ANIRIDIA : L’OCCHIO INDIFESO
Anna Piccioni
Abstract:
L’articolo delinea i principali aspetti clinici e funzionali dell’aniridia, una delle
patologie genetiche che determinano ipovisione.[fine abstract]
All’indomani della costituzione dell’Associazione Aniridia Italiana, promossa da un
gruppo di famiglie coinvolte e finalizzata alla migliore conoscenza e divulgazione di
informazioni su questa rara patologia oculare, ritengo utile dedicare uno spazio al chiarimento
dei suoi principali aspetti clinici e funzionali.
L’ipovisione in età evolutiva riconosce spesso una origine da patologie genetiche,
alcune delle quali assai rare e sempre più oggetto di studio da parte dei ricercatori a livello
internazionale.
L’aniridia fa parte delle patologie rare perché mostra una incidenza da 1:40000 a
1:100000 nati sani in relazione ai diversi campioni di popolazione presi in esame.
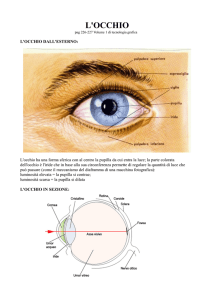
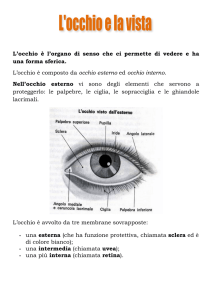
Il quadro clinico è caratterizzato da uno sviluppo parziale e/o anomalo dell’iride, la
struttura che dona all’occhio un colore caratteristico per ogni persona e che, di fatto, risulta
essere un diaframma con al centro la pupilla, dall’aspetto di un pallino nero, ma che è un foro
attraverso il quale la luce entra nell’occhio per raggiungere e stimolare la retina.
L’iride non svolge puramente un ruolo estetico, rappresenta infatti uno schermo opaco
necessario a selezionare il fascio luminoso che entra nell’occhio attraverso il foro pupillare, il
cui diametro muta in relazione alla luce presente nell’ambiente.
L’anomalia dell’iride è quasi sempre bilaterale e la variabilità fenotipica è ampia, vale a
dire che la stessa anomalia del gene si presenta con caratteristiche diverse nei diversi soggetti
affetti. L’iride può essere totalmente o parzialmente assente fino a casi nei quali ad una sua
apparente presenza corrisponde una profonda alterazione morfologica tale per cui l’iride è
“trasparente”, presente come impalcatura di base, ma priva nel suo spessore di strutture e
pigmento utili alla sua funzione di schermo.
La trasparenza iridea è facilmente diagnosticabile in corso di visita oculistica per mezzo
della retroilluminazione del bulbo. In questi casi è necessario porre una diagnosi differenziale
con l’albinismo oculare, che presenta la medesima caratteristica.
All’anomalia di sviluppo embrionale dell’iride si associa ipoplasia della macula, zona
centrale della retina, fondamentale area per la visione del dettaglio fine.
Ipoplasia è sinonimo di ridotto sviluppo e funzione, ma non di degenerazione e morte
cellulare, per cui si comprende come vi sia una ridotta acuità visiva (1-2/10), stabile nel
tempo salvo complicazioni. Caratteristico dell’aniridia è il nistagmo, di solito orizzontale a
scosse più intense nella prima infanzia, con possibilità di componenti rotatorie. La sensibilità
al contrasto e la visione dei colori si presentano nei limiti della norma.
Con il tempo alla sola anomalia iridea bilaterale si possono associare altre patologie
oculari quali: la cataratta (opacamento del cristallino), il glaucoma (aumento della pressione
endobulbare, sofferenza di retina e nervo ottico), alterazioni della trasparenza corneale
(opacità e neovascolarizzazione per alterato ricambio delle cellule dello strato superficiale
della cornea). Le alterazioni corneali sono dovute principalmente ad una insufficienza di
cellule staminali normalmente presenti al limbus, ovvero alla periferia della cornea. In
carenza di cellule staminali, che sono le progenitrici delle cellule dell’epitelio corneale, questo
è più fragile, si danneggia con facilità, ripara lentamente e risulta molto sensibile agli agenti
esterni e all’uso di lenti a contatto.
La cataratta ed il glaucoma di rado si possono manifestare anche in età pediatrica; la
prima come opacità polare posteriore ed il secondo con le caratteristiche simili al glaucoma
congenito.
In presenza di innalzamento della pressione endobulbare in bambini piccoli è necessario
porre diagnosi differenziale con le anomalie congenite del segmento anteriore dell’occhio ,
che presentano ipertensione endobulbare, ma hanno alterazioni morfologiche diverse dall’
aniridia.
Nel dubbio diagnostico ci può essere di aiuto in molti casi la genetica attraverso le
indagini di biologia molecolare per la ricerca del gene anomalo, che è all’origine dell’aniridia.
L’aniridia è una patologia genetica trasmessa con modalità autosomica dominante, vale
a dire che in una coppia di genitori, uno dei quali affetto da aniridia, ad ogni concepimento si
presenta un rischio del 50% di generare un figlio affetto dalla stessa patologia.
Di fatto poi vi sono casi di aniridia anche in famiglie dove mai si era presentata questa
anomalia. Tale possibilità è prevista dalla genetica umana e si definisce come effetto di una
“mutazione de novo”, ovvero l’alterazione di un gene che si manifesta occasionalmente in un
embrione.
