Tappe storiche che hanno portato alla
regolamentazione tossicologica per i Farmaci
in fase pre-clinica
-1930 (USA): gravi reazioni tossiche oculari per uso di
cosmetici con i p-fenilendiammina
-1937 (USA): un centinaio di morti per somministrazione
di sulfamidici in glicol etilenico
-1938 (USA): nascita della FDA (Food and Drug
Administration)
-anni ’60 (Europa): la talidomide antiemetico
somministrato in gravidanza provoca una decina di
migliaia di bambini affetti da focomelia
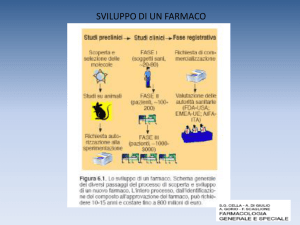
Studi tossicologici nello sviluppo di un farmaco in fase preclinica sono di
importanza fondamentale perché un'inaspettata tossicità del farmaco
può compromettere il suo sviluppo e anche gli investimenti
dell'azienda. Quindi tutte le figure professionali che, nell’industria
farmaceutica, devono affrontare problematiche legate allo sviluppo
non clinico di un nuovo principio attivo e di un farmaco devono avere
conoscenze in campo tossicologico per poter adeguatamente:
-programmare lo sviluppo tossicologico in accordo alle linee guida
più recenti;
-confrontarsi con Organizzazioni di Ricerca a Contratto (CRO);
-discutere protocolli, analizzare, correggere e approvare i report;
-interpretare i dati e proporre eventualmente soluzioni agli imprevisti; verificare se l'analisi rischio/beneficio supporta lo sviluppo di un nuovo
farmaco;
-proporre i dosaggi per i primi studi clinici.
La differenziazione tra Farmacologia e Tossicologia inizia con i
lavori scientifici di Philippus Aureulus Theophrastus
Bombastus Von Hohenheim Paracelso (1493-1541)
Si deve a Paracelso la moderna classificazione delle
sostanze tossiche. “Tossico” è una sostanza chimica
primaria e non una mescolanza di principi. Inoltre il
tossico è un unicum con il farmaco.
Paracelso sosteneva i seguenti corollari tuttora validi:
• La sperimentazione è essenziale nell’esame delle
risposte dell’ organismo alle sostanze chimiche
• Si deve poter distinguere tra proprietà
terapeutiche e proprietà nocive delle sostanze
chimiche ma
– Nessuna sostanza di per sé è un veleno, è la
dose che fa di una sostanza un veleno
– Si dovrebbe accertare un grado di specificità
delle sostanze chimiche e dei loro effetti
terapeutici o tossici.
Nel passato in medicina si faceva gran uso di sostanze tossiche
come l’arsenico, basandosi sull’erronea convinzione che basse
dosi di una sostanza tossica potessero aiutare l’organismo
nella guarigione, stimolando, come fanno alcuni agenti
infettivi, le difese naturali.
Anche sostanze decisamente tossiche e non selettive
per le cellule neoplastiche come la cicuta erano usate
nella terapia antitumorale.
Tossicità= rottura di equilibri biologici
Per capire la tossicità di un fattore
chimico o fisico bisogna conoscere:
1. La sua reattività con le strutture
biologiche.
2. Se tale reattività supera le capacità
omeostatiche del sisema biologico
interessato.
La natura degli effetti tossici
Il meccanismo e la gravità degli effetti tossici delle
sostanze dipende da numerosi fattori biologici
interconnessi in maniera complessa:
• Le proprietà chimico-fisiche della sostanza
• Le condizioni ambientali e quelle patologiche
dell’organismo esposto
• L’eventuale biotrasformazione della sostanza
• Le capacità di bioprotezione dell’organismo
Rapporto Salute / Dose
SALUTE
Omeostasi
Compensazione
Normalità
Sbilanciamento
Patologia
Soglia di
RISCHIO
Rottura di
equilibrio
Morte
NOEL Benchmark o
similari
Dose
SALUTE
Elemento non
essenziale
Elemento essenziale o Farmaco
Normalità
Patologia
Mortecarenza
Intervallo terapeutico
Dose
sovradosaggio
Fase di
esposizione
Disponibilità
all’assorbimento
Escrezione
Fase Tossicicinetica
Assorbimento e
distribuzione
Accumulo o
deposito
Biotrasformazione
Biodisponibilità*
Iterazione con le strutture
bersaglio
Fase Tossicodinamica
EFFETTO
* Biodisponibilità= concentrazione in forma attiva in grado di interagire con le
strutture bersaglio
ANALISI STATISTICHE APPROPRIATE
Dati Quantitativi
Distribuzione normale?
