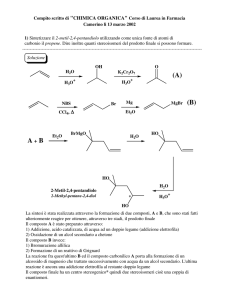
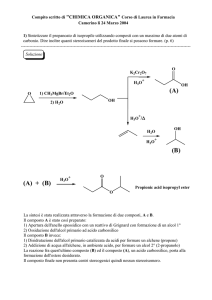
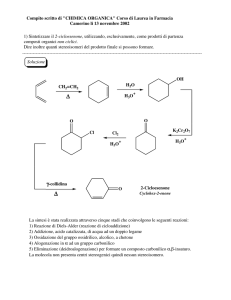
03/12/2011
CINETICA CHIMICA
Ogni reazione chimica ha una sua cinetica cioè una certa velocità ed una certa
sequenza di atti reattivi.
L’atto reattivo può essere uno solo (reazione elementare); oppure possono
avvenire più reazioni intermedie prima di giungere ai prodotti.
Per semplicità tratteremo il meccanismo del singolo atto reattivo anche se
questo fa parte di una serie di atti che insieme conducono ai prodotti.
Es:
A + B → C + D
Perché A e B reagiscano occorre:
1)
che A e B si urtino
2) che l’urto sia efficace, cioè che (I) le molecole posseggano energia
sufficiente e che (II) l’urto avvenga in zone reattive (effetto sterico).
Il punto (I) è molto importante: la trasformazione chimica ha luogo solo se le
molecole reagenti nell’insieme posseggono un’ energia cinetica superiore ad un
valore minimo, detta energia di attivazione Ea.
Quando due reagenti si urtano in una collisione efficace hanno luogo i seguenti
passaggi: collidono, rallentano, si fermano, quindi si staccano allontanandosi e le
particelle risultanti saranno chimicamente diverse da quelle che si sono urtate.
Quando rallentano Ecin diminuisce e per la conservazione dell’energia deve
aumentare la loro energia potenziale.
Energia potenziale
Ea = energia di attivazione
∆H = calore di reazione
Andamento della reazione
Solo le molecole che hanno una energia > Ea sono in grado di superare il
dislivello energetico per giungere ai prodotti.
1
03/12/2011
collisione non efficace
collisione efficace
I grafici precedenti si riferiscono a reazioni esotermiche.
reazione endotermica.
Una reazione esotermica in una direzione
è endotermica nella direzione opposta.
Si possono avere informazioni su cosa realmente avviene durante una collisione
efficace.
Es:
B2 che collide con A2.
1) legami covalenti tra gli atomi di A e tra gli atomi di B.
2) legami A – A e B – B parzialmente rotti e legami A – B parzialmente formati.
3) Legami A – A e B – B rotti. Esistono solo legami A – B.
Il momento 2) si chiama stato di transizione e corrisponde al punto più alto del
diagramma dell’energia potenziale. La specie che esiste in questo instante si
chiama complesso attivato.
Si definisce Molecolarità di una reazione elementare il numero delle molecole
che è necessario che si urtino per dar luogo ad essa. Coincide con il numero di
molecole che costituisce il complesso attivato. E’ difficile da determinare.
2
03/12/2011
Es:
2 ICl + H2 → 2 HCl + I2
Non è trimolecolare, infatti è data da due reazioni bimolecolari:
ICl + H2 → HCl + HI
HI + ICl → HCl + I2
2 ICl + H2 → 2 HCl + I2
Fattori che influenzano la velocità di reazione
1) Natura chimica dei reagenti
2) Loro capacità di venire in contatto
3) Concentrazione dei reagenti
4) Temperatura del sistema reagente
5) Disponibilità di agenti (catalizzatori) che influenzano la velocità, ma che non
vengono consumati dalla reazione
Come si esprime la velocita’
3
03/12/2011
VELOCITA’ DI REAZIONE
E’ la misura essenziale per poter risalire al meccanismo di reazione.
A → B
Es:
A scompare con velocità v, che si può esprimere con variazione (diminuzione) del
numero di moli nell’unità di tempo.
a volume costante se C è la concentrazione molare di A:
v=−
dC
dt
cioè durante il tempo dt la concentrazione
diminuisce di dC (segno negativo).
v=
dC B
dt
se avessimo seguito il formarsi di B.
In genere la velocità è espressa in moli di sostanza consumata (o formata) per
litro e per unità di tempo (frazione di secondo, minuti, ore o anni).
I valori numerici di v si acquisiscono sperimentalmente seguendo la specie che
scompare o che si forma nel modo più opportuno.
Es:
2 N2O5 → 2 N2O4 + O2
per
Si può misurare in tempi
successivi la quantità di O2 sviluppata ad una certa temperatura. Oppure la
scomparsa di N2O5.
dC è dato dalla pendenza delle
dt tangenti (tratteggiate).
La velocità diminuisce all’aumentare
del tempo.
Per trovare la relazione tra v e la
concentrazione si procede per tentativi.
Il caso più semplice prevede che la velocità
con cui N2O5 scompare sia proporzionale a [N2O5]. Cioè:
dC
−
che integrata nella forma:
−
dC
= kdt
C
fornisce
- lnC = kt + cost.
dt
= kC
(I)
Se Co è la concentrazione nota di N2O5 al tempo t = 0 la (I) diventa:
- ln Co = costante
Sostituendo in (I) e passando ai logaritmi decimali:
log
C
k
=−
t
Co
2.303
I dati sperimentali sono costituiti da una
serie di misure di C ai vari tempi t.
4
03/12/2011
C
Riportando in grafico log
contro il tempo t, otteniamo una retta con
Co
k
pendenza −
solo se la nostra ipotesi di partenza è esatta (cioè velocità
2.303
proporzionale a [N2O5]).
Possiamo quindi scrivere:
v=
d [N 2O5 ]
= k [N 2O5 ]
dt
K è quindi calcolato dal coefficiente angolare
della retta e prende il nome di costante di velocità.
Si definisce ordine di reazione il numero dato dalla somma degli esponenti con
cui le concentrazioni delle singole specie chimiche compaiono nell’equazione
della velocità di reazione.
Nell’esempio precedente, la reazione è del primo ordine. In generale se:
aA + bB → cC
e se la velocità può esprimersi
v = k[A]α [B]β
la reazione è di ordine α + β; è di ordine α rispetto ad A
e di ordine β rispetto a B. Generalmente α e β sono diversi da a e b.
L’ordine di una reazione non può essere quindi stabilito in base all’equazione
chimica, ma solo con misure di velocità.
Se a = α e b = β: reazione elementare.
VELOCITA’ DI REAZIONE E TEMPERATURA
Abbiamo visto che:
1) La velocità di reazione aumenta col numero di urti efficaci nell’unità di tempo.
