UNIVERSITÀ DEGLI STUDI DI MILANO
FACOLTÀ DI FARMACIA
ISTITUTO DI ENDOCRINOLOGIA
CORSO DI DOTTORATO DI RICERCA IN
SCIENZE ENDOCRINOLOGICHE E METABOLICHE
Ciclo XVIII
SISTEMI PEPTIDERGICI NELLA PROGRESSIONE DEL CARCINOMA
PROSTATICO UMANO: NEUROPEPTIDE Y E SOMATOSTATINA
Tutor: Chiar.mo Prof. Paolo MAGNI
Coordinatore: Chiar.mo Prof. Flavio PIVA
Tesi di dottorato:
dott. Massimiliano RUSCICA
Matricola N. R04897
Anno Accademico: 2004/2005
1
INDICE
1
LA PROSTATA
pag. 1
1.1
Sviluppo e differenziazione della ghiandola prostatica
pag. 1
1.2
Anatomia della prostata
pag. 2
1.3
La capsula prostatica
pag. 4
1.4
Rapporti stroma epitelio: ruolo degli ormoni e dei fattori di crescita
pag. 5
1.5
Influenze ormonali sulla crescita della prostata
pag. 6
1.6
Controllo molecolare del ciclo cellulare
pag. 8
1.7
Eziologia e fattori di predisposizione per il PCa
pag. 8
1.8
Fattori di crescita e carcinoma prostatico
pag. 14
1.9
Prostatic intraepithelial neoplasia (PIN)
pag. 17
1.10
Progressione verso l’ormono-indipendenza
pag. 19
1.11
h (human) ZAC: un nuovo fattore di trascrizione antiproliferativo
pag. 20
1.12
Geni hZAC e mZAC
pag. 21
2.
NEUROPEPTIDE Y (NPY)
pag. 23
2.1
NPY: sintesi e processing
pag. 24
2.2
Distribuzione anatomica
pag. 27
2.3
Recettori NPY
pag. 28
2.4
La sottofamiglia Y1: Y1, Y4, y6
pag. 29
2.5
La sottofamiglia Y2: Y2 e Y7
pag. 31
2.6
La sottofamiglia Y5: Y5
pag. 31
2.7
Il recettore Y1
pag. 32
2.8
Il recettore Y2
pag. 34
2.9
Il recettore Y3
pag. 35
2.10
Il recettore Y4
pag. 36
2.11
Il recettore Y5
pag. 37
2.12
Il recettore y6
pag. 39
2
2.13
Vie di trasduzione del segnale
pag. 40
2.14
NPY e suoi recettori in modelli transgenici e knockout
pag. 43
2.15
Animali transgenici per NPY
pag. 44
2.16
Animali knockout per NPY
pag. 45
2.17
Topi knockout per il recettore Y1
pag. 46
2.18
Topi knockout per il recettore Y2
pag. 48
2.19
Topi knockout per il recettore Y4
pag. 48
2.20
Topi knockout per il recettore Y5
pag. 49
2.21
NPY: controllo dell’espressione genica e proteica
pag. 49
2.22
Effetti sul comportamento alimentare e sul metabolismo
pag. 53
2.23
Regolazione dei meccanismi neuroendocrini
pag. 54
2.24
NPY: cancro della mammella e della prostata
pag. 56
3
LA SOMATOSTATINA
pag. 58
3.1
Aspetti molecolari
pag. 58
3.2
Localizzazione anatomica e caratteristiche delle cellule che la
producono
pag. 60
3.3
Attività biologiche
pag. 64
3.4
Azioni a livello dei sistemi nervoso centrale e periferico
pag. 64
3.5
Regolazioni delle funzioni ipotalamo-ipofisi
pag. 66
3.6
Regolazione endocrina ed esocrina del pancreas
pag. 68
3.7
Regolazione delle funzioni gastriche, intestinale ed epatiche
pag. 69
3.8
Somatostatina e controllo dell’omeostasi nutrizionale
pag. 70
3.9
Somatostatina: Modulazione delle funzioni renali, polmonari
e del sistema immunitario
pag. 71
3.10
I recettori per la somatostatina
pag. 73
3.11
Distribuzione
pag. 77
3.12
Attivazione e meccanismi intracellulari
pag. 79
3.13
Effetti mediati dal sistema adenilato ciclasi-protein chinasi A
pag. 80
3
3.14
Effetti mediati dal sistema PLC/IP3
pag. 82
3.15
Effetti mediati da variazioni delle concentrazioni ioniche intracellulari
pag. 82
3.16
Effetti mediati da modificazioni dell’attività fosfatasica
pag. 83
3.17
Dimerizzazione dei recettori della somatostatina
pag. 86
3.18
Analoghi della somatostatina
pag. 87
3.19
Somatostatina e suoi analoghi: controllo della crescita cellulare
pag. 89
3.20
Effetto antiproliferativo in cellule non trasformate
pag. 92
3.21
Effetto antiproliferativo in linee cellulari neoplastiche
pag. 93
3.22
Effetto antiproliferativo in linee cellulari neoplastiche di prostata umana pag. 95
4
SCOPO DEL LAVORO
pag. 97
5
MATERIALI E METODI
pag. 100
6
RISULTATI
pag. 108
7
DISCUSSIONE
pag. 113
8
TABELLE
pag. 118
9
FIGURE
pag. 119
10
BIBLIOGRAFIA
4
1. LA PROSTATA
Il termine prostata derivò originariamente dalla parola greca “prohstami” che significa “stare di fronte a”,
termine attribuito a Herophilus di Alessandria che usò il termine nel 335 A.C. per descrivere l’organo
collocato di fronte alla vescica. Descrizione anatomiche dettagliate non comparvero prima del
Rinascimento. Mentre l’esistenza della prostata è riconosciuta da più di 2300 anni, descrizioni accurate
della struttura interna della ghiandola, della fisiologia e della patologia si sono avute solo in tempi
relativamente recenti. Nell’ultimo decennio vi è stato molto interesse nei confronti delle patologie
connesse alla prostata: iperplasia benigna della prostata (BPH) e carcinoma della prostata (PCa). Questo
è parzialmente il risultato dei cambiamenti demografici che hanno portato ad un aumento della
popolazione maschile che raggiunge un’età dopo la quale è particolarmente sensibile a queste patologie.
Inoltre è dovuto, in parte, all’introduzione in queste aree patologiche di nuovi metodi di diagnosi e
trattamento. Infine, l’anatomia della zona e soprattutto la conoscenza della capsula e dei fasci adiacenti
è fondamentale per la comprensione della patologia prostatica (1).
1.1 Sviluppo e differenziazione della ghiandola prostatica
Lo sviluppo del tratto riproduttivo maschile inizia durante lo stadio bipotenziale. In questo stadio gli
embrioni maschili e femminili sono indistinguibili anatomicamente poiché hanno un paio di gonadi
indifferenziate, due set di dotti (Wolffiano e Mǘlleriano) e il seno urogenitale che deriva dalla suddivisione
della cloaca endodermale dal setto uro-rettale. Nei maschi, la differenziazione gonadica in testicoli
determina la produzione di testosterone e di Mǘllerian inhibiting substance (MIS). Gli androgeni testicolari
fetali, quindi, mantengono i dotti Wolffiani (2), ne stimolano la differenziazione in epididimi, dotti deferenti
e vescicole seminifere (Jost 1965); inducono lo sviluppo della prostata dal seno urogenitale, prevengono
la morte cellulare programmata dei tubuli mesonefrici attaccati alla parte finale dei dotti Wolffiani, che
susseguentemente differenziano nei dotti efferenti. In assenza di testosterone o di recettori funzionali
degli androgeni (AR), i dotti Wolffiani e i tubuli mesonefrici degenerano (3). Al contrario i dotti Mǘlleriani
regrediscono in risposta al MIS (4).
Nei maschi il seno uro-genitale differenzia nell’uretra maschile, nella prostata, nelle ghiandole peri-uretrali
e bulbo-uretrale. Gli androgeni prodotti dai testicoli fetali stimolano lo sviluppo dell’epitelio dei seni
5
urogenitali dalla parte caudale al collo della vescica, per formare la prostata e quindi più caudalmente per
formare le ghiandole bulbouretrali. Il seno urogenitale femminile formerà anche la prostata se è stimolato
da androgeni durante i periodi critici dello sviluppo (5). Lo sviluppo prostatico non avviene o è fortemente
compromesso in assenza di androgeni, in assenza del recettore degli androgeni (AR) funzionale, o
quando l’azione degli androgeni è bloccata dalla somministrazione di antiandrogeni (4).
1.2 Anatomia della prostata
La descrizione dell’anatomia e dello sviluppo della prostata umana fatto da Lowsley nel 1912 diede inizio
ad un lungo dibattito sulla nomenclatura usata per descrivere l’organo (6). Il lavoro di Lowsley era basato
su studi della ghiandola fetale. Egli descriveva un lobo dorsale o posteriore, un lobo mediano e due lobi
laterali, e in aggiunta un lobo ventrale, che atrofizzava dopo la nascita. Nella prostata umana adulta
questi lobi sono uniti e non possono essere separati o delineati da dissezione. Questa difficoltà di
delineare la suddivisione in lobuli nella prostata umana ha dato luogo a numerose differenti visioni sulla
sua suddivisione anatomica (7). La nomenclatura ora più comunemente usata per descrivere la prostata
umana è quella di McNeal (8). Egli divide la prostata in aree separate anatomicamente e distinte
istologicamente: lo stroma fibromuscolare non ghiandolare che circonda l’organo, e tre regioni ghiandolari
definite zone periferiche, di transizione e centrale. Queste zone contengono sistemi duttali complessi, ma
distinti istologicamente (9).
La zona periferica è la più ampia della ghiandola prostatica. I dotti della zona periferica si svuotano
nell’uretra lateralmente ai recessi postero-laterali della parete uretrale. I dotti e gli acini sono rivestiti
internamente da semplice epitelio colonnare. Questa area è il principale sito di prostatiti e PCa, sebbene
non del BPH (10).
La zona di transizione rappresenta i rami delle due coppie di dotti maggiormente sulla linea laterale
distale (i dotti immediatamente prossimali alla vescica e più vicini allo sfintere pre-prostatico). Questi dotti
si svuotano nell’uretra alla base del verumontanum. I dotti della zona di transizione sono al confine dello
sfintere prostatico. Essi fuoriescono dal confine dello sfintere e danno luogo ad una massa ghiandolare,
che generalmente si unisce all’uretra prossimale. Il PCa si sviluppa raramente all’interno della zona di
transizione che invece è notoriamente luogo di sviluppo del BPH (11).
I dotti della zona centrale seguono il corso dei dotti eiaculatori e si svuotano nell’uretra prossimamente al
verumontanum. Questi dotti e acini sono più larghi e di contorno irregolare. Gli acini sono poliedrici in
6
sezione crociata. Lo stroma muscolare è molto più compatto che non nella zona periferica. La zona
centrale è virtualmente, ma non completamente, esente da patologie. Il lobo craniale della prostata dei
primati è coinvolta principalmente nella coagulazione del seme e si trova vicino alla vescicola seminale.
Sembra essere analogo alla zona centrale umana, e McNeal ha proposto che esso abbia la stessa storia
embrionale (11). Si è dimostrato che le secrezioni della ghiandola coagulante del ratto, che occupa una
posizione analoga al lobo craniale, sono in grado di sostituire quelle del lobo craniale nell’induzione della
coagulazione del fluido della vescicola seminale nella scimmia rhesus. Il lobo caudale della prostata dei
primati assomiglia alla zona periferica umana sia anatomicamente che biochimicamente (12).
