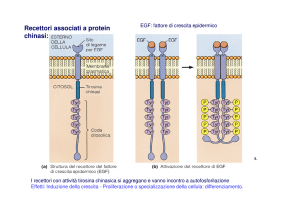
1
ANNO I I
n° 2
MARZO - APRILE 2006
Periodico di informazione ed aggiornamento su tematiche di
diagnostica di laboratorio redatto a cura della:
U.O. Complessa di laboratorio analisi
chimico cliniche e microbiologiche
Direttore : Dott. Ernesto Marco Scafidi
Azienda Ospedaliera “Santa Corona”
di Pietra Ligure (SV)
U.O. complessa di Laboratorio analis
2
In questo numero, senza pretese oltremodo didattiche, si è ritenuto
rivisitare un argomento fra i più discussi e discutibilmente affrontati dalla
pratica clinica quotidiana al di fuori di uno stretto ambito specialistico.
E’ necessario riconoscere anche che spesso solo nell’area professionale
specifica di laboratorio la coagulazione ed i suoi meccanismi ed interazioni
cliniche sono più approfondite.
La così detta “coagulazione”, molto spesso, proprio perché branca “di
nicchia” del laboratorio clinico viene a soffrire di una gestione e di una
comprensione abbastanza approssimativa affidata più spesso al clinico,
come per la endocrinologia, che al patologo.
Esempio di questa sotto attenzione della patologia clinica per la
coagulazione è l’attuale e del tutto recente ricerca di riappropriarsi della
gestione dei centri TAO (centri per lo studio della terapia con
anticoagulanti orali) da parte dei patologi clinici o meglio dei medici di
laboratorio che per anni si sono limitati alla produzione di informazioni,
anche sempre più approfondite ed accurate ma, da affidare alla gestione
del clinico.
Questo fenomeno, in eguale misura si è manifestato anche nella categoria
dei medici di medicina generale (ex medici di famiglia) che, per anni
hanno visto il proprio ruolo limitarsi a prescrittori di terapie ed indicazioni
erogate dallo “specialista”.
Non si deve poi sottostimare come l’offerta di mercato gaggettistico e lo
sconfinamento di alcune “arti” in altre, abbia posto le basi di una quasi
autogestione del monitoraggio della terapia anticoagulante che, lentamente
3
ma inesorabilmente sta passando, attraverso “il banco della farmacia” per
approdare al tavolo della cucina di casa.
Senza nulla togliere alle strumentazioni near patient che oggi, almeno
alcune, hanno raggiunto livelli elevati di affidabilità diagnostica, ritengo
sia necessario, ove gradito, fornire alla stazione “capolinea” del percorso
sanitario, cioè al medico di medicina generale, un piccolo ma costruttivo
“bignami” per rinverdire alcuni concetti ed alcune nozioni che lo possano
rendere più autonomo e clinicamente presente nella gestione delle
problematiche coagulative.
Ernesto Marco Scafidi M.D.
4
La coagulazione del sangue
Sommario
1. Generalità sulla coagulazione del sangue
2. Gli inibitori della coagulazione
3. Cenni sul meccanismo della coagulazione
4. Patologia della coagulazione
5. I principali esami per lo studio della coagulazione del sangue
Generalità sulla coagulazione del sangue
Forse nessun altro sintomo colpisce l’immaginazione di noi tutti come una emorragia. Fin
dall’antichità la fuoriuscita di sangue dal letto vascolare è stata considerata come qualcosa di grave,
in quanto associata con la perdita del fluido vitale. Non stupisce quindi come, nel corso
dell’evoluzione si siano perfezionati dei meccanismi sofisticati in grado di arrestare prontamente
un’emorragia.
Lo scopo dell’emostasi è quello di formare un tappo a partire dai costituenti stessi del sangue,
inizialmente piastrine e poi fibrina, che si ottiene dal fibrinogeno alla fine della cosiddetta cascata
coagulativa (fig.1).
Affinché sia assicurata un’efficiente emostasi è necessario che siano perfettamente funzionanti tre
compartimenti che, agendo in sintonia fra di loro, portano alla rapida riparazione di una ferita e
mettono fine alla fuoriuscita del sangue.
