetone)
L'acetone è un liquido incolore e infiammabile con un odore caratteristico (fruttato); è miscibile con
acqua, etanolo e etere e trova principalmente impiego come solvente.
Il 2-butanone a temperatura ambiente si presenta come un liquido incolore dall'odore di solvente. È
molto infiammabile e irritante. Viene utilizzato in gascromatografia e viene aggiunto nell'alcool
etilico come denaturante, in soluzione con tiofene, denatonium benzoato e colorante inorganico
Reactive Red 24, in quanto la miscela etanolo-2-butanone non è separabile per distillazione.
Il cicloesanone a temperatura ambiente si presenta come un liquido incolore dall'odore pungente. È
un composto infiammabile e dotato di proprietà solventi verso diverse sostanze organiche, inclusa la
gomma naturale (caucciù). Insieme al toluene, al cloroformio e all'etere di petrolio, infatti, è stato
usato per gli studi compiuti sull'isolamento dell'unità strutturale del caucciù (isoprene).
L'acetofenone è il più semplice chetone aromatico. È un liquido viscoso incolore che fonde a 20 °C,
usato principalmente per la sintesi di resine e fragranze.
•
Ammine
Le ammine sono derivate dall’ammoniaca per sostituzione di uno, due o tre atomi di idrogeno con
radicali alchilici o arilici. Il gruppo funzionale di queste molecole è il gruppo amminico.
Quando i radicali sono alchilici l’ammina è alifatica, quando è presente anche un solo radicale
arilico l’ammina è aromatica.
Le ammine si classificano in primarie, secondarie e terziarie a seconda del numero di atomi di
carbonio legati direttamente all’azoto.
R
|
R – NH2
R – NH – R
R–N–R
ammina primaria
ammina secondaria
ammina terziaria
Nella nomenclatura tradizionale si usa il suffisso –ammina, posto di seguito all’elenco dei
sostituenti presenti. La nomenclatura IUPAC delle ammine è molto complessa.
54
CH3 – NH2
CH3
|
CH3 – N – CH3
CH3 – NH – CH3
metilammina
dimetilammina
CH3 – NH – CH2 – CH2 – CH3
trimetilammina
metilpropilammina
La metilammina è la più semplice ammina primaria. La sua struttura chimica è quella di una
molecola di ammoniaca in cui un atomo di idrogeno è stato sostituito da un gruppo metile CH3. A
temperatura ambiente è un gas incolore dal tipico odore ammoniacale; con l’acqua forma soluzioni
basiche. Trova impiego quale intermedio per la produzione di coloranti, insetticidi, anticrittogamici,
prodotti farmaceutici. industrialmente si prepara insieme con la dimetilammina e la trimetilammina
dalle quali poi si separa per distillazione frazionata a bassa temperatura, facendo reagire a 400 ºC il
metanolo con ammoniaca su catalizzatori a base di ossidi di alluminio. La dimetilammina viene
utilizzata nella concia delle pelli. La trimetilammina è utilizzata nella produzione di fattori di
regolazione della crescita delle piante e di resine a scambio ionico.
fenilammina o anilina
metilanilina
difenilammina
trifenilammina
L’anilina, detta anche fenilammina o benzenammina, è usata per la preparazione di coloranti,
medicinali, resine, profumi, esplosivi, solventi per vernici; è cancerogena e velenosa. La
difenilammina a temperatura ambiente si presenta come un solido giallo chiaro dall'odore tenue, è
un intermedio per la preparazione di coloranti. È un composto tossico, pericoloso per l'ambiente.
•
Acidi carbossilici
Il gruppo funzionale degli acidi carbossilici è il carbossile, – COOH, derivato dalla fusione tra il
gruppo ossidrile e il gruppo carbonile.
Il carbossile presenta una elevata reattività a causa dei quattro componenti, forniti di carica parziale,
e dalla propria geometria, tipica degli atomi di carbonio ibridati sp2.
Il gruppo carbossilico è legato a radicali alchilici o arilici R – COOH, fa eccezione l’acido
metanoico o formico, H – COOH.
Nella nomenclatura IUPAC il nome degli acidi carbossilici si ottiene aggiungendo il suffisso –ico al
nome dell’idrocarburo corrispondente alla più lunga catena di atomi di carbonio in cui è presente il
gruppo –COOH. In presenza di due gruppi carbossilici si utilizza il suffisso -dioico.