si
no
Confronti tra medie, deviazioni standard,
errori standard
Dati semi-quantitativi
Dati qualitativi
Analisi per dati non
parametrici
Misura
graduale
Effetto che non necessita interazione specifica
[T]
[T] Reattività
Chimica/Fisica
Bersaglio Biologico
f Effetto
Misura
graduale
Effetto che necessita interazione specifica
[T] + Bersaglio Biologico [B]
K2/K1
[TB]
f Effetto
[T]
Rrisosta cumulativa
1927 Trevan
Frequenz di risosta
Resistenti
Frequenz di risosta
Dose o
Concentrazione
µ-2σ
μ
µ+2σ
LOG Dose o
Concentrazione
μ
LOG Dose o
Concentrazione
Frequenz di risosta
Frequenz di risosta
σ =1
σ =2
µ-2σ
μ
σ =3
μ
µ+2σ
Tra µ-2σ e µ +2 σ è sempre compresa la
risposta del 95,5% della popolazione e
tra µ-1σ e µ +1σ è compresa la risposta
del 68,2 %
σ =1
Rrisosta cumulativa
LOG Dose o
Concentrazione
σ =2
μ
1933 Gaddum introduce la scala NDE= Deviazione normale equivalente
Tale scala trasforma le frequenze di risposta espresse in % in Unità di deviazione
standard la sasala va da -3 a +3 e comprende lo O che corrisponde alla risposta
del 50% della popolazione. Questo permette di trasformare la sigmoide % di
risposta sul Log della concentrazione in una funzione pressoché lineare
100
NDE
Percentuale di occorrenza
+3
8
Scala probit
50
0
-3
0
µ-3σ
µ
µ+3σ
1944 Bliss introduce la scala Probit : NDE+5
(in questa sacla non esiste valore per 0% e 100%)
5
2
100
% Risposta (scala Probit)
99%
75
50%
50
25
1%
0
0
2
4
6
8
10
Log dose (mg/Kg)
Comparazione di:
ED= dose efficace
TD= dose tossica
LD= dose letale
Indice Terapeutico=IT=LD50/ED50
Margine di sicurezza= LD1/ED99
Compito degli Studi Tossicologici in fase
preclinica
1. Definizione della dose massima che non
induce alcun effetto diretto o indiretto su
organi e sistemi
2. Definizione della dose che induce effetto
nocivo e il tipo dell’effetto
3. Definizione della relazione dose terapeutica e
dose tossica
4. Individuare il bersaglio dell’effetto (molecola,
struttura cellulare, organo o sistema) sia del
composto originale che dei suoi metaboliti
5. Definire la reversibilità o meno degli effetti
La Tossicità dei Farmaci
TOSSICITÀ PER ECCESO DI EFFETTO TERAPEUTICO :
EFFETTI FACILMENTE PREVEDIBILI DA:
ECCESSO DI USO, ABUSO, USO IMPROPRIO, ERRORE
TERAPEUTICO
TOSSICITÀ DIFFERENTE DALL’ EFFETTO TERAPEUTICO
(EFFETTI COLLATERALI):
EFFETTI POTENZIALMENTE PREVEDIBILI IN BASE A :
• STUDI DI TOSSICOCINETICA APPROFONDENDO LE
BIOTRASFORAZIONI (METABOLISMO)
• STUDI DI TOSSICODINAMICA:
CONOSCENZA DEGLI EFFETTI, DELLE INTERAZIONI PIÙ PROBABILI
TRA FARMACI /TOSSICI SIA IN FASE TOSSICODINAMICA CHE
TOSSICOCINETICA
COMUNQUE L’EFFETTO TOSSICO (ET) DI UNA MOLECOLA (o
Fattore fisico) DIPENDE DA:
La sua concentrazione (o intensità-energia) (QT)
La durata d’esposizione (t)
ET = QT* t
• TOSSICITÀ ACUTA= Effetto/i nocivo/i derivato/i da
esposizione unica (improvvisa) o di breve durata a
una relativamente elevata quantità di agente
tossico.
• TOSSICITÀ CRONICA= Effetto/i nocivo/i derivato/i
da esposizione ripetuta o molto prolungata nel
tempo a relativamente piccole quantità di agente
tossico(Tossicità subacuta e Tossicità subcronica)
Altre classificazioni e terminologie dell’effetto tossico:
• EFFETTO LOCALE o EFFETTO SISTEMICO
• Effetto immediato o Effetto ritardato (con
latenza)
• Effetto reversibile o Effetto persistente
(irreversibile)
• Effetto specifico o Effetto aspecifico
• Allergie e Idiosincrasie
Un tipo particolare di effetto sono l’Allergia e
l’Idiosincrasia
L’allergia è una risposta indesiderata su base
immunologica ed è il risultato di una percedente
esposizione alla medesima sostanza o a sostanze
analoghe.
