UNIVERSITÀ DEGLI STUDI DI GENOVA
FACOLTÀ DI MEDICINA E CHIRURGIA
Corso di Laurea in Tecniche di Laboratorio Biomedico
Anno Accademico 2008/2009
Tesi di Laurea
MUTAZIONI DI WT1: ANALISI DEL SIGNIFICATO BIOLOGICO
E ASPETTI TECNICI
Relatore
Prof. Maurizio Miglino
Candidata
Simona Roveta
INDICE
INTRODUZIONE
1. LEUCEMIA MIELODE ACUTA
1.1 Patogenesi ……………………………………………………..……..
pag 1
1.2 Clinica ……………………………..……………………..…..……….
pag 1
1.3 Epidemiologia ed eziologia …………………………………………..
pag 2
1.4 Laboratorio …… ………………………………………….………….
pag 2
1.5 Classificazione ……………………………..…………………………
pag 3
1.6 Fattori prognostici ……..……………………………………………..
pag 6
2. Il PROFILO GENETICO NELLE LEUCEMIE ACUTE MIELOIDI
2.1 Le alterazioni citogenetiche più frequenti nelle LAM...........................
pag 7
2.2 Ricerca di nuovi fattori prognostici nelle LAM ……..……………….. pag 11
3. METODI DI INDAGINE DELL’ESPRESSIONE GENICA E DI
ANALISI DEI POLIMORFISMI GENICI
3.1 Polimorfismi genici …………………………………………………… pag 12
3.2 Analisi dell’espressione genica ………………………………………..
pag 13
4. ALTERAZIONI DELL’ ESPRESSIONE GENICA: SIGNIFICATO
PROGNOSTICO
4.1 WT1: il gene del tumore di Wilms …………………………………….
pag 14
4.2 Altri marcatori molecolari …………………..……………………….... pag 16
5. MUTAZIONI GENICHE: SIGNIFICATO PROGNOSTICO
5.1 Mutazioni dei principali geni di interesse prognostico………..……….. pag 18
5.2 Cenni sulle mutazioni di altri geni di possibile interesse clinico………. pag 19
5.3 Mutazioni di WT1………………………………………………………. pag 19
6. SCOPO DELLA TESI………………………………………………… pag 21
MATERIALI E METODI
7. ALLESTIMENTO E AMPLIFICAZIONE DEI CAMPIONI
7.1 Estrazione degli acidi nucleici................................................................
pag 22
7.2 PCR (Polymerase Chain Reaction) ………………..….……………….
pag 24
7.3 Elettroforesi del DNA ……………………………………………..…..
pag 29
8. SSCP: SINGLE STRAND CONFORMATIONAL
POLYMORPHISM ………………….…………………………………… pag 31
9. ANALISI DI SEQUENZA
9.1 Reazione di sequenza…………………………………………………..
pag 34
9.2 Elettroforesi capillare su sequenziatore automatico ….……………….. pag 34
9.3 Analisi dei dati…………………………………………………………. pag 35
RISULTATI
10. FREQUENZA E TIPOLOGIA DELLE MUTAZIONI DI WT1
10.1 Single nucleotide polymorphism (SNP) nell’hot spot dell’esone 7….
pag 37
10.2 Frameshifts dell’esone 7…………………………………………….. pag 38
10.3 Mutazioni puntiformi dell’esone 7 e 9 per sostituzione di una singola
base…………………………………………………………………………. pag 38
DISCUSSIONE
11. SCREENING DELLE MUTAZIONI PUNTIFORMI MEDIANTE
SSCP………………………………………………………………………..
pag 42
12. SIGNIFICATO PROGNOSTICO E BIOLOGICO DELLE MUTAZIONI
DI WT1
12.1
Valore prognostico delle variazioni di sequenza di WT1…………… pag 42
12.2
Significato biologico delle mutazioni di WT1………………………. pag 43
13. CONCLUSIONI………………………………………………………. pag 44
INTRODUZIONE
1. LEUCEMIA MIELODE ACUTA
1.1 Patogenesi
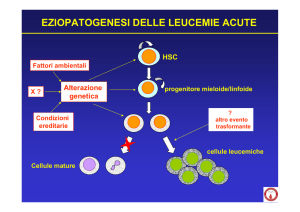
Le leucemie mieloidi acute (LAM) sono un gruppo eterogeneo di malattie neoplastiche
del sistema emopoietico caratterizzate da una difettiva produzione di granulociti,
monociti, piastrine ed eritrociti. Le LAM originano in seguito ad alterazioni dei
meccanismi che regolano la proliferazione e la differenziazione delle cellule staminali
emopoietiche. Le cellule staminali che hanno subito trasformazione neoplastica
proliferano senza differenziarsi o vanno incontro ad una differenziazione anomala.
Attualmente non sono completamente note le ragioni per cui viene soppressa
l'emopoiesi normale; tale deficit può essere ricondotto ad una insufficiente produzione
di stimolatori della crescita cellulare o alla liberazione di inibitori della proliferazione e
differenziazione delle cellule emopoietiche normali. Le sindromi mieloproliferative
acute sono quindi caratterizzate da un accumulo di cellule blastiche mieloidi incapaci,
parzialmente o totalmente, di dare origine a una progenie di cellule mature (eritrociti,
granulociti neutrofili, piastrine). Queste cellule immature si accumulano inizialmente
nel midollo osseo, inibendo la normale ematopoiesi, e in seguito nel sangue periferico,
infiltrando anche organi e tessuti come fegato, milza, polmoni e cute (1).
1.2 Clinica
I sintomi e i segni clinici dell’insufficienza midollare (quadro anemico per
insufficiente produzione di eritrociti, manifestazioni emorragiche conseguenti a
piastrinopenia, febbre ed infezioni dovute a neutropenia) possono essere variabilmente
presenti e manifestarsi con differente gravità. Ai sintomi di insufficienza midollare
possono associarsi sintomi generali, secondari all’espansione della massa leucemica e
alla liberazione di mediatori chimici dell’infiammazione: dolori ossei e muscolari,
sudorazioni profuse, calo ponderale, astenia. La stessa febbre può costituire un sintomo
generale e non essere sempre secondaria a un processo infettivo. Relativamente poco
frequente è il riscontro di epatomegalia, splenomegalia, linfoadenomegalie e
interessamento cutaneo (2).
1
1.3 Epidemiologia ed eziologia
Le LAM insorgono ad ogni età, ma la loro frequenza aumenta considerevolmente con il
passare degli anni (età mediana compresa tra 60 e 65 anni).
La distribuzione e la frequenza delle LAM in diverse aree geografiche e in diverse
popolazioni cambia sostanzialmente, sia per fattori genetici che per fattori ambientali
(ad esempio esposizione professionale ad agenti potenzialmente leucemogeni come
derivati del petrolio, pesticidi oppure esposizione iatrogena per
trattamento con
farmaci citotossici). È importante ricordare che le LAM si distinguono sul piano
biologico, clinico, prognostico in tre grandi categorie. Le LAM primarie compaiono
acutamente in soggetti in cui non è dimostrabile un’esposizione significativa ad agenti
leucemogeni. Nelle LAM secondarie è nota l’esposizione ad agenti leucemogeni;
queste leucemie insorgono in particolare nei soggetti precedentemente trattati con
chemioterapia e/o radiazioni per altra neoplasia. Vi sono, infine, le LAM secondarie a
una precedente sindrome mielodisplastica, della quale costituiscono l’evoluzione (2).
1.4 Laboratorio
La diagnosi di LAM si esegue esaminando il sangue periferico e il midollo osseo. Il
livello emoglobinico è ridotto in misura variabile, l’anemia è di solito di tipo
normocitico e normocromico, secondaria a ridotta eritroblastogenesi. Il numero delle
piastrine può essere considerevolmente ridotto, anche se non in tutti i casi. È importante
identificare tramite la formula leucocitaria il tipo di leucociti circolanti: nelle LAM,
infatti, parte di questi sono blasti mieloidi, talora rari (meno del 10%), ma spesso
predominanti (oltre il 50%).
L’esame del midollo osseo viene eseguito su materiale ottenuto tramite ago aspirato
(completato con biopsia ossea quando l’aspirato è povero di cellule): la cellularità
midollare è quasi sempre aumentata con scomparsa del tessuto adiposo, il parenchima
emopoietico è infiltrato in misura variabile, sino alla pressoché completa sostituzione
da parte di cellule blastiche.
La base per la diagnosi di una LAM è costituita dal riconoscimento che i blasti
leucemici hanno caratteristiche fenotipiche di tipo mieloide: tale riconoscimento
avviene da prima su base morfologica (presenza di granulazioni citoplasmatiche) e può
essere perfezionato utilizzando alcune reazioni citochimiche (mieloperossidasi -MPO-,
2
Sudan nero,
cloro-acetato esterasi -CAE-, esterasi non specifica -NSE-, fosfatasi
acida -FA-, acido periodico di Schiff -PAS-) e identificando, tramite anticorpi
monoclonali, alcune strutture antigeniche di membrana caratteristiche (determinanti
antigenici mieloidi come CD13, CD33, HLA-DR, CD34, CD11, CD14, CD15). In
alcuni casi coesistono blasti con fenotipi mieloidi e linfoidi e la leucemia di definisce
ibrida. L’eterogeneità clinico-biologica delle LAM si riflette dunque sia sulla diversità
dei marcatori fenotipici sia sui parametri fisici delle cellule leucemiche. La
caratterizzazione immunofenotipica delle LAM mediante citofluorimetria a flusso
multiparametrica consente di evidenziare simultaneamente diversi antigeni associati a
un particolare citotipo leucemico, risulta pertanto di grande utilità sia nella fase
diagnostica che nel monitoraggio della terapia e nella valutazione della malattia minima
residua al termine del programma terapeutico (2).
1.5 Classificazione
Il sistema classificativo storico delle LAM è fondato sulla morfologia delle cellule
blastiche ed è noto come sistema FAB (Franco-Americano-Britannico) (3). Questa
classificazione identifica differenti sottotipi di LAM in base alla linea differenziativa
della popolazione leucemica ed alla completa o parziale soppressione della capacità
maturativa del clone neoplastico (Tabella 1).
Le indagini citogenetiche hanno acquisito negli anni crescente importanza nella
caratterizzazione e nella prognosi delle LAM facendo emergere una serie di
associazioni
tra
quadri
citomorfologici
ed
aspetti
citogenetici
(alterazioni
cromosomiche numeriche e/o strutturali, presenti come anomalie singole o in
associazione). Per molte anomalie cromosomiche, mediante la tecnica della PCR, è
stata possibile l’amplificazione di sequenze di DNA tumore-specifiche (o mRNA
mediante retrotrascrizione a c-DNA) per caratterizzare anomalie citogenetiche
ricorrenti nelle LAM. L’ utilizzo sempre più ampio e raffinato delle metodiche
citofluorimetriche ha consentito una caratterizzazione sempre più precisa dei vari
sottotipi di LAM, ha individuato numerosissimi fenotipi aberranti utili per il
monitoraggio quantitativo del clone leucemico ed evidenziato nuovi sottotipi (leucemie
indifferenziate, leucemie bifenotipiche, leucemie bilineari). Ben presto è risultato
evidente che l’ eterogeneità morfologica, immunofenotipica e clinica era in relazione
3
con la presenza di una vasta gamma di aberrazioni citogenetiche e/o di mutazioni
geniche o da alterazioni della funzione o dell’ espressione di numerosi geni. Il ruolo
centrale delle alterazioni genetiche è stato
riconosciuto dalla World Health
Organization (WHO) che ha pubblicato nel 1999 una nuova classificazione delle LAM,
basata su caratteristiche cliniche, citomorfologiche, immunofenotipiche, cariotipiche e
molecolari in cui sono elencate diverse entità clinico-ematologiche associate a precise
alterazioni citogenetiche (Tabella 2) (4-5).
Tabella 1: Classificazione FAB delle leucemie acute mieloidi (LAM)
Linea differenziativa
delle cellule leucemiche
Morfologia
Citochimica
M0
MIELOBLASTICA
indifferenziata
Blasti di varie dimensioni privi
di granuli citoplasmatici e corpi
di Auer.
MPO negativa
Sudan nero negativa
M1
MIELOBLASTICA
senza maturazione
Blasti (tipo I e II) > 90%
Perossidasi o Sudan B
(> 3% blasti positivi)
CD13, CD33,
MP07, (CD14)
M2
MIELOBLASTICA
Con maturazione
Blasti (tipo I e II) < 90%
PMC, MC, MMc e PMN > 10%
Cellule monocitarie < 20%
Perossidasi o Sudan B
(> 20% blasti positivi)
CD13, CD33,
MP07
M3
PROMIELOCITICA
TIPICA
Blasti ipergranulati tipo PMC,
con numerosi bastocelli di Auer
MICROGRANULARE
Blasti con nuclei fogliacei bi- o
multilobati con fini granuli.