CH3COOH
acido etanoico
(acido acetico)
CH3CH2COOH
acido propanoico
(acido propionico)
CH3(CH2)4COOH
acido n-esanoico
(acido capronico)
CH2 = CHCOOH
acido propenoico
(acido acrilico)
acido butanoico
(acido butirrico)
OH
|
CH3CHCOOH
HOOC – COOH
acido etandioico
(acido ossalico)
CH3(CH2)2COOH
acido 2-idrossipropanoico
(acido lattico)
55
CH3(CH2)3COOH
acido n-pentanoico
(acido valerianico)
O
||
CH3 – C – CH2 – COOH
acido 3-ossobutanoico
(acido aceto acetico)
L’acido acetico si presenta come un liquido incolore, di odore pungente, è caratteristico dell’aceto;
allo stato puro costituisce un liquido irritante, solidifica a 16,6 °C e prende il nome di acido acetico
glaciale perché ha un aspetto simile al ghiaccio; costituisce il prodotto finale del metabolismo di
microrganismi del genere Acetobacter che fermentano liquidi alcolici.
L’acido propionico viene prodotto dal metabolismo dei batteri del genere Propionibacterium, che
vivono nelle ghiandole sudoripare umane e nello stomaco dei ruminanti; è infatti l’acido propionico
che conferisce il caratteristico odore al sudore e ad alcuni formaggi; l’acido propionico e alcuni suoi
sali (propionati) hanno proprietà fungicide e sono utilizzati come conservanti alimentari.
L’acido acrilico viene utilizzato per la sintesi di materie plastiche come le fibre acriliche
nell’industria tessile.
L’acido butirrico è presente nel burro, in talune piante; è un liquido incolore, volatile, dall’odore
pungente, solubile in acqua; conferisce un aroma tipico ad alcuni alimenti (latticini, formaggi,
birra); la presenza di batteri anaerobi contaminanti (principalmente clostridi) dà luogo a
fermentazione butirrica, processo che fa aumentare in modo eccessivo la concentrazione di tale
acido, cosicché l’alimento assume uno sgradevole odore di rancido.
L’acido valerianico è presente nella Valeriana officinalis; è un blando sedativo del SNC.
Dei diversi isomeri dell’acido capronico il più importante è quello a catena normale: liquido oleoso,
incolore o giallognolo, di odore sgradevole, che, come gliceride, è contenuto in piccole quantità nel
burro di capra e nel burro di cocco; viene utilizzato per la preparazione di essenze e profumi
artificiali e in campo farmaceutico.
L’acido ossalico è molto diffuso nel regno vegetale allo stato di sale di calcio (nelle alghe, nei
funghi, nei licheni e nelle felci) e di magnesio (nelle foglie di alcune Graminacee). Gli ossalati di
ferro e di calcio si ritrovano come minerali. Negli organismi animali l’acido ossalico è presente in
piccole quantità come normale costituente del sangue (dove rappresenta un prodotto del
metabolismo intermedio, che entra a far parte come ossalacetato del ciclo di Krebs), delle urine,
della bile, delle feci. Nell’organismo umano possono anche formarsi calcoli renali e vescicali
costituiti da ossalato di calcio.
L’acido lattico è noto nelle forme D-lattico, L-lattico (levogiro e destrogiro) e D-L-lattico,
corrispondente al racemo; la forma L è presente in piccole quantità nel sangue, nel fluido muscolare
di uomini e animali, dove tende ad aumentare a seguito di attività fisica, nel fegato; la forma racema
è contenuta nel latte ed è prodotta per azione dei fermenti lattici.
acido benzoico
acido 2-idrossibenzoico o acido salicilico
L’acido benzoico è impiegato, come la gran parte dei suoi sali (benzoati), per la conservazione di
prodotti alimentari (oleomargarina, succhi di frutta, marmellate), di preparati farmaceutici, in
cosmetica, nella sintesi di coloranti.
L’acido salicilico è impiegato in medicina, per uso esterno, come antisettico e disinfettante, per uso
interno come antipiretico, antireumatico, antinevralgico e antisettico (viene anche usato come
conservante nei prodotti non alimentari perché è in grado di arrestare le fermentazioni); è impiegato
per la sintesi dell’acido acetilsalicilico, comunemente detto aspirina.