Questi tre compartimenti sono :
1. La parete dei vasi arteriosi e venosi
2. Le piastrine
3. I fattori della coagulazione. Sono proteine circolanti nel sangue e prodotte quasi tutte dal
fegato. Se ne conoscono una dozzina circa, indicati in genere con un numero romano (es
fattore VII, VIII o IX) o con il loro nome proprio (es. fibrinogeno). Essi hanno la
caratteristica peculiare di agire in sequenza, uno dietro l’altro, e ad ogni tappa il fattore, che
circola inattivo nel sangue, viene attivato ed agisce sul fattore successivo, che viene attivato
a sua volta. Ad ogni tappa aumenta notevolmente il numero di molecole formate, cosicché
alla fine di questa cascata coagulativa (fig. 1), partendo da poche molecole dei fattori che
intervengono per primi, si ottiene un numero enorme di molecole di fibrina. Per la
produzione epatica di alcuni di questi fattori è essenziale la vitamina K.
5
L’importanza di ognuno di questi compartimenti è attestata dall’esistenza di
numerose malattie
emorragiche in cui uno solo di essi è alterato.
Gli inibitori della coagulazione
In condizioni normali il meccanismo emostatico è attivato solo localmente, cioè solo dove è
necessario e per il tempo strettamente indispensabile ad arrestare l’emorragia, mentre nelle altre
zone dell’organismo il sangue continua a mantenere la sua abituale fluidità.
In altre parole, affinché non si verificano danni all’organismo, la coagulazione deve essere
perfettamente controllata nello spazio e nel tempo, altrimenti si potrebbe avere un’eccessiva
coagulazione che potrebbe provocare una trombosi.
Il controllo della coagulazione avviene a vari livelli ad opera di altre sostanze presenti nel sangue:
1. Sostanze anticoagulanti, le principali essendo l’antitrombina III (AT III), la Proteina C, la
Proteina S: ognuna di esse inibisce l’attività di diversi fattori della coagulazione
2. La plasmina, che si forma dal plasminogeno circolante nel sangue, come risultato finale
dell’attivazione del meccanismo della fibrinolisi. La plasmina ha il compito di sciogliere il
coagulo di fibrina che si era formato alla fine della cascata coagulativa.
Un aspetto fondamentale da ricordare è che tutti questi meccanismi sono attivi continuamente
nell’organismo in condizioni normali, cosicché la normale fluidità del sangue può essere
considerata come l’equilibrio che si raggiunge fra la naturale tendenza del sangue a coagulare da
una parte e, dall’altra, dell’attività dei meccanismi anticoagulanti e fibrinolitici che vi si
oppongono.
Cenni sul meccanismo della coagulazione
Anche se per comodità le caratteristiche dei tre compartimenti saranno trattati separatamente, in
realtà essi agiscono in sintonia e pressocché contemporaneamente. Sempre per comodità ci rifaremo
a ciò che avviene nell'organismo dopo una ferita che comporti la lesione di un vaso arterioso o
venoso.
Qualsiasi lesione della superficie interna di un vaso comporta l'interruzione dello strato delle cellule
endoteliali, le quali formano una specie di rivestimento liscio e regolare della parete stessa per
permettere al sangue di scorrere regolarmente.
Nella zona lesionata si verifica una vasocostrizione che riduce il calibro del vaso, il quale libera
anche nel sangue il fattore tessutale e delle sostanze che facilitano l'adesione delle piastrine alla
zona lesionata. Si forma così il cosiddetto tappo emostatico primario che ha il compito di arrestare
l'emorragia.
6
Contemporaneamente il fattore tessutale attiva il fattore VII, che a sua volta attiva il fattore X
innescando l'attivazione della cascata coagulativa attraverso la via estrinseca, alla fine della quale,
come abbiamo già detto, si ha la trasformazione del fibrinogeno in fibrina, ad opera del fattore II a o
protrombina.
La fibrina stabilizza e rinforza il tappo emostatico primario, consolidando così in modo definitivo il
coagulo formatasi nella zona lesionata.
Successivamente viene attivata la fibrinolisi che ha il compito di sciogliere il coagulo; questo viene
riassorbito e, contemporaneamente, si avvia il processo di riparazione della ferita, al termine del
quale si ricostituisce lo strato di cellule endoteliali e la parete vasale riacquista la sua normale
struttura.
Come si può vedere dalla figura 1, la coagulazione può essere attivata attraverso due vie:
l'estrinseca e l'intrinseca. L'importanza di quest'ultima in condizioni fisiologiche è probabilmente
minore rispetto alla prima.
Patologia della coagulazione
Numerose sono le malattie che possono risultare da anomalie di uno o più dei tre compartimenti.
Schematicamente possiamo considerare:
1.
