LIPIDI
I lipidi, che nel loro complesso
costituiscono più del 10% del peso
corporeo, assolvono funzioni diverse:
Funzione energetica
Protezione termica
Funzione strutturale
Funzione bioregolatoria
Funzione energetica
Nel corso dell’evoluzione i lipidi sono stati scelti come le molecole più adatte
per il deposito dell’energia perchè:
1) Gli acidi grassi (costituiscono il 95% dei triacilgliceroli) sono composti altamente
ridotti
e possono fornire molta energia con la loro ossidazione con elevata
resa energetica (9 Kcal/g contro le 4 Kcal/g di glucidi e proteine)
2) I triacilgliceroli insolubili in acqua si raccolgono in gocciole oleose a formare una fase
separata priva di acqua, che non aumenta l’osmolarità del citosol.
In uno spazio ristretto si possono accumulare molte più molecole lipidiche di quanto
sarebbe possibile se le molecole prescelte fossero i carboidrati con la loro acqua di
idratazione. Elevato rapporto “energia depositata/peso del deposito” per la loro
scarsa idratazione (11 Kg di trigliceridi dovrebbero essere 66 Kg di glucogeno)
3) Gli acidi grassi non sono molecole reattive e consentono un deposito anche per tempi
lunghi.
Riserve di un uomo normale di 70 Kg:
100.000 Kcal in trigliceridi
25.000 Kcal in proteine (soprattutto muscolari)
1.600-2.000 Kcal nel glicogeno
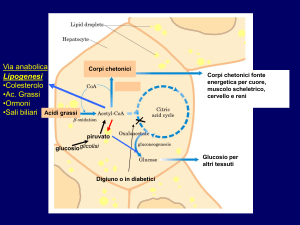
Metabolismo degli acidi grassi
La
funzione
energetica
dei
nell’ossidazione degli acidi grassi
lipidi
si
esplica
La gran parte dei trigliceridi è depositata nel tessuto
adiposo che, idrolizzandoli, fornisce energia ad altri
tessuti, sotto forma di acidi grassi
Il processo di ossidazione degli acidi grassi è detta: beta
ossidazione mitocondriale
L’ossidazione degli acidi grassi può essere
di tipo α, β e d ω.
L’ α-ossidazione si ha nei perossisomi ,
la ω-ossidazione si ha nel reticolo endoplasmatico
la β-ossidazione si verifica sia nei perossisomi che
nei mitocondri
LA PIU’ IMPORTANTE E’ LA β–OSSIDAZIONE
MITOCONDRIALE PERCHE’ E’ L’UNICA AD ESSERE
ACCOPPIATA
CON
LA
FOSFORILAZIONE
OSSIDATIVA.
Matrice mitocondriale
(ac. Grassi C<10)
Catena pari
b- ossidazione
(ac. Grassi C>10)
Perossisomi
Matrice mitocondriale
Matrice mitocondriale
Catena dispari
Ox degli
ac.grassi polinsaturi
Sono richieste ISOMERASI (C16:1)
e EPIMERASI (C 18:2)
a- ossidazione
w-ossidazione
Matrice
Reticolo
endoplasmatico
Acido fitanico
Via secondaria
per azione degli enzimi
IDROSSILASI,(monossigenasi)
citocromo P450 dipendenti
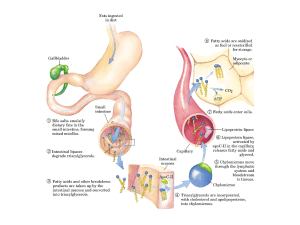
Le fonti principali di acidi grassi per la
beta-ossidazione sono:
la dieta (esogeni) e
le riserve cellulari (endogeni)
(essenzialmente trigliceridi negli adipociti).
• Gli acidi grassi della dieta vengono trasportati
sotto forma di trigliceridi tramite i chilomicroni,
dapprima attraverso il circolo linfatico e poi
ematico, dall'intestino agli organi, dove i
trigliceridi vengono
idrolizzati da specifiche
lipoprotein-lipasi ad acidi grassi e glicerolo.
• Nei tessuti epatico, adiposo e muscolare le
cellule procedono al loro utilizzo o al loro
deposito.
• In particolare negli adipociti gli acidi grassi
sono immagazzinati sottoforma di esteri:
trigliceridi.
•Gli acidi grassi prodotti nel fegato dai carboidrati, e in
eccesso alle richieste energetiche, sono utilizzati per
formare i trgliceridi che vengono trasportati dalle VLDL al
tessuto adiposo.
