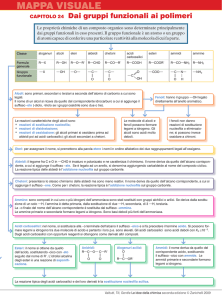
Appunti di chimica organica
Gli alcoli.
Conversione degli alcoli in alogenuri alchilici
Il metodo più comune per la preparazione di un alogenuro alchilico è la reazione di un alcol con un acido
alogenidrico.
Meccanismo della reazione
Il meccanismo della reazione degli alcoli con acidi alogenidrici prevede inizialmente un equilibrio con la
formazione dell'alcol protonato. Questo alcol protonato sottostà ad una reazione SN2 con l'X- o a una SN1
che porta al carbocatione, che viene successivamente attaccato da X-. L'acqua è ilgruppo uscente del
composto protonato C―O+H2. Quale delle vie (SN1 o SN2) venga seguita da un particolare alcol, dipende
dalle condizioni di reazione, dalla stabilità del carbocatione, da fattori sterici e dalle altre considerazioni che
sono state fatte in generale per le reazioni SN1 e SN2.
Di seguito vengono riportati due esempi di reazioni che procedono rispettivamente attraverso un
meccanismo SN2 e attraverso un meccanismo SN1.
SN2
SN1
Disidratazione degli alcoli
Quando un alcol viene trattato con acido solforico (H2SO4), si ottiene un alcol protonato che si può
disidratare facilmente ad alchene attraverso un meccanismo E1.
Gli alcoli terziari si disidratano, via E1, molto facilmente.
Gli alcoli secondari e particolarmente i primari formano il carbocatione intermedio con difficoltà e per
questo motivo la reazione richiede temperature molto più alte.
Meccanismo di reazione
La catalisi acida serve a protonare il gruppo ossidrilico (―OH) dell'alcol, rendendolo un miglior gruppo
uscente:
L'alcol protonato elimina una molecola di acqua con formazione di un carbocatione:
Nello stadio successivo una molecola di acqua presente in soluzione si comporta da base debole e rimuove
un protone in posizione ß con formazione dell'alchene:
Per la catalisi acida è necessario utilizzare acidi come ad esempio H2SO4 o H3PO4 i cui anioni non sono
nucleofili; in tal modo si favorisce la reazione di eliminazione E1 piuttosto che la reazione di sostituzione
nucleofila SN1.
Fenoli
Acidità dei fenoli:
I fenoli sono composti acidi con acidità simile a quella dell'acido solfidrico, e sono, in particolare, molto più
acidi degli alcoli. Questo perché lo ione fenato C6H5O- che si ottiene dal fenolo per perdita di uno ione H+
è stabilizzato per risonanza. La carica negativa di uno ione alcolato (RO-) è localizzata sull'atomo di
ossigeno, nello ione fenato invece tale carica è delocalizzata sull'anello aromatico e questo stabilizza lo ione
stesso:
I sostituenti, in particolare quelli posizionati in orto o para rispetto al gruppo —OH del fenolo, possono
modificare, per effetto induttivo e per effetto mesomerico, l'acidità del fenolo stesso: i gruppi elettronattrattori aumentano l'acidità, i gruppi elettron-donatori diminuiscono l'acidità. Il p-nitrofenolo ad esempio è
circa 600 volte più acido del fenolo. Ciò, come si è detto, è dovuta al fatto che il nitrogruppo è un potente
elettron-attrattore sia per effetto induttivo che per risonanza:
Reazioni dei fenoli
I fenoli sono sostanze che si comportano da nucleofili; in particolare, si comportano da nucleofili bidentati,
nel senso che, sia l'ossigeno del gruppo -OH, che l'anello aromatico, possono essere sede della reattività
nucleofila del fenolo.
I fenoli sono composti che danno facilmente reazioni di sostituzione elettrofila aromatica a causa della
presenza del gruppo -OH. Tale gruppo si comporta da forte attivante e dirige i sostituenti in posizione orto- e
para-.
Simboleggiando con E+ un generico elettrofilo, la reazione può essere rappresentata nel seguente modo:
Acilazione dei fenoli
Come detto i fenoli si comportano da nucleofili bidentati, nel senso che sia l'ossigeno del gruppo -OH, che
l'anello aromatico, possono essere sede della reattività nucleofila del fenolo. L'acilazione del fenolo può
pertanto avvenire:
1.sull'anello aromatico (C-acilazione o acilazione di Friedel-Crafts). Si ha una sostituzione elettrofila
aromatica che porta alla formazione di un arilchetone.