Per l’aniridia nei 2/3 dei casi la patologia si presenta a carattere ereditario e familiare,
mentre la mutazione de novo è responsabile di ca 1/3 dei casi.
Il gene identificato come responsabile dell’aniridia è il PAX6. Esso fa parte di un
gruppo di geni chiamati PAX coinvolti nello stimolare la crescita di organi. Il PAX6 è attivo
nell’occhio, nel cervelletto e nel pancreas.
Ad oggi è possibile individuare nel DNA, con tecniche di biologia molecolare, le
alterazioni del gene PAX6 in un buon numero di casi. Non si può parlare di possibilità
diagnostica al 100%, perché vari sono i modi di mutazione del gene.
Nonostante le speranze maturate in questi anni di dibattito sulle possibilità e i limiti
dell’ingegneria genetica, non è possibile pensare alla sostituzione del gene malato quando
l’occhio è già sviluppato. Una sostituzione sarebbe utile solo all’inizio dello sviluppo
embrionale, che per l’occhio è precocissimo, e ad oggi non è realizzabile.
Il basso residuo visivo presentato dalle persone affette da aniridia può essere fortemente
peggiorato in condizioni ambientali nelle quali la luminosità di fondo o la sua variazione
causano abbagliamento. Questo è causato dall’alterata risposta dei fotoricettori retinici invasi
da una quantità di raggi luminosi non selezionati dal variare dell’apertura pupillare.
L’entrata casuale della luce ed il suo passaggio attraverso il cristallino può subire anche
il meccanismo della diffrazione, che genera una fotofobia sgradevole e funzionalmente
svantaggiosa.
La prescrizione e l’uso delle lenti filtranti offrono protezione contro la luce che causa
abbagliamento e confusione. Tali lenti, montate preferibilmente su montature con alette
laterali che aumentano l’effetto schermatura, operano una selezione sulle lunghezze d’onda
dello spettro visibile abbattendo gli effetti negativi ed enfatizzando i contrasti.
Nei bambini molto piccoli è consigliabile anche l’uso di cappelli con visiera o con falde
larghe.
Da circa un decennio, accanto a tali sussidi, sono state introdotte lenti a contatto di
costruzione artigianale, che riproducono l’iride e l’apertura pupillare, a diametro fisso, la cui
finalità è il prevenire fotofobia e abbagliamento. L’uso di tali lenti a contatto è stato riservato
all’inizio solo all’età adolescenziale, soprattutto per motivi estetici, per poi estendersi anche
alla prima infanzia.
Il dibattito fra gli specialisti è aperto sia per la valutazione dei vantaggi e degli
svantaggi di queste protesi, sia per l’esperienza di counselling alle famiglie richiesto da tali
applicazioni di lenti a contatto.
Sono necessari servizi offerti da professionisti esperti e qualificati che possano seguire
l’intero iter diagnostico ed abilitativo, fornendo istruzioni corrette e supervisione costante.
Ogni lente a contatto rappresenta un corpo estraneo all’interno di una struttura vitale e
variabile quale è il segmento anteriore dell’occhio. Nell’aniridia la conoscenza di una
debolezza delle cellule staminali della cornea al limbus fa di questa una struttura delicata e
reattiva, più che in altre condizioni. La possibilità che la cornea mostri sofferenza e perda la
trasparenza pone dei grossi punti di domanda sul quando e per quanto tempo indossare le lenti
o applicarle al bambino. I vantaggi estetici e di schermatura sono comunque punti a favore.
La scienza ha caratteristiche di universalità, ma dovunque debba essere applicata all’individuo
richiede perizia e prudenza.
Inoltre non si può dimenticare che l’abilitazione del bambino ipovedente va intesa come
intervento globale sullo sviluppo, come attenzione all’integrazione sensoriale per facilitare
l’apprendimento. L’interesse dunque non può essere focalizzato in modo preminente e quasi
esclusivo sul canale visivo, per evitare di lasciare in disparte modalità di supporto, intervento
e conoscenza che, nel tempo, sono indispensabili a creare solide fondamenta per l’evoluzione
delle future strategie di apprendimento e di inserimento.
Riferimenti bibliografici
Cvekl, A., Tamm, E.R. (2004). Anterior eye development and ocular mesenchyme: new
insights from mouse models and human diseases. Bioessays, 26(4), 374-386.
Hanson, I., Churchill, A.,Love, J., Axton, R., Moore, T., Clarke, M., Meire, F., van
Heyningen, V. (1999). Missense mutations in the most ancient residues of the PAX6 paired
domain underlie a spectrum of human congenital eye malformations. Hum. Molec. Genet., 8,
165-172.
Jastaneiah, S., Al-Rajhi, A.A. (2005). Association of aniridia and dry eyes.
Ophthalmology, 112(9), 1535-40.
Neveu, M.M., Holder, G.E., Sloper, J.J., Jeffery, G. (2005). Optic chiasm formation in
humans is independent of foveal development. Eur. J. Neurosci., 22(7),1825-9.
Tzoulaki, I., White, I.M., Hanson, I.M. (2005). PAX6 mutations: genotype-phenotype
correlations. BMC Genet., May 26, 6(1), 27.
Anna Piccioni
oculista, genetista,
consulente di ipovisione