Nei grassi animali e vegetali sono presenti in notevole quantità ed esterificati con il glicerolo, i
seguenti acidi grassi:
acido palmitico: CH3(CH2)14COOH
56
L’acido palmitico o acido esadecanoico (saturo) entra nella costituzione di quasi tutti gli oli e grassi
animali e vegetali in quantità variabili dal 6-7 al 60-70%; è presente anche nella cera d’api; si
presenta come massa incolore insolubile in acqua, solubile in alcol ed etere. I suoi esteri con la
glicerina sono chiamati palmitine. Si usa per preparare esteri nell’industria dei cosmetici e dei
saponi.
acido stearico: CH3(CH2)16COOH
L’acido stearico o acido octadecanoico (saturo) si trova esterificato con la glicerina negli oli e nei
grassi di origine animale (20-35 %) e vegetale (generalmente in minor quantità); è un solido di
aspetto ceroso insolubile in acqua, solubile in alcol ed etere. I sali ed esteri dell’acido vengono detti
stearati e sono sostanze solide usate nella preparazione di unguenti, di creme.
acido oleico: CH3(CH2)7CH=CH(CH2)7COOH.
L’acido oleico o acido 9-octadecenoico (insaturo) è l’acido grasso più diffuso in natura, trovandosi
sotto forma di gliceride in tutti i grassi animali, di origine marina e terrestre, e in notevole quantità,
in tutti gli oli vegetali. Allo stato puro è un liquido oleoso incolore di sapore grasso, insolubile in
acqua; è impiegato nella preparazione degli oleati usati come lubrificanti per fibre tessili, nella
preparazione di detergenti e come solvente in farmaceutica.
•
Esteri
Un estere è il prodotto della reazione di esterificazione (condensazione) tra un acido carbossilico e
un alcol, catalizzata dagli ioni H+.
H+
R – CO OH + H OR’
acido
RCOOR’ + H2O
alcol
estere
O
||
CH3 – C - OH + HO – CH2 – CH3
acido acetico
(acido etanoico)
O
||
CH3 – C – O – CH2 – CH3 + H2O
alcol etilico o etanolo
acetato di etile o etanoato di etile
(estere)
L’acetato di etile si presenta come un liquido volatile, incolore e dal gradevole odore fruttato; viene
utilizzato come solvente per vernici, resine e colle essendo meno tossico di altri solventi aromatici.
Il profumo di molti frutti è dovuto a esteri, spesso sotto forma di miscele, e quelli prodotti
artificialmente vengono utilizzati come aromatizzanti.
L’esterificazione tra acidi grassi e glicerolo porta alla formazione dei trigliceridi, costituenti dei
grassi naturali. La reazione di idrolisi dei trigliceridi con NaOH o KOH è detta saponificazione:
con essa si ottiene sapone e glicerolo.
57
•
Ammidi
Le ammidi sono derivati non secondari degli acidi carbossilici e sono caratterizzate dal gruppo
funzionale ammidico, – CONH2.
Sono sostanze molto polari, solide a temperatura ambiente per l’azione dei legami a idrogeno. Fra le
ammidi si possono ricordare la nicotinammide (vitamina PP) e i derivati dell’acido barbiturico,
utilizzati come sedativi e nel controllo dell’epilessia.
acetammide
nicotinammide
L’acetammide deriva dall’acido acetico; si presenta come polvere igroscopica irritante, solubile in
alcol; viene usata in sintesi organiche, come plastificante; è una sostanza antiacida e solvente.
La nicotinammide è una vitamina importante per la crescita, il buon funzionamento del sistema
nervoso, la pelle, la lingua, l'apparato digerente, essenziale per il metabolismo dei carboidrati, per i
grassi, le proteine.
Le ammidi, a causa di strutture mesomere o di risonanza, che delocalizzano il doppietto elettronico
dell’azoto, sono sostanze neutre e non basiche come le ammine.
•
Anidridi
Le anidridi sono composti che derivano dalla condensazione con eliminazione di una molecola di
acqua di due acidi carbossilici: RCOOH + HOOCR' → RCOOCOR' + H2O
Hanno la seguente struttura generale:
La nomenclatura IUPAC delle anidridi si basa sul nome dei gruppi acilici che la formano. Al
gruppo acilico viene attribuito il suffisso -oica. Se i gruppi acilici sono diversi (in questo caso si
parla di anidridi miste o asimmetriche) vengono citati, in ordine alfabetico, entrambi i gruppi acilici.