L’idiosincrasia è una risposta anomala agli agenti
chimici su base genetica. Un esempio è, in soggetti
geneticamente carenti dell’enzima glucosio-6-fosfatodeidrogenasi, l’ipersensibilità a sostanze ossidanti come
I nitroaromatici, i clorati e gli anti malarici con
conseguente manifestazione di anemia emolitica .
Tipologia dei meccanismi di danno cellulare
Tossico
A
Interazione con
Molecole bersaglio
Disfunzione
cellulare, danno
Riparazione
insufficiente o errata
B
C
TOSSICITÀ
Presenza
nell’organismo
A: Anche la sola presenza provoca
tossicità senza specifiche interazioni
con molecole bersaglio l’azione è
piuttosto dovuta ad alterazione del
microambiente cellulare;
B: l’effetto deriva da interazione con
specifiche molecole bersaglio;
C: il danno e correlato a meccanismi
di riparazione
Quale è lo scopo degli studi di tossicità acuta:
•
•
•
•
•
•
•
•
•
•
Determinare il massimo rischio causato dalla
somministrazione per la via prescelta, dalla
esposizione accidentale (di solito ingestione) o
dalla massima esposizione sistemica possibile
(via endovenosa)
Stabilire grossolanamente l’organo (il sistema)
bersaglio
Definire le dosi con cui partire negli studi di
tossicità subacuta (dosi per gli studi preliminari
di DRF)
TOSSICITÀ ACUTA
•Spesso effetti quantali (tutto o nulla)
•Durata da pochi minuti a qualche giorno ma
con esposizione unica
•Dato espresso in numero di individui che
hanno una risposta: il dato è trasformato in
frequenza di risposta e quindi in risposta
cumulativa
•Calcoli ED50, LC50, LD50
DL50= dose letale in grado di provocare la morte nel 50% della
popolazione
X in Log Y in probit
Probit 50% =5
Y=a+bX
5a
)
b
Significatività χ2 confronto tra % teorica e sperimentale ( non devono essere differenti)
DL50 AntiLog(
Gli studi di tossicità acuta:
Superata per motivi etici la determinazione
della Dose Letale 50% (DL50, linea guida
401 OECD) sono stati proposti metodi
alternativi:
•Fixed dose (OECD 420)
•Acute Toxic Class (OECD 423)
•Up and down procedure (OECD 425)
Tossicità Acuta
Acute Toxic Class:
Start
5mg/kg
3 animals
2-3
50mg/kg
3 animals
0-1
2-3
5mg/kg
3 animals
2-3
GHS
Category 1
> 0-5
5
0-1
2-3
-
2-3
other
30
0-1
50
3 (at 300)
at first step
200
2000mg/kg
3 animals
0-1
2-3
300mg/kg
3 animals
2-3
Category 3
> 50 - 300
Category 2
> 5 - 50
25
0-1
50mg/kg
3 animals
3 (at 50)
at first step
LD50
cut-off
mg/kg b.w.
300mg/kg
3 animals
2000mg/kg
3 animals
0-1
2-3
300
3 at
2000
st
1 step
2 at
2000
st
1 step
500
1000
0-1
Category 5
>2000 - 5000
Category 4
> 300 - 2000
other
0-1
1
2000
per step three animals of a single sex (normally females) are used
0,1,2,3 Number of moribund or dead animals at each step
: unclassified
GHS: Globally Harmonized Classification System (mg/kg b.w.)
2500 5000
0
0
0
Risposte e procedura nel programma
Le tre possibili opzioni scelte in base ai
risultati del trattamento:
•Nessun altro trattamento
•Ripetizione della dose su altri tre
animali
•Trattamento di altri 3 animali con
dose inferiore o superiore in base
al risultato
Classi
DL50 (os Ratto)
Dose letale per
l’uomo (70Kg)
Estremamente tossico
Altamente tossico
Molto tossico
Moderatamente tossico
Debolmente tossico
Praticamente non
tossico
5mg/Kg o meno
1-50mg
5,1-50mg/Kg
51mg-5g o ml
51-500mg/Kg
5,1-30g o ml
501-5000mg/kg
31-500g o ml
5,1-15g/Kg
501-1000g o ml
Oltre15g/Kg
Oltre 1Kg o 1
litro
TOSSICITÀ SubCRONICA
•Spesso effetti graduali
•Durata da qualche giorno a mesi/anni con esposizione continua
o ripetuta
•Obiettivo: individuazione di soglia di tossicità
•Calcoli: NOAEL, NOEL, LOAEL, Benchmark.