Rari blasti ipergranulati
Perossidasi o Sudan B
CD13, CD33,
MP07
M4
MIELOMONOBLASTICA
Blasti (tipo I e II) e altre cellule
granulocitarie più mature < 80%
Perossidasi o Sudan B
Nasde NaF parzialmente
resistenti, Anae
CD13, CD33,
CD14
M5
MONOBLASTICA
M5a-senza maturazione
Monoblasti > 80%
Nasde NaF-sensibili
Anae
CD14, CD13,
CD33
M5b-con maturazione
Monoblasti,
promonociti
monociti > 80%
Nasde NaF-sensibili
Anae
CD13, CD33,
CD14
M6
ERITROBLASTICA
Eritroblasti > 50% e blasti
(tipo I e II) > 30% delle cellule
non eritroidi
Pas (eritroblasti)
Perossidasi o Sudan B
CD42
M7
MEGACARIOBLASTICA
Blasti (tipo I e tipo linfoide)
> 30%, marcata mielofibrosi
Perossidasi piastrinica
CD41
e
Immunofenotipo
di membrana
CD13, CD33
CD13, CD33,
MP07
I blasti tipo I e tipo II sono caratterizzati drispettivamente da assenza e presenza di granulazioni
citoplasmatiche. Per la citochimica e per l’immunofenotipo di membrana sono elencati solo gli elementi
più comuni e più utili per la diagnosi.
4
Tabella 2: Classificazione WHO delle leucemie acute mieloidi (LAM)
______________________________________________________________________
LEUCEMIE ACUTE MIELOIDI CON ANORMALITA’ GENETICHE
RICORRENTI
o Leucemia acuta mieloide con t(8;21)(q22;q22), (AML1/ETO)
o Leucemia acuta mieloide con eosinofilia midollare e inv(16)(p13q22) o
t(16;16)(p13q22), (CBF/MYH11)
o Leucemia acuta promielocitica con t(15;17)(q22;q12), (PML/RAR) e
varianti
o Leucemia acuta mieloide con anomalie della banda 11q23 (MLL)
______________________________________________________________________
LEUCEMIA MIELOIDE ACUTA CON DISPLASIA MULTILINEARE
o Successiva a mielodisplasia (MDS) o mielodisplasia/ sindrome
mieloproliferativa (MDS/MPD)
o Senza MDS o MDS/MPD antecedenti ma con displasia in almeno il 50%
delle cellule o in 2 o più linee mieloidi
______________________________________________________________________
LEUCEMIA ACUTA MIELOIDE E SINDROMI MIELODISPLASTICHE IN
SEGUITO A TERAPIA con:
o Agenti alchilanti; radiazioni ionizzanti
o Inibitori della topoisomerasi III
o Altri
______________________________________________________________________
LEUCEMIE MIELOIDI ACUTE CHE NON APPARTENGONO AI CASI
PRECEDENTI
o LAM minimamente differenziata
o LAM senza maturazione
o LAM con maturazione
o Leucemia acuta mielomonocitica
o Leucemia acuta monoblastica/monocitica acuta
o Leucemia acuta eritroide (eritroide/mieloide o eritroleucemia pura)
o Leucemia acuta megacarioblastica
o Leucemia acuta basofila
o Panmielosi acuta con mielofibrosi
o Sarcoma mieloide
_____________________________________________________________________
5
1.6 Fattori prognostici
Il più importante fattore correlato al miglioramento della sopravvivenza è il
raggiungimento della remissione completa (RC). I principali fattori che ne influenzano
il raggiungimento sono l’età, la conta leucocitaria all’esordio, la risposta iniziale alla
terapia, l’immunofenotipo e le alterazioni citogenetiche.
L’ età alla diagnosi rimane tra i più importanti fattori di rischio pretrattamento; l’età
avanzata (superiore ai 60 anni) è associata a prognosi peggiore principalmente per la
difficoltà a sopravvivere alla terapia di induzione e per la maggiore resistenza ai
citostatici (le cellule leucemiche esprimono più frequentemente il gene di resistenza
plurifarmacologica MDR1 - Multi Drug Resistence 1).
E’ stata segnalata una relazione inversa tra la durata della RC e la conta leucocitaria all’
esordio o il numero assoluto dei blasti circolanti. La rapidità di scomparsa dei blasti
dopo l’ inizio della terapia sembra essere un fattore prognosticamente rilevante nel
predire il rischio di ricaduta.
Sebbene la classificazione FAB non sia solitamente considerata di per sé un fattore
prognostico indipendente, certe caratteristiche del basto come la presenza dei corpi di
Auer e l’immunofenotipo hanno dimostrato di possedere un significato prognostico. In
particolare la co-espressione di antigeni caratteristici di morfologia immatura si associa
ad un esito sfavorevole della malattia.
Per quanto riguarda le alterazioni citogenetiche, esse influenzano la funzione delle
molecole di segnale, dei fattori di trascrizione e dei recettori dei fattori di crescita e
influenzano la risposta al trattamento. Inoltre le molteplici alterazioni genomiche che
spesso coesistono in una singola cellula leucemica riflettono gli eventi trasformanti che
si accumulano nel clone durante lo sviluppo della leucemia (6). L’ identificazione di
sottotipi genetici ha migliorato considerevolmente la stratificazione prognostica,
precedentemente basata unicamente su elementi morfologici e clinico-ematologici e, in
alcuni casi, ha consentito l’ individuazione di terapie adattate sul difetto molecolare,
come nel caso della leucemia acuta promielocitica (LAP).
Il ruolo delle anomalie genetiche appare pertanto fondamentale non soltanto nella
prognosi ma anche nella patogenesi, nella diagnosi e nella terapia delle LAM.
6
2. Il PROFILO GENETICO NELLE LEUCEMIE ACUTE MIELOIDI
2.1 Le alterazioni citogenetiche più frequenti nelle LAM
Nella già menzionata nuova classificazione WHO (4-5) il primo gruppo comprende le
forme di LAM con ricorrenti anomalie genetiche e cioè: la LAM con t(8;21)(q22;q22)
in cui si verifica la formazione del gene ibrido AML1/ETO; la LAM con eosinofilia
midollare e inv(16) o t(16;16)(p13;q22) e formazione del gene ibrido CBFβ/MYH11; la
leucemia acuta promielocitica con t(15;17)(q22;q11-21) e varianti; la LAM con
anomalie 11q23 ( gene myeloid-lymphoid o mixed-lineage leukemia - MLL), ad esempio
t(9;11)(p22;q23) e t(6;11)(q27;q23). Esistono, come è ben noto, numerosissime altre
alterazioni citogenetiche, classificabili come bilanciate (traslocazioni, inversioni) e non
bilanciate (delezioni parziali, monosomie, trisomie …). Le principali sono riportate
nella Tabella 3.
Per quanto concerne il loro contributo alla leucemogenesi le mutazioni geniche possono
essere suddivise nei seguenti gruppi: mutazioni che interferiscono con la trascrizione,
mutazioni di attivazione e mutazioni che interferiscono con il ciclo cellulare.
Le mutazioni che interferiscono con la trascrizione modificano la funzione di fattori di
trascrizione o interferiscono indirettamente con la trascrizione determinando alterazioni
nel processo di differenziazione e/o l’ acquisizione di aberranti proprietà di self-renewal
dei progenitori emopoietici (7). Appartengono a questa classe i geni di fusione derivanti
dalle mutazioni t(8;21), inv(16)/t(16;16), t(15;17) e le mutazioni nei geni
CCAAT/enhancer binding protein alpha (CEBPA) , MLL e runt-related transcription
factor 1 (RUNX1).
Le mutazioni di attivazione attivano vie di trasduzione del segnale, determinando un
aumento della proliferazione o della sopravvivenza dei precursori leucemici.
Appartengono a questa classe le mutazioni di fms-related tyrosine kinase 3 (FLT3), di
neuroblastoma RAS viral oncogene homolog (NRAS) e di JAK2.
Le mutazioni che interferiscono con il ciclo cellulare e l’ apoptosi sono rappresentate
principalmente dalle mutazioni di nucleophosmin 1 (NPM1) e da delezioni di TP53.
Le mutazioni somatiche più frequentemente rilevate in pazienti affetti da LAM con
cariotipo normale interessano i seguenti geni: NPM1, FLT3,CEBPA, MLL, NRAS, WT1,
7
RUNX1. Da segnalare però che queste mutazioni possono essere presenti anche in
pazienti con cariotipo anomalo (8-9).
Numerosi studi retrospettivi e prospettici hanno dimostrato che il cariotipo rappresenta
uno dei più importanti fattori prognostici per risposta all’induzione, rischio di ricaduta e
sopravvivenza (10-15). I cariotipi pre-trattamento sono raggruppati attualmente in tre
gruppi prognostici di rischio (favorevole, intermedio e sfavorevole) e sono riportati
nella Tabella 4. I sistemi proposti dai vari gruppi di studio presentano molti aspetti
comuni ma anche alcune differenze importanti.
Il gruppo a prognosi favorevole include i pazienti che alla diagnosi presentano
t(8;21)(q22;q22), inv(16)(p13;q22), t(15;17); essi sono circa il 20% e hanno più spesso
un’età inferiore a 60 anni, l’85% di possibilità di ottenere una remissione completa
(RC ) ed il 30-40% di andare incontro ad una ricaduta.
Il gruppo definito a prognosi intermedia comprende circa il 45% dei soggetti affetti da
LAM, con outcome molto diversificato. Solo il 25% dei pazienti si può definire come
“lungo-sopravvivente”.
Infine il terzo gruppo è costituito da coloro che hanno un cariotipo complesso (con tre o
più anomalie), delezione del cromosoma 5 o 7, tipiche delle LAM secondarie
all’esposizione a farmaci o sostanze, oppure anomalie dell’11q, t(9;11), t(6;9).Questi
pazienti rispondono in modo deludente a qualsiasi tipo di
terapia e hanno una
probabilità di sopravvivenza a 5 anni inferiore al 5%. Numerose anomalie citogenetiche
incluse nel gruppo a cattiva prognosi [ad esempio -5, -7, del(5q), abn3q, del(7q),
abn11q23 etc] sono spesso osservate assieme ad altre anomalie. Si configura così quello
che è definito un cariotipo complesso, nella cui definizione conta solo il numero
(spesso ≥ 3) e non il tipo di alterazioni cariotipiche. Un recente lavoro in pazienti fino a
60 anni ha dimostrato che le monosomie autosomiche (dei cromosomi 5, 7 o di altri
cromosomi) conferiscono la prognosi peggiore (15). Al contrario trisomie, tetrasomie,
anelli o altre aberrazioni strutturali hanno minor significato prognostico. L’impatto
negativo di due o più monosomie autosomiche o di una monosomia associata ad un’
altra anomalia
è molto forte
e superiore a quello precedentemente indicato dal
cosiddetto cariotipo complesso. I pazienti con cariotipo complesso (anomalie maggiori
o uguali a 3 o a 5) che non soddisfano i criteri del cariotipo monosomico presentano
infatti una prognosi migliore.
8
Tabella 3 : le più frequenti alterazioni citogenetiche nella LAM
Anomalia citogenetica
Alterazione genetica
FAB
Incidenza
t(8;21)(q22;q22)
RUNX1/CBFA2T1
M2
6%
inv(16)(p13q22)
o t(16;16)(p13;q22)
CBFβ/MYH11
M4 eos
7%
t(15;17)(q22;q11-21)
PML/RARα
M3
7%
t(9;11)(p22;q23)
MLL/AF9
M5
2%
t(6;11)(q27;q23)
MLL/AF6
M4 ed M5
1%
inv(3)(q21q26)
o t(3;3)(q21;q26)
EVI1/RPN1
M1,M4,M6,M7
1%
t(6;9)(p23;q34)
DEK/CAN
M2, M4
1%
+8
?
M2, M4, M5
9%
-7/7q-
?
-
7%
-5/5q-
?
-
7%
-17/17p-
TP53
-
5%
-20/20q-
?
-
3%
9q-
?
-
3%
+22
?
M4, M4 eo
3%
+21
?
-
2%
+13
?
M0-M1
2%
+11
MLL
M1,M2
2%
Cariotipo complesso
-
-
10%
-
44%
Traslocazioni / inversioni
Aneuploidie / delezioni
Cariotipo normale
9
Tabella 4: gruppi di rischio in rapporto alle alterazioni citogenetiche (ECOG-SWOG)
Gruppo di rischio
Alterazioni citogenetiche
Favorevole
t(8;21)(q22;q22) inv(16)(p13q22) t(16;16)(p13;q22)
t(15;17)(q22;q11-21)
Intermedio
cariotipo normale; -Y; +8; +11; +13; +21; del(20q)
Sfavorevole
cariotipo complesso; inv(3)(q21q26), t(3;3)(q21;q26)
-7, t(6;9)(p23;q34), t(6;11)(q27;q23), t(11;19)(q23;213.1) -5;
del(5q); del(9q); t(9;11)(p22;q23); del(11q)
In grassetto le alterazioni citogenetiche con significato prognostico largamente condiviso dai vari gruppi
di studio.
10
2.2 Ricerca di nuovi fattori prognostici nelle LAM
In circa il 40% delle LAM sono presenti specifiche alterazioni cromosomiche e
molecolari caratterizzate solitamente dalla formazione di geni ibridi di fusione il cui
trascritto può essere opportunamente amplificato ed utilizzato come marcatore di
malattia alla diagnosi e durante il follow-up. Le proteine di fusione che ne derivano
sono ritenute svolgere un ruolo chiave nel meccanismo di leucemogenesi e
rappresentano quindi un bersaglio ideale di eventuali terapie volte a eradicare il clone
leucemico.