1.3 La capsula prostatica
Una confusione di lunga durata ha circondato non solo la struttura del parenchima prostatico, ma anche
l’esistenza di una “capsula” prostatica. Storicamente, il termine “capsula” ha avuto due differenti
significati. Il primo termine “capsula chirurgica”, nelle ghiandole che sono state alterate dalla BPH, è stato
usato per descrivere la zona periferica residua sottoposta a compressione dall’espansione della zona di
transizione. In questa situazione la zona periferica diventa atrofica ed è formata principalmente da
elementi stromali. Il secondo, più pertinente allo studio del PCa, è il dubbio se il parenchima prostatico è
o non è delimitato da tessuto fibroconnettivo simile alla capsula della milza. La prostata si sviluppa
attraverso una forma di morfogenesi ramificata nella quale le evaginazioni di questi seni urogenitali
invadono un letto di mesoderma splancnico. Lo stroma si organizza intorno alle gemme epiteliali quando
queste penetrano attraverso le cellule mesenchimali indifferenziate. Queste ultime, dalla sedicesima
settimana di gestazione, acquisiscono il fenotipo differenziato del muscolo liscio. E’ questa
differenziazione stroma epitelio che determina gli ultimi confini della prostata. Le cellule mesenchimali
entro questo confine restano fibroblastiche e stabiliscono la zona di perdita di tessuto connettivo
attraverso cui i vasi sanguigni di supporto e i nervi arrivano alla ghiandola. La capsula della prostata è
quindi più simile alla avventizia delle principali arterie che alla capsula fibroconnettiva la quale incapsula il
fegato e la milza (9). Come ci si potrebbe aspettare l’ampia penetrazione delle cellule del PCa attraverso
questa fragile capsula sembra correlarsi con un rischio di un susseguente sviluppo di malattia
metastatica. Curiosamente, una penetrazione capsulare microscopica fornisce scarse informazioni (1).
1.4 Rapporti stroma-epitelio: ruolo degli ormoni e dei fattori di crescita
7
La crescita normale di alcuni tessuti ormono-dipendenti e in cui si sviluppano processi neoplastici
(mammella, prostata, endometrio) dipende dalle particolare relazioni tra l’epitelio e il mesenchima.
Se le cellule mesenchimali e quelli epiteliali vengono messe in coltura separatamente, le prime
continuano a crescere, ma non formano strutture organo-tipiche, mentre le seconde non sopravvivono.
Se poi l’epitelio e il mesenchima sono successivamente ricombinati, il processo morfologico procede
regolarmente. Per quanto riguarda la prostata, è stato dimostrato che qualsiasi epitelio embrionale, a
contatto con mesenchima del seno uro-genitale, si trasforma in epitelio prostatico in presenza di
diidrotestosterone; se invece l’epitelio del seno uro-genitale viene messo a contatto con mesenchima di
diversa provenienza il tessuto ghiandolare prostatico non si sviluppa. Le interazioni epitelio-mesenchima
sono reciproche: così come il mesenchima induce la differenziazione epiteliale, l’epitelio induce a sua
volta la differenziazione mesenchimale (in particolare, nel caso degli organi uro-genitali, la
differenziazione della muscolatura liscia). Lo sviluppo di tali tessuti è peraltro strettamente dipendente
dalla presenza dell’ormone specifico per quel tessuto (androgeni per la prostata, estrogeni per la
mammella e l’endometrio). Un ruolo importante è svolto anche dai fattori di crescita (growth factors) che
mediano i processi della crescita, della differenziazione e della morte cellulare programmata o apoptosi
(13).
1.5 Influenze ormonali sulla crescita della prostata
Sebbene la prostata normale esista strutturalmente circa dalla dodicesima settimana di vita intrauterina,
rimane elementare nell’infanzia e si sviluppa solo dopo la pubertà sotto l’influenza di aumentati livelli di
androgeni circolanti.
Il testosterone (T) è un ormone sintetizzato dalle cellule del Leydig del testicolo sotto l’influenza del
luteinizing hormone (LH). La secrezione di questo ultimo decapeptide è regolata a sua volta dal
luteinizing hormone-releasing hormone (LH-RH) di origine ipotalamica. E’ a questo livello che il
testosterone ha un effetto di feedback negativo che permette ai livelli di testosterone circolante di essere
mantenuti entro i limiti normali (10-35 μg/L). Una variazione diurna nei livelli di testosterone (entro questo
range) è consueta, con picchi di concentrazione plasmatica registrati la mattina presto. Il 95% del
testosterone circolante è legato ad una certa quantità di proteine plasmatiche e di queste quella
predominante è la sex-hormone binding globulin (SHBG). Tutto ciò permette solo ad una piccola parte di
androgeno libero di entrare per diffusione nelle cellule. Un’ulteriore, ma più piccola fonte di androgeni è la
8
corteccia surrenalica. Sotto l’influenza dell’ adrenocorticotropic hormone (ACTH), l’androstenedione viene
rilasciato dalle ghiandole surrenaliche. Quando la funzione testicolare è intatta, questa attività
androgenica suppletiva è di poco impatto, ma dopo castrazione chimica o fisica il loro effetto androgenico
può risultare fondamentale per la sopravvivenza di alcuni cloni delle cellule androgeno dipendenti del
PCa a danno del paziente (14).
Studi recenti sono andati oltre il concetto di PCa inteso come una patologia testosteron-centric come
descritto da Huggins. Poiché è chiaro che i livelli di mortalità e incidenza aumentavano dopo orchiectomia
o terapia di ablazione estrogenica furono presi in considerazione altre vie ormonali. Ad esempio
l’identificazione del follicle-stimulating hormone (FSH) e del suo recettore in cellule di cancro alla prostata
e la capacità di stimolare la proliferazione cellulare in vitro hanno suggerito un meccanismo regolatorio
autocrino/paracrino della crescita (15). Una volta nella prostata, sia il testosterone che gli androgeni
surrenalici vengono rapidamente metabolizzati attraverso l’enzima 5α-reduttasi (un enzima che si trova
principalmente nella membrana nucleare), a diidrotestosterone (DHT). Il DHT è un androgeno più potente
da tre a cinque volte rispetto al testosterone stesso e ha una affinità per il recettore degli androgeni 10
volte superiore al testosterone (16). La sua azione androgenica si compie nel nucleo attraverso una
associazione fisica con i recettori degli androgeni che si trovano principalmente nella membrana
nucleare. Il legame del DHT al recettore degli androgeni libera le heat-shock protein (hsp90) e smaschera
il DNA binding domain del recettore permettendogli di dimerizzare e legarsi alle sequenze specifiche
regolatorie, chiamate hormone response element (HRE). Questo legame avviene a monte dei siti di inizio
della trascrizione dei diversi geni androgeno sensibili e quindi stimola la trascrizione del DNA. L’mRNA
così prodotto codifica per un numero di proteine, compresi diversi fattori di crescita importanti, quali
l’epidermal growth factor (EGF) e il platelet-derived growth factor (PdGF) che modulano e promuovono la
crescita delle cellule prostatiche. Lo sviluppo o il mantenimento della prostata normale dipendono da una
delicata omeostasi fra morte cellulare e sostituzione cellulare. Gli androgeni sono necessari a questa
omeostasi poiché la castrazione sia farmacologica che chirurgica dà un rapido inizio ad un fenomeno di
apoptosi epiteliale o morte cellulare programmata, così come la deplezione stromale. A conferma di ciò,
infatti, il contenuto di DNA delle cellule prostatiche androgeno-deprivate si riduce del 10% in 10 giorni
(17). Le cellule staminali sopravvivono, comunque, poiché una conseguente sostituzione degli androgeni
circolanti sembra essere in grado di far ritornare alla normalità sia la funzione che l’architettura prostatica
concentrandosi sui processi di proliferazione cellulare (18).
9
1.6 Controllo molecolare del ciclo cellulare
La crescita della prostata normale e lo sviluppo dipendono da un particolare pattern di divisione cellulare,
con alcuni tipi cellulari che si dividono molto più spesso che altri e tutte le divisioni avvengono in modo
appropriato e tempestivo.
Le cellule prostatiche epiteliali si dividono regolarmente per mantenere l’integrità strutturale della
ghiandola delle superfici epiteliali dei dotti. La divisione epiteliale maggiormente attiva avviene
all’estremità distale delle ghiandole prostatiche: quelle nella porzione intermedia sono mantenute in uno
stato differenziato, mentre quelle nei dotti prossimali, più vicini all’uretra, subiscono morte cellulare
programmata. Questo processo attivo di apoptosi che controbilancia la nuova formazione di cellule
avviene anche dopo deprivazione androgenica. Due geni che sembrano essere cruciali nell’apoptosi
sono i geni bax e bcl2. La caratteristica morfologica principale dell’apoptosi è la condensazione della
cromatina causata dalla formazione di frammenti di 180-200 paia di basi di
DNA, come risultato
dell’attivazione delle endonucleasi (19). Inibitori della fosfatasi sembrano sinergizzare con il tumour
necrosis factor (TNF) nell’attivare la frammentazione del DNA, in questo sistema di morte cellulare
programmata (20). E’ questo sistema che entra in gioco quando il supporto degli androgeni viene a
mancare alla prostata.
1.7 Eziologia e fattori di predisposizione per il PCa:
A parte un 9% degli uomini con PCa ereditario, il fattore che più è decisivo per lo sviluppo di questa
malattia è l’età. Il PCa è raramente diagnosticato in uomini al di sotto dei 50 anni, ma è comune in modo
crescente con l’avanzare dell’età. Le caratteristiche etniche sono anche abbastanza degli importanti
fattori di predisposizione; ad esempio, i neri del Nord America hanno all’incirca 2 volte il rischio della
malattia rispetto ai bianchi (21).
a) Ormoni
Gli androgeni circolanti sono un prerequisito essenziale per la crescita della prostata normale, per lo
sviluppo del BPH e del PCa. Il ruolo specifico degli androgeni per quanto riguarda la carcinogenesi
all’interno della prostata non è totalmente chiaro, ma sembrano agire promuovendo la crescita cellulare e
la loro divisione. Sebbene la quantità di testosterone circolante sia variabile da un individuo all’altro, non
sembra esserci una correlazione diretta tra il livello sierico di testosterone e il rischio di sviluppo del PCa
10
(22). I livelli serici di testosterone declinano gradualmente con l’aumentare dell’età mentre l’incidenza del
cancro alla prostata aumenta stabilmente.
Questa anomalia apparente potrebbe essere spiegata attraverso il lungo periodo di ritardo tra il
cambiamento neoplastico precoce iniziale e la comparsa del PCa clinicamente evidente, così come
attraverso cambiamenti nei livelli dei recettori per gli androgeni. Il testosterone viene metabolizzata nella
prostata in DHT attraverso l’enzima 5 alpha-reduttasi. Ad oggi è risaputo che il DHT piuttosto che il
testosterone è l’androgeno intracellulare che maggiormente promuove la crescita prostatica (23). Il ruolo
del DHT nella promozione del PCa è ancora poco chiaro, comunque esso sembra non avvenire in uomini
ermafroditi in cui la 5 alpha-reduttasi è assente. La cirrosi epatica può portare ad una diminuzione del
testosterone circolante così come ad un aumento di estrogeni circolanti; entrambi questi cambiamenti
potrebbero essere motivo, in questa condizione, di un ridotto rischio di PCa (24). Il ruolo specifico degli
androgeni nell’induzione e promozione o progressione del PCa non è completamente compreso.
Comunque, una volta stabilita la trasformazione maligna hanno quasi certamente un ruolo nella
stimolazione della divisione e invasione delle cellule maligne
b) Dieta
L’alta incidenza di PCa negli USA sembra essere correlata con un aumento nel consumo di grassi. Aree
degli Stati Uniti in cui il consumo di prodotti caseari e carni rosse è maggiore, anche l’incidenza di PCa è
più alta (25). Lo studio di follow-up della “US Health Professionals” (26) ha valutato oltre 4700 uomini con
i seguenti criteri: dieta e rischio di sviluppo del cancro. Durante il corso di questo studio a 300 uomini è
stato diagnosticato il cancro alla prostata. Il consumo di cibo grasso era direttamente correlato al rischio
di cancro alla prostata ed il rischio era direttamente correlato ai grassi saturi. E’ stato dimostrato inoltre
che il consumo di carne rossa aveva la più alta correlazione con il rischio del cancro (27). L’effetto di una
dieta grassa nei differenti gruppi etnici è stata anche riportata da Whittemor così come dalla World Health
Organization. Ad esempio risultò che l’associazione tra grassi saturi e cancro era maggiore negli asiatici
Americani (28).