2) Gli urti sono efficaci solo se l’energia cinetica è uguale all’energia di attivazione.
3) L’energia cinetica media è proporzionale alla temperatura.
Deve quindi esistere una relazione tra la velocità di reazione e la temperatura.
Misurando i valori di costante di velocità a varie temperature per una certa
reazione si trova che lnk è proporzionale a 1 / T:
ln k ∝ 1 / T
o anche:
k = A eB/T
dove A e B sono due costanti.
Arrhenius e van’t Hoff dimostrarono che B = − E a
R
Ea = E. di attivazione
R = costante dei gas
k = A e-Ea/RT
passando ai log. Decimali log k = −
dove:
Ea
1
⋅ + cos t.
2.303R T
riportando log k contro 1 / T si può determinare Ea.
5
03/12/2011
REAZIONI FOTOCHIMICHE E A CATENA
Sono reazioni che comportano la presenza di specie reattive contenenti elettroni
spaiati. Tendono ad essere molto rapide.
Una specie che contiene uno o più elettroni spaiati viene chiamata radicale libero.
hν
Cl2 → 2 Cl•
Es:
Cl • • Cl
Una volta formati, le reazioni chimiche in cui sono coinvolti sono rapidissime.
In molti casi la reazione porta ad un prodotto molecolare ed un altro radicale che
continua la reazione: reazioni a catena.
Queste hanno uno stadio di inizio in cui vengono prodotti i radicali:
Es:
formazione di H2O da H2 e O2:
H 2 + O2
Superf. calda
→
2 OH •
OH • + H2 → H2O + H •
stadio di inizio
reaz. di propagazione
H • + O2 → OH • + O •
reazioni collaterali
O • + H2 → OH • + H •
H• +
H • → H2
stadio di terminazione
CATALIZZATORI
Sono sostanze in grado di variare la velocità di una reazione, si ritrovano
chimicamente inalterati alla fine di essa, non compaiono nelle equazioni globali
di reazione e non alterano il valore della costante di equilibrio.
Catalizzatori positivi: aumentano la velocità di reazione
Catalizzatori negativi (o inibitori): diminuiscono la velocità
L’azione del catalizzatore positivo è quella di ridurre l’energia di attivazione di
una reazione, facendo seguire alla reazione una via diversa per la quale è
richiesta appunto una minore energia di attivazione.
Catalizzatori omogenei: se si trovano nella stessa fase dei reagenti
Catalizzatori eterogenei: se esistono in una fase diversa dai reagenti.
6
03/12/2011
CATALISI OMOGENEA
Un esempio è dato dall’ossidazione del diossido di zolfo con ossigeno a triossido
di zolfo per la preparazione dell’acido solforico.
La reazione:
2 SO2 + O2 → 2 SO3
(lenta)
è estremamente lenta, mentre diventa rapida per aggiunta di NO:
2 NO + O2 → 2 NO2
(veloce)
2 SO2 + 2 NO2 → 2 SO3 + 2 NO (veloce)
2 SO2 + O2 → 2 SO3
(veloce)
Il catalizzatore è quindi una specie che contemporaneamente reagente e
prodotto di reazione.
SO3 + H2O → H2SO4
CATALISI ETEROGENEA
Un catalizzatore eterogeneo funziona provocando una reazione sulla sua superficie.
Es:
catalizzatore solido in una reazione tra gas:
l’adsorbimento di una specie gassosa avviene di preferenza su centri attivi costituiti
da atomi insaturi per difetti reticolari.
Il processo di adsorbimento è spontaneo (∆G < 0) e poiché c’è un aumento
dell’ordine delle molecole del gas è ∆S < 0. Poiché:
∆G = ∆H - T∆S
deve anche essere ∆H < 0 cioè i fenomeni di adsorbimento sono favoriti a basse
temperature alle quali però è bassa v. di reazione. Si lavora quindi ad una certa T
ottimale dove è massima la resa.
Es:
3 H2(g) + N2(g) → 2 NH3(g)
Giocano un ruolo importante: a) il numero di centri attivi e b) la distanza tra essi
Inoltre i prodotti della reazione devono abbandonare la superficie lasciandola
inalterata.
7
03/12/2011
FINE
EQUILIBRI CHIMICI OMOGENEI
2 NO + 2 H2 ⇔ 2 H2O + N2
Raggiunto l’equilibrio 2 NO e 2 H2 formano 2 H2O e N2 e contemporaneamente,
con la stessa velocità, 2 H2O e N2 formano 2 NO e 2 H2. Le quantità delle singole
specie restano invariate nel tempo.
Se tutti i componenti di un equilibrio fanno parte di una stessa fase, l’equilibrio è
omogeneo, nel caso contrario è un equilibrio eterogeneo.
L’equilibrio è sempre di tipo dinamico.
Infatti perché due o più molecole reagiscano:
a) devono urtarsi.
b) gli urti devono essere urti efficaci cioè con energia e orientazione delle
molecole tali da consentire l’atto reattivo.
A temperatura constante la probabilità d’urto è tanto maggiore quanto
maggiore è la concentrazione delle specie reagenti. Tali concentrazioni
diminuiscono col procedere della reazione per cui la velocità di reazione verso
destra diminuisce col procedere della reazione.
8
03/12/2011
Contemporaneamente aumenta la concentrazione dei prodotti e quindi il numeri
di urti efficaci fra le loro molecole. La velocità di reazione verso sinistra aumenta.
Si raggiunge l’equilibrio quando le due velocità si eguagliano.
La conoscenza dell’equazione chimica non fornisce alcuna indicazione circa i
rapporti all’equilibrio (a variazione ultimata) fra i valori delle concentrazioni dei
reagenti e dei prodotti.
LEGGE DELLE MASSE
αA + βB ⇔ γC + δD
Siano tutte specie gassose a comportamento ideale
A temperatura costante all’equilibrio:
∆G = 0
consideriamo:
G°A, G°B, G°C e G°D le energie libere molari standard (P = 1 atm; 25°C)
GA, GB, GC e GD i valori dell’energia libera molare delle specie all’equilibrio con
pressioni parziali pari a:
pA, pB, pC e pD.