Se i gruppi acilici sono identici (in questo caso si parla di anidridi simmetriche) viene citato l'unico
gruppo presente.
anidride metanoica o anidride formica
anidride etanoica metanoica
anidride etanoica o anidride acetica
L'anidride acetica a temperatura ambiente è un liquido incolore dall'odore irritante, da manipolare
con particolare cautela; è utilizzata per la preparazione dell'aspirina e dell'acetato di cellulosa.
58
Amminoacidi
Gli amminoacidi sono i monomeri delle proteine e, quelli naturali, sono venti. La molecola di un
amminoacido contiene un gruppo amminico – NH2 e un gruppo carbossilico – COOH.
Il gruppo R– è caratteristico di ciascun amminoacido. Con l'eccezione della glicina,
NH2CH2COOH, per la quale R– è un atomo di idrogeno, gli amminoacidi sono molecole chirali, di
ciascuna delle quali esistono due enantiomeri.
Come convenzionalmente avviene per le molecole di interesse biochimico, gli enantiomeri degli
amminoacidi sono contrassegnati dalle lettere D (oppure R = rectus) o L (oppure S = sinister) a
seconda che i sostituenti legati all'atomo di carbonio asimmetrico abbiano disposizione simile a
quella della D-gliceraldeide o a quella della L-gliceraldeide. Queste strutture così denominate (D, L
o R, S) non corrispondono al senso di rotazione (destro o sinistro) operato sulla luce polarizzata
dalla molecola in questione. Infatti, il verso della rotazione viene indicato dai segni (+) e (–) che
nella gliceraldeide, come è stato indicato precedentemente, corrispondono rispettivamente alla
forma D e alla forma L.
La stragrande maggioranza delle proteine sintetizzate da organismi viventi è formata da
amminoacidi L. Qualche amminoacido D è stato trovato in proteine prodotte da organismi che
vivono negli abissi marini, nelle pareti cellulari di alcuni batteri e nel veleno di alcuni animali.
Hanno la seguente struttura generale:
H
|
R – C – COOH
|
NH2
amminoacido
Le proteine sono formate dagli amminoacidi naturali uniti fra loro con legame ammidico chiamato
legame peptidico, – CO – NH – .
R
R’
|
|
H2N – C – COOH + H2N – C – COOH
|
|
H
H
R
R’
|
|
H2N – C – CO – NH – C – COOH + H2O
|
|
H
H
59
Reazioni di chimica organica
Reattività degli alcani
•
Alogenazione o sostituzione
luce o calore
CH4 + Cl2
CH3Cl + HCl
L’alogenazione del metano non si arresta con la formazione di CH3Cl, ma procede dando luogo a
una miscela che contiene anche composti ulteriormente alogenati (CH2Cl2, CHCl3, CCl4).
•
Combustione
CH4
+
2 O2
CO2
+
2 H2O
Reattività degli alcheni
•
Addizione elettrofila
La reazione caratteristica degli alcheni è l’addizione elettrofila. In essa la coppia elettronica π del
doppio legame viene attratta da un elettrofilo, dando luogo alla formazione di un carbocatione:
H
|
+
…….H
+
C=C
C – C+
elettrofilo
|
carbocatione
al quale si attacca un nucleofilo, completando la reazione.
H
H
|
| |
C – C+
+ ClC–C
nucleofilo
|
|
Cl
elettrofilo = reattivo in cerca di elettroni
nucleofilo = reattivo capace di fornire elettroni
Con questo meccanismo procedono le addizioni di:
alogeni
CH2 = CH2 + Br2
CH2Br – CH2Br
acqua, in catalisi acida
CH2 = CH2 + H2O
CH3 – CH2OH
acidi alogenidrici
CH2 = CH2 + HCl
CH3 – CH2Cl
60
Nelle addizioni sopra rappresentate, si ottiene lo stesso prodotto qualunque sia l’orientamento del
reagente nei confronti del doppio legame. Quando, invece, un alchene è asimmetrico, come il
propene, e anche il reagente è asimmetrico, come per esempio HCl, vale la regola di Markovnikov
secondo la quale la parte elettrofila del reagente si attacca al carbonio più ricco di idrogeni.
CH3 – CH = CH2 + HCl
•
CH3 – CHCl – CH3
Addizione di idrogeno: gli alcheni reagiscono con idrogeno molecolare, in presenza di un
catalizzatore metallico (platino o palladio), per formare alcani.