K1
K
[T ] [ B]
2 [TB] EFFETTO
1
NOEC o
NOEL
0,9
0,8
0,7
0,6
0,5
0,4
0,3
0,2
0,1
0
0
0,30
0,4
0,5
0,75
1
2
2,5
3
4
5
7
La pianificazione degli studi di tossicità subacuta (dosi ripetute
per 2 o, di preferenza, 4 settimane) è una delle fasi chiave dello
sviluppo tossicologico di un nuovo prodotto:
• Si tratta, in genere, degli studi che consentiranno di redigere il
dossier per la prima somministrazione nell’uomo
• I risultati di questa fase serviranno come base per la scelta
delle dosi da somministrare negli studi di tossicità subcronica e
cronica
• Gli organi bersaglio saranno definiti in modo preciso
• I risultati in termini di NOAEL (No Adverse Effect Level),
serviranno a stabilire le dosi da cui partire negli studi clinici di
fase I
• A questi studi è delegata la valutazione circa la necessità di
studi specifici di immunotossicità, nonché la determinazione di
eventuali rischi per la funzione riproduttiva (fertilità maschile)
* Significativo
Effetto (Risposta)
rispetto al controllo
*
*
0
1
2
3
4
5
6
7
Controllo
Trattato
*
Concentrazione
NOEL=NO OBSERVED EFFECT LEVEL(CONCENTRATION)
La più alta concentrazione che nel test tossicologico non ha dato differenza significativa di
risposta rispetto al controllo
NOEL e NOAEL (valore più appropriato per Farmaci o Fito-Farmaci)
INCONGRUENZE DELLA NOEL (NOAEL)
1. Non tiene conto dell’intera sperimentazione
2. Deve comunque essere una concentrazione effettivamente
sperimentata
3. Dipende in misura inaccettabile dalla numerosità del
campione e dalla sua variabilità
4. Talvolta la NOEL statisticamente valutata corrisponde ad una
concentrazione che da un incremento del parametro
considerato e del rischio associato anche superiore al 20%
% incremento del valore basale
(controllo) del parametro
40
20
30
10
20
5
1
10
BMR(5)
Benchmark concentration
BMR=Benchmark response
Crump 1984
ED5
BMR(10)
ED10
concentrazione
Valore del parametro
40
Incidenza di risposta
QUALE NOEC ??
NOAEL
LOAEL
NOAEL
LOAEL
NOAEL
LOAEL
Dose
Tossicità Cronica
FDA note on duration of chronic toxicity testing in nonrodents:
The ICH guidance recommends 9-month chronic toxicity studies
in nonrodents. FDA considers 9-month studies in nonrodents
acceptable for most drug development programs, shorter
studies may be equally acceptable in some circumstances and
longer studies may be more appropriate in others, as follows:
· Six-month studies may be acceptable for indications of chronic
conditions associated with short-term, intermittent drug
exposure, such as bacterial infections, migraine, erectile
dysfunction, and herpes.
· Six-month studies may be acceptable for drugs intended for
indications for life-threatening diseases for which substantial
long-term human clinical data are available, such as cancer
chemotherapy in advanced disease or in adjuvant use
Tossicità Cronica
Twelve-month studies may be more appropriate for
chronically used drugs to be approved on the basis of
shortterm clinical trials employing efficacy surrogate
markers where safety data from humans are limited to
short-term exposure, such as some acquired
immunodeficiency syndrome (AIDS) therapies.
· Twelve-month studies may be more appropriate for new
molecular entities acting at new molecular targets where
postmarketing experience is not available for the
pharmacological class. Thus, the therapeutic is the first in
a pharmacological class for which there is limited human
or animal experience on its long-term toxic potential.
La valutazione di potenziali effetti indesiderati a carico del sistema
immunitario deve essere inclusa nello sviluppo standard dei
farmaci
• Immunosoppressione: risposta immune a infezione o cellule
tumorali.
• Immunostimolazione: autoimmunità, ipersensibilità.
Questi effetti possono essere associati a:
1) Farmaci concepiti per modulare il sistema immunitario (per es.
per prevenire il rigetto da trapianto). In questo caso un’esagerata
immunosoppressione è da considerare un’azione farmacologica
esagerata.
2) Farmaci non concepiti per colpire le funzioni immunitarie, ma
che possono causare immunotossicità (necrosi o apoptosi di cellule
immunocompetenti, interazione con recettori condivisi tra il
tessuto target e le cellule immunitarie ).