La variazione di G di 1mole di gas che passa da p1 a p2 a temperatura costante:
∆G = RT ln (p2 / p1)
Nel nostro caso per α moli di A a 25° C che passa da P = 1 atm (stato standard) a pA
(pressione di A all’equilibrio):
∆GA = αGA - αG°A = α RT ln (pA / po)
cioè:
αGA = αG°A + α RT ln (pA / po)
analogamente:
βGB = βG°B + β RT ln (pB / po)
γGC = γG°C + γ RT ln (pC / po)
δGD = δG°D + δ RT ln (pD / po)
ln A + lnB = ln(A.B)
ln A - lnB = ln(A/B)
αlnA = lnAα
Poiché
∆G = (ΣGprod) – (ΣGreag) ognuno moltiplicato per il relativo coeff. Stechiometrico:
∆G = γG°C + δG°D - αG°A - βG°B + RT ln
γ
∆G = ∆G° + RT ln
δ
pC ⋅ p D 1
⋅
p αA ⋅ p Bβ p o
σ
pCγ ⋅ p Dδ 1
⋅
p αA ⋅ p Bβ p o
σ
dove: σ = (γ + δ) – (α + β)
9
03/12/2011
All’equilibrio:
∆G = 0
quindi
γ
δ
∆G° = - RT ln pαc ⋅ p Dβ ⋅ 1
p A ⋅ p B po
σ
A temperatura costante ∆G° = costante per cui:
σ
p Cγ ⋅ p δD 1
⋅ = costante = Kp
p αA ⋅ p Bβ p o
Kp = costante termodinamica di equilibrio espressa in termini di pressioni
parziali.
Un procedimento analogo in termini di concentrazione, anziché di pressione,
ciascuna rapportata alla concentrazione standard Co = 1 molare, porta a:
[C ]γ ⋅ [D ]δ
[ A]α ⋅ [B]β
σ
1
⋅ = costante = KC
Co
Poiché Po = Co = 1, per i calcoli numerici si usa:
Kp =
pCγ ⋅ p δD
KC =
e
p αA ⋅ p Bβ
↑
eq. omog. gassoso
[C ]γ ⋅ [D ]δ
[A]α ⋅ [B]β
↑
eq. omog. gassoso o in soluzione
Legge delle masse:
A temperatura costante in un equilibrio omogeneo è costante il valore del
rapporto fra il prodotto dei valori delle concentrazioni delle specie formate
presenti all’equilibrio, ciascuno elevato al coefficiente stechiometrico proprio, e
l’analogo prodotto relativo alle specie reagenti all’equilibrio.
Quindi vale:
∆G°(p ,T) = - RT ln Kp
0
∆G°(C
0
0
,T)
= - RT ln KC
dove ∆G° va calcolato in base ai valori di ∆G°f delle specie nello stesso stato (gas
o soluzione) in cui sono presenti all’equilibrio.
Poiché PV = n RT
P=
n
RT
V
P = C RT
Dove C = concentrazione molare.
Per cui PA = CA RT
Kp =
;
[C ] [RT ] ⋅ [D] [RT ]
[A]α [RT ]α ⋅ [B ]β [RT ]β
γ
γ
δ
δ
PB = CB RT
=
; ecc.
[C ] ⋅ [D ] ⋅ (RT ) (RT )
[A]α ⋅ [B ]β (RT )α (RT )β
γ
δ
γ
δ
cioè:
Kp = KC (RT)σ
10
03/12/2011
Kp = KC (RT)σ
Kp = KC quando σ = 0
da cui
Cioè quando non si ha variazione di numero di moli gassose.
Es:
2 HI ⇔ H2 + I2
σ=0
2 CO + O2 ⇔ 2 CO2
σ = -1
DIPENDENZA DI K DALLA TEMPERATURA
∆G° = ∆H° - T∆S°
∆G° = - RT ln Kp
∆H ° ∆S °
ln K p = −
+
RT
R
Per piccole variazioni di T ∆H° e ∆S° sono costanti e ln Kp riportato contro 1 /T
ha andamento lineare.
Derivando rispetto a T:
d ln K p ∆H °
=
dT
RT 2
equazione di van’t Hoff
Per ∆H > o < di zero Kp aumenta o diminuisce con l’aumentare della temperatura
(un aumento di Kp vuol dire che la reazione si sposta verso i prodotti).
Reazioni endotermiche (∆H > 0) sono favorite dall’aumento della temperatura
mentre le reazioni esotermiche (∆H < 0) sono ostacolate da tale aumento.
Analogamente:
d ln K C ∆H °
=
dT
RT 2
11
03/12/2011
INFLUENZA DELLA PRESSIONE SU UN EQUILIBRIO GASSOSO
P ha influenza solo sulla composizione di un equilibrio gassoso, ma non sul
valore di K, che dipende solo da ∆G°, che a sua volta dipende solo da T (∆G° è
definito a Po = 1 atm).
Es:
PCl5(g) ⇔ PCl3(g) + Cl2(g)
Kp =
PPCl3 ⋅ PCl2
reazione con aumento del N° di moli
Se: χ = frazione molare; P = pressione totale; pA = P χA
PPCl5
Kp =
Pχ PCl3 ⋅ Pχ Cl2
Pχ PCl5
=P
χ PCl3 ⋅ χ Cl2
χ PCl5
Se P aumenta, poiché Kp deve restare costante, deve diminuire il rapporto :
(χ PCl3 ⋅ χ Cl2 ) e l’equilibrio si sposta a sinistra.
χ PCl5
Se P diminuisce l’equilibrio si sposta a destra.
2 HI(g) ⇔ H2(g) + I2(g)
Es:
Kp =
PH 2 ⋅ PI 2
=
2
HI
P
Pχ H 2 ⋅ Pχ I 2
P χ
2
2
HI
=
χ H2 ⋅ χ I2
2
χ HI
Non compare la P. La sua variazione non altera la composizione dell’equilibrio.
Es:
N2 + 3 H2 ⇔ 2 NH3
Kp =
reazione con diminuzione delle moli gassose
2
P 2 χ NH
3
χ NH 3
1
⋅
2
P χ N 2 ⋅ χ H3 2
2
Pχ N 2 ⋅ P 3 χ H3 2
=
Pertanto:
l’aumento di pressione favorisce le reazioni che avvengono con diminuzione del
volume, ostacola quelle che avvengono con aumento di volume e non ha
influenza su quelle che avvengono senza variazione di volume.
N.B.
particolare attenzione agli equilibri eterogenei:
C(s) + O2(g) ⇔ CO2(g)
reazione senza variazione di volume
12
03/12/2011
Kp e KC : cosa indicano:
2 H2 + O2 ⇔ 2 H2O
KC =
K = 9.1⋅1080 a 25° C
[H 2 O]2
= 9.1 ⋅ 1080
[H 2 ]2 [O2 ]
N2 + O2 ⇔ 2 NO
K = 4.8⋅10-31
K molto grande = la reazione procede quasi completamente.
K≅1
le concentrazioni dei prodotti e dei reagenti all’equilibrio sono simili.
K molto piccolo = a stento si formano prodotti.
Principio di Le Châtelier:
Se un’influenza esterna turba un equilibrio, il sistema risponde nella direzione
contraria all’influenza disturbatrice, in modo da ristabilire l’equilibrio.
1) aggiungendo o sottraendo un reagente o un prodotto.
2) variazione di P.
3) variazione di T.
FINE
13
03/12/2011
DISSOCIAZIONE TERMICA
Viene indicato così il processo, reversibile con T, per il quale all’aumentare di T una
molecola si spezza in due o più molecole più piccole.
