Impossibile visualizzare l'immagine.
La memoria del computer potrebbe
essere insufficiente per aprire
l'immagine oppure l'immagine
potrebbe essere danneggiata.
Riavviare il computer e aprire di nuovo
il file. Se viene visualizzata di nuovo la
x rossa, potrebbe essere necessario
eliminare l'immagine e inserirla di
nuovo.
UNIVERSITA’ DEGLI STUDI DI PAVIA
LABORATORIO DI BIOLOGIA SPERIMENTALE
DIPARTIMENTO DI BIOLOGIA E BIOTECNOLOGIE
I-27100PAVIA(Italia)–ViaFerrata9
Direttore:Prof.LucaFerretti;email:[email protected]
Incontri di Formazione Docenti Scuole del 16 e 17 dicembre 2015
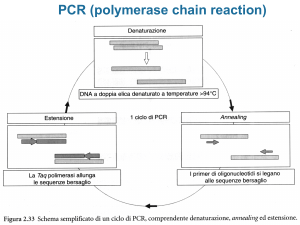
ATTIVITA’ 1 – AMPLIFICAZIONE DI DNA MEDIANTE PCR
Amplificazione di DNA ribosomale di Saccaromyces cerevisiae mediante PCR
In questa attività utilizzeremo la PCR per amplificare dal DNA genomico di S. cerevisiae un
frammento che contiene una porzione di gene per gli RNA ribosomali.
Il tratto di DNA, lungo 841 paia di basi (bp) è indicato di seguito. In verde e in viola sono
evidenziati i tratti utilizzati come inneschi (oligonucleotidi) per la PCR.
5’
TCCGTAGGTGAACCTGCGGAAGGATCATTAAAGAAATTTAATAATTTTGAAAATGGATTTTTTTGTTT
TGGCAAGAGCATGAGAGCTTTTACTGGGCAAGAAGACAAGAGATGGAGAGTCCAGCCGGGCCTGCGCT
TAAGTGCGCGGTCTTGCTAGGCTTGTAAGTTTCTTTCTTGCTATTCCAAACGGTGAGAGATTTCTGTG
CTTTTGTTATAGGACAATTAAAACCGTTTCAATACAACACACTGTGGAGTTTTCATATCTTTGCAACT
TTTTCTTTGGGCATTCGAGCAATCGGGGCCCAGAGGTAACAAACACAAACAATTTTATCTATTCATTA
AATTTTTGTCAAAAACAAGAATTTTCGTAACTGGAAATTTTAAAATATTAAAAACTTTCAACAACGGA
TCTCTTGGTTCTCGCATCGATGAAGAACGCAGCGAAATGCGATACGTAATGTGAATTGCAGAATTCCG
TGAATCATCGAATCTTTGAACGCACATTGCGCCCCTTGGTATTCCAGGGGGCATGCCTGTTTGAGCGT
CATTTCCTTCTCAAACATTCTGTTTGGTAGTGAGTGATACTCTTTGGAGTTAACTTGAAATTGCTGGC
CTTTTCATTGGATGTTTTTTTTCCAAAGAGAGGTTTCTCTGCGTGCTTGAGGTATAATGCAAGTACGG
TCGTTTTAGGTTTTACCAACTGCGGCTAATCTTTTTTTATACTGAGCGTATTGGAACGTTATCGATAA
GAAGAGAGCGTCTAGGCGAACAATGTTCTTAAAGTTTGACCTCAAATCAGGTAGGAGTACCCGCTGAA
CTTAAGCATATCAATAAGCGGAGGA 3’
3’ CGTATAGTTATTCGCCTCCT 5’
Gli oligonucleotidi che utilizzeremo per la PCR sono quindi:
forward ITS1 (5'- TCCGTAGGTGAACCTGCGG - 3')
reverse ITS4 (5'-TCCTCCGCTTATTGATATGC - 3')
Notare che il reverse primer ITS4, proprio perchè deve innescare sul filamento opposto di
DNA (non rappresentato), è “complementare” al tratto di DNA in viola.
Il DNA di 841 bp scelto corrisponde a una porzione del cromosoma XII di S. cerevisiae che
produce un lungo RNA dal quale vengono ricavati per taglio i tre RNA ribosomali 25S, 18S e
5.8S. Questa corrispondenza si può vedere facendo l’analisi della “omologia di sequenza” tra
il nostro tratto di DNA e il database che contiene l’intera sequenza del genoma di S.
cerevisiae. Questa ricerca di omologia si esegue in rete con programmi specifici, uno dei
quali si chiama BLAST.