Es: Agenti anti-proliferativi (antitumorali) possono produrre
immunosoppressioneindesiderata.
Safety Pharmacology Test principali
Cenni di Tossicologia dei farmaci
L’indice della sicurezza del farmaco è talvolta
definito come rapporto LD50/ED50 detto anche
indice terapeutico, ma questo può essere
ingannevole se le curve risposta-Log dose per
effetto desiderato e morte non sono parallele.
Perciò è preferibile calcolalo come LD10/ED90
Il MARGINE DI SICUREZZA è un evoluzione
ulteriore di questi indici ed è NOAEL/ED80
Importanza dei test di genotossicità
Per la gravità e irreversibilità degli effetti genetici, la
verifica dell’attività genotossica è un elemento
fondamentale nella valutazione del rischio delle
sostanze chimiche
Essa è normalmente ritenuta indispensabile per tutte
le sostanze per le quali è prevedibile una esposizione
umana
Obiettivi degli studi di mutagenesi
(OECD, 1986)
Identificazione delle sostanze capaci di indurre danni
genetici nella progenie in seguito all’interazione con il
materiale genetico delle cellule germinali (hazard
identification) e stima quantitativa del danno genetico
trasmissibile (risk characterization)
Predizione dell’attività cancerogena conseguente alla
interazione con il materiale genetico delle cellule
somatiche (hazard identification)
Valutazione del meccanismo di azione (MoA, mode of
action) di cancerogeni chimici (risk characterization)
Modelli sperimentali di mutagenesi
I test di mutagenesi possono essere definiti come test in vitro
e in vivo che hanno il compito di evidenziare composti che
inducono danno genetico attraverso meccanismi diversi:
- Mutazioni geniche
- Aberrazioni cromosomiche
- Modifiche nel numero di cromosomi
Test di mutagenesi a breve termine
(Short term tests, STT)
In vitro
Test di mutazione genica nei batteri
Test di mutazione genica su cellule di mammifero
Test citogenetici (Aberrazioni cromosomiche, Micronucleo)
Test di danno/riparazione (UDS, Comet)
In vivo
Test citogenetici (Aberrazioni cromosomiche, Micronucleo)
su cellule somatiche (midollo osseo,
sangue) e germinali (spermatogoni, spermatociti)
UDS nel fegato, Comet in vari tessuti
Test a breve termine di uso routinario
In vitro
Ames test (OECD 471)
Test di mutazione genica su cellule di mammifero (HPRT o tk)
(OECD 476)
Test di aberrazioni cromosomiche (OECD 473)
Test del micronucleo (OECD 487)
In vivo
Test di aberrazioni cromosomiche (OECD 475)
Test del micronucleo (OECD 474)
Test dell’UDS nel fegato del ratto (OECD 486)
Comet assay in vivo
Strategie di saggio
STANDARD BATTERY
Si possono seguire indifferentemente due opzioni:
Opzione 1
Un test di mutazione genica nei batteri
Un test citogenetico in vitro per evidenziare il danno cromosomico (test
di aberrazioni cromosomiche o del micronucleo) oppure il test di
mutazione genica su cellule di linfoma di topo (mouse lymphoma test)
Un test in vivo per evidenziare il danno cromosomico (in genere
micronuclei o aberrazioni cromosomiche in cellule ematopoietiche di
roditore)
Mutazione genica nei batteri Test di Ames
PRINCIPIO DEL TEST
(OECD No. 471)
Nel test di Ames vengono utilizzati dei ceppi batterici auxotrofi per alcuni
aminoacidi essenziali (incapaci di sintetizzare l’aminoacido e quindi di
crescere e formare colonia)
Il test di Ames permette di evidenziare qualsiasi mutazione in grado di
ripristinare la corretta funzionalità dei geni precedentemente mutati
(reversione) e che permette ai batteri di crescere
I batteri revertenti vengono evidenziati in base alla loro capacità di
crescere in assenza dell’aminoacido che era invece richiesto dal ceppo
parentale
Test di Ames
GROW ON MINIMAL AGAR PLATE
CONTROL
+ MUTAGEN DRUG
Spontaneous mutations
Induced mutations
Test di Ames
CEPPI BATTERICI UTILIZZATI
•
Salmonella typhimurium (mutazione in uno dei geni dell’operone per la
biosintesi dell’istidina)
TA 1535 e TA 100: SOSTITUZIONE DI BASE
TA 1537 e TA 98: INSERZIONE / DELEZIONE DI BASI
•
Escherichia coli (mutazione in un gene dell’operone per la biosintesi del
triptofano)
WP2 uvrA: SOSTITUZIONE DI BASE
* Mutazioni addizionali sono state introdotte in questi ceppi per renderli più
sensibili ad un’ampia varietà di sostanze
Test di Ames
PROTOCOL
Strains: TA1535, TA1537, TA98, TA100, WP2 uvrA
Test article: five dose levels (max. 5 mg/plate)
Metabolic Activation: +/- S9 microsomal fraction
Experimental Procedures: Preincubation Test or standard plate incorporation
assay
Positive and negative controls:
– Positive (-S9): Sodium Azide, 9-aminoacridine,2-nitrofluorene,
methylmethanesulphonate
– Positive(+S9) : 2-aminoanthracene, Benzopyrene
One experiment is generally sufficient with clearly negative or positive
compounds. Equivocal or weak positive results should be confirmed in a
second experiment.