Es:
PCl5
2 HI
N2O4
⇔ PCl3 + Cl2
⇔ H 2 + I2
⇔ 2 NO2
Si indica col nome di grado di dissociazione (α) la frazione di mole che, all’equilibrio,
ha subito dissociazione.
0 ≤ α ≤ 1 α = (moli dissociate/moli iniziali) ; oppure 0% e 100% di dissociazione
Se il gas è costituito da n moli e ν è il numero di particelle provenienti dallo
spezzamento di una molecola, il numero totale di moli all’equilibrio è:
ntot. = (n - αn) + αnν = n (1 - α + αν) = n [1 + α(ν - 1)]
[1 + α(ν - 1)] = fattore di dissociazione
n - αn = numero di moli rimaste indissociate
αn
= moli dissociate
α nν = numero di moli delle specie provenienti dalla dissociazione delle αn moli
COSTANTE DI EQUILIBRIO E GRADO DI DISSOCIAZIONE
E’ possibile esprimere K in funzione di α.
Es:
PCl5
1 mole contenuta in volume V e alla temperatura T.
α = grado di dissociazione
PCl5 ⇔ PCl3 + Cl2
(1- α)
α
α
← moli all’equilibrio
[PCl5 ] = (1 − α )
[PCl3 ] = [Cl 2 ] = α
V
V
Se C è la concentrazione iniziale di PCl5:
[PCl5 ] = C (1 − α )
[PCl3 ][Cl2 ] = α = α C
[PCl5 ] V (1 − α ) 1 − α
2
KC =
[PCl3 ] = [Cl2 ] = Cα
e
2
Per Kp:
all’equilibrio ntot = 1 - α + α + α = 1 + α
le frazioni molari n / ntot e le pressioni parziali diventano:
14
03/12/2011
PCl2 = PPCl3 = P ⋅
α
PPCl5 = P
(1 + α )
(1 − α )
(1 + α )
2
Kp =
PPCl3 ⋅ PCl2
PPCl5
P ⋅α
(1 + α )
2
= α P
=
1−α ) 1−α 2
(
P
(1 + α )
La legge delle masse è valida per qualunque equilibrio omogeneo comunque
esso sia raggiunto:
2 NO + 2 H2 ⇔ N2 + 2 H2O
Es:
Può instaurarsi unendo quantità a piacere di:
1)
2)
3)
4)
NO
N2
NO
NO
+
+
+
+
H2
H2O
H2 + H2O
H2 + N 2
5)
6)
7)
H2 + N2 + H2O
NO + N2 + H2O
NO + H2 + N2 + H2O
In tutti i casi le specie unite reagiscono fra loro per formare la o le specie
mancanti e la Kc:
KC =
[N 2 ][H 2 O]2
[NO]2 [H 2 ]2
Avrà sempre lo stesso valore (a parità di temperatura).
2 NO + 2 H2 ⇔ N2 + 2 H2O se unisco tra loro a due a due:
NO
H2O
NO
H2
+
+
+
+
N2
H2
H2O
N2
Non possono condurre all’equilibrio considerato
15
03/12/2011
EQUILIBRI ETEROGENEI
Sono equilibri relativi a reazioni tra specie chimiche in fasi diverse.
Es:
A(s) ⇔ B(g) + C(g)
(a)
Per applicare la legge delle masse dobbiamo fare alcune considerazioni:
Anche le fasi condensate hanno una loro pressione di vapore che è costante a
temperatura costante; quindi nella fase gassosa (a) esisterà l’equilibrio
omogeneo:
PB ( g ) ⋅ PC ( g )
K 'p =
A(g) ⇔ B(g) + C(g)
PA( g )
Poiché in (a) è presente A solido il valore di PA(g) è il valore di una pressione di
vapore saturo, che essendo costante può essere inglobato in Kp:
Kp = PB(g) ⋅ PC(g)
Cioè a questi equilibri può applicarsi la legge delle masse non considerando nella
formula di K le specie solide.
Lo studio di un sistema eterogeneo all’equilibrio consiste nel determinare il
numero di parametri intensivi che intervengono nell’equilibrio e che possono
essere variati senza alterare l’equilibrio cioè senza alterare il numero delle fasi
presenti all’equilibrio.
Un sistema eterogeneo è costituito da fasi cioè da porzioni di materia fisicamente
e chimicamente omogenee.
Si definisce grado di libertà o varianza di un sistema il numero di parametri
intensivi (T, P, concentrazione) i cui singoli valori possono essere variati a piacere e
indipendentemente l’uno dall’altro senza alterare l’equilibrio del sistema cioè
senza alterare il numero delle fasi presenti.
Il sistema ha 2 gradi di libertà
cioè è bivariante (punto A)
Il sistema ha un solo grado di libertà
cioè è monovariante (punto A)
16
03/12/2011
Nel punto triplo il sistema è zero variante: non
è possibile variare alcun parametro senza
provocare la scomparsa di almeno una fase.
REGOLA DELLE FASI
Sistema in equilibrio costituito da f fasi e C specie chimiche indipendenti e le uniche
variabili sono T e P, il valore della varianza è dato da:
V=C+2–f
La varianza di un sistema costituito da C componenti indipendenti comunque
distribuiti tra le f fasi e per il quale le uniche variabili sono T e P, è data dalla
differenza tra il numero dei componenti aumentato
di due ed il numero delle fasi.
punto P = varianza = 2
punti P’ = varianza = 1
punti B, C, D = varianza = 0
Nei sistemi isotermi (T = costante) o isobari
(P = costante) un solo parametro fisico è variabile.
V=C+1–f
FINE
17
03/12/2011
SOLUZIONI ELETTROLITICHE
Elettroliti: specie chimiche che in soluzione si scindono totalmente o parzialmente
in ioni. (cationi e anioni). – Dissociazione –
a) Elettrolita forte: se totalmente dissociato in ioni.
b) Elettrolita debole: se parte di esso è presente in soluzione sotto forma di
molecole indissociate.
Fanno parte di a) tutti i sali, alcuni acidi e alcune basi. In b) troviamo solo acidi e basi
(deboli) e alcuni sali di Cd.
Es:
aq
a)
KI → K+aq + I-aq
b)
HCN ⇔ H3O+ (aq) + CN- (aq)
aq
Il grado di dissociazione, α, definito come la frazione di moli che si dissocia, è α = 1
per gli elettroliti forti. Nel caso di elettroliti deboli:
CA ⇔ C+ + Ac
0
0
c – cα cα cα
legge di diluizione di Ostwald
t=0
equilibrio
[A ][C ] = α
−
K=
+
[ AC ]
2
c
1−α
Applicazione della legge delle masse a equilibrio di dissociaz. di elettrolita debole.