Il risultato della ricerca è illustrato nella Figura 1 dove nel pannello principale il nostro
frammento di 841 bp è indicato in basso, in grigio, con il nome Query_54433; in viola sono
indicati vari geni per RNA ribosomali tra i quali quello che dà origine al lungo trascritto dal
quale si formano gli RNA 25S, 18S e 5.8S (codice RDN37-1). Si può vedere come il nostro
tratto di DNA di 841 bp “corrisponda” (più correttamente sia omologo) al lungo trascritto
RDN37-1 e anche a trascritti di geni più piccoli, che rappresentano il prodotto finale della
1
frammentazione del lungo trascritto nei differenti RNA ribosomali. Il tratto di DNA di 841 bp
che amplificheremo contiene, nell’ordine, da sinistra a destra, una piccola porzione del gene
25S, l’intero gene 5.8S e una piccola porzione del gene 18S.
Infine, notare che in alto nella figura è rappresentato schematicamente l’intero cromosoma
XII; la porzione evidenziata da una freccia bianca su fondo blu è quella contenente i geni
ribosomali che sono ingranditi nel pannello principale della Figura 1, come appena descritto.
Figura 1. Omologia di sequenza tra il prodotto di PCR e i geni ribosomali sul cromosoma XII di S. cerevisiae
1-1) Estrazione rapida di DNA da colonia di S. cerevisiae
Questa procedura estrae velocemente dalle cellule il DNA ed ma non lo purifica in alcun
modo, quindi affinchè la PCR seguente funzioni si deve prestare attenzione a non esagerare
con il prelievo di cellule (ecco perché mezza colonia). Viceversa, se si esagera, si aumenta il
rischio che le molte componenti cellulari solubili che rimarranno presenti nel campione
assieme al DNA inibiscano la reazione di PCR (in particolare gli RNA di piccole dimensioni).
Procedura
• Preparare un tubo eppendorf da 1.5 ml (per banco) e contrassegnarlo in modo
univoco usando il numero del vostro bancone (da 1 a 8, vi verrà detto quale bancone
siete). Pipettate nel tubo 200 microlitri di H2O
• Prendere la piastra con le colonie di lievito S. cerevisiae che vi verrà data; con on
un’ansa prelevare circa mezza colonia e stemperare la massa di cellule nel tubo
eppendorf contenente 200 microlitri di H2O
• Vortexare il tubo per assicurarsi di avere risospeso bene le cellule
• Incubare i tubi a 100°C per 15 minuti in modo da distruggere le cellule
• Centrifugare i tubi in una centrifuga Eppendorf al massimo della velocità per 2 minuti
• Rimuovere i tubi dalla centrifuga e tenere da parte il tubo perché il DNA che contiene
servirà tra poco per la reazione di PCR.
1-2) Preparazione delle reazioni di PCR
2
Materiale necessario che vi verrà messo a disposizione sul banco e che si utilizza in comune
con i colleghi del banco: H2O, oligonucleotidi ITS1 e ITS4 (entrambi 10 µM), sali GoTaq 5X,
MgCl2 25 mM, dNTPs 2 mM. In più avrete il tubo con il DNA estratto da colonia (punto 1-1).
L’enzima GoTaq Polymerase, 5 U/microlitro vi verrà fornito a reazione assemblata, banco per
banco, dagli istruttori.
Gli istruttori vi spiegheranno come usare pipettatrici e puntali monouso.
Ricordate sempre che quando si utilizzano tubi in comune bisogna procedere con
attenzione per evitare contaminazioni. Per questo cambiate sempre i puntali della
pipettatrice ogni volta che passate a un tubo in comune da cui prelevare una aliquota
per il vostro utilizzo. Questo vale anche nei casi in cui utilizzate tubi per componenti
non in comune, ma diversi tra di loro.
Procedura
• Preparate un tubo Eppendorf da 0.5 ml e contrassegnatelo in modo univoco usando il
numero del vostro bancone (da 1 a 8, vi verrà detto quale bancone siete) e poi le
lettere A, B e C a seconda che siate da soli al bancone (1A), in due (1A, 1B) o tre (1A,
1B, 1C).