Nella LAP, ad esempio, la traslocazione t(15;17)
ed il riarrangiamento genico
PML/RARα che ne deriva sono presenti nella quasi totalità dei casi. Numerosi studi di
malattia minima residua (MRD) in corso di LAP mediante PCR qualitativa del
trascritto ibrido PML/RARα ne hanno dimostrato il valore predittivo al termine dei cicli
di consolidamento.
Tuttavia, più del 50% delle LAM mancano di anomalie genetiche o marcatori clonali
noti e quindi adatti per il monitoraggio della MRD. Sono stati pertanto intrapresi
numerosi studi nel tentativo di identificare anomalie citogenetiche e molecolari
associate con la trasformazione leucemica (16-17).
Negli ultimi anni le indagini molecolari si sono concentrate proprio sulle LAM a
cariotipo normale (CN),
producendo una mole considerevole di dati non sempre
concordi. Nella fase attuale la disponibilità di tecniche di biologia molecolare capaci di
studiare (per mutazione o espressione) svariati geni ha ingenerato nell’ ematologo
clinico una certa incertezza. Il significato prognostico dei vari profili genetici è in
qualche caso ancora incerto, come il rapporto con particolari alterazioni citogenetiche e
opzioni terapeutiche (ad esempio il trapianto di cellule staminali allogeniche).
11
3. METODI DI INDAGINE DELL’ESPRESSIONE GENICA E DI ANALISI DEI
POLIMORFISMI GENICI
3.1 Polimorfismi genici
È ben noto come l’espressione genica, ed in parte la sua funzione, possa essere alterata
da mutazioni, delezioni, inserzioni, duplicazioni anche di una singola base. Svariati
sono i metodi per analizzare tali polimorfismi. La comparative genomic hybridization
(CGH) permette di evidenziare polimorfismi anche a carico di un singolo nucleotide
(18). Accanto a questa esistono altre metodiche basate sull’amplificazione mediante
PCR del segmento di DNA o RNA specifico e su particolari elettroforesi in grado di
evidenziare l’alterata corsa del segmento mutato. La corsa sul gel di elettroforesi, in
determinate condizioni, dipende non solo dal peso molecolare, ma anche e soprattutto
dalla sequenza nucleotidica del segmento in questione: come nell’SSCP (19-22), un
elettroforesi ad amperaggio o voltaggio e temperatura costante su un gradiente di
acrilamide; nel DGGE, tecnica elettroforetica per la separazione di frammenti di DNA
in base alle loro differenti proprietà di dissociazione o“melting” (23-24); nel TGGE,
tecnica in cui viene formato un gradiente di temperatura per la separazione in una
seconda dimensione e in cui la separazione avviene in base a differenze di
conformazione (25).
Molto usata è la DHPLC (Denaturing High Performance Liquid Chromatography) una
tecnica che, in condizioni parzialmente denaturanti e sotto un diretto controllo della
temperatura, permette di discriminare all’interno di prodotti eterogenei di PCR
molecole di DNA eteroduplex rispetto alle molecole omoduplex. La metodica si basa
sulla differente velocità di eluizione in una colonna cromatografica degli eteroduplex e
degli omoduplex. per la rilevazione di mutazioni del DNA. I duplex si formano quando
frammenti amplificati di DNA vengono denaturati termicamente e lasciati ricombinare.
Una qualsiasi variazione (mutazione o polimorfismo) tra le due forme alleliche di un
frammento porta alla formazione di un eteroduplex (combinazione di due catene di
DNA a singola catena, non perfettamente corrispondenti, caratterizzata dalla presenza
di una “bolla” a livello della quale si trova il mismatch). L’eteroduplex si comporta
12
cromatograficamente in modo differente sia dall’omoduplex non mutato che
dall’omoduplex mutato: l’eteroduplex è solitamente più veloce (meno trattenuto) degli
omoduplex e da ciò si può caratterizzare la presenza di una variazione nucleotidica in
un campione. La presenza di una mutazione o di un polimorfismo si evidenzia quindi,
mediante picchi ulteriori o con un profilo diverso rispetto al “wild type” (26).
Tutti questi metodi sono in grado di identificare la presenza di sequenze mutate. Il
passo successivo d’obbligo è il sequenziamento diretto della sequenza mutata, al fine di
identificarne la natura. Molto scarsi sono i dati in letteratura sul significato e sulla
valenza prognostica delle differenti mutazioni dei vari geni marker. Allo stesso tempo
molto importante è identificare alterazioni di sequenza non random e correlarle
clinicamente e biologicamente.
3.2 Analisi dell’ espressione genica
A tal fine viene comunemente utilizzata la Real-Time PCR su cDNA ottenuto per
trascrizione inversa dall’RNA totale del paziente. La Real Time PCR è una PCR in
cinetica in cui l’amplificazione ed il rilevamento dell’amplificato avvengono nello
stesso momento. Questo è possibile grazie all’introduzione all’interno della reazione di
una molecola fluorescente, che ci dà la possibilità di seguire la reazione da un punto di
vista visivo, grazie all’ausilio di appositi software. Sono generalmente utilizzate sonde
Taqman (Taqman probes), oligonucleotidi lineari di 25-28 pb marcate al 5’ con il
Reporter ed al 3’ con il Quencer. Il Quencer estingue la fluorescenza del reporter solo
quando la sonda è integra; quando la sonda viene tagliata, il Quencer ed il Reporter si
liberano in soluzione e si manifesta la fluorescenza. (27-30).
13
4.
ALTERAZIONI
DELL’
ESPRESSIONE
GENICA:
SIGNIFICATO
PROGNOSTICO
4.1 WT1: il gene del tumore di Wilms
Il gene del tumore di Wilms (WT1), isolato per la prima volta nell’omonima neoplasia
renale pediatrica, è formato da dieci esoni localizzati sul cromosoma 11p13 e codifica
per un fattore di trascrizione con un dominio regolatore al N-terminale (esoni 1-6) e un
dominio al C-terminale tipo 4-Cys2-His2 zinc-finger (esoni 7-10) coinvolto nella
proliferazione, differenziazione ed apoptosi cellulare (31).
WT1 è espresso durante il periodo dell’organogenesi in cui svolge un ruolo cruciale:
topi knock-out per il gene WT1 muoiono a livello embrionale e presentano alterazioni a
livello dell’apparato urogenitale. Il gene è infatti espresso precocemente nelle cellule
durante lo sviluppo dell’apparato genitourinario ed ematopoietico. Con il differenziarsi
di entrambi i sistemi WT1 viene silenziato. Inizialmente a questo gene è stato attribuito
un ruolo onco-soppressore, tuttavia, al contrario di altri oncosoppressori come Rb e p53
che sono espressi in tutti i tessuti, l’espressione del gene WT1, oltre ad avere valori
molto bassi, è limitata solo alle cellule delle gonadi, dell’utero, dei reni, della milza, dei
mesoteli e dei progenitori ematopoietici. Il ruolo di WT1 nelle neoplasie renali appare
ormai abbastanza chiaro, mentre il significato preciso della sua espressione nella
ematopoiesi (normale e maligna) resta ancora controverso. WT1 risulta particolarmente
espresso nei precursori CD 34+ del sistema ematopoietico mentre va incontro ad un
rapido processo di down-regulation nel corso del processo di differenziamento cellulare
ed è assente nei leucociti maturi.
Il ruolo di WT1 nella leucemogenesi è ancora molto dibattuto. Il gene WT1 è
iperespresso non solo nella LAM, ma anche nella leucemia linfoblastica acuta, nella
leucemia
mieloide
cronica,
nelle
sindromi
mielodisplastiche,
nei
disordini
mieloproliferativi Ph negativi e in alcuni linfomi non-Hodgkin suggerendo che questo
gene oncosoppressore possa avere paradossalmente un’attività oncogenica nelle cellule
ematopoietiche. Alcuni lavori hanno dimostrato che linee cellulari transfettate in modo
permanente con WT1 mostrano difetti nella risposta ad agenti differenzianti e questo
fenomeno potrebbe contribuire alla genesi della leucemia. Alcuni modelli sperimentali
14
riportano una tendenza all’aumentata proliferazione cellulare, altri un arresto di
crescita. Questa diversità funzionale può essere spiegata con differenti isoforme
prodotte nei diversi apparati. (31-34).
Al momento esistono sostanzialmente due ipotesi contrastanti sul ruolo di WT1 nelle
leucemie: secondo una teoria WT1 agisce come un oncogene e rappresenta la tappa
finale di diverse vie di “trasformazione” attivate all’interno della cellula; una seconda
teoria parte dall’assunto che WT1 agisca come oncosoppressore. La sua
overespressione, pertanto, costituirebbe semplicemente un epifenomeno in risposta ai
segnali trasformanti attivati all’intero della cellula. Questa dualità funzionale al
momento non è ben interpretata, ma è opinione diffusa che il ruolo di WT1 possa
variare da cellula a cellula anche solo per il grado di differenziazione di queste (35-41).
Aldilà del suo significato biologico nella leucemogenesi, dopo l’introduzione delle
tecniche di RT-PCR , WT1 è diventato un utile marker molecolare . I livelli di WT1 e la
loro variazione in corso di terapia possono infatti essere utilizzati come indici di
malattia residua minima e sembrano assumere un significato prognostico in alcune
neoplasie ematologiche ( 42). Il ruolo prognostico dei livelli di espressione di WT1 alla
diagnosi nelle LAM non è in realtà ancora ben definito. Le prime segnalazioni in
letteratura sembravano dimostrarne una correlazione fra elevata espressione e prognosi
negativa, come già dimostrato nelle sindromi mielodisplastiche (42-44). Più
recentemente tale ruolo negativo non è stato confermato ed in un recente studio del
gruppo spagnolo l’espressione di WT1 alla diagnosi non riveste alcun ruolo prognostico
(45). Esistono infine segnalazioni in cui si dimostra un associazione fra elevata
espressione di WT1 e cariotipo favorevole (46). Nelle core binding factor LAM inoltre
elevati valori di WT1 sono stati associati ad una maggiore probabilità di raggiungere la
remissione completa (47). Come si vede non vi è ancora chiarezza, e questo in parte è
legato a motivi statistici. Parliamo di valori elevati, ma non è chiaro se si debba porre
un valore cut-off
che
identifichi due distinti gruppi prognostici o se si debba
considerare WT1 come variabile continua o ancora se i vari laboratori debbano
condurre una analisi suddividendo in percentili i vari valori. Un parziale chiarimento
potrà essere raggiunto nel momento in cui l’analisi verrà standardizzata, e saranno
definiti i valori normali di espressione e le fasce di rischio. Al momento ciascun
laboratorio deve costruirsi la propria curva di normalità analizzando l’espressione in
15
soggetti normali, utilizzando reagenti e macchinari non codificati. Per questi motivi i
risultati presentati dai vari gruppi al momento possono essere confrontati con qualche
difficoltà.
4.2 Altri marcatori molecolari
Oltre a WT1 vi sono anche altri geni la cui espressione viene utilizzata come elemento
d’indagine nelle LAM. Ad esempio il gene ERG risulta overespresso in LAM a
cariotipo complesso, con alterazioni a carico del cromosoma 21 anche criptiche, ma
anche in LAM a cariotipo normale (48-49). L’overespressione di ERG è stata descritta
anche in leucemie linfoblastiche acute soprattutto a fenotipo T. Alcuni studi dimostrano
come la overespressione di ERG alla diagnosi di LAM sia associata a prognosi
sfavorevole, inoltre elevati livelli di espressione di ERG si associano spesso ad elevata
espressione del gene brain and acute leukemia, cytoplasmic (BAALC), configurando un
sottotipo di LAM a prognosi particolarmente sfavorevole (50-53).
Il gene BAALC risulta overespresso nelle cellule CD34+. Tale espressione viene down
regolata negli stadi maturativi successivi: di qui l’ipotesi che BAALC rappresenti un
marcatore molecolare specifico dei progenitori emopoietici più immaturi. Essendo
BAALC
marcatore
molecolare
specifico
di
cellule
emopoietiche
alquanto
indifferenziate, la sua overespressione si associa a LAM a fenotipo immaturo (54-55).
Una elevata espressione di BAALC alla diagnosi nelle LAM ha valenza prognostica
negativa in termini di inferiore percentuale di RC e ridotta sopravvivenza. (56-57).
Elevate espressioni di tale gene si rinvengono in pazienti che già presentano fattori
prognostici negativi (cariotipo sfavorevole FLT3-ITD, NPM1 wild-type o elevata
espressione di ERG), pertanto, BAALC potrebbe rappresentare un indicatore generale
della presenza di alterazioni sfavorevoli. Contraddittori sono, infatti, i risultati che si
ottengono quando si analizza il valore prognostico di BAALC in coorti di pazienti che,
per altri marcatori, vengono considerati a basso rischio. Esperienze condotte su pazienti
a cariotipo favorevole (NPM1 mutati, FLT3-ITD negativi) non dimostrano alcun valore
prognostico per i livelli di espressione di BAALC. D’altro canto quando si analizzano i
profili genici di LAM ad alto rischio, il gene BAALC risulta sempre overespresso.
16
BAALC non sarebbe, quindi, un fattore prognostico autonomo, ma un mero indicatore
di particolare instabilità genica della cellula staminale leucemica.