Più rigorosamente dovrebbe scriversi:
CA( sol ) + ( m + n )H 2O ⇔ A(−H 2O )m + C(+H 2 O )m
K' =
[A
−
( H 2 O )m
][C
+
( H 2 O )n
m+ n
]
[CAsol ][H 2O ]
Per soluzioni acquose diluite di un elettrolita debole il numero di moli d’acqua
impegnate nell’idratazione è trascurabile rispetto alle moli totali del solvente, per
cui:
A− C +
K=
[ ][ ]
[CA ]
cioè è solo funzione della concentrazione molare degli ioni e delle molecole
indissociate.
Ogni soluzione elettrolitica possiede una concentrazione analitica e più
concentrazioni ioniche: Es:
CaCl2
10-3 M
CaCl2 → Ca2+ + 2 Cl-
[CaCl2] = 10-3 M
[Ca2+] = 10-3 M
[Cl-]
= 2 x 10-3 M
Questo perché alcune volte interessa solo la concentrazione di una determinata
specie ionica.
Es:
Ag+ + ClLa soluzione è 10-3 M in CaCl2
Le concentrazioni ioniche
:
:
18
03/12/2011
ATTIVITA’ E COEFFICIENTE DI ATTIVITA’
Per poter utilizzare le relazioni ottenute per le soluzioni ideali anche per le
soluzioni reali, dove esistono interazioni tra le particelle del soluto, si deve
considerare l’attività.
TEORIA DEGLI ELETTROLITI FORTI
Al crescere della concentrazione di una soluzione è sempre più probabile che gli
ioni si dispongano non casualmente: cioè nella nube ionica è più probabile trovare
ioni di segno opposto. Il sistema è dinamico.
Se si effettua una misura di conducibilità il valore
trovato per alte concentrazioni risulta minore di quello
teoricamente calcolabile in quanto lo ione viene
rallentato dalla nube ionica (gli ioni sono meno attivi)
che tende a muoversi in senso opposto.
Anche la formazione di coppie ioniche riduce la conducibilità.
Lo studio di soluzioni elettrolitiche concentrate è complesso.
Debye e Hückel proposero per il calcolo del coefficiente di attività medio di un
elettrolita in soluzione:
log f ± = −0.51z + z − µ
log f ± = −0.51z + z − µ
µ = ½ Σi Ci zi2
valida a 25° C e per concentrazioni < 0.01 M
f± = coefficiente di attività medio
z+,z- = numero delle cariche del catione e dell’anione
µ = forza ionica definita come :
dove Ci = concentrazione molare dello ione iesimo presente in soluzione.
0.51 = fattore che comprende: temperatura (25° C), carica elettrone, costante
dielettrica dell’acqua, costante di Boltzmann, numero di Avogadro, passaggio da
ln naturali a log decimali.
Per soluzioni di elettroliti più che 1 – 1 valenti e per soluzioni più concentrate di
0.01 M l’equazione diventa sempre meno valida.
In questi casi i valori dei coefficienti di attività vanno calcolati sperimentalmente
mediante, ad esempio, misure di tensione di vapore, abbassamento crioscopico, o
da misura di forza elettromotrice.
19
03/12/2011
Es:
Coefficiente di attività dell’acetone?
Fraz. mol. acet. χ = 0.710
e
P°acet. = 344.5 a 35°C
In soluzione di cloroformio.
Pacet. = 230.8 Torr. A 35° C
valore misurato sperimentalmente
Per una soluzione ideale a 35° C la pressione di vapore dell’acetone sarebbe:
P = χacet. ⋅ P°acet. = 0.710 ⋅ 344.5 = 244.6 Torr
Da cui
f± =
230.8
= 0.944
244.6
Attività e coefficiente di attività sono validi nel campo dei gas, dei liquidi e
dei solidi
ACIDI E BASI
Definizione secondo Lewis
Acido: specie chimica in grado di accettare una o più coppie elettroniche
(lone pairs).
Base: specie chimica in grado di fornire uno o più lone pairs.
La reazione acido – base consiste quindi nella formazione di uno o più legami
dativi.
F3B
Acido
+
:NH3
F3B NH3
base
addotto
Le basi secondo Brönsted rientrano fra le basi di Lewis.
Es:
NH3
addizione un H+ per mezzo del lone pair.
Meno evidente è l’inquadramento degli acidi:
Es:
HCl e H2SO4
non hanno la possibilità di accettare lone pairs. Ma
fornendo H+, che è un acido di Lewis, questi possono essere fatti rientrare
indirettamente fra gli acidi di Lewis. Egli li chiamò acidi secondari.
H+ è un acido primario.
20
03/12/2011
FORZA DEGLI ACIDI E DELLE BASI
Faremo riferimento alla definizione di Brönsted e definiremo un sistema acido –
base come costituito da una specie in grado di accettare protoni (base) a da una in
grado di fornirli (acido).
Di due acidi il più forte è quello che più facilmente dona protoni ad una stessa
base.
Tra due basi è più forte quella che più facilmente accetta protoni da uno stesso
acido.
Per tutti gli acidi solubili in H2O si può determinare il valore della costante di
dissociazione e riportarli in una scala per rappresentare la forza degli acidi (rispetto
alla base H2O).
CH3COOH + H2O ⇔ CH3COO- + H3O+
Es:
(C1)
HCOOH
+ H2O ⇔ HCOO-
(C2)
C1 e C2 note e piccole
attività = concentrazione
[H O ][CH COO ] = α C
+
K a .acet . =
K a . form. =
+ H3O+
3
−
3
[CH 3COOH ]
2
1
1
1 − α1
α 22 C2
1 −α2
C1 e C2 sono note
α1 e α2 si possono determinare (es. con misure di conducibilità)
si ottiene
Ka.for. > Ka.acet.
Ka.ac. = 1.8⋅10-5
Ka.for. = 1.8⋅10-4
cioè la reazione è più spostata verso i prodotti per l’acido
formico.
L’acido formico è più forte dell’acido acetico.
21
03/12/2011
Gli acidi forti sono completamente dissociati in soluzione acquosa poco
concentrata (< 0.1 M).
Aumentando la concentrazione aumenta la percentuale di molecole indissociate.
Le considerazioni viste sin qui per gli acidi possono essere fatte anche per le basi.
Scala forza delle basi. Vedi tabella
Es:
per l’acido Arsenico:
H3AsO4 + H2O ⇔ H2AsO4H2AsO4- + H2O ⇔ HAsO42HAsO42- + H2O ⇔ AsO43-
K1 =
K2
[H AsO ][H O ] = 5.0 ⋅ 10
2
−
4
+
3
[H 3 AsO4 ]
[HAsO ][H O ] = 1.8 ⋅ 10
=
[H AsO ]
[AsO ][H O ] = 3.9 ⋅ 10
=
[HAsO ]
2−
4
+
3
−
4
2
K3
3−
4
+
3
2−
4
+ H3O+
+ H3O+
+ H3O+
−3
−7
−12
E’ un equilibrio multiplo e le
espressioni per K1, K2 e K3 sono
sempre contemporaneamente
soddisfatte in ogni soluzione di acido
arsenico
CH3COOH + H2O ⇔ CH3COO- + H3O+
HCl +
H2O → Cl- + H3O+
Quanto più un acido è forte tanto più è debole la sua base coniugata
(analogo per le basi).