Test di Ames
PLATE INCORPORATION ASSAY
+
2 mL
Soft Agar
+ essential a.a.
0.1 mL
Bacterial
Strain
+
0.1 mL
Test compound
or solvent
+
Glucose minimal
Agar plate
Count number
of revertants
37C° for 48 or 72 hr
0.5 mL
S9 mix
or buffer
Test di Ames
VALUTAZIONE ED INTERPRETAZIONE DEI RISULTATI
Un composto viene considerato mutageno se determina un incremento
dose-dipendente del numero di colonie revertenti in uno o più ceppi ( 2
volte)
Un composto viene considerato non mutageno se non induce un aumento
dose-dipendente del numero di colonie revertenti in tutti i ceppi impiegati
I risultati del test si definiscono inconclusivi quando non permettono di
identificare il composto in esame come mutageno o non mutageno
Test di aberrazioni
cromosomiche
TEST DI ABERRAZIONI CROMOSOMICHE IN CELLULE DI MAMMIFERO (OECD No.
473)
Lo scopo del test di aberrazioni cromosomiche in vitro è di identificare
sostanze che causano aberrazioni cromosomiche strutturali
Le cellule più comunemente usate sono i linfociti umani di sangue periferico
e le linee cellulari di Hamster cinese
Le colture cellulari vengono esposte alla sostanza da testare (max conc. 1
mM o 0.5 mg/ml) in assenza e in presenza di un sistema di attivazione
metabolica (S9)
Ad intervalli di tempo predeterminati, le cellule vengono trattate con una
sostanza che le arresta in metafase, raccolte, colorate ed analizzate al
microscopio per la presenza di aberrazioni cromosomiche.
Test di aberrazioni
cromosomiche in linfociti umani
SCHEMA SPERIMENTALE
Le cellule vengono trattate con il composto da testare per 3 - 6 ore in
presenza e in assenza di attivazione metabolica
Dopo aver allontanato il terreno contenente il composto, le cellule
vengono coltivate in terreno di coltura fresco per un periodo di tempo
corrispondente a circa 1.5 volte il ciclo cellulare dall’inizio del trattamento
Un trattamento continuo senza attivazione metabolica è necessario solo
in caso di risultati negativi o equivoci nei trattamenti brevi
I controlli positivi devono sempre essere inclusi per verificare la sensibilità
del test system utilizzato
Test di aberrazioni
cromosomiche in linfociti umani
SCHEMA SPERIMENTALE
TRATTAMENTO CONTINUO SENZA ATTIVAZIONE METABOLICA
T=0h
T = 48 h
T = 72 h
Test di aberrazioni
cromosomiche in linfociti umani
ABERRAZIONI DI TIPO CROMATIDICO
Test di aberrazioni
cromosomiche in linfociti umani
ESEMPI DI ABERRAZIONI CROMOSOMICHE
Test di aberrazioni
cromosomiche in linfociti umani
VALUTAZIONE ED INTERPRETAZIONE DEI RISULTATI
Un prodotto viene considerato positivo in questo test se induce un
aumento del numero di cellule con aberrazioni cromosomiche rispetto al
controllo negativo, che sia correlato con la dose e riproducibile
Generalmente vengono utilizzati dei metodi statistici come aiuto
supplementare nella valutazione dei risultati
Un aumento del numero di cellule poliploidi può indicare che la sostanza
può inibire i processi mitotici e di indurre aberrazioni cromosomiche di
tipo numerico.