Recentemente è stato descritta overespressione del gene meningioma 1 (MN1) in LAM
che presentavano inv(16). In un’altra segnalazione MN1 risultava coinvolto nella
formazione del gene chimerico derivante dalla t(12;22). In generale, sebbene non sia
ancora nota la precisa funzione di MN1 nei processi di oncogenesi, pare dimostrato il
fatto che alti livelli di MN1 alla diagnosi connotino un gruppo di LAM a cattiva
prognosi e siano associati a bassa incidenza di NPM1 mutato e alta incidenza di elevati
valori di espressione di BAALC. A tal riguardo, considerata la stretta corrispondenza fra
elevata espressione di MN1 e di BAALC, si può ipotizzare una cooperazione fra questi
due geni nei processi di leucemogenesi. (58-62).
17
5. MUTAZIONI GENICHE: SIGNIFICATO PROGNOSTICO
Vi sono svariati geni le cui mutazioni rivestono un interesse prognostico nelle LAM.
Da un punto di vista clinico lo studio di queste mutazioni è importante anche per
l’individuazione di nuovi bersagli per lo sviluppo di terapie mirate.
5.1 Mutazioni dei principali geni di interesse prognostico
Il gene NPM1 risulta frequentemente traslocato o mutato in malattie oncoematologiche. I meccanismi leucemogenetici delle mutazioni NPM1 (NPM1mut) non
sono pienamente compresi in quanto la proteina NPM1 è coinvolta in molti processi
cellulari, come il trasporto di sostanze tra nucleo e citoplasma, la regolazione di geni
oncosoppressori, il controllo della duplicazione del centrosoma durante il ciclo cellulare
e l’attivazione dell’apoptosi. Le mutazioni del gene della NPM1 sono state trovate in
circa il 35% dei pazienti adulti affetti da LAM e nel 60% di coloro che presentano alla
diagnosi un cariotipo normale (63-64). L’espressione di NPM1 mutata alla diagnosi è
stata associata in molti studi ad una prognosi favorevole in termini di maggiore
percentuale di remissione completa (RC) dopo terapia di induzione, più lunga
sopravvivenza libera da eventi avversi (EFS) e sopravvivenza totale (OS) (65-69).
Circa il 40% dei pazienti con mutazioni di NPM1 è portatore anche di mutazioni a
carico di FLT3 (70-72).
Il gene FLT3 viene espresso precocemente dai progenitori emopoietici e gioca un ruolo
importante nella proliferazione delle cellule staminali emopoietiche, nella loro
differenziazione e sopravvivenza. Mutazioni che inducono un’attivazione costitutiva di
FLT3 sono presenti in circa il 30% dei pazienti con LAM-CN, spesso si tratta di
internal tandem duplication (ITD), più raramente di mutazioni puntiformi. Numerosi
studi hanno dimostrato che i pazienti con
FLT3-ITD (FLT3-ITDpos) hanno una
prognosi peggiore in termini di RC, EFS e OS rispetto ai soggetti che non presentano
questa mutazione (FLT3-ITDneg) (73-74). Questo tipo di mutazione ha un ruolo
prognostico dominante rispetto ad altri marcatori molecolari: ad esempio la già citata
mutazione di NPM1 è un fattore prognostico positivo, ma solo nei pazienti FLT3ITDneg (75-77). Il gruppo di pazienti con genotipo NPM1mut/FLT3-ITDneg e cariotipo
18
normale è quello collegato alla migliore prognosi assoluta, e sembra non beneficiare del
trapianto (71).
Le mutazioni del gene CCAAT/enhancer binding protein alpha (CEBPA), riscontrabili
nel 5-14% delle LAM, si osservano più frequentemente nei pazienti con CN e sono
state associate con EFS o OS buone (78-80) L’impatto favorevole sulla prognosi delle
mutazioni di CEBPA è osservato solamente in assenza di cariotipo complesso e di
FLT3-ITD (81).
Il gene MLL può essere coinvolto in riarrangiamenti sia intercromosomici che
intragenici, in questo caso si hanno mutazioni definite partial tandem duplications
(PTD). Alcuni studi condotti su pazienti LAM-CN con MLL-PTD evidenziano una
minore durata della RC rispetto ai pazienti senza MLL-PTD mentre l’OS non differisce
significativamente (82-83).
5.2 Cenni sulle mutazioni di altri geni di possibile interesse clinico
Vi sono geni di cui sono state descritte le rispettive mutazioni in una percentuale
significativa di LAM ma che attualmente hanno ancora un significato prognostico
dubbio, controverso o sconosciuto.
Mutazioni in NRAS, che determinano un’attivazione costitutiva della proteina RAS, si
osservano nel 9-14% dei giovani adulti con LAM-CN (70, 84-85). Nessun studio ha
sinora dimostrato una rilevanza prognostica per queste mutazioni (70, 85) che, tuttavia,
potrebbero essere importanti come bersaglio di terapia orientata da un punto di vista
molecolare.
RUNX1 codifica per un fattore trascrizionale che è coinvolto nella differenziazione
emopoietica normale. Mutazioni di RUNX1 sono state recentemente osservate nel 10%
dei pazienti con LAM-CN (86) ma il loro significato prognostico è ancora sconosciuto.
5.3 Mutazioni di WT1
Le mutazioni di WT1 (WT1mut) consistono in sostituzioni o delezioni dell’esone 7 o
dell’esone 9 che annullano le sue funzioni promuovendo la proliferazione e il blocco
della differenziazione delle cellule staminali. Mutazioni di WT1 si ritrovano in circa il
12 % dei pazienti affetti da LAM, più spesso di giovane età, con elevata blastosi
19
periferica ed elevati livelli serici di LDH (87). Esse rappresentano in questo gruppo di
pazienti una delle forme più comuni di mutazione dopo quelle relative a NPM1 e le
internal tandem duplication (ITDs) del gene fms-like tyrosine kinase 3 (FLT3) (16).
Sebbene le mutazioni di WT1 nelle LAM siano state segnalate più di dieci anni fa, il
loro ruolo nel determinarne la prognosi è ancora controverso.
Summers et al.
nel 2007 dimostrarono in 70 pazienti con LAM-CN che queste
mutazioni erano associate al fallimento della terapia di induzione (88).
In modo simile Virappane et al, analizzando 470 pazienti affetti da LAM (89),
affermarono che le mutazioni di WT1 sono un indicatore prognostico negativo: i
pazienti che esprimono WT1mut hanno una ridotta percentuale di RC rispetto a quelli
che hanno WT1 normale (79% vs 90%), di sopravvivenza libera da eventi avversi (22%
vs 44%) e di sopravvivenza a 5 anni (26% vs 47%). L’impatto negativo delle mutazioni
di WT1 è risultato indipendente sia da FLT3-ITDs che dalla presenza di mutazioni di
NPM1.
Nel recente lavoro di Paschka et al. (90) dei 196 pazienti affetti da LAM-CN i 21 che
presentavano WT1mut avevano una percentuale di RC simile a quella del gruppo con
WT1 normale (76% vs 84%), ma andavano incontro a ricaduta più frequentemente
(88% vs 51%). Il rischio di morire si è rivelato tre volte superiore nei soggetti con
WT1mut
rispetto a quelli senza la mutazione. Lo stesso autore, con
un’analisi
multivariata, ha dimostrato che l’impatto negativo di WT1mut sulla prognosi è
indipendente dall’espressione di altri marcatori molecolari prognostici (mutazioni di
NPM1 e FLT3-ITD negativo) e dalle caratteristiche cliniche alla diagnosi.
Il lavoro di Gaidzik et al. (91) condotto su 617 pazienti con LAM-CN evidenzia invece
che i soggetti che presentano WT1mut e positività FLT3-ITD (FLT3-ITDpos ) hanno una
percentuale di RC inferiore rispetto a quelli con WT1mut e FLT3-ITD negativo (63% vs
92%) mentre non vengono riscontrate differenze significative in termini di
sopravvivenza libera da malattia e sopravvivenza globale fra i pazienti con o senza
mutazioni di WT1. Pertanto la presenza di WT1mut in associazione al genotipo FLT3ITDpos sembra avere un impatto negativo.
20
6.
SCOPO DELLA TESI
Questo lavoro si propone di
verificare se la Single Strand Conformational
Polymorphism Polymerase Chain Reaction (SSCP-PCR) è una valida metodica per lo
screening delle alterazioni genetiche dell’esone 7 e dell’esone 9 di WT1 per poi andare
a confermarne l’effettiva presenza con il sequenziamento. La scelta di questi esoni è
dovuta al fatto che corrispondono a regioni segnalate in studi precedenti come hot spots
di mutazione nelle LAM-CN.
Una volta accertata sia la presenza che l’esatta natura delle mutazioni puntiformi di
WT1 si potrà quindi verificare se i campioni WT1mut presentano anche altre mutazioni di
geni di cui è noto il valore prognostico o un’espressione significativa di altri marcatori
molecolari di interesse clinico (ad esempio i livelli di espressione dello stesso WT1)
per poter stabilire se vi è una correlazione tra le mutazioni di WT1, i suddetti parametri
e l’outcome clinico.
21
MATERIALI E METODI
7.
ALLESTIMENTO E AMPLIFICAZIONE DEI CAMPIONI
Sono stati analizzati 200 casi di LAM-CN (di cui 18 evolute da MDS) diagnosticati
presso la Clinica Ematologica dell’Università di Genova nel periodo 2008– 2009.
7.1 Estrazione degli acidi nucleici
Gli acidi nucleici sono stati estratti da campioni di sangue midollare e periferico
durante le indagini diagnostiche di routine.
I campioni di sangue intero sono stati privati degli eritrociti mediante separazione su
gradiente di densità Ficoll-Hypaque: il sangue viene diluito 1:1 con Phosfate Buffered
Saline (PBS) e stratificato molto lentamente sopra il Ficoll e successivamente
centrifugato per 30’ a 1600 rpm. Questo permette la stratificazione delle cellulle in base
alla loro densità: alla fine della centrifugazione gli eritrociti si depositano sul fondo
della provetta, in quanto grazie alla loro maggiore densità riescono ad attraversare lo
strato di Ficoll, i polimorfonucleati si raccolgono nello strato al di sopra degli eritrociti,
le cellule mononucleate (linfociti e monociti) invece vanno a formare un anello
biancastro nell’interfaccia tra il Ficoll e il plasma (a causa della loro bassa densità non
riescono a migrare attraverso lo strato di Ficoll) (Figura 1). Queste sono le cellule di
nostro interesse, che possono essere recuperate per mezzo di una pipetta di tipo Pasteur.
Le cellule così isolate vengono successivamente lavate con PBS buffer e centrifugate
per 10’ a 3000 rpm.. Il pellet di cellule ottenuto dopo la centrifugazione viene vortexato
in una soluzione di estrazione (TNE1X: TRIS 50mM, NaCl 0,1M, EDTA 5mM),
contenente sostanze tensioattive (Sodium Dodecyl Sulfate - SDS 2%) che lisano le
cellule e proteasi (proteinasi K 100 µg/ml) che digeriscono le proteine. Le proteine
vengono ulteriormente rimosse dalla preparazione trattando la soluzione acquosa
contenente gli acidi nucleici con una miscela di fenolo, cloroformio e alcol isoamilico.
Il
fenolo è un forte denaturante delle proteine che le lega mediante legami H
alterandone la struttura: le proteine denaturate, con i gruppi idrofobici esposti,
diventano solubili nella fase fenolica o precipitano all’interfase fenolo-acqua. Il
cloroformio completa la denaturazione delle proteine, rimuove i lipidi e grazie alla
22
sua elevata densità facilita la separazione della fase acquosa (contenente il DNA
deproteinizzato) da quella organica (fenolica) stabilizzando l’interfaccia tra le due fasi.
L’ alcol isoamilico riduce la schiuma che si forma nel corso dell’estrazione e crea
un’interfase tra la fase acquosa ed il fenolo.
I campioni incubati a 37° C per 12 ore vengono trattati con una soluzione di fenolocloroformio-alcool isoamilico (25:24:1) e dopo agitazione per 10 minuti sono
centrifugati per 30’ a 3000 rpm. Dopo la centrifugazione si ottengono tre fasi: quella
superiore (fase acquosa) contenente la soluzione di acidi nucleici, l’interfase in cui si
trovano le proteine denaturate e la fase inferiore (fase fenolica) in cui sono presenti
lipidi e proteine ricche di aminoacidi idrofobici. Dopo aver recuperato la fase acquosa
superiore creatasi per gradiente di densità si effettua un’altra volta il trattamento con
fenolo-cloroformio-alcol isoamilico. La fase finale dell'estrazione è costituita dalla
precipitazione alcolica in presenza di cationi monovalenti: all’ultimo sovranatante
recuperato viene aggiunto un volume di alcol isopropilico pari a 6/10 del campione e
un volume di sodio acetato pari a 2/10 e dopo agitazione il DNA è fatto precipitare a
4°C per 10-15 minuti. Con una pipetta di vetro si recupera il filamento di acido
nucleico che viene poi “lavato” in etanolo 70% per rimuove i sali precipitati e
ricentrifugato. Il pellet dopo essiccazione all’aria è risospeso in adeguato tampone a
bassa forza ionica TE (TRIS 10mM, EDTA 0,1mM) a pH 7.6-8.0. Per favorire
ulteriormente la risospensione è possibile lasciare il campione a 4° C in agitazione e nei
casi di risospensione non ottimale il campione può essere lasciato a 65° C per 10
minuti.