Ci sono diversi modi potenziali di azione dei grassi nella patologia del cancro alla prostata. In alcuni tipi di
ratti predisposti geneticamente, alcuni grassi riducono il tempo di induzione del testosterone nello
sviluppare il cancro alla prostata (29). Un altro possibile meccanismo in cui alti livelli di consumo di
grasso aumentano il rischio di cancro alla prostata è la riduzione dell’assorbimento di vitamina A. Un
aumento del livello circolante di beta-carotene, che dipende dalla quantità di assorbimento della vitamina
11
A, sembra essere protettivo contro lo sviluppo di alcuni tumori (30). I livelli di testosterone circolanti
diminuiscono sino al 30% in uomini che seguono una dieta vegetariana a basso contenuto di grassi e
potrebbero quindi ridurre l’induzione del cancro alla prostata (31). In Giappone, dove la dieta è
tradizionalmente povera di grassi, l’incidenza di cancro alla prostata ha iniziato ad aumentare in
associazione ad un aumento del fenomeno dell’occidentalizzazione. Un trend simile sta avvenendo
anche in Cina (32). La dieta tradizionale in Giappone e altri paesi Asiatici è composta da quantità di
verdure gialle e verdi. In questa dieta, alti livelli di vitamina A potrebbero proteggere contro l’induzione al
cancro alla prostata. Fito-estrogeni quali la genisteina e daidaina che si trovano in verdure quali la soia
possono alterare il milieu ormonale e opporsi agli effetti del testosterone sulla prostata. Inoltre questi
hanno attività antiossidante e agiscono come fagociti di radicali liberi. Altri meccanismi possibili di attività
anticarcinogenica comprendono l’inibizione dell’angiogenesi e della protein-tirosin chinasi specifica dei
recettori del fattore di crescita. La variazione sostanziale a livello mondiale del cancro alla prostata
sembra dipendere da fattori genetici, ma differenze a livello di dieta possono essere la causa di variazioni
nell’incidenza di studi di migrazione. Sembra verosimile che la dieta occidentale che è più ricca di grassi
animali e più povera di verdure comporti un più alto rischio di cancro alla prostata.
c) Vitamine
Supplementi dietetici di vitamina E e selenio potrebbero possibilmente offrire protezione contro lo
sviluppo di cancro alla prostata. Uno studio ha riportato che più alti livelli di selenio nelle unghie dei piedi
di uomini furono associati a più bassi rischi di incremento di cancro alla prostata (33) e il rischio di PCa
era ridotto di 1/3 in uomini che assumevano 200 mcg di selenio al giorno. E’ stato suggerito quindi, un
rapporto fra i livelli di alpha-tocoferolo e il beta-carotene assunti con la dieta. La supplementazione a
lungo termine di alpha-tocoferolo è stata dimostrata ridurre l’incidenza di cancro alla prostata in soggetti
fumatori; al contrario il beta-carotene sembra aumentarne l’incidenza (34). Sebbene vi siano
considerevoli dati in letteratura, uno studio finlandese su soggetti fumatori ha mostrato un livello più alto
di cancro alla prostata in un gruppo a cui veniva somministrato beta-carotene. Ciò supporta le precedente
osservazioni di un aumentato rischio di cancro alla prostata in uomini che assumono questa sostanza.
Una scoperta interessante di questo studio è stata una associazione negativa tra cancro alla prostata e
assunzione di vitamina E (35). Fra quei soggetti che ricevevano alpha-tocoferolo vi è stata una
significativa diminuzione percentuale di cancro alla prostata. Va tenuto comunque in considerazione che
12
lo studio non fu fatto specificatamente per dimostrare cambiamenti nell’incidenza del cancro alla prostata
e quindi le scoperte vanno considerate come preliminari.
Schwartz e Hulka hanno notato un aumento dell’incidenza del PCa con l’aumento della distanza
dall’equatore, infatti la patologia è più comune negli stati più a Nord degli USA e dell’Europa rispetto a
quelli a SUD. Su questa base, la deficienza di vitamina D è una potenziale causa contribuente. E’ stato
dimostrato che la vitamina D ha un effetto antiproliferativo e differenziante sulle linee cellulari di PCa negli
essere umani sebbene studi epidemiologici abbiano mostrato dati conflittuali. Questa osservazione ha
portato alla possibilità che la vitamina D (1,25 diidrocolecalciferolo) potrebbe in qualche modo essere una
protezione contro la protezione da questa patologia (36).
d) Fattori ambientali
Nel corso degli ultimi decenni l’attenzione è stata sempre più focalizzata su un possibile ruolo
dell’inquinamento ambientale nella promozione di numerose patologie neoplastiche. Una esposizione a
sostanze chimiche usate largamente nell’industria è dimostrata contribuire allo sviluppo di particolari
tumori maligni. Il cancro alla prostata si sviluppa con minore probabilità in uomini che vivono in un
ambiente rurale rispetto a quelli che vivono in città, i quali sono a più alto rischio di morte per queste
patologie (32). I fattori responsabili potrebbero essere l’inquinamento ambientale da agenti chimici, così
come l’esposizione a sostanze nel luogo di lavoro. I lavoratori esposti a sostanze chimiche in industrie
della gomma, tessili, chimiche, farmaceutiche, di fertilizzanti, e che sfruttano energia atomica hanno un
aumentato rischio di sviluppo di cancro alla prostata. Le precise sostanze chimiche che inducono tale
patologia non sono conosciute, sebbene la scoperta di aumentati livelli di cadmio in pazienti col PCa
suggerì che ciò poteva essere una dei possibili fattori di induzione. Ci sono anche prove che
suggeriscono che l’esposizione al trizio,
51Cr, 59Fe, 60Co
e
65Zn
potrebbero aumentare il rischio di cancro
alla prostata (37).
e) Fumo
Sebbene il tabacco è stato implicato come un fattore eziologico nei tumori maligni del polmone e della
vescica e di molte altre neoplasie, tale associazione non è stata ancora trovata nel cancro alla prostata
(38).
f) Virus
Agenti infettivi, particolarmente i virus, sono conosciuti per essere la causa di carcinogenesi in alcune
forme di tumore maligno ai genitali, così come il cancro alla cervice. I virus sono certamente una
13
potenziale promotore ambientale per il PCa, ma ciò è difficile da provare poiché molti virus sono ubiquitari
e altri sono impossibili da isolare dalle cellule tumorali. Un possibile candidato per l’eziologia del PCa è il
virus dell’ herpes simplex di tipo 2. Anticorpi per questo virus sono stati trovati nel siero del 71% di
soggetti con cancro alla prostata, ma in solo il 66% di soggetti controllo con ipertrofia prostatica benigna
(39). Come ulteriore supporto sono stati anche identificati frammenti di RNA virale in cellule di cancro alla
prostata (40). E’ anche interessante notare che le mogli di uomini a cui è stato diagnosticato il cancro alla
prostata avevano un’incidenza maggiore di sviluppare il cancro alla cervice uterina (41). Il ruolo
eziologico degli RNA virus è rappresentato dalla presenza dell’oncogene H-ras p21 in cellule tumorali di
cancro alla prostata, anche se ciò sembra essere associato a tumori alla prostata non ben differenziati
(42).
1.8 Fattori di crescita e carcinoma prostatico
L’EGF e il TGFα sono polipeptidi tra loro correlati che si legano allo stesso recettore cellulare; il primo è in
genere secreto sia dalle cellule normali che da quelle tumorali, mentre il secondo viene prevalentemente
prodotto dalle cellule tumorali (43). Il TGFα è sintetizzato e secreto nel mezzo di cultura sia in linee
cellulari androgeno-dipendenti (LNCaP) (44) che androgeno-indipendenti (PC3, DU145) (45). Nelle
cellule androgeno-indipendenti, il trattamento con DHT stimola la proliferazione cellulare senza un
contemporaneo aumento di EGF, ma con un aumento dell’espressione del TGFα e del recettore
dell’EGF; in tal modo innescano una stimolazione continua di tipo autocrino delle cellule LNCaP. E’ stato
altresì dimostrato che il TGFα da solo stimola la crescita cellulare, ma che TGFα e DHT, insieme
inducono una stimolazione significativamente più grande. Inoltre, bloccano il recettore dell’EGF, si arresta
sia la crescita indotta dal DHT che quella indotta dal TGFα e questo dato suggerisce che l’effetto del DHT
è modulato dal sistema autocrino TGFα-recettore dell’EGF. A tali evidenze sperimentali fanno riscontro i
risultati di studi immunoistochimici effettuati in campioni tissutali ottenuti da tumori primitivi di pazienti
sottoposti a prostatectomia radicale e in campioni provenienti da metastasi di pazienti sottoposti a terapia
endocrina. Nei tumori prostatici primitivi, il recettore dell’EGF era localizzato nelle cellule epiteliali e il
TGFα nelle cellule stremali, mentre la coespressione del TGFα e del recettore dell’EGF è stata osservata
in quali l’80% dei campioni prelevati da metastasi che avevano perduto la responsività ormonale. Questi
risultati suggeriscono che nei tumori primitivi predomina un modello di stimolazione paracrina mentre, nel
momento in cui il tumore perde la dipendenza ormonale, predomina un meccanismo autocrino.
14
La famiglia del fattore di crescita di fibroblasti (FGF) è costituita da almeno 7 diversi fattori; l’FGF acido
(aFGF) e l’FGF basico (bFGF) sono costituiti da 155 aminoacidi e hanno in comune un’omologia di
sequenza degli aminoacidi di circa il 55%. Uno di questi fattori, l’EGF7 è un fattore di crescita dei
cheratociti e per tale motivo denominato KGF; nella prostata è largamente espresso nei fibroblasti
stromali e agisce specificatamente sulle cellule di origine epiteliale come mediatore paracrino. I fattori di
crescita che appartengono a questa famiglia possono essere coinvolti nella proliferazione cellulare del
carcinoma prostatico. Nel carcinoma prostatico umano il bFGF è presente ma a bassi livelli; tale fattore
può stimolare la sintesi di DNA in linee cellulari androgeno-dipendenti (LNCaP) e alcune linee androgenoindipendenti (DU145) ma non in altre (PC3). Nonostante tale frammentarie evidenze, il ruolo del KGF nel
carcinoma prostatico resta tuttora non chiaro.
Un altro fattore prodotto dallo stroma prostatico è l’Insulin Growth Factor I (IGF-I) che si lega a un
recettore localizzato, invece, nello strato basale dell’epitelio prostatico, dove si trova anche il recettore
dell’EGF (tale sub-compartimento epiteliale è responsabile della proliferazione cellulare, mentre il subcompartimento secretorio - dove sono localizzati prevalentemente i recettori androgenici – produce una
varietà di secrezioni proteiche come la fosfatasi acida e l’antigene prostatico). Nel nostro laboratorio
abbiamo dimostrato che nel tessuto prostatico ipertrofico di pazienti ce erano stati trattati con analoghi del
GnRH si verifica un aumento dei recettori dell’IGF-I (e dell’EGF); inoltre – dopo privazione androgenica –
tali recettori sono presenti anche nelle cellule dell’epitelio secretorio dove, al contrario, si riduce la
concentrazione dei recettori androgenici e dei prodotti secretori (fosfatasi acida prostatica, PSA).
Per quanto riguarda il carcinoma prostatico, è stato dimostrato che linee di carcinoma prostatico umano
metastatico (PC-3 e DU145) esprimono in grande quantità il recettore dell’IGF-I e che gli androgeni
perdono la capacità di modularne l’espressione. L’IGF-I è stato identificato nel medium condizionato delle
cellule DU145 e questo dato ha fatto supporre che l’IGF-I, a differenza del TGF- α e del bFGF, continui
ad agire, nel carcinoma prostatico avanzato, come un fattore di crescita paracrino.