• Assemblate la reazione di PCR nel tubo contrassegnato seguendo il protocollo e
rispettando l’ordine delle componenti così come è rappresentato più avanti.
• Per l’utilizzo della pipettatrice seguite le istruzioni che vi sono state fornite. In
particolare, nel prelevare le aliquote dai tubi stock e nel trasferirle all’interno del vostro
tubo di reazione, cercate di non fare bolle e di svuotare completamente il puntale.
• Come già detto sopra cambiate il puntale della pipettatrice dopo ogni prelievoaggiunta e spuntate ogni volta – sul vostro protocollo di reazione - la riga del
componente che avete aggiunto per essere sicuri di procedere correttamente
• Prima dell’aggiunta di enzima, se necessario, potete mescolare le componenti
picchiettando delicatamente il tubo con un dito e poi centrifugando brevemente in una
centrifuga eppendorf.
1-3) Protocollo della reazione di PCR - Aggiunta delle componenti (rispettare l’ordine)
1) H2O
2) Sali GoTaq 5X
3) MgCl2 25 mM
4) dNTPs 2 mM
5) Primer ITS1 10 µM
6) Primer ITS4 10 µM
7) DNA estratto (punto 1-1 )
6.5 microlitri
4
1
2
2
2
2
8) Enzima GoTaq Polimerasi 5U/microlitro
Totale
0.5
20 microlitri
E’ buona pratica fare sempre un tubo di reazione PCR di controllo (il cosiddetto – “meno” o
controllo negativo) che si chiama così perché è preparato come sopra con l’unica differenza
che NON contiene DNA. Nel nostro caso verrà predisposto dagli istruttori.
1-4) Trasferimento delle reazioni di PCR sul Termociclatore
Finita la preparazione delle reazioni PCR i tubi vengono trasferiti nel termociclatore e
assoggettati all’amplificazione secondo il seguente programma ciclico che è stato
precedentemente impostato e memorizzato nel termociclatore:
3
95°C 2 minuti
30X (30 cicli) fatti ciascuno di tre step: 95°C 20 sec, 53°C 20 sec, 72°C 1 minuto
72°C 10 minuti (estensione finale)
Fine programma
Alla fine del programma è opportuno rimuovere subito i campioni e procedere alle successive
analisi (ad es. elettroforesi su gel di agarosio; taglio con enzimi di restrizione). Altrimenti è
bene congelarli a -20°C in attesa di essere analizzati.
Il termociclatore può essere programmato in modo che alla fine del programma di PCR
abbassi la temperatura a 4°C e la mantenga fino al recupero dei campioni. Peraltro questa
pratica sottopone a stress il blocco termico del termociclatore e alla lunga abbassa la “vita”
dello strumento quindi, se possibile, è meglio rimuovere i campioni finita la PCR.
ATTIVITA’ 2 – DIGESTIONE DI DNA CON ENZIMI DI RESTRIZIONE
Gli enzimi di restrizione (tipo II) sono prodotti dalla stragrande maggioranza dei
microorganismi procariotici e riconoscono sul DNA sequenze specifiche lunghe da 4 a 8 e più
bp. Tipicamente tagliano in corrispondenza della sequenza riconosciuta, ma non sempre.
Nella nostra attività utilizzeremo l’enzima ApaI (“Apa uno”). ApaI è un enzima di restrizione
tipico, nel senso che taglia internamente alla sequenza riconosciuta:
I triangoli indicano la posizione dove il DNA di ciascun filamento viene tagliato. Il risultato
finale è la formazione di frammenti di DNA con estremità sporgenti come indicato di seguito
5’……GGGCC 3’
3’……C 5’
5’ C……3’
3’ CCGGG……5’
Con “estremità” 5’ e 3’ ci si riferisce alla nomenclatura/numerazione degli atomi di Carbonio
dello zucchero sul DNA (il desossiribosio) e tipicamente dopo il taglio ogni filamento di DNA
espone un Carbonio 5’ con attaccato un gruppo fosfato (-P) e un Carbonio 3’ con attaccato
un gruppo ossidrile (-OH). Esistono centinaia di enzimi di restrizione, ciascuno con la propria
specificità di taglio. Ad es. uno tra i primi scoperti e molto usati è EcoRI (“Eco R uno”) che
riconosce e taglia la sequenza
Nota Bene: DNA tagliati con gli enzimi di restrizione possono essere saldati nuovamente
usando enzimi che si chiamano DNA ligasi, che saldano covalentemente estremità 5’-P con
estremità 3’-OH. Infatti le estremità prodotte da enzimi di restrizione sono “coesive” vedi
figure dei siti di taglio. Questo fatto si sfrutta quando si creano molecole di DNA
ricombinante, cioè prodotte dalla unione (saldatura con DNA ligasi) di DNA diversi, tagliati
con lo stesso enzima di restrizione, ad esempio il DNA di un plasmide tagliato con EcoRI
saldato al DNA di un gene di nostro interesse tagliato a sua volta con EcoRI. Con questa
operazione si “clona” il DNA del gene nel plasmide.