Emorragie che possono essere dovute ad:
o
alterazioni congenite o acquisite della parete vascolare
o
Piastrinopenie o piastrinopatie, cioè anomalie delle piastrine il cui numero può essere
anche normale
o
Deficit congeniti o acquisiti di uno più fra i fattori della coagulazione (per es.
emofilia, malattia di Von Willebrand)
o
Eccessiva attività del meccanismo della fibrinolisi
2. Trombosi, che possono essere dovute a:
o
Alterazioni, in genere acquisite della parete vasale
o
Deficit congeniti o acquisiti degli inibitori naturali della coagulazione (per es. deficit
di AT III, di Proteina C o proteina S
o
Aumento notevole e persistente delle piastrine
o
Deficit del meccanismo fibrinolitico
7
I principali esami di laboratorio per lo studio della coagulazione
•
Esame emocromocitometrico: permette di conoscere il numero delle piastrine
•
Esame del sangue periferico al microscopio: permette di valutare grossolanamente il numero
delle piastrine e, soprattutto la loro forma e dimensione.
•
Tempo di emorragia: permette di valutare, dopo aver punto il polpastrello o il lobo di un
orecchio, il tempo necessario per l'arresto dell'emorragia
•
Tempo di Quick: permette di valutare in laboratorio il tempo necessario per la coagulazione
del sangue. Valuta soprattutto le tappe finali della cascata coagulativa. Questo esame è
conosciuto anche come tempo di protrombina o PT o INR.
•
Tempo di tromboblastina parziale, noto anche come PTT o aTTP che valuta la via intrinseca
e le tappe finali della coagulazione
•
Dosaggio dei singoli fattori della coagulazione. Generalmente è disponibile sono in
laboratori specializzati, e viene effettuato per confermare il sospetto di una carenza di uno o
più fattori, in seguito al riscontro di alterazioni a carico del PT o del PTT.
Dosaggio di ATIII o degli altri inibitori della coagulazione: è effettuato soprattutto nel
sospetto di trombosi familiare o in giovani soggetti senza cause predisponenti a trombosi
venose e/o arteriose.
8
Le vie della coagulazione del sangue (fig.1)
9
La via intrinseca
Lesione cellulare con esposizione di una superficie con carica negativa (anche
vetro)
XII + superficie > XIIa
Prekallikreina > kallicreina
XI > XIa
IX > IXa
X > Xa
Test clinico: Tempo di tromboplastina
•Tempo necessario per la coagulazione dopo contatto con una superficie standard:
caolino e cefalina (fosfolipide)
•Normale: 30-50 s
10
La via estrinseca
III (proteina transmembrana) + Ca++ > VIIa
X > Xa
Test clinico: tempo di protrombina
•Tempo necessario per la coagulazione dopo esposizione a un fattore tissutale
standard: caolino e cefalina (fosfolipide)
•Normale: 10-15 s
11
Via comune
Fattore X converte protrombina in trombina
Trombina converte fibrinogeno (solubile) in fibrina (insolubile)
Test clinico: tempo di trombina
•Tempo necessario per la coagulazione dopo contatto con trombina
•Normale: 10-15 s
12
Protrombina > Trombina
150 mg/l in plasma, sintetizzata in fegato
T1/2=24 h
Fattore Xa converte trombina
•Fattore limitante del processo di coagulazione
•Tempo di protrombina (11-15 s): test di funzionalità epatica
Dipende da Vitamina K
13
Fibrinogeno > fibrina
3 g/l in plasma, sintetizzato in fegato, (gba)2 T1/2=4 g
Trombina converte fibrinogeno solubile in fibrina insolubile
Stabilizzazione con XIII
14
Stabilizzazione del coagulo di fibrina con
Fattore XIII (transglutamidasi), attivato da
trombina
Vitamina K
Liposolubile
Non passa attraverso placenta
K1: vegetali verdi
K2: deriva da K1 ad opera di batteri intestinali
K3 o menadione: preparato industriale
15
Reazione della Vitamina K
Carbossilazione di glutammato (Glu) a carbossiglutammato (Gla) in molti dei
fattori della coagulazione (incluso trombina, II, VII, IX e X)
Deficit di Vitamina K (rari), associati al malassorbimento dei grassi:
•Infanti sottoalimentati
•Ittero ostruttivo
•Lunghe cure antibiotiche