FATTORI STRUTTURALI
la forza acida aumenta
da 1.8⋅10-5
a
2.0⋅10-1
a causa dell’effetto induttivo esercitato dai Cl più elettronegativi dell’H.
Analogamente:
forza dell’acido in aumento
22
03/12/2011
Altri fattori strutturali sono legati a :
1) fenomeni di risonanza
Maggiore è il numero di forme di risonanza di una specie più essa è stabile.
2) interazioni con il solvente
Legami ad idrogeno più forti con gli ioni che derivano dalla dissociazione.
3) separazione di carica
Es:
H2SO4 → HSO4HSO4- → SO42-
+ H+
+ H+
È più difficile strappare il secondo protone perché lo si deve allontanare da una
specie carica negativamente.
ELETTROLITI ANFOTERI
Si comportano da acidi in presenza di basi e da basi in presenza di acidi.
Es:
H2O
acido
base
NH3 + H2O ⇔ NH4+ + OHbase
acido
CH3COOH + H2O ⇔ CH3COO- + H3O+
Altri elettroliti anfoteri sono:
+ 2 H3O+ → Zn2+ + 4 H2O
Zn(OH)2
Al(OH)3
Cr(OH)3
Be(OH)2
+ 2 OH- → ZnO22- + 2 H2O
Sn(OH)2
As(OH)3
ecc.
Sono generalmente idrossidi poco solubili.
Un generico composto XOH può dissociarsi:
X+ + OH-
base
XO- + H+
acido
XOH
L’una o l’altra dissociazione dipende dalla competizione della forza dei due
legami X – O e O – H. Dipende dall’elettronegatività di X:
Se X >> di H
lo spostamento di carica da X a O è modesto:
XOH → XO- + H+
si comporta sempre da acido
Se l’elettronegatività di X è molto inferiore a quella di H è l’O che addensa su di
se la carica negativa. XOH → X+ + OHsi comporta da base
23
03/12/2011
Se il valore dell’elettronegatività di X è paragonabile a quella di H l’elettrolita
risulta anfotero.
H2O in cui O è legato a due H è l’anfotero per eccellenza.
FINE
24
03/12/2011
PRODOTTO IONICO DELL’ACQUA
L’H2O pura presenta una piccola conducibilità. Ciò vuol dire che vi sono ioni.
Essendo un anfotero:
2 H2O(l) ⇔ H3O+(aq) + OH-(aq)
Kw =
a
H 3O +
a
⋅a
OH −
poiché aH2O = 1
2
H 2O
Kw = aH3O+ ⋅ aOH-
e può essere determinato termodinamicamente
∆G° = [∆G°f(H3O+) + ∆G°f(OH-) ] - 2 ∆G°f(H2O)
sostituendo i valori tabulati
∆G° = -237.19 – 157.30 + 474.38 = 79890 J poiché
∆G° = - RT ln K
79890 = - 8.3144 ⋅ 298.16 ⋅ 2.303 log Kw
da cui
Kw = 1.00⋅10-14
Per cui a 25° C:
Kw = aH3O+ ⋅ aOH- = 1.00⋅10-14
H3O+ e OH- sono in uguale numero per cui:
aH3O+ = aOH- = 1.00 ⋅ 10 −14 = 1.00⋅10-7
i valori di aH3O+ e aOH- sono piccoli per cui possiamo sostituire le concentrazioni:
Kw = [H3O+][OH-]= 1.00⋅10-14
[H3O+] = [OH-]= 1.00⋅10-7 M
Kw = prodotto ionico dell’acqua. E’ una costante di equilibrio, per cui è valida
qualunque sia la provenienza degli ioni H3O+ e OH-.
A 25 ° C
[H3O+] > 10-7 M
[H3O+] = 10-7 M
[H3O+] < 10-7 M
soluzione acida
soluzione neutra
soluzione basica
La [H3O+] in soluzione, viene espressa come: (log decimali)
25
03/12/2011
pH = colog aH3O+ = log (1/ aH3O+) = - log aH3O+
p = - log
pH = colog [H3O+] = log (1/ [H3O+]) = -log [H3O+]
Per cui a 25° C:
Soluzione acida:
pH < 7
(aH3O+ > 10-7 M ; aOH- < 10-7 M)
Soluzione neutra:
pH = 7
(aH3O+ = aOH- = 10-7 M)
Soluzione basica:
pH > 7
(aH3O+ < 10-7 M ; aOH- > 10-7 M)
p Kw = pH + pOH = 14
il pH = 0 per aH3O+ = 1M
e
pH = 14 per aOH- = 1M
piò assumere valori negativi e superiori a 14, ma si perde significato.
pH DI SOLUZIONE DI ACIDO O BASE FORTE
Per soluzioni diluitissime: [H3O+] < 10-6 M
E’ importante anche il contributo a [H3O+] derivante dalla dissociazione dell’H2O.
[H3O+]ac = ioni provenienti dell’acido
[H3O+]aq = ioni provenienti dall’acqua
X = diminuzione di [H3O+]aq e [OH-]aq dovuta allo spostamento dell’equilibrio della
dissociazione dell’H2O in seguito all’aggiunta di [H3O+]ac .
Kw = ([H3O+]ac + [H3O+]aq - X)([OH-]aq - X) = 10-14
Dove abbiamo:
[H3O+]aq = [OH-]aq = 10-7
;
[H3O+]ac = conc. analitica acido
Si ricava quindi X che si sottrae dalla somma [H3O+]ac + [H3O+]aq
26
03/12/2011
Es:
in 1 l di H2O si aggiungono 10-7 moli di HCl.
pH?
Kw = (10-7 + 10-7 – X)(10-7 – X) = 10-14
X = 0.38⋅10-7
[H3O+] = 10-7 + 10-7 - 0.38⋅10-7 = 1.62⋅10-7
pH = 6.79
Per soluzioni diluite: [H3O+]ac
tra 10-5 e 10-3
Gli H3O+ provenienti dall’H2O sono trascurabili rispetto a quelli dell’acido.