2-1) Preparazione delle reazioni di digestione ApaI
4
Materiale necessario in comune che trovate sul banco:
H2O, sali ApaI 10X, BSA 1 mg/ml, tubo con il DNA da tagliare.
Come DNA si utilizza il prodotto della reazione di PCR descritto nell’Attività 1, ma per
mancanza di tempo (i prodotti della amplificazione saranno disponibili solo domani) vi verrà
fornito sul banco un DNA identico preparato precedentemente dagli istruttori (vedi “Punto 2-2
più avanti). L’enzima ApaI 10 U/microlitro verrà fornito al momento, banco per banco, dagli
istruttori.
Procedura
• Preparate un tubo Eppendorf da 0.5 ml e contrassegnatelo in modo univoco usando il
numero del vostro bancone (da 1 a 8, vi verrà detto quale bancone siete) e poi le
lettere A, B e C a seconda che siate da soli al bancone (1A), in due (1A, 1B) o tre (1A,
1B, 1C).
• Assemblate la reazione di digestione nel tubo contrassegnato seguendo il protocollo e
rispettando l’ordine delle componenti così come è rappresentato più avanti.
• Per l’utilizzo della pipettatrice seguite le istruzioni che vi sono state date. In particolare
nel prelevare le aliquote dai tubi stock e nel trasferirle all’interno del vostro tubo di
reazione cercate di non fare bolle e di svuotare completamente il puntale.
• Come già detto sopra cambiate il puntale della pipettatrice dopo ogni prelievoaggiunta e spuntate ogni volta – sul vostro protocollo di reazione - la riga del
componente che avete aggiunto per essere sicuri di procedere correttamente
• Prima dell’aggiunta di enzima, se necessario, potete mescolare le componenti
picchiettando delicatamente il tubo con un dito e poi centrifugando brevemente in una
centrifuga eppendorf.
2-2) Protocollo del taglio di DNA con ApaI - Aggiunta delle componenti (rispettare
l’ordine)
1) H2O
2) Sali ApaI 10X
3) BSA 1 mg/ml
4) DNA per Apa (prodotto PCR di 841 bp (*))
11.5 microlitri
2
2
4
Enzima ApaI 10 U/microlitro
Totale
0.5
20 microlitri
La digestione con ApaI va incubata alla temperatura di 25°C, quindi i tubi di reazione
verranno trasferiti in un blocco termostatato a temperatura controllata impostato a 25°C. I tubi
verranno lasciati a 25°C per tutta la notte (O/N).
Alla fine dell’incubazione recuperare i tubi e procedere alla visualizzazione dei prodotti su gel
di agarosio (Attività 3).
ATTIVITA’ 3 – ELETTROFORESI DI DNA SU GEL DI AGAROSIO
L’elettroforesi su gel di agarosio serve a visualizzare DNA (ma anche RNA). Le applicazioni
più tipiche di questa procedura sono la visualizzazione dei prodotti di una reazione di PCR e
la visualizzazione di frammenti di DNA che derivano dal taglio con enzimi di restrizione. Le
due cose possono essere combinate (taglio di un prodotto di PCR).
Nell’attività descritta di seguito utilizzeremo due gel di agarosio per:
1) visualizzare i risultati della PCR fatta sui geni ribosomali di lievito (Attività 1)
5
2) Visualizzare i prodotti della digestione dello stesso prodotto di PCR con l’enzima di
restrizione ApaI (Attività 2)
3-1) Preparazione del gel di agarosio
• Preparare in una bottiglia (o beuta) di vetro Pirex 100 ml di tampone TAE 1X (40 mM
Tris, 20 mM Acetato, 1 mM EDTA) diluito dallo stock 50X. Per 100 ml di gel usare una
bottiglia (o beuta) da almeno 250 ml.