Antimetaboliti di Vitamina K
Dicumarolo
Warfarin
16
Dissoluzione del coagulo (fibrinolisi)
Nel coagulo di fibrina è presente plasminogeno
Plasminogeno > plasmina
•Catalizzatori: Tissue Plasminogen Activator (tPA) e urokinasi
•Inibito da a2-antiplasmina Plasmina > fibrina (proteolisi)
Farmaci fibrinolitici (infarto del miocardio)
•tPA ricombinante
•Streptokinasi
•Urokinasi
Struttura dei vasi sanguigni
Cellule endoteliali
•Se intatto: attività antitrombotica
>Rilascio di prostaciclina e NO
•Se leso: vasocostrizione
>Rilascio di serotonina e trombossano A2
Tessuto connettivo - intima•Ricco in collageno
•Se esposto alla circolazione: attivazione della via intrinseca e estrinseca
•Attivazione delle piastrine (vWF)
Muscolatura liscia
•Più pronunciata sul versante arterioso
Tessuto connettivo - adventitia
17
Piastrine
Cellule discoidi, anucleate
•Diametro 2-3 mm•Vita media 9-12 gg
Livello normale: 150 000 – 400 000 /nl
Contengono actina
Forte tendenza ad aggregarsi
•Misura dell’adesività: conta delle piastrine prima e dopo il contatto con una
superficie di vetro
18
Attivazione delle piastrine
Attivatori:
•ADP, epinefrina, collageno, trombina, PAF (platelet activating factor), complessi
Ab-Ag, shear stress
Le piastrine aderiscono al collageno
•Fattore di von Willebrand (vWF): glicoproteina plasmatica, media fra piastrine e
collageno
Polimerizzazione di actina
•Discoide > sferico
Aggregazione
Rilascio di numerosi fattori
19
Le piastrine nei difetti coagulativi
Anomalie congenite
•Sindrome di von Willebrand
•Sindrome di Bernard-Soulier: deficit del recettore
Anomalie acquisite
•Trombocitopenia (basso livello di piastrine)
•Distruzione delle piatrine circolanti: leucemia, sindromi mieloproliferative, eccesso
di aspirina etc)
Farmaci antipiastrinici:
•Aspirina (inibisce cicloossigenasi e riduce TXA2
•Dipiramidolo (inibisce effetto di ADP
Coagulazione: evento autocatalitico
che deve essere controllato strettamente
Controllo del livello di trombina (protrombina >> trombina)
•Antitrombina III (inibisce anche IXa, Xa, XIa e XIIa)
•a2-macroglobulina
•a1-antitripsina
Prostaciclina (endotelio): inibisce aggregazione piastrinica
Proteine C e S: disattivano V e VIII
Eparina: potenzia azione di antitrombina III
Antimetaboliti di vitamina K: Glu ® Gla in protrombina
Aspirina:
•Potenzia azione di antitrombina III
•Inibisce cicloossigenasi e formazione di trombossano
•Inibisce prostaciclina
20
- Patologie della coagulazione Emofilia A o emofilia classica
Deficit di Fattore VIII (150 differenti mutazioni descritte)
X-linked, frequenza 1:5 000-10 000 nati maschi
Emorragie
Trattamento con infusione di VIII
- Patologie della coagulazione Emofilia B, deficit di Fattore IX (300 mutazioni
conosciute)
Deficit di Fattore XIII (autosomici recessivi)
von Willebrand Disease, deficit di Fattore VIII
•La patologia ereditaria più diffusa (125 nati per milione, doppio di
emofilia A)
•Adesione piastrinica difettosa
Deficit di Fattore XI, o emofilia C, 3 mutazioni conosciute
•Mancata attivazione da contatto con superfici cariche negativamente
•Identificata in 1953, comune negli ebrei Ashkenazi
Deficit di antitrombina
•Subsintomatico, autosomico dominante (1 per 2000-5000)
•Sintomatico (trombosi venose profonde, embolismo polmonare) in
associazione con interventi chirurgici, traumi e gravidanza
21
Fibrinogeno
Livelli alti di fibrinogeno
•Pazienti con ipertensione, diabete, iperliproteinemia, ipertrigliceridemia e
coronaropatie
•Gravidanza, ipercoleresterolemia, contraccettivi orali, menopausa, fumo
Malattie genetiche
•Afibrinogenemia (aborto spontaneo da emorragie dal cordone ombelicale e interne)
•Ipofibrinogenemia, acquisita o ereditaria
•Disfibrinogenemia (fibrinogeno non funzionale, con emorragie, aborti e
tromboembolismo)
22
Coagulazione intravasale disseminata
(CID o DIC)
Coagulopatia da consumo.