Il calcolo del pH è immediato:
pH = colog [acido]
Es:
[HCl] = 10-3
pH = colog 10-3 = 3
[NaOH] = 10-4
pH = colog Kw/[OH-] = colog 10-14/10-4 = 10
Per soluzioni concentrate: 10-1 ÷ 2 M si deve conoscere il coefficiente di attività:
Es:
(f± = 0.795)
pH di soluzione di HCl 0.1 M
pH = colog (0.1⋅0.795) = 1.1
pH DI SOLUZIONE DI ACIDO DEBOLE (O BASE DEBOLE)
Soluzioni diluitissime:
< 10-4 M
Caso complicato: si ha bisogno di un sistema di equazioni
Soluzioni diluite: 10-1 ÷ 10-3 M
Si devono conoscere concentrazione dell’acido e costante di dissociazione.
Es:
CH3COOH + H2O ⇔ CH3COO- + H3O+
[CH COO ][H O ] = 1.8 ⋅10
−
K 25°C =
3
+
3
[CH 3COOH ]
−5
concentrazione analitica dell’acido :
si è trascurata la frazione dell’acido dissociata.
Non sempre lecita come approssimazione. Può essere fatta solo per soluzioni
non troppo diluite ( > 10-2 M) di acidi con Ka < 10-3.
27
03/12/2011
Nel nostro esempio se conc. acido = 10-2 M
Ka = 1.8⋅10-5
poiché [CH3COO-] = [H3O+]:
1.8 ⋅ 10
−5
[H
]
2
O+
=
[CH 3COOH ]
3
da cui [H3O+] = 4.243⋅10-4 M
pH = 3.37
Quindi il pH della soluzione di un acido debole (Ka ≤ 10-3) non troppo diluito;
cioè di concentrazioni di Ca tale che sia lecito trascurare [H3O+] provenienti
dall’H2O, si calcola da:
Kac ≅ [H3O+]2 / Ca
pH = colog K ac C a
cioè
Se l’acido è poliprotico Kac si riferisce alla prima dissociazione.
per una base debole:
pOH = colog K baseCb
Per soluzioni concentrate: 10-1 ÷ 2 M. Vale quanto detto per le soluzioni
diluite, ma si devono considerare le attività.
FINE
28
03/12/2011
INDICATORI DI pH
Sono acidi o basi deboli che sciolte in soluzione acquistano colorazioni diverse a
seconda del pH acido o basico della soluzione.
Es:
indicatore acido RH.
RH + H2O ⇔ R- + H3O+
rosso
giallo
[R ][H O ]
−
K=
+
3
oppure
[RH ]
(I)
]
[H O ] = K [[RH
R ]
+
3
−
(II)
la concentrazione di H3O+ non varia con l’aggiunta di un indicatore.
Quindi [H3O+] = concentrazione nella soluzione prima dell’aggiunta.
[H3O+] determina il colore in base all’equilibrio (I)
soluzione basica
soluzione acida
da: (II) :
equilibrio (I) spostato a destra: colore giallo
equilibrio (II) spostato a sinistra: colore rosso
pH = pK + log [R-]/[RH]
quando [R-] = [RH]
pH = pK
soluzione color arancio.
Cioè l’indicatore è al punto di viraggio. Si conosce il pH della soluzione.
Se la soluzione appare rossa pH < pK. Non si conosce il pH della soluzione.
Se la soluzione appare gialla pH > pK. Non si conosce il pH della soluzione cioè la
soluzione è basica rispetto all’indicatore usato.
Ogni indicatore indica solo il valore pH = pK per cui occorre una serie di indicatori
con diversi valori di pK per misurare il pH di una soluzione con gli indicatori.
Trovano largo uso nelle titolazioni Acido – base e per misure grossolane del pH.
In pratica più che punto di viraggio (pH = pK) difficilmente apprezzabile ad occhio
nudo, si considera il campo di viraggio: cioè l’intervallo di pH che separa le due
colorazioni estreme distinguibili ad occhio nudo.
Se RH ed R- sono le due forme colorate:
quando [RH] ≅ 10 [R- ]
rossa
[R- ] ≅ 10 [RH]
gialla
poiché [H 3O + ] = K
[H3O+] = K⋅10
e
[RH ]
[R ]
−
avremo le due condizioni:
[H3O+] = K⋅10-1
cui corrisponde
∆pH = pK ± 1
29
03/12/2011
EQUILIBRI IONICI IN SOLUZIONE
Idrolisi salina, sistemi tampone, prodotto di solubilità.
Abbiamo già visto che:
1) nell’acqua e nelle soluzioni acquose esiste l’equilibrio:
2H2O ⇔ H3O+ + OHKw (25° C) = aH3O+ ⋅ aOH- = 1.00⋅10-14
2) un equilibrio A + B ⇔ C + D si può realizzare partendo da A + B o da C + D in
rapporti qualsiasi e a temperatura costante.
[C ][D ]
K=
[ A][B]
3) in soluzione acquosa acidi e basi forti sono totalmente dissociati, mentre acidi
e basi deboli danno luogo ad equilibrio di dissociazione acido – base.
4) in H2O i sali (elettroliti forti) sono totalmente dissociati.
5) Se in soluzione acquosa coesistono più equilibri con almeno uno ione comune,
la variazione di concentrazione di una specie che fa parte di uno di questi
equilibri, si ripercuote sulla composizione degli altri equilibri, anche se in essi non
compare tale specie.
Es:
CH3COOH + H2O ⇔ H3O+ + CH3COO2 H2O ⇔ H3O+ + OH-
H3O+
in comune ai due equilibri
[H O ][CH COO ]
+
K ac =
3
−
3
[CH 3COOH ]
Kw = [H3O+][OH-]
Se aggiungiamo alla soluzione ioni OH- (es. NaOH) la dissociazione dell’acqua
retrocede per cui diminuisce [H3O+] altro acido si dissocia per produrre H3O+ in
modo che restino costanti Kac e Kw .
Dunque: OH- aggiunti spostano l’equilibrio dell’acido acetico pur non facendone
parte.
IDROLISI SALINA
Si indica con questo termine la reazione di un soluto col solvente acqua. In
particolare il soluto deve essere un sale.
Si distinguono quattro casi:
30
03/12/2011
a)
Sali che provengono dalla reazione tra un acido forte e una base debole
es. NH4Cl
In questa soluzione abbiamo gli ioni:
NH4+ Cl- H3O+ OH-
H3O+ e Cl- non possono reagire
NH4 + e OH- invece reagiscono e per il punto 2):
NH4 + + OH- ⇔ NH4OH
Questa reazione consuma OH- e poiché a 25° C:
[H3O+][OH-] = 1⋅10-14 dell’acqua si dissocia per mantenere costante il Kw.
Esistono quindi due equilibri
NH4 + + OH- ⇔ NH4OH
2 H2O ⇔ OH- + H3O+
eq. multiplo
la cui somma:
NH4 + + 2 H2O ⇔ NH4OH + H3O+
Si chiama reazione di idrolisi dello ione NH4 + e mostra che il pH è < 7.
La soluzione di un sale descritto in a) é acida.
b) Sale proveniente da acido debole e base forte.