Per dosare gli acidi nucleici estratti è stato utilizzato il metodo spettrofotometrico che
sfrutta la capacità degli acidi nucleici di assorbire la luce UV con un massimo di
assorbimento alla lunghezza d'onda di 260 nm. Per determinare la concentrazione ed il
grado di purezza del campione estratto viene misurata la densità ottica (O.D.),
considerando l’assorbanza (A) alle lunghezze d’onda di 260 e di 280 nm. Il rapporto
A260/A280 è usato per stimare la purezza della preparazione degli acidi nucleici, in
quanto le proteine, che sono la principale fonte di contaminazione, assorbono a 280 nm.
Lo spettrofotometro viene azzerato a 260 e 280 nm utilizzando come “bianco” la
soluzione TE. Il valore letto viene quindi moltiplicato per la diluizione (in genere
1:100).
23
Preparazioni pure di DNA hanno valori di A260/A280 uguali a 1.8: un rapporto
inferiore a 1.8 è indice di contaminazione proteica, mentre un rapporto superiore a 2 è
indice di frammentazione del DNA o contaminazione di RNA.
Figura 1: Stratificazione delle cellule su gradiente di densità Ficoll-Hypaque
7.2 PCR (Polymerase Chain Reaction)
La PCR è una tecnica che consente, in poche ore, di amplificare milioni di volte una
sequenza definita di acido nucleico sfruttando la reazione di sintesi in vitro del DNA,
catalizzata dall’enzima DNA polimerasi (si usa la Taq polimerasi ricavata dal batterio
“Thermophylus aquaticus” resistente alle alte temperature). Questo enzima richiede per
il suo funzionamento un tampone contenente il cofattore MgCl2,
la presenza di
deossinucleosidi 5’-trifosfato (dNTPs) e uno stampo (template), rappresentato da un
filamento di DNA a cui deve trovarsi appaiato un primer (corto oligodeossinucleotide
che funge da innesco fornendo un 3’-OH libero). La reazione di PCR si basa sull’uso di
due primer (forward e reverse, diretti in direzione opposta ma convergente, definiscono
le estremità del futuro prodotto dell’amplificazione) di lunghezza pari a una ventina di
24
nucleotidi, che sono disegnati in modo da essere esattamente complementari alle
corrispondenti sequenze fiancheggianti il tratto di DNA da amplificare, nel caso in
esame l’esone 7 e l’esone 9 del gene WT1 (Tabella 5) (89). Per ogni campione da
analizzare sono state allestite 2 reazioni di amplificazione: una per l’esone 7 e una per
l’esone 9.
Tabella 5: Sequenze dei primers utilizzati nello studio.
esone
primer
sequenza
7
forward
5’ –GACCTACGTGAATGTTCACATG- 3’
7
reverse
5’ –ACCAACACCTGGATCAGACCT- 3’
9
forward
5’ –TGCAGACATTGCAGGCATGGCAGG- 3’
9
reverse
5’ –GCACTATTCCTTCTCTCAACTGAG- 3’
La metodica si compone di tre principali passaggi (Figura 2) che vengono ripetuti per
30-40 cicli utilizzando un termociclatore automatico (Figura 3): la prima tappa è la
denaturazione che viene effettuata a temperatura di 94-95°C per separare i due
filamenti della molecola stampo. Sono infatti i primer che, nella seconda tappa della
PCR (annealing), appaiandosi ai filamenti denaturati determinano il punto di innesco
della sintesi di DNA. La reazione di annealing avviene a temperatura inferiore a quella
di denaturazione in modo da consentire ai primer di appaiarsi alle sequenze
complementari. La temperatura di annealing è un parametro variabile e critico nel
determinare la specificità della PCR: di norma questa temperatura è compresa tra 5060°C, in questo caso è stata utilizzata una temperatura di annealing di 60°C. La tappa
successiva (polimerizzazione o estensione) è condotta a 72°C, temperatura ottimale per
la Taq DNA polimerasi. Questa tappa dura in funzione della lunghezza del tratto da
sintetizzare (la Taq DNA polimerasi in media sintetizza 1kb/min) (Figura 3). Come si
può osservare, la reazione di polimerizzazione avviene a temperature inferiori rispetto
alla denaturazione, ma maggiore rispetto all’appaiamento: questo ne aumenta la
specificità riducendo gli appaiamenti errati e aspecifici degli oligonucleotidi e la
25
formazione casuale di strutture secondarie. I due primer determinano la specificità della
reazione, la quale a sua volta è funzione anche della temperatura e della durata delle
reazioni di appaiamento e di polimerizzazione, nonché della molarita' degli ioni
magnesio e della concentrazione della polimerasi.
In questo studio la PCR è stata preparata in un volume totale di 50 µl contenente:
1 g DNA
200 mmol/L di ciascun nucleotide (dNTPs)
1,5 mmol/L di MgCl2
170 g/ml di Bovine Serum Albumine (BSA)
2,5 U di Taq polimerasi
10 pmol/l di ciascun primer (Tib Molbiol Genova)
I campioni sono stati amplificati con 35 cicli con una denaturazione iniziale di 3’ a
94°C e una elongation finale di 10’ a 72 °C. Ciascun ciclo comprendeva:
denaturazione: 30’’ a 95 °C
annealing: 1’ a 60 °C
estensione: 1’ a 72 °C
In ogni batteria di amplificazione è stato compreso un campione di controllo contenente
tutti i reagenti escluso l’acido nucleico per poter evidenziare l’eventuale
contaminazione dei reagenti.
26
Figura 2: Le fasi di un ciclo di PCR
27
Figura 3: (A) Termociclatore automatico che consente la variazione della temperatura
durante i cicli di PCR. (B) Apposite provette predisposte per l’inserimento nel
termociclatore.
A
B
28
7.3 Elettroforesi del DNA
Al termine della PCR, per verificare che l’amplificazione sia effettivamente avvenuta, i
prodotti sono stati sottoposti a corsa elettroforetica su gel di agarosio (polisaccaride
purificato dell’agar capace di polimerizzare) 2% che consente di separare i frammenti
di DNA in funzione del loro peso molecolare.
Per preparare il gel di agarosio si versano in una beuta 100 ml di tampone Tris-BoratoEDTA (TBE: TRIS 900mM; H3BO3 900mM; EDTA 25mM) 0.5 X a pH 8.3 e vi si
solubilizzano 2 g di agarosio. Si riscalda nel forno a microonde impostando 3’ con
temperatura medio alta e in seguito si lascia raffreddare fino a quando diventa tiepido.
A questo punto si aggiungono 5 ml di etidio-bromuro agitando delicatamente per
solubilizzarlo (facendo attenzione a non inalarne i vapori) e si versa la soluzione
ottenuta (facendo attenzione che non si formino bolle) nell’apposita “slitta” per gel già
predisposta con il pettine per i pozzetti. Il preparato rimane allo stato liquido fino a
40°C ma per raffreddamento al di sotto di questa temperatura diventa gel. Quando il gel
solidifica si toglie il pettine e lo si inserisce nella camera elettroforetica (Figura 4)
Figura 4: Camera elettroforetica collegata all’alimentatore
29
5 µl dell’amplificato vengono mischiati a 5 µl di colorante orange G e caricati sul gel
procedendo come segue: con una micropipetta Gilson si pone il colorante in un punto
su di un foglio di parafilm e si aggiunge il DNA del campione unendolo al colorante
cercando di mescolare con cura. A questo punto i campioni sono pronti per essere
caricati nei relativi pozzetti (facendo attenzione a non forare il gel sottostante col
puntale). Sul gel si depone anche una miscela di frammenti a peso molecolare noto che
serve come riferimento per facilitare la determinazione del peso molecolare
dell’amplificato. Terminata l'operazione di caricamento si accende l'alimentatore a 130
Volt: dopo la corsa elettroforetica il gel viene visualizzato su transilluminatore a UV,
l’etidio-bromuro intercalato tra le basi azotate degli acidi nucleici evidenzia i frammenti
come bande color arancio brillante (Figura 5).
Figura 5: Esempio di
visualizzazione delle bande
(M: marker a peso molecolare
noto, 1-2-3-4-5: campioni
amplificati).
30
8. SSCP: SINGLE STRAND CONFORMATIONAL POLYMORPHISM
L’ SSCP è un metodo di separazione elettroforetica di singoli filamenti di acidi
nucleici, capace di rilevare variazioni nella sequenza anche molto piccole. Questo è
dovuto al fatto che, data la natura relativamente instabile del singolo filamento di DNA,
variazioni anche di una sola base determinano un differente accoppiamento
intramolecolare e perciò una variazione nella struttura secondaria del singolo filamento.
La conseguenza di questo sarà una variabilità nella mobilità elettroforetica, poiché le
interazioni differenziali delle molecole con le fibre del gel faranno si che i vari isomeri
della sequenza migrino differentemente. Queste modificazioni risulteranno visibili
come bande spostate.
Per la migrazione elettroforetica dell’amplificato, la metodica si avvale dell’ausilio di
gel a gradiente di poliacrilamide preparato e poi colorato con le modalità di seguito
riportate.
Il gel è composto di due soluzioni al 5 ed al 20% preparate al momento dell’uso e così
composte:
SOLUZIONE 20%
SOLUZIONE 5%
10 ml Poliacrilamide 49:1
2.5 ml Poliacrilamide 49:1
2.5 ml TBE 5X
2.5 ml TBE 5X
1.25 ml glicerolo
1.25 ml glicerolo
11.25 ml H2O
18.75 ml H2O
20 μl temed
20 μl temed
25 μl APS 30%
25 μl APS 30%
------------------
-------------------
25 ml
25 ml
31
Il gel viene preparato in un sistema orizzontale di due vetri delle dimensioni di
260x220x1 mm. Dopo aver pulito entrambi i vetri, su quello che non presenta i pozzetti
viene fatto aderire un apposito foglio di plastica che serve da supporto per il gel.
Successivamente viene montato su di esso il vetro contenente i pozzetti, quindi,
posizionando verticalmente i due vetri, si colano tra di essi le due soluzioni a diverso
gradiente con l’ausilio di una pompa peristaltica (Figura 6).
Fatto ciò, per consentire una polimerizzazione uniforme del gel, si pongono
all’estremità superiore di esso circa 3 ml di isopropanolo. Una volta avvenuta la
polimerizzazione, il gel viene collocato sulla piastra della camera di corsa contenente il
tampone TBE 0.5X e due fogli di carta bibula imbevuta di tampone vengono posti alle
estremità del gel con la funzione di elettrodi.
Nei pozzetti vengono caricati 5µl di amplificato denaturato al calore (95°C per 5
minuti) e diluito con 5µl di buffer denaturante (98% formammide deionizzata, 0.025%
xilene cianolo FF, 0.025% blu di bromofenolo e 10mM EDTA). A questo punto viene
fatta iniziare la corsa elettroforetica che avviene ad un voltaggio costante di 500Volt
con raffreddamento a 23°C ed ha una durata di circa 15-16 ore (Figura 7).
Il protocollo originario faceva uso di marcatura radioattiva, ma in seguito è stata messa
a punto una metodologia basata sul riconoscimento delle bande di migrazione tramite
colorazione con argento.
La colorazione del gel si articola nei seguenti passaggi:
-
lavaggio del gel con soluzione di Etanolo al 10% per 5’ in agitatore
-
immersione del gel in soluzione di HNO3 all’1% per 3’ in agitatore (il colorante
cambia colore e diventa verde)
-
lavaggio in acqua per 5’
-
aggiunta di AgNO3 12mM (0.2% in 500ml) lasciando in agitazione per 20’
-
lavaggio in acqua per 5’
-
spostamento del gel in nuova vaschetta ed aggiunta di soluzione di NaCO3
0.28M e formalina 0.019%. L’agitazione è manuale e quando la soluzione
diventa scura si cambia (almeno 3 volte)
-
blocco della reazione, quando le bande sono abbastanza scure, con acido acetico
al 10% agitando manualmente per 2’
-
essiccamento per 1h e 30’-2h.
32
Figura 6: Apparecchiatura per l’allestimento del gel per SSCP.
Figura 7: Gel nella fase iniziale della corsa elettroforetica.
33
9. ANALISI DI SEQUENZA
I campioni che presentavano profili SSCP anomali sono stati sequenziati utilizzando i
prodotti di amplificazione della PCR previa purificazione (QIAquick PCR Purification
Kit; QIAGEN) per rimuovere i nucleotidi non incorporati e i primer residui.
9.1 Reazione di sequenza
La reazione di sequenza è stata effettuata mediante il kit commerciale “Big Dye
Terminator Cycle Sequencing Ready Reaction” (Applied Biosystems) che utilizza il
metodo dei didesossi-nucleotidi (ddNTPs) o di Sanger: tale metodica si basa sulla
sintesi enzimatica di una nuova catena di DNA su un’elica stampo, utilizzando, oltre ai
normali dNTPs, analoghi ddNTPs marcati con diversi fluorocromi. L’assenza di un 3’OH nei ddNTPs impedisce la formazione di un legame fosfodiesterico con il successivo
precursore e, quindi, l’incorporazione di ddNTP (“terminatore”) porta all’arresto della
reazione polimerasica. Nella reazione di sequenza la concentrazione dei ddNTPs deve
essere circa 1/100 di quella dei dNTPs, in quanto l’incorporazione dei terminatori deve
essere del tutto casuale e garantire, comunque, una certa sintesi di DNA.
Per ciascuna reazione di sequenza sono stati utilizzati 2μl di Terminator Ready
Reaction Mix e 1.6 pmoli di primer (gli stessi primers usati per l’amplificazione)
(Figura 8).