Contro tale ipotesi ci sono altri dati sperimentali ottenuti con le stesse linee cellulari trattate con analoghi
peptidici dell’IGF-I, con funzioni di antagonisti recettoriali, in cui è stato osservato un rallentamento della
crescita. Alcuni dati più recenti sembrano aver dato una spiegazione a questo fatto con la dimostrazione
che le linee cellulari di carcinoma prostatico metastatizzato producono IGF-II; tale fattore agisce sul
recettore dell’IGF-I e pertanto, anche in questo caso, sarebbe attivo un meccanismo autocrino IGFII/recettore IGF-I.
15
Una serie di dati sembra, infine, indicare l’esistenza di interazioni tra alcuni fattori di crescita (EGF/TGFα, IGF-I, KGF) e i meccanismi a cascata di trasduzione del segnale androgenico. La famiglia del TGFβ è
costituita da 5 isoforme di TGFβ e molti peptidi correlati (attivine, inibine). Sono stati descritti 3 tipi di
recettori, tipo I, II e III; essi legano il TGFβ1, il TGFβ 2 e il TGFβ 3 ma non la forma latente del TFGβ Il
controllo dell’espressione e dell’attività del TGFβ è complesso e comprende la regolazione della
trascrizione/traduzione del TGFβ, l’attivazione della forma latente nella forma matura e il sequestro del
TGFβ attivato da parte della matrice extracellulare e delle proteine leganti. Il suo ruolo è bi-funzionale e il
prevalere degli effetti stimolatori o inibitori è strettamente dipendente dal tipo cellulare e dalla
concentrazione del TGFβ stesso. La maggior parte delle cellule stromali, infatti, viene stimolata dal TGFβ
in senso mitogenico, mentre la maggior parte delle cellule di origine epiteliale viene inibita. Inoltre, il
TGFβ regola la sintesi e il turnover della matrice extracellulare, stimola gli inibitori della proteasi e inibisce
l’attivazione del plasminogeno tissutale. Ad alte concentrazioni, infine, il TGFβ stimola l’angiogenesi,
l’invasione e la diffusione metastatica e sopprime la risposta immune.
Nella prostata umana, il TGFβ può agire come fattore di crescita positivo o negativo e influenzato
dall’azione androgenica. Nel tessuto prostatico normale prevalgono gli effetti negativi e, nelle cellule
epiteliali in coltura, il TGF- β è il principale antagonista dei fattori di crescita mitogenici e svolge un ruolo
di primaria importanza nel fenomeno della morte cellulare programmata (apoptosi). L’espressione del
TGF- β è sotto il controllo negativo degli androgeni; nel ratto, dopo castrazione si osserva insieme
all’involuzione della prostata, un’aumentata espressione del TGF- β e del suo recettore. Se,
successivamente si somministra un androgeno, i livelli ritornano alla norma entro 4 giorni. Non si dispone
comunque ad oggi di dati che correlino l’espressione del TGF- β con lo stadio, il grado e i marker della
proliferazione cellulare. Il TGF- β potrebbe avere un ruolo nella formazione delle metastasi osteoblastiche
del carcinoma prostatico dal momento che tale fattore inibisce il riassorbimento osseo e stimola la
neoformazione ossea. Un fattore correlato con la famiglia del TGF- β, la proteina DVR-6 che induce la
formazione ossea nell’embriogenesi, è espressa in notevole quantità nel carcinoma prostatico nel ratto,
ma, al momento non sono noti dati di questo genere nell’uomo (13)
1.9 Prostatic intraepithelial neoplasia (PIN)
La precoce identificazione e la cura del cancro sono spesso dipese dall’abilità di riconoscere il
cambiamento pre-maligno. La definitiva identificazione dei precursori del carcinoma della prostata è
16
rimasta indietro rispetto ad altri organi equivalenti, così come quello della cervice uterina. Questo è
dovuto in parte alle difficoltà tecniche nel condurre biopsie ripetute sullo stesso esatto punto della
prostata, ad esempio quando paragonato a una ripetizione del PAP test o della colposcopia cervicale.
Anche con biopsia guidata transrettale ad ultrasuoni (TRUS-guidata) è impossibile essere sicuri che una
re-biopsia sia presa da un campo microscopico identico. Per questo motivo l’interpretazione delle
mutazioni all’interno della prostata da stadi displastici a maligni è deficiente a causa della mancanza di
dati riguardo la progressione di lesioni specifiche nel tempo.
Progressi riguardanti l’identificazione di precursori furono fatti a metà degli anni ’80 quando McNeal e
Bostwick (20) riportarono l’esistenza di una lesione che definirono “displasia intraduttale”. Essa fu
proposta quale precursore del tumore maligno sulla base di una atipia citologica, mostrando un
pleiomorfismo e una prominenza nucleare simile a quella vista nel cancro alla prostata (tab 2.1). In
seguito ad una conferenza nel 1989, il termine PIN fu adottato come il più appropriato per questa lesione
(tab 2.2) allineandolo alle altre lesioni maligne di altri organi quali la neoplasia intraepiteliale cervicale
(CIN). I cancri della zona di transizione non sono generalmente associati al PIN, ma nei tumori della zona
periferica il PIN è frequentemente trovato adiacente ai carcinoma (21, 22). Un PIN grave è spesso più
rilevante nei tumori multifocali (21). A parte i pleiomorfismi nucleari già menzionati, le lesioni PIN dividono
anche altre caratteristiche col tumore maligno, come la perdita della continuità dello strato basale (23), la
produzione di acido mucinico (24), tracce di differenziazione come evidenziato da un diminuito
immunostaining del PSA (26, 27) e anormalità dei ploidi.
La diagnosi di un “highgrade” PIN è stabilita da un’aumentata proliferazione e da cambiamenti citologici. Il
PIN è comunemente associato con il cancro alla prostata e con una disgregazione dello strato basale
come identificato dall’analisi immunoistochimica della citocheratina ad alto peso molecolare. Un’altra
lesione che è stata proposta rappresentare un cambiamento pretumorale e una atipica iperplasia
adenomatosa. Quest’ultima simile in apparenza al carcinoma basso grado può anche essere associata
ad una disgregazione dello strato basale sebbene evidenze recenti suggeriscono che questo non sia un
cambiamento pre-maligno. Come negli altri sistemi di organi, il grado di malignità o il suo livello di
partenza si è stabilito essere un importante marker prognostico nella prostata. Diversi sistemi di
classificazione sono stati proposti ma quello applicato più ampiamente è quello di Gleason (28) per
definire il grado del tumore. In particolare, dall’esame bioptico e dall’analisi al microscopio dell’istologia
del campione di carcinoma alla prostata prelevato, esiste la possibilità di stabilire il grado di aggressività
17
del tumore facendo riferimento al sistema di Gleason che si basa esclusivamente sull’architettura delle
ghiandole considerando il livello di organizzazione strutturale che i campioni tissutali analizzati
presentano. Si distinguono 5 diversi gradi di differenziamento, da 1 a 5: i gradi 1 e 2 Gleason non si
discostano molto dalla prostata normale; nel grado Gleason 3 ancora si riconosce l’unità ghiandolare, ma
si osserva un’invasione nello stroma muscolare da parte delle ghiandole; nel grado Gleason 4 si ha una
notevole perdita di organizzazione con distruzione della normale unità ghiandolare, fenomeno questo che
si accentua nel grado Gleason 5. Importante poi per stabilire la severità del tumore e la prognosi è il
cosiddetto Gleason score che può variare da 2 a 10 e che risulta dalla somma dei gradi attribuiti a due
diversi elementi del campione tissutale. Maggiore è il Gleason score, peggiore risulta essere la prognosi,
come confermato da uno studio di Chodak e collaboratori del 1994 (46).
1.10 Progressione verso l’ormono-indipendenza
La disponibilità di farmaci che inibiscono a vari livelli e con meccanismi diversi l’azione ormonale ha
consentito di sostituire le vecchie terapie ablative chirurgiche con i medesimi soddisfacenti risultati in
termini di regressione del carcinoma prostatico in fase avanzata. Comunque, in un lasso di tempo
variabile dall’inizio della terapia endocrina, tale tumore diventa resistente alle terapie iniziali. Il fenomeno
della progressione verso l’ormono-indipendenza costituisce pertanto un problema biologico e clinico di
notevole rilevanza. Una delle ipotesi alla base di questo fenomeno è che in un tessuto tumorale possano
esistere cloni che mantengono la dipendenza ormonale (e che quindi risentono positivamente dei
trattamenti che rimuovono lo stimolo trofico) e cloni cellulari indipendenti che esprimono altri segnali
oncogenici e possono, col tempo, diventare dominanti e incontrollabili. Un’altra causa della perdita
dell’ormono-responsività è l’espressione costitutiva di uno o più geni coinvolti nella crescita cellulare. Una
serie di geni regolatori della crescita sono iper espressi, amplificati, mutati o parzialmente deleti (c-myc,
p53 e il recettore dell’FGF). Diverse mutazioni somatiche sono state determinate in campioni tumorali di
pazienti affetti di carcinoma prostatico. Tali mutazioni sono state localizzate nell’hormone binding domain
del recettore androgenico; sebbene il numero delle mutazioni riportate dai vari gruppi sia estremamente
variabile, il più alto numero è nel gruppo dei tumori in stadio avanzato (stadio D) che non rispondono più
alle terapie ormonali. In un limitato numero di casi è stata dimostrata una aumentata risposta del
recettore mutato agli androgeni surrenalici, ai progestinici e agli antiandrogeni. Altri tipi di mutazione del
18
recettore androgenico, di tipo disattivante, sono stati riscontrati nel carcinoma prostatico latente e
potrebbero essere responsabili della mancata progressione del tumore.
1.11 h (human) ZAC: un nuovo fattore di trascrizione antiproliferativo
hZAC è una seven Zinc-finger protein di 463 aminoacidi (50 kDa) appartenente alla famiglia C2H2 che
controlla contemporaneamente l’apoptosi e la progressione del ciclo cellulare attraverso pathways
separati, è l’unico gene umano a condividere con p53 e m (murino) Zac1 questa capacità funzionale.
Secondo la mappa del genoma umano hZAC è localizzato tra i markers D6S308 e D6S978 sul 6q24-25
(47). Analisi Northern blots mostrano diverse varianti dell’mRNA del gene hZAC, con un peso compreso
tra le 3 e le 8 kb, suggerendo che il messaggero possa andare incontro a diversi splicing alternativi e/o
sia soggetto a differenti poliadenilazioni (48). A causa delle sue proprietà funzionali, della sua
localizzazione cromosomica e della sua mancanza di espressione in modelli di cellule trasformate si
ipotizza un ruolo di ZAC come tumor suppressor gene (TSG) (49).
Recentemente è stato dimostrato che mZac1 interagisce con membri della superfamiglia dei recettori per
gli steroidi e loro co-attivatori: mZac1 sembrerebbe quindi avere un ruolo nel controllo della proliferazione
dei tessuti ormone-dipendenti (50). Il gene mZac1 è stato anche isolato da Abdollahi e collaboratori (51), i
quali erano alla ricerca di geni la cui espressione fosse persa in modelli in vitro di cellule ovariche a
seguito di trasformazione neoplastica: questi ricercatori hanno dato allo stesso gene il nome di LOT1 (da
Lost On Transformation). Dei 9 esoni che costituiscono il gene di hZAC i primi sette sono esoni non
codificanti: sono questi ad andare incontro a splicing alternativo. Il primo esone si trova a circa 60 kb a
monte del primo esone codificante (l’esone 8) (49).
1.12 Geni hZAC e mZac1
Mentre l’organizzazione genomica dei due geni è essenzialmente uguale (l’analogo murino mappa nella
regione prossimale del cromosoma 10), le proteine umana e di topo differiscono significativamente nella
sequenza aminoacidica. mZac1 ha due domini assenti rispetto dal suo omologo umano: una regione
contenente 34 triplette ricche in prolina (PLE, PMQ, PML) e una regione costituita prevalentemente da
prolina, acido glutammico e glutammina. Nonostante queste differenze strutturali, però, le due proteine, in
saggi di apoptosi e differenziamento del ciclo cellulare, sono funzionalmente indistinguibili (47, 52).