2.sull'ossigeno del gruppo -OH (O-acilazione). Si ha una sostituzione nucleofila che porta alla formazione di
un estere. Tale reazione è favorita da una catalisi acida o basica.
Come agenti acilanti è possibile utilizzare:
1.cloruri acilici
2.anidridi degli acidi carbossilici (in presenza di AlCl3 per la C-acilazione)
Alogenazione
L'alogenazione dei fenoli (clorurazione e bromurazione) è una reazione che avviene velocemente anche in
assenza di catalizzatori.
La sostituzione avviene in posizione -para rispetto al gruppo -OH, a meno che tale posizione non sia già
impegnata da un sostituente. In tal caso la sostituzione avviene in posizione -orto.
Eteri
Conversione di alcoli in eteri
Gli alcoli possono essere convertiti in eteri. Se un alcol viene trattato con H2SO4 a temperatura più bassa di
quella necessaria per disidratare l'alcol stesso ad alchene, e se è presente un eccesso di alcol, si può infatti
avere condensazione dell'alcol con formazione dell'etere.
La reazione complessiva può essere rappresentata nel seguente modo:
Se si mescolano in acido solforico due alcoli si ottengono tre eteri:
Ammine
Basicità delle ammine
Le ammine (così come l'ammoniaca) hanno un debole carattere basico. Grazie al doppietto solitario
sull'atomo di azoto, ogni ammina può strappare un protone all'acqua e trasformarsi nel suo acido coniugato
(R3NH+):
La basicità delle ammine dipende sensibilmente dai sostituenti legati all'atomo di azoto.Le
alchilammine (R—NH2) ad esempio sono più basiche dell'ammoniaca: in particolare la metilammina
(CH3—NH2) è circa 22 volte più basica dell'ammoniaca. Il gruppo metilico della metilammina è infatti un
gruppo elettron-repulsore e per effetto induttivo stabilizza la carica positiva presente sull'acido coniugato
(R3NH+) e sposta l'equilibrio precedente verso destra.
Le ammine secondarie sono leggermente più basiche delle ammine primarie, mentre per le ammine
terziarie c’è un inversione di tendenza e sono pertanto meno basiche delle ammine secondarie. Il motivo
di questo comportamento è da ricercarsi nell’ingombro sterico dei tre sostituenti che, tra l'altro, diminuisce
anche l’effetto di solvatazione dell’acqua sullo ione alchilammonio (R3NH+), rendendolo meno stabile.
Le ammine aromatiche invece sono basi molto più deboli delle ammine alifatiche.
Preparazione delle ammine
Alchilazione di ammoniaca
L'ammoniaca reagisce facilmente con gli alogenuri alchilanti, quali gli alogenuri alchilici.
la reazione può essere schematizzata nel seguente modo:
Nitrosazione delle alchilammine
Formazione di N-nitrosammine e reazione di deamminazione
Quando una soluzione di nitrito di sodio (NaNO2) viene acidificata con acido cloridrico diluito o con acido
solforico diluito, vengono generate numerose specie chimiche che possono agire come sorgenti del catione
nitrosile, tra le quali la più importante è l'acido nitroso.
L'acido nitroso viene solitamente preparato in situ poichè è altamente instabile e reagisce anche con
l'umidità presente nell'atmosfera.
La sequenza di reazioni che portano alla formazione del catione nitrosile può essere riassunta come qui di
seguito:
Le ammine secondarie reagiscono con l'acido nitroso portando alla formazione di N-nitrosammine.
La formazione della N-nitrosammina avviene per attacco nucleofilo dell'ammina all'atomo di azoto dello
ione nitrosile.
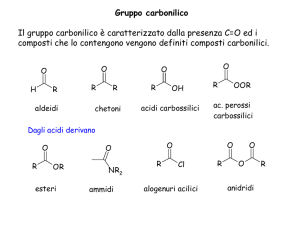
Aldeidi e chetoni
L'atomo di ossigeno del gruppo carbonilico ha una elettronegatività molto più elevata dell'atomo di
carbonio. Di conseguenza l'ossigeno attrae a sé gli elettroni del doppio legame C=O con una forza maggiore
e il legame risulta fortemente polarizzato.