Completata la reazione di sequenza, si procede con la purificazione del prodotto per
rimuovere l’eccesso di ddNTPs marcati con fluorocromi mediante colonnine a base di
sephadex che trattengono i nucleotidi singoli ma non i frammenti.
9.2 Elettroforesi capillare su sequenziatore automatico
I prodotti di reazione purificati (3.5 μl) sono risospesi in formamide (10 μl), denaturati
a 95°C per 5’ e sottoposti a corsa elettroforetica con lo strumento ABI PRISM® 310
Genetic Analyzer (Applied Byosistems). I frammenti, che differiscono tra loro per una
sola base, vengono separati in funzione della loro lunghezza tramite elettroforesi
capillare dal sequenziatore automatico e le bande sono rilevate attraverso un sofisticato
sistema ottico che, rilevando la differenza tra le lunghezze d’onda emesse dai quattro
34
fluorocromi legati ai ddNTPs, permette l’identificazione dell’esatta successione delle
basi nel segmento di DNA in esame (Figura 9).
Per una corretta interpretazione dei dati i picchi di fluorescenza devono essere ben
definiti, con sfondo scarso o nullo.
9.3 Analisi dei dati
Le sequenze ottenute sono state comparate con le sequenze depositate in banca dati
GenBank (http://www.ncbi.nlm.nih.gov/) tramite il programma Blast dell’NCBI
utilizzando il software ChromasPro.
Figura 8: Schema della reazione di sequenza
35
Figura 9: Determinazione di una sequenza di basi tramite sequenziatore automatico
che rileva le differenti lunghezze d’onda emesse dai quattro fluorocromi legati ai
ddNTPs (ddATPs: verde, ddGTPs : giallo, ddCTPs: blu, ddTTPs: rosso).
Le fasi dell’analisi di sequenza possono essere quindi così riepilogate:
1. Amplificazione del DNA (PCR)
2. Purificazione dell’amplificato
3. Reazione di sequenza
4. Purificazione della reazione di sequenza
5. Denaturazione
6. Corsa elettroforetica
7. Lettura dei dati ottenuti
36
RISULTATI
10.
FREQUENZA E TIPOLOGIA DELLE MUTAZIONI DI WT1
Tra gli amplificati sottoposti a SSCP (Figura 10) sono stati rilevati 41 profili di
migrazione anomali relativamente all’esone 7 e 22 a carico dell’esone 9.
I risultati del sequenziamento (Figura 11) di ciascun profilo anomalo hanno
confermato la presenza di 30 variazioni di sequenza sull’esone 7 localizzate in 27
pazienti (13,5%), mentre una sola mutazione è stata riscontrata sull’esone 9 (0.5%)
(Tabella 6). La frequenza delle suddette variazioni di WT1 nei casi di LAM-CN
analizzati è risultato pertanto del 14 %.
Tra le 30 variazioni di sequenza relative all’esone 7, 23 erano raggruppate in regioni
distinte segnalate come hot-spots nel precedente studio di Virappane et al. (89). La
prima di queste regioni, posta tra i nucleotidi 1301-1307, presentava 20 sostituzioni di
A con G in forma eterozigote e una sostituzione A-G in forma omozigote, mentre la
seconda regione, posta tra i nucleotidi 1333 e il 1341 presentava 2 mutazioni puntiformi
consistenti rispettivamente in una sostituzione C-T e una T-G, entrambi in forma
eterozigote. Altre 4 mutazioni rappresentavano dei frameshift, mentre 3 cadevano in
punti non segnalati in letteratura e sono state riscontrate in pazienti che presentavano
anche altre variazioni di sequenza nelle regioni considerate hot-spots.
L’unica mutazione dell’esone 9 evidenziata in questo studio, consistente in una
sostituzione T-C in forma eterozigote, era situata nell’hot spot tra i nucleotidi 15881591 (89).
10.1
Single nucleotide polymorphism (SNP) nell’hot spot dell’esone 7
Le 21 sostituzioni A-G riscontrare nell’hot spot 1301-1307 rappresentano un
polimorfismo a singolo nucleotide (SNP) già descritto in letteratura da Damm et al.
come SNP rs16754 (92). La presenza dell’allele minore di questo polimorfismo, sia in
eterozigosi (WT1AG) che in omozigosi (WT1GG), avrebbe secondo questi autori un
significato prognostico favorevole rispetto alla presenza dell’allele maggiore (WT1AA).
I pazienti individuati in questo studio come portatori dell’allele minore di SNP rs16754,
pari al 10.5% di quelli analizzati, avevano tutti genotipo FLT3-ITDneg e un terzo di essi
erano anche NPM1mut, presentavano pertanto un quadro genetico favorevole a basso
37
rischio. Tuttavia solamente 4 di essi hanno avuto un outcome clinico favorevole, un
paziente è ricaduto, 9 sono deceduti a causa della malattia (tra questi 1 paziente è stato
sottoposto solo a terapia di supporto a causa dell’età molto avanzata e un altro invece
ha rifiutato il trattamento chemioterapico) e 2 per altre cause, mentre dei rimanenti 5
non è stato possibile reperire notizie cliniche (Tabella 6).
10.2 Frameshift dell’esone 7
I 4 pazienti in cui è stato riscontrato un frameshift nella regione dell’esone 7 di WT1
hanno avuto tutti un outcome clinico negativo (1 non ha risposto alle terapie e 3 sono
ricaduti sviluppando chemioresistenza). Tra questi solamente 1 mostrava un quadro
sfavorevole dovuto al genotipo FLT3-ITDpos e assenza di mutazioni di NPM1.
Un altro paziente presentava invece una prognosi apparentemente favorevole dovuta al
genotipo FLT3-ITDneg/NPM1mut ; tuttavia, nonostante una buona risposta iniziale alla
terapia di induzione con il raggiungimento della remissione a livello clinico ed
ematologico (ma non molecolare), è successivamente ricaduto sviluppando resistenza ai
chemioterapici.
Uno dei pazienti con genotipo FLT3-ITDneg e assenza di mutazioni di NPM1 presentava
valori relativamente elevati di espressione di BAALC e WT1 (Tabella 6) e non ha
risposto alle terapie. Mentre l’altro paziente, anch’esso con genotipo FLT3-ITDneg e
assenza di mutazioni di NPM1, mostrava invece valori normali di BAALC e WT1
positivo, tuttavia la risposta alla terapia non è stata ottimale ed è deceduto durante
l’induzione pre-trapianto.
10. 3 Mutazioni puntiformi dell’esone 7 e 9 per sostituzione di una singola base
Tutte le sostituzioni di singola base (2 nell’esone 7 e 1 nell’esone 9) situate negli hot
spots erano presenti in forma eterozigote. Tra i pazienti che avevano questo tipo di
mutazione
solamente
uno
presentava
il
genotipo
sfavorevole
FLT3-ITDpos
contemporaneamente ad un valore elevato di BAALC ed è stato avviato al trapianto
eseguito con successo. Gli altri 2 pazienti, che presentavano un profilo genetico e
molecolare a basso rischio, hanno avuto un outcome clinico favorevole.
38
Tabella 6: Variazioni di sequenza degli esoni 7 e 9 e valori degli altri marcatori noti.
mutazione
esone 7
n.
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
regione
1301-1307
1301-1307
1301-1307
1301-1307
1301-1307
1301-1307
1301-1307
1301-1307
1301-1307
1301-1307
1301-1307
1301-1307
1301-1307
1301-1307
1301-1307
1301-1307
1301-1307
1301-1307
1301-1307
1301-1307
1301-1307
1333-1341
1333-1341
-----------------
tipo
A-G
A-G
A-G *
A-G
A-G
A-G
A-G
A-G
A-G
A-G
A-G
A-G
A-G
A-G
A-G
A-G
A-G
A-G
A-G
A-G
G-G*
C-T
T-G *
frame shift
frame shift
frame shift
frame shift
WT1-ELN
694
1884
122
480
41,5
1329,7
0
343
459
1784,1
985
2463
815,5
38,8
138,3
2
553
148
292
851
9,6
1234,6
862
453
797
ITD FLT3
neg
neg
neg
neg
neg
neg
neg
neg
neg
neg
neg
neg
neg
neg
neg
neg
neg
neg
neg
neg
neg
neg
neg
neg
neg
neg
pos
FLT3 20
neg
neg
neg
neg
neg
neg
neg
neg
neg
neg
neg
neg
neg
neg
neg
neg
neg
neg
neg
neg
neg
neg
neg
neg
neg
neg
neg
NPM-A
0
6
0
28
0
0
0
0
53
2
0
0
1
0
0
284
0
0
0
0
8552
0
0
0
15499
0
NPM-B
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
657
0
0
0
13087
0
BAALC
1107
4278
158555
609
3629
375
1916
42690
140
3838
3588
29389
30696
383
240
195
33851
21
117164
62697
23
867
3417
744
222
390
Outcome clinico
----decesso
ricaduta
favorevole
favorevole
decesso
decesso
decesso
decesso (non dovuto alla malattia)
decesso
favorevole
favorevole
decesso
----decesso
decesso (non dovuto alla malattia)
----decesso
decesso
--------favorevole
favorevole
non risposta alla terapia
ricaduta e decesso
ricaduta e decesso
ricaduta e decesso
esone 9
1
1588-1591
T-C
1379
pos
neg
3
0
25030
trapianto
* presenza di un’ulteriore mutazione puntiforme in punti non segnalati in letteratura.
39
Figura 10: SSCP in cui sono evidenziati profili di migrazione anomali visibili come
una triplice banda.
Dettaglio che evidenzia la triplice
banda
40
Figura 11: Esempi di elettroferogrammi con profilo normale (A) e con mutazione
puntiforme (B).
A
N
B
mutazione puntiforme
41
DISCUSSIONE
11. SCREENING DELLE MUTAZIONI PUNTIFORMI MEDIANTE SSCP
L’analisi di sequenza è la metodica di eccellenza per la ricerca di mutazioni puntiformi
(sia già note che non note) in virtù della sensibilità estrema e dell’informazione
completa che fornisce su posizione e natura della mutazione.
La SSCP, pur essendo estremamente suscettibile a piccole variazioni delle condizioni
standard, è una tecnica semplice da usare e l’analisi dei dati ottenuti risulta agevole.
I risultati ottenuti in questo studio hanno dimostrato che, con l’impiego di questa
metodica, è stato possibile rilevare la presenza di tutte le tipologie di variazione di
sequenza degli esoni 7 e 9 di WT1 descritte in letteratura, siano esse frameshift o
polimorfismi di un singolo nucleotide.
Pertanto lo screening dei campioni tramite SSCP può rivelarsi un sistema utile per
ridurre il numero di casi da sottoporre all’analisi di sequenza; si potrebbero infatti
ottenere dei vantaggi in termini di riduzione della mole di lavoro e dei costi.
12. SIGNIFICATO PROGNOSTICO E BIOLOGICO DELLE MUTAZIONI DI
WT1
12.1 Valore prognostico delle variazioni di sequenza di WT1
Sin dalle prime descrizioni delle mutazioni di WT1 nelle LAM (93), risalenti agli anni
’90, è stato ipotizzato che la loro presenza fosse associata con un andamento clinico
sfavorevole, tuttavia la scarsità degli studi disponibili (in particolare nelle LAM
dell’adulto) non consentiva di affermarlo con certezza. Il ruolo di WT1 nella
leucemogenesi non era ancora stato pienamente compreso e il meccanismo con cui le
mutazioni di WT1 potevano conferire alle cellule leucemiche la resistenza ai
chemioterapici non era noto.
Gli studi di Summers (88), Virappane (89) e Paschka (90), condotti nel periodo 20072008 attribuiscono alla presenza di mutazioni frameshift negli esoni 7 e 9 di WT1 un
impatto negativo sulla prognosi, mentre nel lavoro di Gaidzik del 2009 (91) il
significato prognostico sfavorevole delle mutazioni di WT1 viene ridimensionato: esso
sarebbe infatti valido solo in associazione alla presenza del genotipo FLT3-ITDpos.
42
Nel più recente studio di Damm et al. (92), condotto su 249 pazienti con LAM-CN, la
presenza dell’allele minore del polimorfismo a singolo nucleotide rs16754 dell’esone 7
di WT1 viene considerata come un fattore prognostico favorevole indipendente, mentre
le mutazioni di WT1 vengono indicate come prive di impatto prognostico. L’allele
minore di tale polimorfismo, presente nella popolazione sana con una frequenza
analoga a quella riscontrata nei pazienti analizzati, conferirebbe secondo gli autori una
maggiore sensibilità ai farmaci chemioterapici, sebbene i meccanismi con cui questo
avviene non siano noti.
I risultati di questi studi non devono essere necessariamente considerati contradditori
tra loro in quanto mutazioni differenti possono avere varie conseguenze per le funzioni
cellulari e quindi un significato diverso nella patogenesi e un impatto clinico differente.
I dati presentati nel corrente studio contribuiscono all’ampliamento della casistica delle
descrizioni delle variazioni di sequenza di WT1. Tra le 27 alterazioni della sequenza qui
evidenziate nell’esone 7, le 4 mutazioni frameshift si confermano come indicatore
prognostico sfavorevole indipendente, sia per la risposta alla terapia che per la
sopravvivenza. Per quanto riguarda i 21 pazienti che presentavano il polimorfismo SNP
rs16754 nell’esone 7, essi pur avendo un quadro complessivo a basso rischio dal punto
di vista genetico, non hanno mostrato un outcome clinico favorevole. Tuttavia non è
possibile definire se l’outcome negativo sia da imputare a questo solo fattore o anche ad
altri parametri, alcuni pazienti mostravano infatti anche valori elevati di BAALC ed
un’età avanzata.