Si tratta di una patologia che ogni medico deve conoscere e temere, in
considerazione del fatto che solo una diagnosi tempestiva può salvare la vita
del paziente. Essa dipende dall’attivazione della reazione a cascata della
coagulazione con formazione intravasale, cioè dentro i vasi e delle più fine
ramificazioni capillari, di microtrombi di fibrina. A tale processo consegue una
reazione omeostatica di equilibrio di iperfibrinolisi secondaria.
Eziologia.
a)
Immissione in circolo di attivatori della protrombina:
-embolia di liquido amniotico aspirato dopo il parto, distacco precoce di placenta,
atonia con emorragia post-partuum, aborto settico, ritenzione di feto morto, aborto
da cloruro di sodio;
-interventi su organi ricchi di trombochinasi, per es. sul polmone, pancreas,
prostata, placenta (le 4 P!);
-emolisi grave, per es. per incompatibilità in errori trasfusionali;
-veleno di serpente;
-stati
neoplastici
terminali
con
liberazione
in
circolo
di
sostanze
tromboplastinosimili, nelle leucemie.
b)
Attivazione della coagulazione tramite mediatori:
endotossine di batteri gram-negativi in gravide;
sindrome di Waterhouse-Friderichsen=coagulopatia da consumo
con emorragia cutanea, shoch, rigidità nucale ed emorragie
cutanee e surrenali nella sepsi da meningococco TERAPIA
URGENTE CON PENICILLINA G E.V.!!
23
Coagulopatia da consumo nella setticemia (cioè quando i batteri
sono nel sangue) da batteri gram negativi;
Porpora fulminante: affezione acuta, con microtrombi vasale
post-infettiva con emorragie cutanee estese e simmetriche della
cute e necrosi centrale e DIC.
Decorso.
Nella Coagulopatia Intravasale Disseminata o CID o DIC avremo 3 fasi:
1.
fase pre DIC: presenza di malattie e stati a rischio;
2.
fase della DIC: alterazioni di laboratorio e diatesi emorragica
3.
fase post-DIC: ipercoagulabilità reattiva , ma via via non si rilevano i prodotti
di degradazione del fibrinogeno che in un primo momento sono presenti ad alto
dosaggio.
Clinica.
Il malato presenta shock, con polso piccolo e frequente, agitazione, pallore, sudorazione
fredda e labbra cianotiche; possibilmente ha delle petecchie e/o porpora emorragica, cioè
delle macchie che sembrano “piccoli nevi puntiformi” diffusi in tutto il corpo, mentre si
manifestano emorragie (per es. in un caso clinico una paziente aveva avuto una “ripresa
del ciclo”, secondo quanto appreso all’anamnesi, emissione di sangue nelle urine, lacrime
miste a sangue, insufficienza cardio-respiratoria acuta, fino al coma irreversibile ed
all’exitus, con consumo delle piastrine, aumento dei PDF e del D-dimero (vedi avanti).
Diagnosi.
E’ la fase più delicata della CID e presuppone che un medico abbia il sospetto
clinico di ciò che cerca , che presuppone l’attenta ed umile valutazione del malato.
LABORATORIO:
1. all’emocromo avremo calo continuo delle piastrine, anche nel giro di 30- 40 minuti!!
2. fibrinogeno ed ANTI TROMBINA III che si consumano e si riducono velocemente;
3. dimostrazione di monomeri di fibrina;
4. Prodotti di degradazione del Fibrinogeno
5. D-Dimero (!!!!)
6. Tempo di Quick che si riduce;
7. PTT che aumenta
24
8. Riduzione dei fattori V = Trombina
9. Riduzione dei fattoti VIII
TERAPIA (!!)
E’ una terapia spesso e volentieri drammatica; il trattamento della malattia
scatenante è imperativo, per es. l’infezione che la sottende, la ritenzione di placenta, la
rimozione del tessuto neoplastico ecc.
Nella fase del pre-DIC, cioè come profilassi si impiegano eparina a 500 UI/ ora in pompa
infusionale, riducendo se necessario, monitorizzando la coagulazione;
Durante la DIC o CID:
si impiega un prodotto concentrato a base di ANTI TROMBINA III, circa
3000-5000 UI di ATIII;
plasma fresco per integrare il fibrinogeno ridotto, se il tempo di quick è
ridotto e se il PTT sale, almeno 500 ml nelle prime 2 ore;qui non si
impiega eparina!
Post-DIC
eparina per ridurre l’ipercoagulabilità, con PTT che deve salire X 1,5 -2
volte la norma
ANTITROMBINA III – se la sua attività scende al di sotto dell’80%.
LABINFORMA
anno II°
- 2006 n°2 marzo - aprile