19
2. NEUROPEPTIDE Y (NPY)
Il Neuropeptide Y (NPY) fu per la prima volta isolato dal cervello di suino da Tatemoto e Mutt utilizzando
una metodica specifica per l’identificazione di peptidi con residui carbossi-terminali amidati (53). L’NPY fa
parte di una famiglia di peptidi costituiti dallo stesso NPY e da due ormoni intestinali, il peptide YY (PYY)
(54) e il polipeptide pancreatico (PP) (55). Mentre l’NPY è localizzato solamente a livello neuronale e il
PP si trova principalmente nelle cellule endocrine del pancreas, differenti da quella che producono
insulina, glucagone o somatostatina, il PYY è sintetizzato prevalentemente nel tratto digestivo (54) ad
opera sia di cellulare endocrine che di neuroni, facendo quindi ipotizzare possibili funzioni di
neurotrasmettitore (56). PYY e NPY originarono, presumibilmente, da una duplicazione di un gene
ancestrale comune nei vertebrati, ma i lori geni sono collocati su cromosomi differenti. In modo simile, il
gene PP origina, probabilmente, da una duplicazione del gene PYY ed i loro geni sono posti uno vicino
all’altro nello stesso segmento cromosomiale (57). PYY e PP hanno una sequenza omologa a quella di
NPY rispettivamente per il 70 e 50%. Ognuno di questi peptidi è composto da 36 aminoacidi,
caratterizzati da un considerevole numero di residui tirosinici amidati in posizione C-terminale. Basandosi
sulla struttura cristallografica del PP aviario, è stato proposto un modello tridimensionale di NPY, PYY e
PP che fa riferimento a un modello definito “PP-fold”. Le caratteristiche comuni della famiglia dei peptidi
“PP-fold” consistono in un’elica poliprolinica (residui 1-8) seguita da una struttura a forcina rigida che
ruota e un’elica antiparallela (aminoacidi 13-32) che terminano in un corto ammide tetrapeptidico
spazialmente vicino alla porzione N-terminale (58, 59). NPY è uno dei peptidi più conservati tra quelli
conosciuti (57). Infatti, la sequenza aminoacidica di NPY nella specie Torpedo marmorata è identica a
quella dei mammiferi in 33 dei 36 aminoacidi che lo compongono; le sequenze aminoacidiche dell’NPY,
in diversi mammiferi, differiscono solo in due dei 36 aminoacidi. Al contrario, PYY ha una variabilità di otto
aminoacidi tra i diversi ordini di mammiferi e poiché PP si è evoluto molto rapidamente, è considerato uno
dei meno conservati (56).
2.1 NPY: sintesi e processing
Nell’uomo, il gene NPY si trova sul cromosoma 7 in posizione 7p15.1 ed è composto da quattro esoni che
codificano per un trascritto delle dimensioni di 0.8 kb (60). Il promotore presenta la tipica struttura della
20
regione regolatrice di un gene eucariotico; infatti, procedendo in direzione 5' dal sito d’inizio della
trascrizione, si incontrano diverse sequenze consenso putative per fattori di trascrizione quali: una
sequenza TATA canonica, una consenso per SP-1 (un fattore di trascrizione capace di attivare i
promotori di molti geni ad espressione costitutiva), una per AP-1, una per CAAT, altre cinque sequenze
consenso per SP-1 (61).
L’NPY è il peptide più espresso a livello del sistema nervoso centrale (SNC); viene sintetizzato
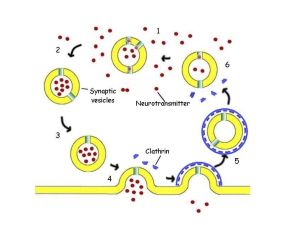
principalmente negli interneuroni GABA-ergici e immagazzinato in vescicole a livello dei terminali nervosi.
Una stimolazione neuronale prolungata e associata a stimoli motori causa il rilascio di NPY e di altri
neuropeptidi quali somatostatina, encefaline, tachichinine e colecistochinina, oltre ai classici
neurotrasmettitori (62, 63). Mentre i depositi tissutali dei trasmettitori classici sono ricostituiti rapidamente
dopo stimoli acuti attraverso un re-uptake e una sintesi locale a livello dei terminali nervosi, i neuropeptidi
vengono degradati dopo il loro rilascio (64). I livelli tissutali dei neuropeptidi, quindi, diminuiscono spesso
per 2-3 giorni dopo un episodio acuto di stimolo intenso (65). Affinché il neuropeptide si riaccumuli nei
depositi è necessaria una sintesi ribosomiale del rispettivo prepro-peptide a livello del pericarion. I propeptidi vengono poi processati all’interno del nucleo delle vescicole via reticolo endoplasmatico e
apparato del Golgi per poi essere conseguentemente convertiti da enzimi proteolitici (pro-ormone
convertasi) nei rispettivi peptidi maturi (66). Le vescicole contenenti il pro-peptide e/o il peptide maturo
vengono trasportati alla terminazione nervosa prima che il peptide sia disponibile per il proprio rilascio.
Questo processo complicato e lungo spiega la prolungata deplezione tissutale dei neuropeptidi,
conseguente al loro rilascio, dopo stimoli acuti indotti (66).
L’NPY biologicamente attivo deriva da un precursore di 97 aminoacidi, il pre-proneuropeptide Y
(proNPY), che subisce almeno quattro eventi enzimatici post-trasduzionali (67). Dopo la rimozione della
sequenza segnale, si forma il proNPY, composto da 69 aminoacidi, il quale subisce una successiva
reazione di proteolisi, a livello di un singolo paio di aminoacidi basici (Lys 38-Arg39) ad opera degli enzimi
proconvertasi PC1/3 e/o PC2, da cui ha origine la forma NPY1-39 e un peptide carbossi-terminale di 30
aminoacidi, che è il peptide C-flanking dell’NPY (CPON) (58). Studi di cinetica hanno dimostrato che
l’enzima PC1/3 è molto più efficiente di PC2 nel clivaggio del proNPY (68). NPY1-39 è ulteriormente
processato da un enzima carbossipeptidasi simile che porta alla formazione di NPY 1-37, che viene
amidato nella parte C-terminale dall’enzima peptidil-α amidasi monossigenasi, in un processo che,
rimuovendo un singolo amminoacido, porta alla formazione di NPY 1-36 (forma biologicamente attiva). Esso
21
può essere degradato da specifici enzimi generando NPY2-36 e NPY3-36. La dipeptidil peptidasi IV (DPPIV)
è una proteasi di membrana che ha una attività di clivaggio sulla prolina in penultima posizione (69). La
rimozione del dipeptide Try-Pro nella parte N-terminale di NPY, da parte di DPPIV genera la forma NPY336,
un frammento con una ridotta affinità per il recettore NPY Y1, ma che mantiene la sua capacità di
legare i recettori NPY Y2 e NPY Y5 (70). DPPIV è presente nei reni, nel fegato, nell’ileo ed è inoltre
abbondante a livello delle cellule endoteliali, epiteliali, e degli astrociti così come a livello dei linfociti T e B
attivi (71). E’ stata anche trovata una forma solubile di questo enzima (72). Nell’uomo un altro enzima,
l’ammino-peptidasi P (AmP), rimuove a livello renale e del digiuno, una tirosina N-terminale dai peptidi
PYY e NPY generando la forma NPY2-36 (73), un potenziale agonista semiselettivo per i recettori NPY Y2
e NPY Y5. L’AmP è presente nei polmoni (74), negli astrociti e nei neuroni, così come a livello della
muscolatura liscia e delle cellule endoteliali (75). Anche per questo enzima esiste una forma solubile
circolante (74).
NPY può essere degradato anche dalle proteasi presenti a livello del sistema nervoso centrale (SNC); ad
esempio, nei sinaptosomi dell’ippocampo, attraverso un singolo clivaggio tra i residui Leu 30 e Ile31, NPY
viene metabolizzato efficacemente nei frammenti NPY1-30 (troncato a livello C-terminale terminale) e
NPY31-36. Si ipotizza che gli enzimi coinvolti in questo processo abbiano proprietà aspartil proteasica. In
aggiunta, la endopeptidasi neutra che si trova nella membrana sinaptica del nervo striato e a livello renale
ha una maggiore attività idrolizzante su NPY. Al contrario, NPY è resistente all’azione enzimatica di altre
due amminopeptidasi di membrana, la amminopeptidasi N e W, e all’azione dell’enzima angiotensina
convertasi (76).
Nell’uomo, le concentrazioni plasmatiche di NPY sono normalmente comprese in un range tra 0.25 e 129
pM (77, 78). Questa variabilità nei valori delle concentrazioni sieriche di NPY circolante è probabilmente
dovuta ad una cross reattività dell’antisiero NPY-specifico verso il pro-NPY, i frammenti di NPY, così
come verso i peptidi PYY e PP. Nei ratti, i valori delle concentrazioni sieriche di NPY variano da 25 pM a
3000 pM. In modo particolare, in queste specie il prelievo potrebbe rappresentare un problema, poiché le
piastrine contengono alti livelli di NPY e l’emorragia causa il rilascio di NPY dalle terminazioni dei nervi
simpatici (79).
2.2 Distribuzione anatomica
22
Nei mammiferi sono state identificate elevate quantità di NPY sia in molteplici aree cerebrali che nel
sistema nervoso periferico (80). A livello del sistema nervoso centrale è stata evidenziata un'analogia per
quanto riguarda la distribuzione del peptide nell'uomo e nel ratto. In quest’ultimo, le più alte
concentrazioni sono state ritrovate a livello del sistema limbico (amigdala e nucleo accumbens) e
dell'ipotalamo (81, 82), mentre più basse quantità sono state rilevate nei gangli della base, nel globo
pallido, nell’ippocampo e nella corteccia (81). Al contrario, nell'uomo, studi post-mortem hanno mostrato
che la quota maggiore è in corrispondenza dei gangli della base (caudato e putamen), del nucleo
accumbens e dell’amigdala, mentre un’immunoreattività moderata è stata rilevata nell'ipotalamo,
nell’ippocampo, nel nucleo settale ed in alcune aree corticali (83); molto bassa è invece la concentrazione
di NPY a livello del talamo e della substantia nigra (84). Sia nell’uomo che nel ratto, l’NPY è stato
identificato a livello della colonna vertebrale, in particolare nella regione delle corna dorsali (85, 86).
Un'immunoreattività NPY specifica (NPY-IR) in corrispondenza delle terminazioni nervose simpatiche
associate ai vasi sanguigni fa supporre la presenza di NPY anche a livello di alcuni organi endocrini. A
conferma di ciò, Lundberg e Terenius hanno osservato che a livello periferico NPY è selettivamente
coespresso con la noradrenalina e la norepinefrina (87), suggerendo interazioni funzionali reciproche tra
questi agenti (88). Una NPY-IR è stata anche rilevata nella tiroide, nella cute, nella milza, nei reni, e
anche a livello dell'apparato genitale femminile, dove esplica un effetto inibitorio, in modo dosedipendente, sulla contrazione delle tube di Falloppio indotta dalle catecolamine (89). L’NPY, secondo uno
studio condotto da Bagnoli e collaboratori (90), potrebbe avere un ruolo fondamentale anche nello
sviluppo della retina dei mammiferi visto che se ne osserva una elevata espressione, associata ad altri
peptidi, proprio in corrispondenza dell’apertura degli occhi.
2.3 I recettori NPY
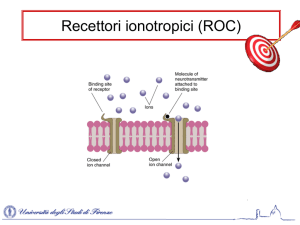
I peptidi NPY, PYY e PP interagiscono tutti con una famiglia di recettori accoppiati alle G-protein,
appartenenti alla superfamiglia dei recettori simili a quelli di classe 1 della rodopsina. Nei mammiferi sono
stati clonati cinque sottotipi recettoriali per NPY (Y1, Y2, Y4, Y5, y6), mentre il sottotipo recettoriale Y3,
non ancora clonato, è stato postulato sulla base di studi farmacologici effettuati su tessuti di mammifero.