I trigliceridi degli adipociti vengono idrolizzati dalla lipasi
ormono-sensibile nelle fasi di digiuno ed esercizio fisico
(glucagone
ed
adrenalina)
ad
acidi
grassi
e
monoacilgliceroli; l’ulteriore idrolisi dei monoacilgliceroli
a glicerolo ed acidi grassi è catalizzata probabilmente da
una più specifica e attiva monoglicerolo-lipasi anche se la
lipasi ormono-sensibile è anch’essa in grado di operare
questa conversione.
.
Il glicerolo e gli acidi grassi escono liberamente
dall’adipocita attraverso la membrana plasmatica.
Il glicerolo verrà trasportato al fegato, in cui sarà
riconvertito a glucosio (gluconeogenesi).
Gli acidi grassi liberi sono trasportati agli organi
complessati all'albumina.
Giungeranno ai diversi organi, ove verranno
ossidati: muscolo scheletrico, cuore, fegato,
I triacilgliceroli sono riserve di energia
molto concentrate essendo ridotti ed
anidri.
La completa ossidazione degli acidi
grassi è caratterizzata da una resa
energetica pari a circa 9 kcal/g,
quasi doppia rispetto alle circa 4
kcal/g
dei carboidrati e delle
proteine.
Grande deposito
di trigliceridi
circondato da una
piccola striscia di
citoplasma.
Fotografia al microscopio elettronico di un adipocita
Mobilizzazione dei triacilgliceroli
Azione delle lipasi pancreatiche
nel muscolo e nel tessuto adiposo
ApoC-II
CATABOLISMO DEI TRIGLICERIDI
TRIGLICERIDI
trigliceride
lipasi
ACIDI GRASSI
sottoposta a regolazione ormonale
GLICEROLO
citoplasma carnitina mitocondrio
ACIDI GRASSI
glicolisi
beta-ossidazione
ACETIL - CoA
pag. 22
b-OSSIDAZIONE DEGLI
ACIDI GRASSI
pag. 27
b-OSSIDAZIONE DEGLI
ACIDI GRASSI
La b-ossidazione è l'insieme dei processi che hanno luogo sul
carbonio in b al carbonile.
Il primo enzima del processo è l'acil coenzima A deidrogenasi che si
trova sulla membrana mitocondriale interna e ha come cofattore il
FAD che si riduce a FADH2 e cede il suo potere riducente al
coenzima Q (catena respiratoria);
l'acil coenzima A deidrogenasi catalizza la reazione:
Acil-coenzima A
enoil -coenzima A
(trans 2,3 enoil coenzima A; molecola α-b insatura; alchene)
Ossidazione degli acidi grassi saturi
Acil-CoA deidrogenasi ha tre isozimi:
VLCAD agisce su acidi grassi da 12 a 18 atomi di carbonio
MCAD agisce su catene da 4 a 14 atomi di carbonio
SCAD agisce su acidi grassi da 4 a 8 atomi di carbonio
Le ultime tappe di questa sequenza sono catalizzate da
due gruppi di enzimi a seconda della lunghezza delle
catene degli acidi grassi.
Per gli acidi grassi con 12 o più atomi di carbonio le
reazioni sono catalizzate dalla proteina trifunzionale, TFP.
Quando TFP ha accorciato la catena dell’acido grasso fino
a 12 atomi di carbonio, l’ulteriore ossidazione viene
catalizzata da un gruppo di quattro enzimi presenti nella
matrice mitocondriale.
Malattie metaboliche ereditarie
Il più comune difetto genetico del catabolismo degli acidi grassi
nella popolazione caucasica è causato da una mutazione nel
gene che codifica per la acil-CoA deidrogenasi a catena
intermedia (MCAD).
La patologia è associata a ricorrenti episodi caratterizzati da un
accumulo di grassi nel fegato, ipoglicemia, sonnolenza vomito
e coma.
Nella prima infanzia la mortalità per questa malattia varia dal
25% al 60%.
Se il difetto genetico viene diagnosticato tempestivamente
dopo la nascita si ricorre ad una terapia alimentare a basso
contenuto di acidi grassi ed elevato contenuto di carboidrati.
pag. 31
b-OSSIDAZIONE DEGLI
ACIDI GRASSI
Il secondo enzima della b-ossidazione è:
enoil coenzima A idratasi
che catalizza la reazione:
enoil coenzima A
L-b idrossi acil coenzima A
questo enzima è assolutamente stereospecifico per l'isomero L-b idrossi acil
coenzima A
pag. 33
b-OSSIDAZIONE DEGLI
ACIDI GRASSI
La reazione successiva è catalizzata dalla L-b idrossi acil coenzima A deidrogenasi
(enzima NAD dipendente):
L-b-idrossi acil-coenzima A
b-cheto acil-coenzima A
contemporaneamente avviene la riduzione del NAD+ a NADH
pag. 35
b-OSSIDAZIONE DEGLI
ACIDI GRASSI
Infine, interviene una tiolasi (b-cheto acil coenzima A tiolasi)
il coenzima A funge da agente litico:
si forma un frammento a due atomi di carbonio (cioè l'acetil coenzima A)
il rimanente scheletro carbonioso è un acil-coenzimaA con due atomi di carbonio in
meno rispetto a quello di partenza
L'acil-coenzimaA ottenuto con la b-ossidazione, ripete il processo finché non si
ottiene solamente acetil-coenzima A.