• Pesare 1.5 grammi di agarosio e aggiungerli alla bottiglia. Pesare la bottiglia con tampone
e agarosio dentro e prendere nota del peso.
• Sciogliere l’agarosio mettendo la bottiglia nel forno a microonde, impostato a 700-900 W
(se avete tenuto il tappo sulla bottiglia assicuratevi che non sia chiuso altrimenti il
vapore che si forma durante il riscaldamento nel forno può fare ESPLODERE la
bottiglia).
• Iniziare riscaldando per circa un minuto, poi per periodi più brevi (ad es 15-20 sec)
sorvegliando sempre la beuta/bottiglia in modo da poter interrompere l’incubazione se
necessario per evitare che la soluzione bolla fuori dalla bottiglia.
• Alla fine di ogni riscaldamento agitare delicatamente la beuta (usare guanti protettivi:
attenzione alla temperatura della bottiglia!) e controllare la progressione dello
scioglimento dell’agarosio che sarà completo quando la sospensione diventa
completamente trasparente.
• Una volta sciolto l’agarosio ripesare la bottiglia e aggiungere H2O a compensare la perdita
avvenuta per evaporazione.
• Aggiungere Bromuro di Etidio (EtBr) alla concentrazione finale di 0.5 migrogrammi/ml (nel
nostro caso lo stock è a 10 milligrammi/ml, quindi diluiremo 20000 volte, cioè 5 microlitri di
EtBr stock nei 100 ml del gel). Prestare attenzione perché il EtBr è un mutageno;
indossare sempre i guanti; per la nostra attività questa operazione la eseguiranno
gli istruttori. In alternativa al EtBr si possono usare coloranti che non richiedono
precauzioni e non presentano rischi; ne esistono molti e nel caso si seguano le istruzioni
d’uso fornite.
• EtBr e coloranti alternativi servono come intercalanti del DNA per visualizzarlo nel gel una
volta illuminato con il transilluminatore a lunghezze d’onda variabili a seconda dei coloranti
usati (260-310 nm per il EtBr)
• Attendere che la soluzione di agarosio si raffreddi per poterla versare nel vassoio dove si
formerà il gel. La temperatura a cui si può versare è circa 55°C, non di più, ma in pratica ci
si regola su quando si può tenere la bottiglia nelle mani senza scottarsi.
• Versare l’agarosio nel vassoio preparato precedentemente e contenente il pettine (o i
pettini a seconda delle necessità). E’ importante che non si formino bolle nel gel e
soprattutto “sotto” i denti del pettine. Se c’è qualche bolla che “gallegia” sul gel la si può
trascinare delicatamente verso i bordi con un puntale per pipettatrice pulito.
• Lasciare riposare il gel almeno 30 minuti o fino a quando la gelificazione non è completa.
A questo punto si può rimuovere il pettine (delicatamente, per evitare che si rompa
l’agarosio e si rovinino i pozzetti nel gel. Allo scopo si può - prima di rimuovere il pettine usare una spruzzetta con acqua per bagnare il gel in corrispondenza del pettine).
Rimuovere poi lo scotch che sigilla le estremità del vassoio e infine alloggiare il gel nella
vaschetta elettroforetica riempita di tampone TAE 1X + EtBr 0.5 microgrammi/ml come il
gel.
• Prima di procedere al caricamento dei campioni controllare che il gel sia completamente
coperto dal tampone nella vaschetta, ma senza esagerare; circa 2-3 millimetri è la quantità
giusta.
3-2) Preparazione dei campioni e caricamento del gel
Nella nostra attività faremo due gel identici, entrambi 1.5% agarosio in TAE 1X.
Sul primo caricheremo i prodotti di PCR, sull’altro i prodotti della digestione ApaI.
6
Ciascuno di voi preparerà due campioni da caricare sui due gel
Preparazione del campione della digestione ApaI
• Prendere il tubo in cui è stata fatta la digestione ApaI (20 microlitri totali di miscela,
attività 2)
• Aggiungere al tubo 4 microlitri di “Gel Loading Buffer” 6X
• mescolare bene con la pipettatrice
Preparazione del campione della reazione di PCR
• Prendere il tubo della reazione PCR e aggiungervi 4 microlitri di Gel Loading Buffer 6X
• mescolare bene con la pipettatrice.