Modelli sperimentali di mutagenesi
TEST DEL MICRONUCLEO IN CELLULE DI MAMMIFERO
(OECD No. 487 DRAFT)
Permette di identificare sostanze che causano perdita di cromosomi o
rotture cromosomiche, per cui un micronucleo può contenere interi
cromosomi o un frammento acentrico
Le cellule più comunemente usate sono i linfociti umani di sangue
periferico e le linee cellulari di Hamster cinese
Le colture cellulari vengono esposte alla sostanza da testare (max conc. 1
mM o 0.5 mg/ml) in assenza e in presenza di un sistema di attivazione
metabolica (S9)
Ad intervalli di tempo predeterminati, le cellule vengono trattate con una
sostanza che blocca la divisione cellulare, raccolte, colorate ed analizzate
al microscopio per la presenza di micronuclei
Test del micronucleo
Human lymphocytes
purified from whole blood
by gradient centrifugation
72h of culture
+ PHA, genotoxic agent, Cytochalasin B
Harvested
on glass
slides by
cytospin
Giemsa staining
Scoring
Human Lymphocytes
Modelli sperimentali di
mutagenesi
TEST DEL MICRONUCLEO NEGLI ERITROCITI DI RODITORI (OECD No. 474)
Rivela i danni sia cromosomici che dell’apparato mitotico che una sostanza
o un suo metabolita può provocare dopo somministrazione ad un roditore
(topo o ratto)
Il metodo è basato sull’aumento, nel midollo osseo, degli eritrociti
policromatici micronucleati negli animali trattati, rispetto agli animali di
controllo
I micronuclei sono costituiti da frammenti cromosomici o da cromosomi
interi che durante la mitosi non raggiungono i poli del fuso e non vengono
inclusi nel nucleo delle cellule figlie
I micronuclei rimangono nel citoplasma dell’eritrocita policromatico,
mentre il nucleo principale è espulso
Test del micronucleo negli
eritrociti di roditori
SCHEMA SPERIMENTALE
Topi di 5-7 settimane o ratti di 7-9 settimane
5 maschi e 5 femmine per gruppo sperimentale
3 dosi della sostanza in esame + un controllo negativo e un controllo
positivo
La dose più alta dovrebbe essere la massima dose tollerata (MTD) o la
massima tecnicamente somministrabile
Singola somministrazione, anche se si possono eseguire trattamenti
ripetuti, in base ad indicazioni tossicologiche
Il prelievo del midollo osseo viene eseguito 24 e 48 ore dopo la
somministrazione della sostanza.
Test del micronucleo negli eritrociti di
roditori
Test del micronucleo negli
eritrociti di roditori
VALUTAZIONE DEI RISULTATI
La percentuale degli eritrociti policromatici micronucleati dei gruppi trattati
con la sostanza in esame viene confrontata con il guppo di controllo
utilizzando test statistici
La sostanza viene considerata positiva se almeno due gruppi di trattamento,
con una relazione biologica tra di loro, inducono un aumento statisticamente
significativo degli eritrociti policromatici micronucleati rispetto al controllo
negativo.
Test della cometa
Single Cell Gel Electrophoresis
Può essere condotto sia in vitro che in vivo
Permette di evidenziare rotture al DNA sia a singolo che doppio filamento
Le cellule, sospese in agarosio liquido, vengono stratificate su un vetrino,
lisate con detergenti e soluzioni saline, e il DNA viene fatto correre in un
campo elettroforetico
I vetrini, colorati con bromuro di etidio, vengono osservati al microscopio a
fluorescenza
In assenza di danno, il nucleo risulta compatto e rotondeggiante
Se vi sono rotture al DNA, i frammenti migrano verso l’anodo, conferendo al
materiale nucleare la morfologia di una cometa
Test della cometa
Single Cell Gel Electrophoresis
Valutazione dei risultati dei
tests di mutagenesi
I tests di mutagenesi (in vitro e in vivo) individuano cancerogeni che
agiscono attraverso un meccanismo che implica un danno genetico
diretto. Non individuano cancerogeni non genotossici.
Se i risultati della batteria di tests indicano l’assenza di un potenziale
genotossico, i trials clinici possono essere condotti sia in soggetti sani che
in pazienti.