pag. 37
b-OSSIDAZIONE DEGLI
ACIDI GRASSI (mitocondrio)
CH3
CH2
COO-
CH2
CH2
CH2
CoA-SH
CH2
CH3
CoA
COOCH3
CH2
CH3
ATP
ACETIL-CoA NADH+H+ FADH2
ACETIL-CoA NADH+H+ FADH2
CoA
ACETIL-CoA NADH+H+ FADH2
COO-
CH2
CH2
CoA
CH3
CoA
ACETIL-CoA NADH+H+ FADH2
COO-
COOH
CH3
CoA
ACETIL-CoA NADH+H+ FADH2
COOCO
S-CoA
CICLO DI KREBS
3 ATP
2 ATP
pag. 38
b-OSSIDAZIONE DEGLI
ACIDI GRASSI
Regola generale:
quando la deidrogenazione avviene tra due atomi adiacenti con elevata
differenza di affinità elettronica, il cofattore dell'enzima deidrogenasi, è quasi
sempre il NAD
se la deidrogenazione si ha tra due atomi adiacenti con bassa differenza di
affinità elettronica, il cofattore è il FAD
Regolazione della degradazione di acidi grassi
1. Sito di controllo primario
Carnitina aciltransferasi I blocca l’ingresso degli Acil-Coa nei
mitocondri. E’ inibita dal malonil-CoA, uno dei primi
intermedi della sintesi degli acidi grassi.
2. Siti di controllo secondario
Quando il rapporto NADH/NAD+ è elevato, la beta-idrossiacilCoA deidrogenasi viene inibita. Concentrazioni elevate di
acetil-CoA inibiscono la tiolasi.
3. Regolazione a lungo termine
La famiglia dei recettori nucleari PPAR comprende fattori di
trascrizione che regolano molti processi metabolici. PPAR alfa
agisce nel muscolo, nel tessuto adiposo e nel fegato dove
attiva una serie di geni essenziali per la beta ossidazione.
pag. 47
pag. 49
OSSIDAZIONE
IDRATAZIONE
viene riossidato dalla catena di
trasporto degli elettroni
mitocondriale:PRODUCE 2 ATP
Ciclo della
b-ossidazione
(C16:0)
OSSIDAZIONE
TIOLISI
Ac. Grasso + corto di 2atomi di C
Ac. Grassi a
Catena pari
Degradazione degli acidi
grassi a gruppi di unità
bicarboniose per volta, il
residuo rientra nel ciclo fino
alla completa degradazione.
Ciclo della
b-ossidazione
(C16:0)
Ad ogni ciclo
l’acido palmitico
perde 2 atomi di C sottoforma di AcetilCoA
Prodotto finale della b-ossidazione:
Acetil-CoA (Ac.grassi catena pari)
Propionil-CoA + Acetil-CoA (Ac. Grassi catena dispari)
b- ossidazione
Ac. Grassi a
Catena dispari
Via catabolica
del
Propionil-CoA
Prodotto finale
del ciclo
Propionil-CoA
Ciclo
di Krebs
Un’attività difettosa dell’enzima determina un accumulo di L-metilmalonil-CoA
che esce dalle cellule sottoforma di Acido, metilmalonico
ACIDOSI
(pH ematico )
Danno al SNC
(Acidemia metilmalonica)
Terapia: Ingente somministrazione
attivare la metilmalonil-CoA mutasi.
di Vitamina B12 che sembra
Distribuzione e fabbisogno
La vitamina B12 è sintetizzata esclusivamente dai microrganismi.
Nell’uomo la sintesi di B12 operata dai batteri intestinali è del tutto insufficiente,
per cui la vitamina deve essere introdotta con la dieta.
Particolarmente ricchi di B12 sono il fegato ed rene.
La carne, il latte, il formaggio e le uova contengono quantità modeste di
vitamina, mentre gli alimenti di origine vegetale non contengono
tale vitamina.
La sua carenza nell’uomo ha come principale manifestazione
l’anemia perniciosa, caratterizzata in circolo dalla presenza di elementi
immaturi della serie eritrocitaria.
In più, la degenerazione delle fibre nervose e altre anomalie del
sistema nervoso.