A questo punto ciascuno avrà due tubi per il caricamento su gel, il tubo digestione ApaI e il
tubo prodotto PCR.
3-3) Caricamento su gel ed elettroforesi
• Caricare sul gel dei prodotti PCR 12 microlitri del tubo prodotto PCR + Gel Loading
Buffer appena preparato
• Caricare sull’altro gel, quello dei prodotti di digestione ApaI, 12 microlitri del tubo della
digestione ApaI + Gel loading Buffer appena preparato.
• Su entrambi i gel verranno caricati dagli istruttori marcatori di peso molecolare e altri
campioni di controllo.
Per la separazione dei DNA su gel ci si regola caso per caso e in funzione delle dimensioni
dei frammenti di DNA attesi e/o della concentrazione del gel (percentuale di agarosio).
Nel nostro caso faremo una corsa elettroforetica a 80 Volts, ca 100 mA per il tempo
necessario, 30-60 minuti.
Dopo la corsa elettroforetica i gel verranno fotografati ponendoli su un transilluminatore e
utilizzando una apparato fotografico dotato di CCD (Charged Coupled Device, la stessa
tecnologia usata nelle macchine fotografiche digitali, compatte e Reflex, e nelle fotocamere
dei telefoni cellulari) che digitalizza l’immagine.
3-4) Analisi dei risultati
L’analisi dei risultati ottenuti verrà fatta assieme agli istruttori.
Cosa ci si aspetta di vedere?
Nel caso dei prodotti di PCR abbiamo amplificato un tratto di DNA dei geni ribosomali di S.
cerevisiae (Attvità 1) lungo 841 bp, quindi ci aspettiamo di vedere su gel frammenti come
quelli mostrati nella figura che segue
12345
1000bp
700bp
7
PCR da colonia di S. cerevisiae realizzata con i primers ITS1-ITS4. 1) MARKER 1Kb Plus Fermentas; 2)
Campione A; 3) Campione B; 4) Campione C; 5) PCR di Controllo (-)
E per quanto riguarda il taglio del prodotto di PCR con ApaI cosa ci aspettiamo di vedere?
Questo dipende da quanti siti di taglio possiede ApaI sul nostro prodotto di PCR. Dal
momento che conosciamo la sequenza intera del frammento
5’
TCCGTAGGTGAACCTGCGGAAGGATCATTAAAGAAATTTAATAATTTTGAAAATGGATTTTTTTGTTT
TGGCAAGAGCATGAGAGCTTTTACTGGGCAAGAAGACAAGAGATGGAGAGTCCAGCCGGGCCTGCGCT
TAAGTGCGCGGTCTTGCTAGGCTTGTAAGTTTCTTTCTTGCTATTCCAAACGGTGAGAGATTTCTGTG
CTTTTGTTATAGGACAATTAAAACCGTTTCAATACAACACACTGTGGAGTTTTCATATCTTTGCAACT
TTTTCTTTGGGCATTCGAGCAATCGGGGCCCAGAGGTAACAAACACAAACAATTTTATCTATTCATTA
AATTTTTGTCAAAAACAAGAATTTTCGTAACTGGAAATTTTAAAATATTAAAAACTTTCAACAACGGA
TCTCTTGGTTCTCGCATCGATGAAGAACGCAGCGAAATGCGATACGTAATGTGAATTGCAGAATTCCG
TGAATCATCGAATCTTTGAACGCACATTGCGCCCCTTGGTATTCCAGGGGGCATGCCTGTTTGAGCGT
CATTTCCTTCTCAAACATTCTGTTTGGTAGTGAGTGATACTCTTTGGAGTTAACTTGAAATTGCTGGC
CTTTTCATTGGATGTTTTTTTTCCAAAGAGAGGTTTCTCTGCGTGCTTGAGGTATAATGCAAGTACGG
TCGTTTTAGGTTTTACCAACTGCGGCTAATCTTTTTTTATACTGAGCGTATTGGAACGTTATCGATAA
GAAGAGAGCGTCTAGGCGAACAATGTTCTTAAAGTTTGACCTCAAATCAGGTAGGAGTACCCGCTGAA
CTTAAGCATATCAATAAGCGGAGGA 3’
la possiamo analizzare con dei “tools” gratuiti disponibili in rete per stabilire come viene
tagliato questo tratto di DNA mediante una digestione “virtuale”. Di tools del genere ne
esistono tantissimi, tra di essi Watcut che si trova a questo indirizzo web:
http://watcut.uwaterloo.ca/template.php?act=restriction_new
Una volta aperta la pagina
facciamo un copia/incolla della nostra sequenza (solo le basi, non i numeri) nel riquadro
centrale, poi aggiungiamo un nome (opzionale; nella figura PCR lievito) ottenendo questo:
8
A questo punto facciamo clic sul pulsante “Submit new sequence” e verrà visualizzata una
finestra che elenca, enzima per enzima, in ordine alfabetico, tutti i tagli che si possono
produrre sul frammento. Tra i molti enzimi c’è anche ApaI che taglia una sola volta (vedi
figura).