Al recettore y6 è stata assegnata questa nomenclatura minuscola (nomenclatura della International Union
of Pharmacology (IUPAR) (91) poiché la proteina del recettore è tronca nella maggior parte dei
mammiferi, compreso l’uomo. Il recettore y6 è funzionale nei topi e ciò va considerato per interpretare
23
alcuni dei risultati nei topi knockout. I recettori Y1, Y2, Y4, Y5 clonati sono tutti associati alle proteine G i
che mediano l’inibizione della sintesi di cAMP (92-95). L’attivazione dei recettori per l’NPY, da parte di un
segnale extracellulare, coinvolge la fosforilazione, sensibile alla tossina della pertosse, mediata dalle
protein chinasi (PKC) 1 e 2, confermando che questi recettori sono accoppiati a proteine G i o Go. Mentre
PKC media la fosforilazione connessa principalmente al recettore Y5 e al Y1, una via PKC indipendente
potrebbe anche essere coinvolto nei segnali mediati dai recettori Y1, Y2, Y4 (96). I recettori per l’NPY
sono associati anche alla fosfoliposi C (PLC) determinando il rilascio di Ca 2+ intracellulare (92, 93, 97,
98). I recettori NPY mostrano molte delle caratteristiche strutturali dei recettori rodopsina-simili, incluse
due cisteine extracellulari che si ritiene formino un legame disulfide tra il primo e il secondo loop
extracellulare, come descritto per la struttura della rodopsina bovina ottenuta per cristallografica a raggi X
(99).
Y1, Y4, y6 hanno inoltre due cisteine aggiuntive, una nella coda ammino-terminale e una nel terzo loop
extracellulare, che può quindi formare potenzialmente un secondo legame disulfide. Tutti i recettori per
l’NPY hanno almeno una cisteina nella coda citoplasmatica che probabilmente, attraverso
palmitoilazione, ancora la coda all’interno della membrana. I recettori Y1 e Y4 hanno tre siti di
glicosilazione del tipo (Asn-X-Ser/Thr) localizzati sul lato extracellulare del residuo ammino-terminale e
uno nel secondo loop extracellulare, mentre i recettori Y2 e Y5 hanno rispettivamente uno e due nella
porzione ammino terminale (99).
Nei mammiferi, i cinque recettori Y possono essere divisi in tre sottofamiglie distinte sulla base della loro
omologia aminoacidica. Queste sottofamiglie sono state chiamate come i loro primi membri Y1, Y2, Y5, i
quali presentano una omologia solo del 27-31% che raggiunge il 40-43% nella regione transmembrana
(100). Poiché ognuno di questi recettori si evolve lentamente, si può presumere che esistano da molto
tempo e ciò è stato peraltro confermato dal ritrovamento nella lampreda di fiume (Lampreta fluvialis) di
geni Y1 (101) e Y2 simile (102). La sottofamiglia Y1 comprende i recettori Y1, Y4, y6 dei mammiferi i
quali presentano una identità aminoacidica del 50%, che raggiunge il 60% nelle regioni transmembrana
(100). Nella sottofamiglia Y1 sono incluse anche tre recettori dello zebrafish a cui furono inizialmente
attribuiti i nomi provvisori di Ya, Yb, Yc (103). La sottofamiglia Y2 comprende il recettore Y2 e Y7
scoperto di recente nello zebrafish e identificato anche nelle rane (104).
2.4 La sottofamiglia Y1: Y1, Y4, y6
24
Il primo recettore Y ad essere clonato fu il sottotipo Y1 (93, 105, 106), un tipico recettore peptidico
appartenente alla superfamiglia della rodopsina e accoppiato alle proteine G. Studi condotti in diverse
specie di mammiferi (107) ed anche nel pollo (108) hanno evidenziato una caratteristica peculiare, di
questo recettore, che è quella di legare le forme attive sia del PYY che dell’NPY, ma non le forme
tronche. Il recettore Y1 è stato clonato anche nella rana Xenopus laevis (109) e nello spinarolo imperiale
(Squalus acanthias) (110), sebbene su di essi non sia stato condotto alcuno studio funzionale.
Sorprendentemente nessuna sequenza del Y1 è stata identificata nel genoma sia dello zebrafish che del
pesce palla (102).
Il secondo membro della sottofamiglia Y1 è il sottotipo Y4, il quale presenta un’affinità maggiore per il
peptide PP (98, 111) rispetto ai peptidi NPY e PYY. Tale affinità è più accentuata nel ratto e nel topo
rispetto ad altre specie di mammiferi, quali l’uomo e il porcellino d’India, suggerendo che vi è stata una
più rapida evoluzione del PP e dell’Y4 nei roditori (57, 112). Il sottotipo Y4 è stato identificato e
caratterizzato anche nel pollo dove, sorprendentemente, mostra affinità uguale per NPY, PYY e PP (113).
Nell’uomo, studi in vivo condotti con PP suggeriscono che Y4 potrebbe avere un effetto di riduzione
sull’appetito (114).
Interessante è stata la scoperta di un terzo recettore Y1-simile, che dopo l’identificazione nel topo e nel
coniglio e la caratterizzazione in vitro, è stato chiamato Y6 (115). Secondo la nomenclatura IUPAR,
poichè non è stata dimostrata alcuna attività fisiologica, il suo nome dovrebbe essere y6 (con la y
minuscola); tuttavia essendo il recettore apparso molto precocemente nell’evoluzione dei vertebrati, ciò
ha indotto Larhammar (102) a considerarlo al pari degli altri sottotipi e a scriverlo con la Y maiuscola (Y6).
Il recettore y6 è codificato da un pseudo gene nell’uomo e in alcuni primati (111, 116, 117), manca dal
genoma di ratto (118) e non è funzionale neanche nel porcellino d’India (119, 120). Si è però scoperto un
y6 funzionale in un parente lontano del suino, il pecari (102). Ciò fa sorgere il quesito se il gene y6 non
sia funzionale in tutti i mammiferi ed abbia quindi perso la sua funzione negli antenati dei mammiferi o se
sia stato inattivato indipendentemente in diversi generi di mammiferi, come suggerito dall’osservazione di
alcune distinte mutazioni inattivanti nei recettori y6 degli esseri umani, dei suini e dei porcellini d’India.
Nonostante y6 non sia importante in nessun mammifero, esso ha chiaramente un’origine evolutiva antica,
essendone state clonate le sequenze nei polli e nella rana come negli spinaroli imperiali (110).
2.5 La sottofamiglia Y2: Y2 e Y7
25
Il sottotipo Y2 fu descritto per la prima volta nel 1986 soltanto a livello farmacologico, come un recettore
presinaptico con affinità predominante per i peptidi NPY e PYY (Wahlestedt 1986) e solo
successivamente fu clonato nell’uomo. (97, 121, 122). Y2 ha una sequenza aminoacidica abbastanza
diversa da quella del Y1 con circa il 30% d’identità. Nel ratto e nel topo, cosi come nel porcellino d’India
(123) e nel pollo (124) Y2 possiede la peculiarità farmacologica di legare diversi frammenti di peptidi
tronchi quali, ad esempio, i peptidi NPY e PYY mancanti nella parte ammino-terminale degli aminoacidi
13, 18 o 22 (91). Il recettore Y2 è presente anche nelle rane, nello zebrafish, nella trota e nello spinarolo.
In queste specie è stato inoltre scoperto un nuovo recettore definito Y7. Le sue proprietà farmacologiche
sono state studiate solo nella zebrafish, dove si è evidenziato che, rispetto al sottotipo Y2, ha una
maggiore affinità ai ligandi peptidici tronchi (104).
2.6 La sottofamiglia Y5: Y5
La scoperta del recettore Y5 fu anticipata da diverse pubblicazioni che descrivevano la possibile
esistenza di un recettore coinvolto nella stimolazione dell’appetito, ma che fosse distinto dal Y1 (125127). La sua sequenza aminoacidica è correlata a quella dei recettori Y1 e Y2 e sebbene leghi gli stessi
ligandi, ha con loro una identità aminoacidica di solo il 30% (128, 129). La peculiarità farmacologica di Y5
è quella di avere un’alta affinità per le forme tronche (mancanti dei primi due aminoacidi) dei peptidi NPY
e PYY, ma di avere, a differenza di Y2, una bassa affinità nei confronti di frammenti più corti (91). La
stessa proprietà è stata osservata nel Y5 del porcellino d’India (130), mentre il Y5 del pollo lega anche
frammenti peptidici molto corti (108).
2.7 Il recettore Y1
Il recettore Y1 è stato il primo recettore NPY ad essere clonato. Nel ratto inizialmente fu pubblicato come
recettore orfano (105), e successivamente sulla base della distribuzione dell’mRNA nel cervello di ratto fu
identificato come recettore Y1 (93, Larhammar, 1992 #451). Il gene che codifica per Y1 si trova sul
cromosoma umano 4q (31.3-32) (131), insieme a quelli che codificano per Y2 e Y5. In tutte le specie
studiate, contrariamente agli altri recettori NPY, nella sequenza genica di Y1, gli esoni codificanti 2 e 3
sono separati da un piccolo introne di circa 100 paia di basi e nel topo le tre varianti (1a, 1b, 1c) di questo
recettore sono da attribuirsi ad una trascrizione alternativa di esoni non codificanti. Tra le diverse specie
26
in cui è stato identificato, il recettore Y1 mostra un’identità aminoacidica superiore al 95% nelle regioni
transmembrana (100).
Studi in vivo hanno portato all’identificazione nel topo di due varianti di questo recettore (132) la forma
corta di 307 aminoacidi, con pochi aminoacidi dopo il terzo loop extracellulare, forma un recettore a 7
domini transmembrana incompleto che lega l’NPY con un’affinità simile a quella per la proteina completa,
composta da 384 aminoacidi. Comunque, il signaling associato alla forma corta del recettore è
compromesso, indicando che i sette domini transmembrana e la coda carbossi-terminale non sono
essenziali per l’interazione ligando-recettore, bensì per l’attivazione della proteina G. Come molti recettori
accoppiati alle proteine G, il recettore Y1 può essere internalizzato insieme al ligando dopo stimolazione
con l’agonista; infatti dopo stimolazione con PYY il recettore Y1 è internalizzato rapidamente negli
endosomi e riciclato in superficie in 60 minuti, come mostrato da studi condotti utilizzando radioligandi
(133) o ligandi fluorescenti analizzati al microscopio confocale (134). Diversi promotori, alcuni dei quali
posizionati tra gli esoni, sono attivati alternativamente e permettono quindi una espressione recettoriale
tessuto-specifica (135). Y1 è presente in diversi tessuti quali il cervello, il cuore, i reni, il tratto
gastrointestinale ed è espresso costitutivamente in alcune linee cellulari come SK-N-MC (136), una linea
di neuroblastoma umano, e le cellule HEL (137), una linea cellulare di leucemia eritrocitaria. Analisi di
binding condotte in linee cellulari transfettate stabilmente hanno permesso di stabilire il seguente ordine
d’affinità per questo recettore: NPY=PYY=[Leu31,Pro34]NPY>frammenti C-terminali di NPY>PP (106,
Magni, 2003 #2003). Comunque, dopo che sono stati clonati i recettori Y4 e Y5, si notò, ad esempio, che
[Leu31,Pro34]NPY, un classico agonista Y1, era un ligando più selettivo per questi altri due recettori
piuttosto che per il Y1. Nuovi agonisti quali [D-Arg25]NPY, [D-His26]NPY e DesAA11-18[Cis7,21,D-Lys9(Ac),DHis26,Pro34]NPY, [Phe7,Pro34]NPY possiedono un’alta affinità per il recettore Y1, inibendo la produzione di
AMP ciclico (cAMP) indotta da forskolina già a concentrazioni picomolari (138). Questi agonisti, nei
roditori, somministrati per via intracerebroventricolare stimolano il pasto in maniera dose dipendente,
suggerendo un ruolo, per il recettore Y1, di mediatore del comportamento alimentare.