Pt
CH2 = CH2 + H2
•
CH3 – CH3
Ossidazione del propene: sintesi del propanone (dimetil chetone o acetone)
2 CH2 = CHCH3
+ O2
2 CH3COCH3
Reattività degli alchini
Gli alchini effettuano reazioni di addizione come gli alcheni anche se sono più lente. Le addizioni
più comuni sono di alogeni, di acidi alogenidrici e di idrogeno (idrogenazione con catalizzatori).
Reattività degli areni
La reazione più comune dei composti aromatici è quella di sostituzione di uno o più idrogeni con
altri atomi o gruppi (sostituzione elettrofila aromatica).
Clorurazione
+
δ+
δ-
+ Cl – Cl ……….FeCl3
H
catalizzatore
Cl
+ FeCl4-
ione benzenonio
+
Cl
Cl + H+
H
reazione complessiva
H
|
Cl
+
Cl
+ FeCl4-
+ Cl2 + FeCl3
61
+ HCl + FeCl3
Reattività degli alcoli
•
Disidratazione
Formazione di alcheni o eteri per disidratazione con H2SO4:
CH3CH2OH
2 CH3CH2OH
•
H2SO4
H2SO4
CH2 = CH2 + H2O
CH3CH2OCH2CH3 + H2O
Ossidazione
Gli alcoli primari per ossidazione blanda danno aldeidi e per ossidazione più energica danno acidi.
Ox
R – CH2 – OH
Ox
R – CHO
R - COOH
Gli alcoli secondari producono chetoni.
Ox
R – CH – OH
|
R
•
R–C=O
|
R
Sintesi della trinitroglicerina
CH2 – OH
|
CH – OH + 3 HNO3
|
ac. nitrico
CH2 – OH
H2SO4
glicerina
CH2 – O – NO2
|
CH – O – NO2 + 3 H2O
|
CH2 – O – NO2
trinitroglicerina
Questo potente esplosivo, scoperto da Ascanio Sobrero (Casale Monferrato1812 – Torino 1888), è
talmente instabile da esplodere per effetto di un semplice urto. Ad Alfred Bernhard Nobel
(Stoccolma 1833 – Sanremo 1896) si deve l’invenzione della dinamite che può essere usata con
maggior sicurezza perché costituita da nitroglicerina mescolata a farina fossile (terra di diatomee),
materiale inerte che rende l’esplosivo più stabile.
•
Esterificazione del glicerolo con tre molecole di acidi grassi: i trigliceridi
CH2 – OH + HOOC(CH2)14CH3
|
CH – OH + HOOC(CH2)14CH3
|
CH2 – OH + HOOC(CH2)14CH3
glicerolo
CH2 – O – OC(CH2)14CH3
|
CH – O – OC(CH2)14CH3 + 3 H2O
|
CH2 – O – OC(CH2)14CH3
acido palmitico
trigliceride tripalmitina
62
Reazione di saponificazione
CH2 – O – CO – (CH2)16 – CH3
|
CH – O – CO – (CH2)16 – CH3 + 3 NaOH
|
idrossido di sodio
CH2 – O – CO – (CH2)16 – CH3
CH2 – OH
|
CH – OH + 3 CH3 – (CH2)16 – COONa
|
stearato di sodio (sapone)
CH2 – OH
tristearato di glicerile (tristearina)
glicerina
Se si usa KOH si ottiene sapone “molle” da barba.
•
Sostituzione elettrofila nei fenoli
Cl
OH + 3 Cl2
Cl
OH + 3 HCl
Cl
La clorurazione del fenolo avviene velocemente e senza la presenza di un catalizzatore. La
clorurazione del benzene avviene invece con la presenza di un catalizzatore.
Reattività degli eteri
Gli eteri vengono impiegati come solventi perché presentano modesta reattività. La reazione più
significativa è quella con l’acido iodidrico.
calore
CH3CH2 – O – CH2CH3 + HI
CH3CH2OH + CH3CH2I
dietil etere
etanolo
ioduro di etile
Reattività di aldeidi e chetoni
La reazione caratteristica del carbonile è quella di addizione nucleofila al doppio legame, in
seguito al quale il carbonio assume ibridazione sp3 formando composti a struttura tetraedrica.