Modelli sperimentali di
cancerogenesi
STUDI DI CANCEROGENESI
Mentre gli studi di mutagenesi devono essere condotti
durante lo sviluppo clinico della maggior parte dei
farmaci, la conduzione degli studi di cancerogenesi
dipende dalla durata del trattamento nell’uomo
Linee Guida per la Conduzione degli
Studi di Cancerogenesi
•
ICH S1A, S1B, S1C(R2): CARCINOGENICITY STUDIES (1995, 1997, 2008)
•
EMEA “NOTE FOR GUIDANCE ON CARCINOGENICITY POTENTIAL” (2003)
•
EMEA SWP “CONCLUSIONS AND RECOMMENDATIONS ON THE USE OF
GENETICALLY MODIFIED ANIMAL MODELS FOR CARCINOGENICITY
ASSESSMENTS” (2004)
•
OECD GUIDELINES FOR TESTING OF CHEMICALS
Carcinogenicity studies: 451
•
ICH M3 (R2): TIMING OF TOXICOLOGY STUDIES (2009)
Modelli sperimentali di
cancerogenesi
STUDI DI CANCEROGENESI
Gli studi di cancerogenesi devono essere condotti per ogni prodotto
farmaceutico che si prevede usare in modo continuo per almeno 6
mesi
Gli studi di cancerogenesi sono generalmente necessari anche per
prodotti usati di frequente in maniera intermittente nel trattamento di
malattie croniche o ricorrenti
Prodotti somministrati non di frequente o per periodi brevi non
necessitano di studi di cancerogenesi a meno che non ci siano motivi di
preoccupazione (cause for concern)
Modelli sperimentali di
cancerogenesi
“CAUSE FOR CONCERN”
Precedente evidenza di potenziale cancerogeno nella classe chimica a
cui il prodotto appartiene
Relazione struttura-attività che suggerisce un rischio cancerogeno
Evidenza di lesioni preneoplastiche in studi di tossicologia a dose
ripetuta
Ritenzione del prodotto o dei suoi metaboliti nei tessuti per periodi
lunghi, dando luogo a reazioni tissutali locali o ad altre reazioni
patofisiologiche.
Modelli sperimentali di
cancerogenesi
PRODOTTI GENOTOSSICI
Prodotti che sono inequivocabilmente genotossici non
necessitano di studi di cancerogenesi a lungo termine.
Comunque, se il prodotto deve essere somministrato all’uomo
cronicamente, uno studio di tossicità cronica nell’animale (fino ad
1 anno) può essere necessario per evidenziare effetti cancerogeni
precoci
Modelli sperimentali di
cancerogenesi
“TIMING” DEGLI STUDI DI CANCEROGENESI
Quando è necessario condurre gli studi di cancerogenesi, questi devono
essere completati prima della registrazione del prodotto
Per prodotti sviluppati per il trattamento di malattie gravi, gli studi di
cancerogenesi possono essere condotti dopo la registrazione, per
anticipare la disponibilità del prodotto
Per malattie in cui l’aspettativa di vita è breve (meno di 2-3 anni), gli
studi di cancerogenesi non sono necessari (per es. antitumorali); al
contrario, in caso di terapia adiuvante in pazienti senza tumore, gli studi
di cancerogenesi sono generalmente richiesti
Modelli sperimentali di
cancerogenesi
APPROCCIO DA SEGUIRE PER LA VALUTAZIONE DEL
POTENZIALE CANCEROGENO DI PRODOTTI FARMACEUTICI
1.
Uno studio di cancerogenesi a lungo termine nei roditori
2.
Un test a breve termine che utilizza modelli transgenici o roditori neonati,
oppure uno studio a lungo termine in una seconda specie roditrice
Studi di cancerogenesi a
lungo termine nei roditori
SELEZIONE DELLA SPECIE
La specie viene selezionata sulla base dei dati di farmacologia,
tossicologia, farmacocinetica e metabolismo della sostanza in esame
In assenza di chiare evidenze sperimentali in favore di una determinata
specie, il ratto è la specie raccomandata
Studi di cancerogenesi a
lungo termine nei roditori
DURATA DELLO STUDIO
In genere 24 mesi per il ratto ed almeno 18 mesi per il topo e hamster
Comunque, lo studio può essere terminato quando la sopravvivenza
delle dosi più basse e del gruppo di controllo raggiunge il 25 %.
Studi di cancerogenesi a
lungo termine nei roditori
OSSERVAZIONI
Segni clinici e mortalità
Peso corporeo
Consumo di cibo
Oftalmoscopia
Masse palpabili
Parametri biochimici ed ematologici
Analisi delle urine
Esposizione sistemica
Studi di cancerogenesi a
lungo termine nei roditori
PATOLOGIA
Necroscopia: su tutti gli animali morti durante lo studio o sacrificati in
extremis e sugli animali sopravvissuti alla fine dello studio
Istopatologia:
- tutti i tumori visibili e lesioni sospette
- tutti gli organi e tessuti previsti dal protocollo
a) di tutti gli animali morti o sacrificati durante lo studio
b) degli animali della dose più alta e di controllo
- se si osservano differenze significative nelle lesioni neoplastiche tra la
dose più alta e il controllo, l’esame microscopico degli organi/tessuti
coinvolti deve essere condotto su tutti gli animali