In termini di risonanza, il gruppo carbonilico può essere rappresentato tramite due forme limite, di cui la
prima (A) è più stabile della seconda (B):
Addizione nucleofila al gruppo carbonilico
La conseguenza alle considerazioni appena fatte è che il gruppo carbonilico può dare facilmente reazioni
di addizione nucleofila.
In generale, i chetoni sono meno reattivi delle aldeidi per due ragioni. La prima è per ragioni steriche:
l'atomo di carbonio carbonilico è più ingombrato nei chetoni che nelle aldeidi. Il secondo è dovuto ai gruppi
alchilici che tendono a diminuire, per effetto elettron-donatore, la carica positiva del carbonio carbonilico.
La formaldeide, completamente priva di gruppi alchilici, è l'aldeide più reattiva.
L'addizione nucleofila può essere rappresentata come segue:
In generale, indicando con Nu: un generico nucleofilo, potremmo scrivere:
E' l'attacco nucleofilo al carbonio carbonilico a indurre la rottura del legame π.
A causa dei doppietti elettronici presenti sull'ossigeno del gruppo carbonilico, la reazione di addizione
nucleofila può essere iniziata anche da un attacco elettrofilo sull'atomo di ossigeno del gruppo carbonilico.
Gli acidi catalizzano così l'addizione nucleofila al carbonio carbonilico per protonazione dell'atomo di
ossigeno:
In questo modo il carbonio assume una carica positiva maggiore e l'attacco nucleofilo è reso più semplice.
Preparazione
Aldeidi e chetoni possono essere ottenuti per ozonolisi di alcheni seguita da idrolisi.
Idratazione degli alchini
I chetoni possono essere ottenuti per idratazione degli alchini. Dalla reazione si ottiene un alcol vinilico
(enolo) che, per tautomeria cheto-enolica, riarrangia rapidamente nel corrispondente chetone.
Reazioni
Riduzione di aldeidi e chetoni ad alcani
La reazione di Clemmensen prevede la riduzione del gruppo carbonilico di un un'aldeide o di un chetone a
gruppo metilenico (―CH2―) mediante l'utilizzo di acido cloridrico e di un amalgama di zinco-mercurio.
Riduzione di Wolff-Kishner
La reazione di Wolff-Kishner prevede la riduzione di aldeidi e chetoni a idrocarburi in presenza di un
eccesso di idrazina (NH2NH2) e di basi forti ad alte temperature. Come solvente si utilizza glicole etilenico.
Riduzione ad alcoli
Aldeidi e chetoni possono essere ridotti ad alcoli. La riduzione di aldeidi porta alla formazione di alcoli
primari mentre la riduzione di chetoni chetoni porta alla formazione di alcoli secondari.
Ossidazione ad acidi carbossilici
E' possibile ossidare aldeidi e chetoni ad acidi carbossilici. Però, mentre l'ossidazione delle aldeidi avviene
anche in condizioni blande, l'ossidazione dei chetoni avviene solo in condizioni estremamente drastiche.
Le aldeidi sono fra le classi di composti organici che si ossidano con maggiore facilità. Esse vengono
convertite in acidi carbossilici da numerosi agenti ossidanti, inclusi non solo gli usuali reagenti come
permanganato e bicromato, ma anche reagenti relativamente blandi come ioni argento e rame.
Alogenazione
Aldeidi e chetoni che possiedono idrogeni in posizione α (aldeidi e chetoni enolizzabili) reagiscono con gli
alogeni per dare reazioni di sostituzione al carbonio in α.
Acidi carbossilici
L'acidità degli acidi carbossilici
In soluzione acquosa gli acidi carbossilici si dissociano per formare un anione carbossilato(RCOO-) ed
un idrogenione (H+) secondo il seguente equilibrio:
RCOOH <==> RCOO- + H+
La spiccata acidità degli acidi carbossilici è dovuta a due fattori: l'effetto di risonanza e l'effetto induttivo.