Addizione di acqua con formazione di dioli geminali
O
OH
+
||
H
|
R – C – R + H2O
R–C–R
|
OH
• Addizione di alcoli con formazione di emiacetali e acetali nel caso delle aldeidi o di
emichetali e chetali nel caso dei chetoni
O
OH
OR’
||
H+
|
R’OH, H+
|
R – C – H + R’OH
R – C – OR’
R – C – OR’ + H2O
|
|
H
H
•
emiacetale
acetale
63
Addizione di ammoniaca con formazione di immine
O
NH
||
||
R – C – H + NH3
R – C – H + H2O
•
immina
Reazioni di polimerizzazione
La polimerizzazione si verifica mediante reazioni di addizione o di condensazione tra le molecole
semplici chiamate monomeri.
•
Polimeri di addizione: si ottengono per addizione di monomeri insaturi in seguito all’apertura
dei loro doppi legami.
L’etilene polimerizza a caldo e a pressioni elevate, in presenza di un catalizzatore, in genere
costituito da un perossido organico. Dalla reazione si ottiene il polietilene, costituito da lunghissime
catene risultanti dall’unione di decine di migliaia di molecole di etilene.
n CH2 = CH2
( - CH2 – CH2 - )n
Polipropilene: si ricava dal propilene per poliaddizione. Una fibra commerciale che si ottiene
filando per estrusione il polimero fuso è il meraklon.
............CH2 = CH + CH2 = CH………
|
|
CH3
CH3
…….CH2 – CH – CH2 – CH – CH2 – CH – CH2 – CH – CH2 – CH.........
|
|
|
|
|
CH3
CH3
CH3
CH3
CH3
Poliacrilonitrile: le fibre acriliche sono ottenute dalla polimerizzazione del nitrile acrilico o
acrilonitrile (prodotto di addizione dell’acido cianidrico all’acetilene). I nomi commerciali delle
fibre acriliche sono : orlon, leacril, dralon.
............CH2 = CH + CH2 = CH………
|
|
CN
CN
…….CH2 – CH – CH2 – CH – CH2 – CH – CH2 – CH – CH2 – CH.........
|
|
|
|
|
CN
CN
CN
CN
CN
Polivinilcloruro: è un polimero del cloruro di vinile. Una tipica fibra in PVC è il movil.
............CH2 = CH + CH2 = CH………
|
|
Cl
Cl
…….CH2 – CH – CH2 – CH – CH2 – CH – CH2 – CH – CH2 – CH.........
|
|
|
|
|
Cl
Cl
Cl
Cl
Cl
64
•
Polimeri di condensazione: derivano dalla reazione di due diverse funzioni portate da un
unico monomero o da due diversi reagenti; in entrambi i casi vengono eliminate come
sottoprodotto molecole di piccole dimensioni (HCl, H2O ecc.).
Poliammidi: sono fibre sintetiche di condensazione, di cui la prima assai nota è il nylon 6,6,
costituito dall’acido adipico e dall’esametilendiammina.
Le poliammidi sono polimeri lineari caratterizzati dalla presenza del gruppo ammidico –NH–CO– .
L’applicazione più studiata è la fibra. La produzione industriale di nylon 6,6 iniziò nel 1939 negli
Stati Uniti, in un impianto della Du Pont (calze e derivati).
Successivamente, il nylon 6,6 venne utilizzato anche come materiale plastico e resina. Durante la
seconda guerra mondiale fu impiegato per la produzione di paracaduti, pneumatici da aeroplani e
altri manufatti.
n H2N – (CH2)6 – NH2 +
esametilendiammina
n Cl – CO – (CH2)4 – CO – Cl
+ 2n NaOH
cloruro di adipoile
----- (NH – (CH2)6 – NH – CO – (CH2)4 – CO)n ----- + 2n NaCl
nylon 6,6
+ 2n H2O
Questa reazione è caratterizzata dalla formazione di polimero all’interfaccia, cioè alla superficie di
separazione di due fasi liquide immiscibili di cui generalmente una acquosa e l’altra organica.
Poliestere: è una fibra sintetica di condensazione. La più importante è il polietilentereftalato (PET),
polimero del glicole etilenico con l’acido tereftalico. I nomi commerciali delle fibre poliesteri sono
numerosi: terital, dacron, terilene, trevira.
O
||
n HO – CH2 – CH2 – OH + n HO – C –
O
||
– C – OH
glicole etilenico
acido tereftalico
--------(
O
||
O – CH2 – CH2 – O – C –
O
||
–C–O
polietilentereftalato (PET)
65
)n---------
+ n H2O