Selezionando in alto a sinistra il pulsante “In table” sotto Display Results e poi facendo clic
clic a destra nella finestra su “Update display”; otterremo la seguente figura dove si vede che
ApaI taglia il frammento in posizione 302.
9
Ciò significa che ApaI taglia il frammento di 841 bp in due frammenti, uno di 539 bp e uno di
302 bp.
Quindicomerisultatodellaattività2(tagliodelprodottodiPCRconApaI)ciaspettiamodi
visualizzaresulgeldiagarosiodueframmentidiDNAcorrispondentiilpiùgrandelungo539bp
eilpiùpiccololungo302bp.
----------------------------TAMPONI-------------------------------
TAE50X
Per1litrodibufferTAE50Xusare:
• 242gTrisbase(Tris-OH)(2-ammino-2-idrossimetil-propan-1,3-diolo)
• 57.1mLacidoaceticoglaciale(=100%acidoacetico)
• 100mL0,5MNa2EDTA(pH8.0)
• VersareH2Ofinoaraggiungereunvolumetotaledi1litro.
Perpreparare0,5MNa2EDTA(pH8.0)aggiungere186,1gdietilenediamminotetraacetatodi
disodioinH2Ofinoa800mlofH2O.Agitarevigorosamente.PortareilpHa8,0conNaOH(20g
ca.diNaOH).Sterilizzareinautoclave.Avvertimento:L'Na2EDTAnonsisciogliefinoachela
soluzionenonèapHcirca8.0tramitel'aggiuntadiNaOH.
SaliApaI10X(SaliNEBBuffer4)
500mMPotassioAcetato
200mMTris-Acetato
100mMMagnesioAcetato
10
10mMDTT
pH7.9@25°C
GELLOADINGBUFFER6X
10mMTris-HCl(pH7.6)
0.03%BludiBromofenolo(BPB)
0.03%XilenCianoloFF(XC)
60%glicerolo
60mMEDTA
Ingelall’1%diagarosioilBPBmigraconlastessamobilitàdiDNAlunghi300bp,loXCconDNA
dica4000bp
UnaformulazionepiùsemplicediGelLoadingBuffer–chevacomunquebeneperigeldi
agarosio-èlaseguente:
GELLOADINGBUFFER10X
0.9%SDS(SodioDodecilSolfato)
50%Glicerolo
0.05%BludiBromofenolo(BPB)
notarechecontienesoloBludiBromofenoloedè10X
11
RisultatodellaPCR.Fotografiadigitalizzatadellaelettroforesisugeldiagarosioal1.5%inTAE
1X.Icodicisonoquellidavoiusati(posizionisulbanco).Mèunmarcatoredipesimolecolari,Cè
unaPCRdicontrollo
12
RisultatodelladigestioneconApaI.Fotografiadelgelall’1.5%inTAE1X.
Icodicisonocomesoprariferitiaibanchidilavoro.Nonsonosicuroche6Csiacorretto,forseera
6B.
Mèunmarcatoredipesimolecolari.Lefrecceindicanoiprodottideltaglioconleloro
dimensioni.Nellacorsia3Bèvisibileunframmentodi841bpcherappresentailprodottonon
tagliato.InquestocasoladigestioneconApaInonèavvenutacompletamente.
13








![mutazioni genetiche [al DNA] effetti evolutivi [fetali] effetti tardivi](http://s1.studylibit.com/store/data/004205334_1-d8ada56ee9f5184276979f04a9a248a9-300x300.png)