Anemia perniciosa
Solo eccezionalmente causata da carenza di vitamina nella dieta,
è generalmente causata da un blocco del suo assorbimento intestinale
(avitaminosi condizionata) per assenza
di una specifica glicoproteina del succo gastrico,
detta “Fattore intrinseco di Castle”,
che ha il compito di legare la vitamina, trasportarla all’ileo,
legarsi a siti specifici presenti sui microvilli ed
immetterla negli eritrociti per azione un fattore di rilascio.
Gli elettroni liberatisi durante il
ciclo ossidativo vengono trasferiti
alla catena respiratoria
mitocondriale per la formazione
dell’ATP utilizzando sistemi FAD- e
NAD- dipendenti
(trasporto elettronico).
Benchè la maggior parte della ossidazione degli acidi grassi
abbia luogo nei mitocondri, una parte di essa ha
luogo anche in organelli cellulari detti
PEROSSISOMI,
caratterizzati da elevate concentrazioni dell’enzima Catalasi,
che catalizza la dismutazione del perossido di idrogeno
in acqua ed ossigeno molecolare.
nb. L’ossidazione degli acidi grassi che, in questi organelli,
si arresta a livello dell’ottanil CoA,
può servire ad accorciare le lunghe catene per renderle migliori
substrati della b ossidazione.
L’organello contiene un
cristallo di urato
ossidasi
ed
è circondato da una
singola membrana
bistratificata.
Le strutture granulari
scure all’esterno del
perossisoma sono
particelle di glicogeno.
Fotografia al microscopio elettronico di un
perossisoma in un epatocita
(ottanil-CoA)
Prima tappa della degradazione degli acidi grassi nei perossisomi
La b-ossidazione perossisomiale
Benchè la maggior parte della ossidazione degli acidi grassi abbia luogo nei
mitocondri, una parte di essa ha luogo anche in organelli cellulari detti
PEROSSISOMI, caratterizzati da elevate concentrazioni dell’enzima Catalasi, che
catalizza la dismutazione del perossido di idrogeno in acqua ed ossigeno
molecolare.
Negli animali la b-ossidazione perossisomiale serve ad accorciare acidi grassi a
catena molto lunga (C>18-22), i quali entrano poi nel sistema della bossidazione mitocondriale per essere degradati.
Gli acidi grassi a lunga catena diffondono nel perossisoma senza richiedere la
CARNITINA.
L’attivazione avviene ad opera di una Acil-CoA sintetasi a lunga catena.
1° OX diversa
La tiolasi si blocca se
presenti acil-CoA con
catena C8 o minore
si arresta a livello dell’Ottanil CoA
Ossidazione a trans D-enoil-CoA (I Step)
Acil- CoA
Come cofattore è stato usato il FAD, ma gli elettroni sottratti vengono trasferiti direttamente
all’O2 invece che passare attraverso la catena respiratoria e fosforilazione ossidativa. Quindi
rispetto all’ossidazione mitocondriale per ogni ciclo si producono in meno 2 ATP
a- ossidazione e la sindrome di Refsum
Degradazione degli acidi grassi per eliminazione di un carbonio alla volta
dall’estremità carbossilica della molecola.
Avviene nei tessuti cerebrali e non richiede CoA e non forma fosfati ad alta
energia
Ossidazione avviene in posizione a su acidi grassi con ramificazioni in posizione
dispari
L’a-ossidazione è stata scoperta studiando pazienti affetti da Sindrome di Refsum: una
malattia neurologica molto rara e grave dovuta ad accumulo di Acido Fitanico derivante dal
Fitolo, costituente della clorofilla e abbondante nei vegetali e nei cibi derivati dagli erbivori
(prodotti lattiero-casearei)
a-ossidazione
Il gruppo CH3 in posizione 3
impedisce la b-ox
a-ossidazione
ACIDO PRISTANICO
Riprende la b-ossidazione
a-ossidazione e
malattia di Refsum
Via difettosa nella sindrome di
Refsum. Quindi non si forma
ac.pristanico
NON Riprende la b-ossidazione
Terapia: dieta senza
ω-ossidazione
La ω-ossidazione i cui enzimi sono presenti nel reticolo endoplasmatico del fegato e
del rene è una via solitamente poco praticata nei mammiferi ma diventa importante
quando la β per mutazione o carenza di carnitina funziona male.
Nella prima tappa viene inserito un gruppo ossidrilico e l’ossigeno di questo gruppo
proviene dall’O2 molecolare per azione di una ossidasi a funzione mista P450 e
NADPH dipendente.
Sul carbonio ω intervengono altri due enzimi l’alcol deidrogenasi, che ossida il gruppo
ossidrilico ad aldeide, e l’aldeide deidrogenasi che ossida il gruppo aldeidico ad acido
generando un acido grasso con doppia estremità carbossilica, ognuna delle quali può
legare il CoA, ora la molecola può entrare nel mitocondrio dove subisce una normale
βossidazione.