12.2 Significato biologico delle mutazioni di WT1
Mutazioni frameshift analoghe a quelle riscontrate in questo studio, già descritte in
studi precedenti (87-89, 93-94), comportano la sintesi di proteine WT1 tronche. Non è
ancora chiaro se queste proteine tronche siano stabili all’interno dell’ambiente cellulare
oppure instabili in quanto bersaglio di enzimi di degradazione, in ogni caso non
sarebbero in grado di traslocare nel nucleo a causa della perdita del segnale di
localizzazione nucleare. Va inoltre considerato che le proteine tronche vengono ad
essere prive anche dei domini di legame per l’interazione con le proteine regolatorie
delle funzioni di WT1, tra cui l’oncosoppressore p53 (32).
43
Per quanto riguarda le mutazioni nell’esone 9, quelle frameshift sono meno frequenti
rispetto all’esone 7: in questo studio non ne sono state riscontrate, mentre è stata
evidenziata 1 mutazione missenso. Mutazioni di WT1 di questo tipo erano già state
descritte da Virappane et al nelle LAM e anche in altre patologie (95-96): la
sostituzione amminoacidica sembra tradursi nella perdita della capacità di legame al
DNA da parte della proteina (97). Le mutazioni di WT1 di questo tipo, dunque, sono in
grado di impedire o modificare il legame della proteina WT1 ai propri geni bersaglio,
inclusi quelli che codificano per proteine coinvolte nella regolazione della normale
ematopoiesi (RARA, CSF1), dell’apoptosi (BCL2, BCL2A1, BAK1), del ciclo cellulare
(CCNE1, CDKN1A), della trascrizione (MYC, PAX2, MYB, EGR1) e della
proliferazione cellulare (TGFB1, PDGFA) (34).
L’altra tipologia di alterazione di sequenza di WT1 individuata in questo studio,
descritta da Damm et al. nel 2010 come l’allele minore di SNP rs16754, non ha ancora
un chiaro significato biologico. I meccanismi con cui esso determinerebbe
un’aumentata sensibilità delle cellule leucemiche ai farmaci chemioterapici potrebbero
essere molteplici: alterazioni nella stabilità, nel ripiegamento o nello splicing dell’RNA,
differenze nella selezione del tRNA, oppure vi potrebbe essere un’interazione con le
isoforme di altri geni coinvolti nel controllo del metabolismo, del trasporto o
dell’eliminazione dei farmaci (92).
13. CONCLUSIONI
I protocolli terapeutici attuali per la leucemia mieloide acuta si basano su fattori
prognostici come l’età, il conteggio dei leucociti, il cariotipo e le mutazioni genetiche
che contribuiscono alla stratificazione terapeutica. Il monitoraggio della MRD tramite
il rilevamento di target specifici come i trascritti dei geni di fusione o le mutazioni
consente di
individuare i pazienti a elevato rischio di recidiva. La risposta alla
chemioterapia di induzione offre un ulteriore fattore predittivo per avvalorare le
decisioni relative alla terapia di consolidamento, in particolare per quanto riguarda il
trapianto allogenico. Vi è tuttora l’esigenza di raffinare la stratificazione del rischio
tramite lo sviluppo di approcci alternativi per consentire il rilevamento di MRD per
individuare in modo più affidabile i pazienti che possono trarre effettivamente
vantaggio dal trapianto: circa la metà dei pazienti con LAM presenta infatti una carenza
44
di target specifici adeguati. In questo contesto le mutazioni di WT1 potrebbero
rappresentare un utile nuovo marcatore di MRD.
Lo studio del profilo molecolare di un ampio numero di casi clinici di cui sia possibile
disporre di un adeguato follow-up sarà un elemento essenziale per poter determinare la
rilevanza delle variazioni di sequenza di WT1 (siano esse mutazioni o polimorfismi di
un singolo nucleotide) nello sviluppo di nuovi modelli che consentano di migliorare la
stratificazione del rischio e aumentare l’efficacia del trattamento tramite la messa a
punto di terapie mirate.
La valutazione delle variazioni di sequenza del gene WT1 sembra, pertanto, un utile
elemento da aggiungere agli altri parametri (espressione di WT1, BAALC, alterazioni
FLT3, mutazione NMP1) all’esordio di una LAM: esso infatti può rappresentare un
fattore prognostico indipendente che potrebbe anche rivelarsi determinante nella scelta
del trattamento terapeutico più appropriato.
45
BIBLIOGRAFIA
1)
Lowenberg B, Downing JR and Burnett A. Acute myeloid leukaemia. N Eng J Med 1999; 341:
1051-1062.
2)
Tura S, Baccarani M. Malattie del sangue e degli organi emolinfopoietici. Ed. Esculapio srl
2007; 136-152.
3)
Bennet JM, Catowsky D, Daniel MT, et al. Proposals for the classification of acute myeloid
leukaemia: a report of the French-American-British Cooperative Group. Br J Haematol 1976; 33:
451-458.
4)
Harris NL, Jaffe ES, Diebold J, et al. World Health Organization classification of neoplastic
diseases of the hematopoietic and lymphoid tissues: report of the Clinical Advisory Committee
meeting-Airlie House, Virginia, November 1997. J Clin Oncol. 1999;17:3835–3849.
5)
Vardiman JW, Harris NL, Brunning RD. The World Health Organization (WHO)
classification of the myeloid neoplasms. Blood. 2002;100:2292–2302.
6)
Löwenberg B. Diagnosis and prognosis in acute myeloid leukemia--the art of distinction. N Engl
J Med. 2008; 358:1960-1962.
7)
Gilliland DG, Jordan CT, Felix CA. The molecular basis of Leukemia. Hematology Am Soc
Hematol Educ Program. 2004;80-97.
8)
Dohner K, Dohner H. Molecular characterization of acute myeloid leukemia. Haematologica.
2008; 93:976-982.
9)
Mrozek K, Heerema NA, Bloomfield CD. Cytogenetics in acute leukemia. Blood Rev.
2004;18:115-136.
10)
Leith C, Kopecky K, Godwin J, et al. Acute myeloid leucemia in the elderly: assessment of
multidrug resistance (MDR1) and cytogenetics distinguishes biologic subgroups with remarkably
distinct responses to standard chemotherapy. A Southwest Oncology Group study. Blood 1997;
89: 3323-3329.
11)
Mrozek K, Heinonen K, de la Chapelle A, et al. Clinical significance of cytogenetics in acute
myeloid leukemia.Semin Oncol. 1997; 24:17-31.
12)
Grimwade D, Walker H, Oliver F, et al. The importance of diagnostic cytogenetics on outcome
in AML: analysis of 1.612 patients entered into the MRC AML 10 trial. Blood 1998; 7:2322 2333.
13)
Slovak ML, Kopecky KJ, Cassileth PA, et al. Karyotypic analysis predicts outcome of preremission and post-remission therapy in adult acute myeloid leukemia: a Southwest Oncology
Group/Eastern Cooperative Oncology Group study. Blood. 2000;96:4075–4083.
14)
Byrd JC, Mrózek K, Dodge RK, et al. Pre-treatment cytogenetic abnormalities are predictive of
induction success, cumulative incidence of relapse, and overall survival in adult patients with de
novo acute myeloid leukemia: results from Cancer and Leukemia Group B (CALGB 8461).
Blood. 2002;100:4325–4336.
46
15)
Breems DA, Van Putten WL, De Greef GE, et al. Monosomal karyotype in acute myeloid
leukemia: a better indicator of poor prognosis than a complex karyotype. J Clin Oncol. 2008;
26:4791.
16)
Mrozek K, Dohner H, Bloomfield C. Influence of new molecular prognostic markers in patients
with karyotypically normal acute myeloid leukemia: recent advances. Curr Opin Hematol.
2007;14:106-114.
17)
Schlenk RF, Do¨hner K, Krauter J, et al. Mutations and treatment outcome in cytogenetically
normal acute myeloid leukemia. N Engl J Med. 2008; 358:1909-1918.
18)
Rucker FG, Bullinger L, Schwaenen C, et al. Disclosure of candidate genes in acute myeloid
leukemia with complex karyotypes using microarray-based molecular characterization. J Clin
Oncol. 2006;24:3887–3894.
19)
Chen PM, Yen CC, Wang WS, et al. Down-regulation of Notch-1 expression decreases PU.1mediated myeloid differentiation signaling in acute myeloid leukemia. Int J Oncol. 2008;32:13351341.
20)
Meyer S, Barber LM, White DJ, et al. Spectrum and significance of variants and mutations in
the Fanconi anaemia group G gene in children with sporadic acute myeloid leukaemia. Br J
Haematol. 2006;133:284-292.
21)
Lee JW, Kim YG, Soung YH, et al. The JAK2 V617F mutation in de novo acute myelogenous
leukemias. Oncogene. 2006; 25:1434-1436.
22)
Wojcik I, Szybka M, Golanska E, et al. Abnormalities of the P53, MDM2, BCL2 and BAX genes
in acute leukemias. Neoplasma. 2005; 52:318-324.
23)
Aggerholm A, Grønbaek K, Guldberg P, et al. Mutational analysis of the tumour suppressor
gene MMAC1/PTEN in malignant myeloid disorders. Eur J Haematol. 2000; 65:109-113.
24)
Visser M, Hofstra RM, Stulp RP, et al. Absence of mutations in the RET gene in acute myeloid
leukemia. Ann Hematol. 1997; 75:87-90.
25)
Hestekin CN, Barron AE. The potential of electrophoretic mobility shift assays for clinical
mutation detection. Electrophoresis. 2006; 27: 3805-3815. Review.
26)
Bianchini M, Ottaviani E, Grafone T, et al. Rapid detection of FLT3 mutations in acute
myeloid leukemia patients by denaturing HPLC. Clin Chem. 2003; 49:1642-1650.
27)
Bacher U, Zander AR, Haferlach T, et al. Minimal residual disease diagnostics in myeloid
malignancies in the post transplant period. Bone Marrow Transplant. 2008; 42:145-157.
28)
Kern W, Haferlach C, Haferlach T, et al. Monitoring of minimal residual disease in acute
myeloid leukemia. Cancer. 2008;112:4-16. Review.
29)
Sakhinia E, Farahangpour M, Tholouli E, et al. Comparison of gene-expression profiles in
parallel bone marrow and peripheral blood samples in acute myeloid leukaemia by real-time
polymerase chain reaction. J Clin Pathol. 2006; 59:1059-1065.
30)
Picard C, Silvy M, Gabert J. Overview of real-time RT-PCR strategies for quantification of
gene rearrangements in the myeloid malignancies. Methods Mol Med. 2006;125:27-68.
47
31)
Yang L, Han Y, Suarez Saiz F, et al. A tumor suppressor and oncogene: the WT1 story.
Leukemia. 2007;21:868-876.
32)
Hohenstein P, Hastie ND. The many facets of the Wilms' tumour gene, WT1. Hum Mol Genet.
2006; 15: R196-201.
33)
Nishida S, Hosen N, Shirakata T, et al. AML1-ETO rapidly induces acute myeloblastic leukemia
in cooperation with the Wilms tumor gene, WT1. Blood. 2006 ;107:3303-3312.
34)
Ariyaratana S, Loeb DM. The role of the Wilms tumour gene (WT1) in normal and malignant
haematopoiesis. Expert Rev Mol Med. 2007 ;9:1-17.
35)
Park S, Schalling M, Bernard A, et al. The Wilms tumour gene WT1 is expressed in murine
mesoderm-derived tissues and mutated in a human mesothelioma. Nat Genet. 1993; 4:415-420.
36)
Pritchard-Jones K, Fleming S. Cell types expressing the Wilms' tumour gene (WT1) in Wilms'
tumours: implications for tumour histogenesis. Oncogene. 1991; 6: 2211-2220.
37)
Baird PN, Simmons PJ. Expression of the Wilms' tumor gene (WT1) in normal hemopoiesis.
Exp Hematol. 1997; 25: 312-320.
38)
Maurer U, Brieger J, Weidmann E, et al. The Wilms' tumor gene is expressed in a subset of
CD34+ progenitors and downregulated early in the course of differentiation in vitro. Exp
Hematol. 1997; 25: 945-950.
39)
Menssen HD, Renkl HJ, Entezami M, et al. Wilms' tumor gene expression in human CD34+
hematopoietic progenitors during fetal development and early clonogenic growth. Blood. 1997;
89:3486-3487.
40)
Menssen HD, Renkl HJ, Rodeck U, et al. Presence of Wilms' tumor gene (WT1) transcripts and
the WT1 nuclear protein in the majority of human acute leukemias. Leukemia. 1995 ;9:10601067.
41)
Miwa H, Beran M, Saunders GF. Expression of the Wilms' tumor gene (WT1) in human
leukemias. Leukemia. 1992; 6: 405-409.
42)
Inoue K, Sugiyama H, Ogawa H, et al. WT1 as a new prognostic factor and a new marker for
the detection of minimal residual disease in acute leukemia Blood. 1994; 84:3071-3079.