Es. CH3COONa
In soluzione abbiamo:
CH3COO-
Na+
H3O+
OH-
Na+ e OH- non reagiscono (base forte)
CH3COOH invece è un acido debole
CH3COO- + H3O+ ⇔
CH3COOH + H2O
Che consuma H3O+. Poiché Kw deve restare costante H2O si dissocia:
CH3COO- + H3O+ ⇔ CH3COOH + H2O
2 H2O ⇔ H3O+ + OHla cui somma.
equilibrio multiplo
CH3COO- + H2O ⇔ CH3COOH + OHRappresenta la reazione di idrolisi di CH3COO- e mostra che il pH è > 7.
La soluzione di un sale descritto in b) è basica.
31
03/12/2011
c) Sale proveniente da acido forte e base forte:
Es:
NaCl :
Na+ Cl- H3O+ OHSono ioni corrispondenti a elettroliti forti. Il sale non subisce idrolisi.
La soluzione risulta neutra.
d) sale proveniente da acido debole e base debole.
Es: CH3COONH4
CH3COO- NH4+ H3O+
OH-
Sia CH3COO- che NH4+ idrolizzano.
Si possono verificare tre casi:
1) Se l’acido è più debole della base si formano più molecole indissociate di acido
per cui vengono consumati più ioni H3O+ e la soluzione risulta basica.
2) Se la base è più debole dell’acido, la soluzione risulta acida.
3) Se acido e base sono ugualmente deboli (Ka ≅ Kb) la soluzione risulta neutra.
Le reazioni di idrolisi sono reazioni acido – base.
COSTANTE DI IDROLISI
Es:
NaCN
proveniente da acido debole (HCN) e base forte (NaOH)
Na+CN- + H2O ⇔ HCN + Na+ + OHCioè:
CN- + H2O ⇔ HCN + OHKi =
[HCN ][OH
−
[CN ]
]
= reazione di idrolisi
[H2O] = costante
−
Ki = costante di idrolisi
Poiché
Kw = [H3O+][OH-]
e
Ki =
[HCN ]Kw
Kw
[H O ][CN ] = K
+
−
3
a
dove Ka = costante di dissociazione dell’acido debole.
Per un sale proveniente da acido forte e base debole:
Ki = Kw / Kb
Se entrambi i sono costituenti sono deboli: Ki = Kw / Ka⋅Kb
32
03/12/2011
pH DI IDROLISI
Es:
NaCN
Ki =
Cs ≅ 0.1 M
di concentrazione
[HCN ][OH
[CN ]
−
]
che idrolizza:
CN- + H2O ⇔ HCN + OH-
−
[OH-] proveniente da H2O è trascurabile rispetto a quelli provenienti dall’idrolisi:
[HCN] = [OH-]
[OH ]
[CN ]
− 2
Ki =
[OH ] =
−
da cui
−
[
K i CN −
]
La reazione di idrolisi è generalmente spostata verso sinistra per cui:
[CN-] ≅ [NaCN] = Cs
− 2
Ki ≅
[OH ]
[OH ] ≅
−
Cs
K iCs
da cui si calcola il pOH e il pH.
Analogamente: (per l’idrolisi di sali provenienti da acido forte e base debole)
Ki ≅
[H
3O
]
+ 2
[H O ] ≅
+
3
Cs
K iCs
Le relazioni viste possono anche scriversi:
1) per sale proveniente da acido forte e base debole:
[H O ] =
+
3
K i Cs =
Kw
Cs
Kb
2) sale proveniente da acido debole e base forte:
[OH ] =
−
K i Cs =
Kw
Cs
Ka
33
03/12/2011
EQUILIBRI IN SOLUZIONE COSTITUITA DA ACIDO DEBOLE
E DA UN SUO SALE CON BASE FORTE
CH3COOH + H2O ⇔ CH3COO- + H3O+
Es:
CH3COO- + H2O ⇔ CH3COOH + OHDate da CH3COOH e CH3COONa
[CH COO ][H O ]
−
Ka =
+
3
Ki =
3
[CH 3COOH ]
[CH 3COOH ][OH − ]
[CH COO ]
−
3
Acido e sale sono contemporaneamente nella stessa soluzione per cui
[CH3COOH] è uguale nelle due espressioni così come [CH3COO-]. E poiché tra
[H3O+] e [OH-] vale Kw = [H3O+][OH-]
Le due espressioni sono equivalenti per rappresentare i due equilibri.
Dalla prima abbiamo:
COOH ]
[H O ] = K [[CH
CH COO ]
+
3
3
−
a
3
indicando con
CA = conc. analitica dell’acido
CS = conc. analitica del sale
[H O ] = K
+
3
a
CA
CS
pH = pK a + log
CS
CA
Nel caso invece di soluzione costituita da base debole e da un suo sale con acido
forte, si ha:
[OH − ] = K b CC B
S
C
pOH = pK b + log S
CB
Sono note come equazioni di Henderson e Hasselbach.
Le soluzioni tampone sono soluzioni acquose di opportune specie chimiche che,
per aggiunta o sottrazione di ioni H3O+ o OH- mantengono il loro pH praticamente
invariato.
34
03/12/2011
Es:
Acido acetico + Acetato di Sodio.
Se le concentrazioni di acido [CH3COOH] e della sua base coniugata [CH3COO-]
vengono rese uguali (o quasi). La soluzione acquista caratteristiche di soluzioni
tampone.
Meccanismo di funzionamento:
1) aggiungiamo 1 milliequivalente di HCl a 1000 cc di H2O.
2) aggiungiamo 1 millieq. di HCl a 1000 cc si soluzione 0.1 M di CH3COOH
e 0.1 M in CH3COONa.
1) In H2O pura [H3O+] = [OH-] = 10-7 per cui pH = 7 e per aggiunta di 10-3 eq. di
HCl: [H3O+] = 10-3 M per cui
pH = 3 e ∆pH = 7 – 3 = 4
2) L’aggiunta di un millieq. di HCl provoca la formazione di 1 millieq. di CH3COOH
e al scomparsa di 1 millieq. di CH3COO-.
Prima dell’aggiunta: [H3O+] = 1.8⋅10-5
Dopo l’aggiunta : [H3O+] = 1.8⋅10-5(
pH = 4.735
0.1
= 1.8⋅10-5
0.1
e pH = 4.745
0.1 + 0.001
) = 1.84⋅10-5 e
0.1 − 0.001
pertanto il
∆pH = 0.01
Perché una soluzione sia tamponante il rapporto CA/CS o CB/CS deve mantenersi
praticamente costante dopo l’aggiunta di acido o di base.
Le aggiunte di acido o di base devono essere non superiori a 1/50 ÷ 1/100 delle
quantità di specie tamponanti presenti.
Il massimo della capacità tamponante si ha quando CA/CS = 1 cioè quando il pH ≅
pKa oppure pOH ≅ pKb.
FINE
35