Effetto di risonanza
Se confrontiamo l'acidità dell'acido acetico (CH3COOH) con quella dell'etanolo (CH3CH2OH), notiamo
che il primo è cento miliardi di volte più acido dell'etanolo:
CH3COOH <==> CH3COO- + H+ (Ka= 10-5)
CH3CH2OH <==> CH3CH2O- + H+ (Ka= 10-16)
Nello ione etilato (CH3CH2O-) infatti la carica negativa è localizzata sull'atomo di ossigeno, mentre
nello ione acetato (CH3COO-) la carica negativa è delocalizzata per effetto di risonanza tra i due atomi di
ossigeno:
Lo ione acetato viene così stabilizzato per risonanza e ciò lo rende particolarmente stabile. Pertanto
l'equilibrio dell'equazione
CH3COOH <==> CH3COO- + H+
è più spostato a destra rispetto a quello dell'equazione
CH3CH2OH <==> CH3CH2O- + H+
I dati fisici confermano la risonanza dello ione acetato. Nell'acido acetico infatti i due legami carbonioossigeno hanno lunghezze diverse, mentre nello ione acetato entrambi i legami carbonio-ossigeno sono
identici e la loro lunghezza ha un valore intermedio tra quella di un singolo e quella di un doppio legame
carbonio-ossigeno:
Effetto induttivo
Un fattore molto importante che determina variazioni anche notevoli nel valore dell'acidità degli acidi
carbossilici, è il tipo di gruppo legato al carbossile: i gruppi elettronattrattori determinano un aumento
di acidità mentre i gruppi elettrondonatori la fanno diminuire.
Prendiamo come esempio l'acido cloro-acetico: il cloro legato al carbonio in α (alfa) al gruppo carbossilico,
essendo un elemento molto elettronegativo, ha un effetto elettronattrattore che stabilizza lo ione
cloroacetato:
Viceversa, i gruppi alchilici (–CH3) sono debolmente elettronrepulsori e quindi accentuano la carica
negativa sugli ossigeni dello ione carbossilato e determinano quindi una diminuzione di acidità. Per tale
motivo l'acido acetico (CH3COOH) è circa 12 volte più debole dell'acido formico (HCOOH).
Preparazione
Carbonatazione dei reattivi di Grignard
Un metodo importante per la preparazione degli acidi carbossilici è il trattamento di un composto organometallico
con anidride carbonica. La carbonatazione dei reattivi di Grignard converte un alogenuro alchilico (o arilico)
dal quale il reattivo di Grignard proviene in un acido carbossilico la cui catena carboniosa contiene un
atomo di carbonio in più rispetto a quella dell'alogenuro alchilico (o arilico).
La reazione può essere rappresentata nel seguente modo:
Esteri
Gli esteri sono composti organici che derivano dagli acidi carbossilici per sostituzione del gruppo —OH
con il gruppo —OR, pertanto hanno fomula RCOOR:
Un estere ciclico viene definito lattone.
La reazione di un estere con una base forte si chiama saponificazione.
La saponificazione di un estere segue una cinetica di secondo ordine
Due meccanismi differenti sono plausibili per una cinetica di questo tipo: il primo coinvolgente la rottura
del legame acilico C-O; il secondo quella del legame alchilico O-C.
Scissione acilica (scissione del legame C-O)
Viene spezzato il legame tra il gruppo acilico e l'ossigeno.
Scissione alchilica (scissione del legame O-C)
Viene spezzato il legame tra il gruppo alchilico e l'ossigeno.
Idrolisi acido-catalizzata degli esteri
Eseguendo l'idrolisi di un estere in una soluzione acquosa acida diluita, si ottiene un acido carbossilico e un
alcol.
Ammidi
Le ammidi sono composti che derivano dagliacidi carbossilici e hanno la seguente struttura generale:
Quella appena descritta è una ammide primaria, ma esistono anche, a seconda del numero di sostituenti
presenti sull'azoto,ammidi secondarie e ammidi terziarie:
Preparazione delle ammidi
Le ammidi si possono ottenere in vari modi:
- Per reazione tra ammine (R2NH) ed esteri (R'COOR'').
- Per reazione tra ammine (R2NH) e cloruri acilici (R'COCl)
- Per reazione di ammine con anidridi (R'COOCOR'')
- Per reazione tra ammine e acidi carbossilici.
Reazioni
Idrolisi delle ammidi
Per idrolisi delle ammidi si ottengono acidi carbossilici:
RCONH2 + H2O → RCOOH + NH3
La reazione solitamente viene condotta tramite riscaldamento prolungato e in presenza di catalizzatori acidi
o basici.
Riduzione delle ammidi
Utilizzando l'idruro di litio alluminio è possibile ridurre le ammidi ad ammine.