43)
Barragán E, Cervera J, Bolufer P, et al. Prognostic implications of Wilms' tumor gene (WT1)
expression in patients with de novo acute myeloid leukemia. Haematologica. 2004; 89:926-933.
44)
Garg M, Moore H, Tobal K, et al. Prognostic significance of quantitative analysis of WT1 gene
transcripts by competitive reverse transcription polymerase chain reaction in acute leukaemia.
British Journal of Hematology 2003; 123: 49-59.
45)
Santamaría CM, Chillón MC, García-Sanz R, et al. Molecular stratification model for
prognosis in cytogenetically normal acute myeloid leukemia.Blood. 2009;114:148-152.
46)
Rodrigues PC, Oliveira SN, Viana MB, et al. Prognostic significance of WT1 gene expression in
pediatric acute myeloid leukemia. Pediatr Blood Cancer. 2007; 49:133-138.
47)
Lasa A, Carricondo M, Estivill C, et al. WT1 monitoring in core binding factor AML:
Comparison with specific chimeric products. Leuk Res. 2009; 33:1643-1649
48
48)
Moore SD, Offor O, Ferry JA, et al. ELF4 is fused to ERG in a case of acute myeloid leukemia
with a t(X;21)(q25-26;q22). Leuk Res.2006;30:1037-1042.
49)
Marcucci G, Maharry K, Whitman SP, et al. High expression levels of the ETS-related gene,
ERG, predict adverse outcome and improve molecular risk-based classification of cytogenetically
normal acute myeloid leukemia: a Cancer and Leukemia Group B Study. J Clin Oncol. 2007;
25:3337-3343.
50)
Marcucci G, Baldus CD, Ruppert AS, et al. Overexpression of the ETS-related gene, ERG,
predicts a worse outcome in acute myeloid leukemia with normal karyotype: a Cancer and
Leukemia Group B study. J Clin Oncol. 2005; 23: 9234-9242.
51)
Eid MA, Attia M, Abdou S, et al. BAALC and ERG expression in acute myeloid leukemia with
normal karyotype: impact on prognosis. Int J Lab Hematol. 2009.
52)
Baldus CD, Liyanarachchi S, Mrózek K, et al. Acute myeloid leukemia with complex
karyotypes and abnormal chromosome 21: Amplification discloses overexpression of APP, ETS2,
and ERG genes. Proc Natl Acad Sci USA. 2004; 101:3915-3920.
53)
Metzeler KH, Dufour A, Benthaus T, et al. ERG expression is an independent prognostic factor
and allows refined risk stratification in cytogenetically normal acute myeloid leukemia: a
comprehensive analysis of ERG, MN1, and BAALC transcript levels using oligonucleotide
microarrays. J Clin Oncol. 2009;27:5031-5038.
54)
Qi X, Shen Y, Cen J, et al. Up-regulation of BAALC gene may be an important alteration in
AML-M2 patients with t(8;21)translocation. J Cell Mol Med. 2008; 12: 2301-2304.
55)
Baldus CD, Tanner SM, Ruppert AS, et al. BAALC expression predicts clinical outcome of de
novo acute myeloid leukemia patients with normal cytogenetics: a Cancer and Leukemia Group B
Study. Blood. 2003; 102:1613-1618.
56)
Baldus CD, Thiede C, Soucek S, et al. BAALC expression and FLT3 internal tandem duplication
mutations in acute myeloid leukemia patients with normal cytogenetics: prognostic implications. J
Clin Oncol. 2006; 24: 790-797.
57)
Langer C, Radmacher MD, Ruppert AS, et al. High BAALC expression associates with other
molecular prognostic markers, poor outcome, and a distinct gene-expression signature in
cytogenetically normal patients younger than 60 years with acute myeloid leukemia: a Cancer and
Leukemia Group B (CALGB) study. Blood. 2008; 111:5371-5379.
58)
Langer C, Marcucci G, Holland KB, et al. Prognostic importance of MN1 transcript levels, and
biologic insights from MN1-associated gene and microRNA expression signatures in
cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin
Oncol. 2009; 27: 3198-3204.
59)
Meester-Smoor MA, Janssen MJ, Grosveld GC, et al. MN1 affects expression of genes
involved in hematopoiesis and can enhance as well as inhibit RAR/RXR-induced gene expression.
Carcinogenesis. 2008; 29: 2025-2034.
60)
Carella C, Bonten J, Sirma S, et al. MN1 overexpression is an important step in the
development of inv(16) AML. Leukemia. 2007; 21: 1679-1690.
61)
Dohner K, Hinrichsen T, Rudolph C, et al. MN1 overexpression induces acute myeloid
leukemia in mice and predicts ATRA resistance in patients with AML. Blood. 2007; 110: 16391647.
49
62)
Heuser M, Beutel G, Krauter J, et al. High meningioma 1 (MN1) expression as a predictor for
poor outcome in acute myeloid leukemia with normal cytogenetics. Blood. 2006; 108: 3898-3905.
63)
Falini B, Nicoletti I, Bolli N, et al. Translocations and mutations involving the nucleophosmin
(NPM1) gene in lymphomas and leukemias. Haematologica 2007; 92: 519–532.
64)
Falini B, Mecucci C, Tiacci E, et al. GIMEMA Acute Leukemia Working Party. Cytoplasmic
nucleophosmin in acute myelogenous leukemia with a normal karyotype N Engl J Med. 2005;
352: 254-266.
65)
Schnittger S, Schoch C, Kern W, et al. Nucleophosmin gene mutations are predictors of
favorable prognosis in acute myelogenous leukemia with a normal karyotype. Blood. 2005; 106:
3733–3739.
66)
Dohner K, Schlenk RF, Habdank M, et al. Mutant nucleophosmin (NPM1) predicts favorable
prognosis in younger adults with acute myeloid leukemia and normal cytogenetics: interaction
with other gene mutations. Blood. 2005; 106: 3740–3746.
67)
Verhaak RG, Goudswaard CS, van Putten W, et al. Mutations in nucleophosmin (NPM1) in
acute myeloid leukemia (AML): association with other gene abnormalities and previously
established gene expression signatures and their favorable prognostic significance. Blood.
2005;106: 3747–3754.
68)
Thiede C, Koch S, Creutzig E, et al. Prevalence and prognostic impact of NPM1 mutations in
1485 adult patients with acute myeloid leukemia (AML). Blood. 2006; 107: 4011–4020.
69)
Lo-Coco F, Cuneo A, Pane F, et al. Acute Leukemia Working Party of the GIMEMA group.
Prognostic impact of genetic characterization in the GIMEMA LAM99P multicenter study for
newly diagnosed acute myeloid leukemia. Haematologica. 2008; 93: 1017-1024.
70)
Mrózek K, Marcucci G, Paschka P, et al. Clinical relevance of mutations and gene-expression
changes in adult acute myeloid leukemia with normal cytogenetics: are we ready for a
prognostically prioritized molecular classification? Blood. 2007; 109: 431-448.
71)
Schlenk RF, Döhner K, Krauter J, et al. German-Austrian Acute Myeloid Leukemia Study
Group. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N
Engl J Med. 2008; 358:1909-1918.
72)
Gale RE, Green C, Allen C, et al. The impact of FLT3 internal tandem duplication mutant level,
number, size and interaction with NPM1 mutations in a large cohort of young adult patients with
acute myeloid leukemia. Blood. 2008; 111: 2776–2784.
73)
Kottaridis PD, Gale RE, Frew, ME, et al. The presence of a FLT3 internal tandem duplication
in patients with acute myeloid leucemia (AML) adds important prognostic information to
cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients
from the United Kingdom Medical Research Council AML 10 and 12 trials. Blood. 2001; 98:
1752–1759.
74)
Stirewalt, DL & Radich JP. The role of FLT3 in haematopoietic malignancies. Nature Reviews
Cancer. 2003; 3: 650–665.
75)
Döhner K, Döhner H. Molecular characterization of acute myeloid leukemia. Haematologica.
2008; 93: 976-982.
50
76)
Dohner K, Schlenk RF, Habdank M, et al. Mutant nucleophosmin (NPM1) predicts
favorable prognosis in younger adults with acute myeloid leukemia and normal cytogenetics:
interaction with other gene mutations. Blood. 2005; 106: 3740–3746.
77)
Boonthimat C, Thongnoppakhun W, Auewarakul CU. Nucleophosmin mutation in Southeast
Asian acute myeloid leukemia: eight novel variants, FLT3 coexistence and prognostic impact of
NPM1/FLT3 mutations. Haematologica. 2008; 93:1565-1569.
78)
Preudhomme C, Sagot C, Boissel N, et al. Favorable prognostic significance of CEBPA
mutations in patients with de novo acute myeloid leukemia: a study from the Acute Leukemia
French Association (ALFA). Blood. 2002; 100: 2717–2723.
79)
Frohling S, Schlenk RF, Stolze I, et al. CEBPA mutations in younger adults with acute myeloid
leukemia and normal cytogenetics: prognostic relevance and analysis of cooperating mutations.
Journal of Clinical Oncology. 2004; 22: 624–633.
80)
Wouters BJ, Löwenberg B, Erpelinck-Verschueren CA, et al. Double CEBPA mutations, but
not single CEBPA mutations, define a subgroup of acute myeloid leukemia with a distinctive gene
expression profile that is uniquely associated with a favorable outcome. Blood. 2009; 113: 30883091.
81)
Renneville A, Roumier C, Biggio V, et al. Cooperating gene mutations in acute myeloid
leukemia: a review of the literature. Leukemia. 2008; 22: 915-931.
82)
Dohner K, Tobis K, Ulrich R, et al. Prognostic significance of partial tandem duplications of the
MLL gene in adult patients 16 to 60 years old with acute myeloid leukemia and normal
cytogenetics: a study of the Acute Myeloid Leukemia Study Group UIm. Journal of Clinical
Oncology. 2002; 20: 3254–3261.
83)
Munoz L, Nomdedeu JF, Villamor N, et al. Acute myeloid leukemia with MLL rearrangements:
clinicobiological features, prognostic impact and value of flow cytometry in the detection of
residual leukemic cells. Leukemia. 2003; 17: 76–82.
84)
Bowen DT, Frew ME, Hills R, et al. RAS mutation in acute myeloid leukemia is associated with
distinct cytogenetic subgroups but does not influence outcome in patients younger than 60 years.
Blood. 2005; 106: 2113-2119.
85)
Bacher U, Haferlach T, Schoch C, et al. Implications of NRAS mutations in AML: a study of
2502 patients. Blood. 2006; 107: 3847-3853.
86)
Schnittger S, Dicker F, Kern W, et al. Cooperating molecular mutations in AML/RUNX1
mutated AML differ dependent on the cytogenetic subgroup. .Blood. 2007;110:114.
87)
King-Underwood L, Pritchard-Jones K. Wilms' tumor (WT1) gene mutations occur mainly in
acute myeloid leukemia and may confer drug resistance. Blood. 1998; 91: 2961-2968.
88)
Summers K, Stevens J, Kakkas I, et al. Wilms' tumour 1 mutations are associated with FLT3ITD and failure of standard induction chemotherapy in patients with normal karyotype AML.
Leukemia. 2007; 21: 550-551.
89)
Virappane P, Gale R, Hills R, et al. Mutation of the Wilms' tumor 1 gene is a poor prognostic
factor associated with chemotherapy resistance in normal karyotype acute myeloid leukemia: the
United Kingdom Medical Research Council Adult Leukaemia Working Party. J Clin Oncol. 2008;
26: 5429-5435.
51
90)
Paschka P, Marcucci G, Ruppert AS, et al. Wilms' tumor 1 gene mutations independently
predict poor outcome in adults with cytogenetically normal acute myeloid leukemia: a cancer and
leukemia group B study. J Clin Oncol. 2008; 26: 4595-4602.
91)
Gaidzik VI, Schlenk RF, Moschny S, et al. Prognostic impact of WT1 mutations in
cytogenetically normal acute myeloid leukemia: a study of the German-Austrian AML Study
Group. Blood. 2009; 113: 4505-4511.
92)
Damm F, Heuser M, Morgan M, et al. Single nucleotide polymorphism in the mutational
hotspot of WT1 predicts a favorable outcome in patients with cytogenetically normal acute
myeloid leukemia. J Clin Oncol. 2010; 28:578-585.
93)
King-Underwood L, Renshaw J, Pritchard-Jones K. Mutations in the Wilms' tumor gene WT1
in leukemias. Blood. 1996; 87:2171-2179.
94)
Mrózek K, Marcucci G, Paschka P, et al. Clinical relevance of mutations and gene-expression
changes in adult acute myeloid leukemia with normal cytogenetics: are we ready for a
prognostically prioritized molecular classification? Blood. 2007; 109:431-448.
95)
Ruf RG, Schultheiss M, Lichtenberger A, et al. Prevalence of WT1 mutations in a large cohort
of patients with steroid-resistant and steroid-sensitive nephrotic syndrome. Kidney Int. 2004;
66:564-570.
96)
Borel F, Barilla KC, Hamilton TB, et al. Effects of Denys-Drash syndrome point mutations on
the DNA binding activity of the Wilms' tumor suppressor protein WT1. Biochemistry. 1996;
35:12070-12076.
97)
Stoll R, Lee BM, Debler EW, et al. Structure of the Wilms tumor suppressor protein zinc finger
domain bound to DNA. J Mol Biol. 2007; 372:1227-1245.
52