Sebbene la prima azione associata a Y1 fu la mediazione della vasocostrizione (139), esso regola anche
la stimolazione del food intake e l’attivazione dell’asse neuroendocrino. Poiché il recettore Y1 poteva
rappresentare un potenziale target terapeutico, si iniziarono studi di screening di antagonisti non peptidici.
Il BIBP3226, il primo antagonista selettivo per Y1, aveva una Ki nel range delle nanomolare (140) con
una affinità non significativa per gli altri recettori. Sfortunatamente sia la bassa bioattività che solubilità ne
27
hanno precluso un largo impiego in studi in vivo. Tuttavia, una somministrazione periferica del BIBP3226
è in grado di attenuare, nei reni e nella mucosa nasale, sia l’effetto indotto da NPY esogeno, sulla
pressione sanguigna (141), che la vasocostrizione susseguente ad una stimolazione del nervo simpatico
(142). Altri antagonisti Y1, non peptidici, quali 1229U91 e GR231118, pur avendo un’alta affinità per Y1 e
mostrando una buona azione di blocco delle azioni mediate da questo recettore, hanno proprietà
agoniste per il recettore Y4 (121).
2.8 Il recettore Y2
Un cDNA codificante il recettore umano NPY Y2 fu inizialmente clonato nella linea cellulare SMS-KAN, e
successivamente dal cervello umano così come dalla linea cellulare di neuroblastoma umano KNA-TX
(121). Nell’uomo, il gene codificante per Y2 si trova sul cromosoma 4q31, prossimo ai loci dei recettori Y1
e Y5 (143) e codifica per una proteina di 381 aminoacidi (Ammar 1996) altamente conservata fra le
specie e con più del 90% di identità fra i mammiferi. L’mRNA per il recettore Y2 è stato identificato in vivo,
in varie regioni del sistema nervoso centrale (SNC) (144, Sten Shi, 1997 #2782) ed in vitro nelle linee
cellulari umane LN319 e SMS-KAN, così come nella linea di neuroblastoma umano SH-SY5Y (145). La
peculiarità del recettore Y2 è quella di legare il frammento peptidico carbossi-terminale con elevata
affinità; tale estremità deve essere intatta per quanto concerne gli ultimi 12 aminoacidi (25-36) e deve
risultare amidata ai fini dell’attività biologica (146). A differenza del recettore Y1, non sembra
internalizzarsi dopo stimolazione agonista prolungata (Parker 2001, Gicquiaux 2002) o lo fa molto
lentamente. Il recettore Y2 lega gli analoghi dell’NPY con il seguente ordine di potenza:
NPY=PYY=frammenti C-terminali di NPY>[Leu31,Pro34]NPY>PP (97, 147); alcuni frammenti tronchi
dell’NPY, come 1-4-Ahx-25-36 NPY e [Leu28,31]NPY24-36 hanno un’aumentata selettività per Y2 se
comparati all’agonista NPY13-36 (145, 148).
2.9 Il recettore Y3
Il recettore y3 è un recettore putativo che non è stato mai clonato, ma caratterizzato sia
farmacologicamente che funzionalmente utilizzando analoghi tronchi di NPY o NPY simili. Infatti, una
peculiarità di y3 è la possibilità di essere attivato da NPY, ma non da PYY. La potenza degli analoghi di
NPY sui recettori y3 può dipendere dalla specie e dai tessuti studiati (149), seppur solo un piccolo
numero di tessuti sembra esprimerlo. Ad esempio, nei bovini, le cellule surrenaliche cromaffini (150), nei
28
ratti, i neuroni simpatici dei gangli cervicali superiori (151), i nuclei del tratto solitario (152), le membrane
del ventricolo cardiaco (153), il colon distale (154, 155) nel ratto e nelle cellule PC12 differenziate,
feocromocitoma di ratto, con NGF (156), esprimono siti di legame con un profilo farmacologico coerente
con quello associato al recettore y3. Comunque, si è stabilito che l’ordine di potenza degli analoghi
dell’NPY nello stimolare la secrezione di catecolamine da cellule cromaffini è il seguente: NPY> NPY 1336=
NPY3-36=PP>>>[Leu31,Pro34]NPY=PYY (157). Non si può tuttavia escludere che questo pattern di
stimolazione rifletta la presenza di più di un recettore sulla superficie cellulare, che potrebbe quindi legare
indipendentemente differenti ligandi o potrebbe anche formare dimeri con particolare affinità di legame
per gli analoghi di NPY (149).
2.10 Il recettore Y4
Il gene del recettore Y4, clonato da diversi gruppi (98, 158), è localizzato sul cromosoma 10q11.21 (159)
in modo abbastanza diverso rispetto alla localizzazione cromosomica dei recettori Y1 e Y2. Paragonando
le sequenze dell’uomo e del ratto, solo il 75% degli aminoacidi sono uguali, suggerendo che il recettore
Y4 sia, tra i recettori accoppiati alle proteine G, uno di quelli che si è evoluto più rapidamente (160).
Y4 ha un’alta affinità per il peptide PP ed una relativamente più bassa per NPY (121) ed il fenomeno
dell’internalizzazione è controverso, infatti Parker (133) riporta che Y4 si internalizza in seguito ad una
stimolazione con agonista, mentre Voisin (161) asserisce il contrario. Recentemente Berglund (162) ha
dimostrato che il recettore Y4, espresso in cellule di mammifero, forma un omodimero costitutivo che si
dissocia dopo stimolazione con agonista. L’Analisi Northern blot indica che Y4 è espresso, nell’uomo,
principalmente a livello dell’intestino, del colon, della prostata, dei muscoli lisci e scheletrici; in ogni modo
modesti livelli di mRNA di Y4 sono stati evidenziati anche a livello dell’ipotalamo e dell’amigdala (158,
163). Nella famiglia dell’NPY, NPY dimostra un’elevata affinità per i recettori Y1, Y2 e Y5 e solo moderata
per il Y4, mentre il peptide PP lega preferenzialmente Y4 mostrando una bassa affinità per gli altri
sottotipi recettoriali. La peculiarità del recettore Y4 è quindi quella di legare PP con alta affinità (98, 161).
Comunque il PP umano sembra avere un’affinità significativa non solo per il recettore Y4 umano, ma
anche per quello di ratto e di topo. Anche il peptide PYY e alcuni analoghi di NPY con la [Pro 34] sostituita
hanno una alta affinità per il sottotipo recettoriale Y4 (98); da notare che [Leu31, Pro34]NPY è il solo
analogo di NPY che è in grado di interagire significativamente, nel ratto, con il Y4 (160). Comunque, alte
concentrazioni di NPY non influenzano l’accumulo di cAMP indotto da forskolina, mentre concentrazioni
29
nanomolari di NPY sono in grado di spiazzare PP dai suoi recettori in linee cellulari umane che
esprimono il sottotipo Y4 (161). Questi dati suggeriscono che NPY potrebbe legarsi al recettore NPY Y4,
ma non necessariamente attivare vie di segnale intracellulari secondarie. Considerati nel loro complesso,
questi studi indicano che PP è il ligando endogeno principale del recettore Y4 (149).
2.11 Il recettore Y5
Il gene Y5 si trova sul cromosoma umano 4 (4q32), più precisamente nello stesso locus del gene di Y1,
ma è trascritto in direzione opposta (125); esso codifica per una proteina di 446 aminoacidi (126) che ha
un terzo loop citoplasmatico esteso, con circa 100 aminoacidi in più rispetto agli altri recettori dell’NPY.
Comunque la coda carbossi-terminale di Y5 è molto più corta di quella degli altri recettori Y1, Y2 e Y4. Il
recettore Y5 è molto ben conservato (88-90% di identità aminoacidica e 95-98% se il terzo loop
intracellulare non viene considerato) tra le specie di mammiferi (127).
Enormi interessi sono stati fatti convergere sull’identificazione del recettore Y responsabile del controllo
dell’assunzione di cibo; studi fisiologici iniziali suggerivano che questo controllo potesse essere attribuito
al recettore Y1, ma studi sul recettore clonato Y1 rivelarono una bassa affinità per il peptide NPY 2-36, un
ligando con le caratteristiche di un potente fattore in grado di indurre l’assunzione di cibo. Inoltre, studi in
situ in aree ipotalamiche coinvolte nella regolazione dell’appetito, mostrano bassi livelli di mRNA per il
recettore Y1, suggerendo l’esistenza di un unico recettore dell’appetito localizzato nel nucleo
paraventricolare dell’ipotalamo (164, 165). Il recettore Y5 isolato mostra un profilo farmacologico simile a
quello proposto per il recettore dell’appetito e comunque questo profilo condivide diversi aspetti con Y1,
possedendo una sostanziale affinità con gli agonisti Y2 specifici, come NPY2-36 e NPY3-36 (125, 126).
L’iniezione di questi peptidi a concentrazioni nanomolari all’interno di alcuni nuclei dell’ipotalamo, dove è
espresso l’mRNA di Y5, stimola drammaticamente l’assunzione di cibo (125).
Y5 mostra una scarsa identità nei confronti degli altri membri della famiglia dei recettori Y, con una
maggior affinità nei confronti di Y1 (35%), ed è inoltre caratterizzato da alcuni inusuali tratti distintivi, quali
la terza ansa intracellulare molto lunga e una catena carbossi-terminale molto corta. L’ mRNA del
recettore Y5 è stato identificato in alcune aree del cervello, incluse quelle ritenute importanti per la
regolazione dell’assunzione di cibo e, a livello periferico, principalmente nei testicoli (149). In cellule
transfettate per Y5, questo sottotipo recettoriale risulta essere accoppiato con una proteina G inibitoria e
al blocco dell’accumulo di cAMP. Gli analoghi dell’NPY hanno dimostrato il seguente grado di potenza nel
30
legare Y5, nell’inibire l’accumulo di cAMP e nella stimolazione dell’appetito: NPY = PYY >
[Leu31,Pro34]NPY = NPY2-36 = NPY3-36 > NPY13-36 (125).
Il PP ha una affinità molto bassa per i recettori Y5 umani e di ratto, mentre i peptidi PP umano e bovino
hanno un’affinità significativa per Y5. Il primo analogo dell’NPY con una capacità selettiva per il Y5 è
stato [D-Trp32]NPY (166) e seppur descritto come un antagonista, questo composto stimolava il food
intake nei ratti (167, 168), indicando o che il food intake non era mediato da questo recettore o che
questo composto fosse meno selettivo di quanto ci si aspettasse.
Poiché entrambi i recettori Y1 e Y5 si pensa giochino un ruolo importante nella regolazione
dell’assunzione di cibo, l’espressione coordinata dei loro geni specifici potrebbe essere importante nella
modulazione dell’attività di NPY. Basandosi sulla distribuzione dell’mRNA del recettore Y5 e
sull’attivazione del recettore clonato Y5, indotta dagli stessi ligandi che stimolano l’assunzione di cibo nel
modello in vivo, si è proposto che Y5 sia il recettore dell’appetito (125, 126). Comunque questi studi non
sono in grado di distinguere definitivamente Y5 dagli altri sottotipi, conosciuti o sconosciuti, che possono
a loro volta stimolare l’assunzione di cibo. Perché si possa dimostrare l’identità dei sottotipi recettoriali
responsabili dell’induzione dell’assunzione di cibo, sono necessari ulteriori studi, inclusi esperimenti con
modelli knockout per i vari geni e studi funzionali con un repertorio di agonisti e antagonisti selettivi (149).
2.12 Il recettore y6
Un recettore aggiuntivo è stato clonato recentemente in specie differenti e ad esso è stato attribuito il
nome di y6. Studi di ibridazione fluorescente in situ hanno localizzato il gene umano codificante per il
recettore y6 sul cromosoma 5q31. Ad oggi la distribuzione tissutale di questo sottotipo recettoriale non è
molto chiara, anche se l’espressione genica è stata osservata nel cervello di topo, attraverso l’ibridazione
in situ, ma non confermata da analisi Northern blot (169), così come nei conigli, dove y6 è stato
identificato, solo attraverso analisi RT-PCR, in alcune aree cerebrali, incluse l’ipotalamo e l’ippocampo,
nel piccolo intestino e nel surrene, mentre l’analisi Northern blot è risultata positiva solo nel cuore e nei
muscoli scheletrici. Comunque, la scoperta più importante è che le sequenze dell’uomo e della scimmia
contengono una mutazione putativa strutturale recettoriale a livello del terzo loop intracellulare (116) che
introduce un codone di stop aggiuntivo, codificando per una proteina troncata di solo 290 aminoacidi.
Quindi, sebbene i topi esprimano una proteina di lunghezza completa, l’espressione nei primati non
produce una proteina funzionale (119). E’ importante sottolineare come la farmacologia dei recettori y6
31
differisca drasticamente tra queste due specie: nel topo, il recettore ha un’elevata affinità per PP e [Leu 31,
Pro34]NPY e nessuna per il frammento carbossi-terminale dell’NPY; questo ordine è completamente
invertito a livello del recettore y6 del coniglio (111), un dato questo, che ancora una volta pone in
evidenza l’importanza dei ligandi competitori e radioligandi impiegati.
2.13 Vie di trasduzione del segnale
I recettori per l’NPY sono recettori a sette domini transmembrana che appartengono alla famiglia dei
recettori accoppiati alle proteine G eterotrimeriche (la subunità , la e la ). In seguito al legame di un
agonista, le proteine inattive legate a GDP (guanosina difosfato) scambiano GDP con GTP (guanosina
trifosfato) a livello della subunità Gα, che quindi si dissocia dal dimero Gβγ. Entrambe la subunità attiva Gα
legata a GTP e il complesso Gβγ possono individualmente regolare, a valle del recettore, l’attività di
effettori specifici. Studi iniziali dimostrarono che NPY media alcuni dei suoi effetti fisiologici attraverso
l’inibizione dell’adenilato ciclasi (AC) (170, 171); determinando una diminuzione, in molte linee cellulari e
tessuti, dell’incremento di cAMP indotto da forskolina senza alcun effetto significativo sui livelli basali di
cAMP (149). Poiché si è dimostrato che questa risposta è sensibile alla tossina della pertosse, ciò indica
che i recettori sono accoppiati alle proteine eterotrimeriche inibitorie G i e Go. NPY induce, anche, una
mobilitazione del calcio, determinandone un aumento dei livelli intracellulari (137). La variazione della
concentrazione di Ca++, associata alla stimolazione dei recettori dell’NPY, può essere indotta da
meccanismi distinti: attivazione dell’influsso di Ca++ attraverso i canali di tipo L, inibizione dell’influsso
tramite i canali di tipo N e mobilizzazione da depositi intracellulari. In alcuni lavori è stata dimostrata
l’associazione fra l’incremento dei livelli del Ca++ e la generazione di inositolo fosfato (172), mentre in altri
non si è avuto lo stesso riscontro (137). Quindi i recettori dell’NPY possono essere accoppiati alla
mobilizzazione del Ca++ dai depositi intracellulari attraverso vie che dipendono dall’inositolo 1,4,5
trifosfato (IP3) o che sono indipendenti da quest’ultimo, così come avviene a livello delle cellule muscolari
liscie delle arterie polmonari del conoglio e dell’aorta del maiale (173, 174). Entrambe le vie di segnale,
cAMP e Ca++ sono comunque compatibili con le proprietà vasocostrittive dell’NPY e con la sua capacità
di potenziare l’azione di altri vasocostrittori. Studi condotti sempre su cellule muscolari liscie dell’aorta del
maiale, utilizzando un inibitore specifico della fosfolipasi C, hanno evidenziato un blocco non solo della
sintesi di IP3, NPY indotta, ma anche della mobilizzazione del Ca++ (174). Sempre in questo modello
cellulare, NPY induce anche una rapida fosforilazione della catena leggera della miosina, che
32
rappresenta uno step fondamentale per l’inizio della contrazione (175). Usando una serie di analoghi
dell’NPY, è stato dimostrato che l’inibizione dell’AC, così come lo stimolo del rilascio di calcio
intracellulare sono mediati dal recettore Y1 (149). Esperimenti di transfezione, usando cloni di cDNA per
Y1 hanno confermato che questo sottotipo recettoriale è quello in grado di inibire l’accumulo di cAMP
stimolato da forskolina, di aumentare i livelli di calcio intracellulari (93) e di stimolare la via di segnale
intracellulare mitogen-activated protein kinase (MAPK) (176, 177), in particolare di p42/p44 MAPK.
Quest’ultimo effetto sensibile alla tossina della pertosse, (coinvolgimento delle proteine Go e Gi),
potrebbe dipendere dalla fosfatidilinositolo 3 (PI-3) chinasi (176). La fosforilazione della proteina MAPK
da parte del recettore Y1 può essere a seconda del tipo cellulare, protein chinasi C (PKC) dipendente o
indipendente, come nel caso delle linee cellulari di carcinoma prostatico androgeno indipendente DU145
e PC3 (osservazioni personali).
I segnali intracellulari associati al sottotipo recettoriale Y2 sono stati, invece studiati particolarmente nei
neuroni gangliari della radice dorsale di ratto (DRG). In queste cellule, NPY sembra inibire l’influsso del
Ca++ attraverso la modulazione dei canali del Ca++ voltaggio dipendenti (178), anche se è stato descritto,
sempre in queste cellule, un aumento intracellulare di Ca++, NPY indotto, secondario alla produzione di
IP3. L’accoppiamento di Y2 con una proteina inibitoria G i è stato suggerito dalla sensibilità alla tossina
della pertosse (179, 180). Nelle cellule di neuroblastoma umano SMS-KAN, NPY inibisce l’influsso di
Ca++, sensibile alla ω conotossina, modulando i canali di tipo N, così come il rilascio di calcio, dai depositi
intracellulari, stimolato dall’angiotensina II (181). Contrariamente a quanto osservato nei neuroni dorsal
root ganglian (DRG) di ratto, NPY non è in grado, nelle cellule SMS-KAN, di mobilitare il rilascio di calcio
intracellulare, ma di inibire l’accumulo di cAMP indotto da forskolina (149). Questi dati sono stati
confermati utilizzando delle cellule transfettate per Y2, nelle quali l’attivazione di Y2 induceva una
diminuizione dei livelli di cAMP e un aumento dei livelli di Ca++ (182).
Anche la linea cellulare umana di glioblastoma LN319, si è rivelata utile nel definire i meccanismi di
trasduzione intracellulari accoppiati a Y2, poiché esprime esclusivamente questo sottotipo recettoriale. In
queste cellule NPY inibisce l’accumulo del cAMP indotto da forskolina e stimola un aumento del calcio
intracellulare con un meccanismo sensibile alla tossina della pertosse e possibilmente dipendente
dall’attivazione della PLC (183). A conferma di ciò, l’attivazione da parte di NPY delle isoforme di PKC
calcio-indipendente e diacilglicerolo (DAG)-dipendente, attraverso il recettore Y2 (184), è stata anche
33
dimostrata nelle cellule cigliate umane (185). Infine, il recettore Y2 si associa alla via delle MAPK
mediante una proteina Gi e la PI-3K (176).
Le vie di segnale intracellulari associate con gli altri sottotipi recettoriali dell’NPY sono state
principalmente studiate in cellule transfettate; sebbene l’importanza di tali risultati sia fuori discussione,
dati funzionali ottenuti in situazioni fisiologiche, sono limitati. E’ stato mostrato che Y4, Y5 e y6 sono
accoppiati all’inibizione dell’accumulo di cAMP (161, 186), ma non è ancora stato stabilito se modifichino
la concentrazione intracellulare di Ca++. In particolar modo, il recettore NPY Y4 induce, nel ratto, a livello
dei neuroni del nucleo arcuato ipotalamico, oscillazioni di Ca++ (187), regola i canali del calcio e del
potassio nelle cellule HEK293 e negli oociti dello Xenopus (188). Infine sono state riportate, in colture
primarie di cardiomiociti, altre vie di segnale associate con l’attivazione di Y5 di topo quali una rapida
fosforilazione di ERK, JNK, p38 MAPK, come pure l’attivazione di PKC. Questi effetti stimolatori
sembrano essere sensibili alla tossina della pertosse e mediati dal recettore Y5, poiché agonisti Y5
selettivi possono mimare le azioni di NPY e l’attivazione della MAPK è abolita nelle cellule deficenti del
recettore Y5. Tuttavia nella linea cellulare transfettata HEC-1B, questi risultati non sono stati confermati,
poiché NPY non attiva né la MAPK né la via della PKC (189). Ciò porta a concludere che NPY possa,
nelle diverse linee cellulari, associarsi con pathways intracellulari distinte (149).
2.14 NPY e suoi recettori in modelli transgenici e knockout
I modelli sia transgenici che knockout si sono rivelati strumenti importanti per lo studio delle funzioni della
famiglia dell’NPY, che consiste di cinque differenti recettori e di tre ligandi, coinvolti nella regolazione di
numerosi processi fisiologici. I modelli knockout sono anche una preziosa risorsa per valutare la
specificità di agonisti e antagonisti o per determinare la specificità di anticorpi contro i differenti recettori.
E’ stato infatti possibile chiarire, seppur in parte, molte delle funzioni connesse al sistema NPY: il foodintake, l’omeostasi energetica, la riproduzione, l’ansia, l’epilessia e la regolazione cardiovascolare.
Inoltre, nuove azioni del sistema NPY sono state identificate, come ad esempio la regolazione centrale
della formazione ossea e la rigenerazione neuronale (190).
Il gran numero di recettori Y e la disponibilità di pochi agonisti ed antagonisti specifici, quest’ultimi con
problemi per quanto riguarda la solubilità e la disponibilità orale,
che ne riducono il valore per
esperimenti in vivo, ha reso difficile in passato delineare in modo specifico le funzioni del sistema NPY.
34
Negli ultimi otto anni sono stati descritti studi su topi transgenici over-esprimenti NPY e PP, ratti overesprimenti NPY, topi sia knockout che over-esprimenti NPY, Y1, Y2, Y4 e Y5 (190).
2.15 Animali transgenici per NPY
Ad oggi sono stati creati due modelli di topo (191, 192) e uno di ratto (193) entrambi over-esprimenti
NPY. Solo un modesto incremento del 15% è stato riscontrato nella immunoreattività ad NPY nel modello
generato da Inui, che con solo il 18% di incremento netto dei livelli di NPY nel nucleo arcuato,
fenotipicamente non mostra alcun cambiamento nel food intake o nel peso corporeo. Comunque, un
fenotipo obeso con incremento transitorio del food intake può essere indotto in questi topi una volta che
essi sono sottoposti ad una dieta ricca di saccarosio. Approssimativamente ad un anno di età, questi topi
sviluppano iperglicemia e iperinsulinemia senza alterate oscillazioni dei livelli di glucosio, confermando
che gli elevati livelli di espressione di NPY a livello ipotalamico sono importanti per lo sviluppo dell’obesità
(194).
Il modello prodotto da Thiele mostra approssimativamente livelli di mRNA 5 volte più alti rispetto ai topi
wild-type, con un’abbondante espressione proteica nella corteccia, nell’amigdala e nell’ippocampo, ma
non nel nucleo arcuato dell’ipotalamo. L’unico dato fisiologico disponibile, in questo modello, è una
significativa diminuzione volontaria dell’assunzione di alcool associata a maggiore sensibilità all’effetto
dello stesso (192); nessun dato riguardo il feeding ed il peso corporeo è disponibile.
2.16 Animali knockout per NPY
Erickson e colleghi nel 1996 iniziarono l’era dei topi knockout nel campo dell’NPY con la generazione di
una linea di topo knockout per NPY (195). Questo modello, in condizioni normali, non mostrava riduzione
né del food intake né del peso corporeo, sebbene il digiuno induceva un comportamento iperfagico.
Comunque in questi topi NPY-/-, se rapportati ai controlli, i trattamenti con leptina riducevano sia il food
intake che il peso corporeo. Al contrario, studi condotti da (196), su topi knockout, incrociati con topi
C57BL/6, mostravano una riduzione significativa del food intake dopo 24 o 48 ore di digiuno, suggerendo
un importante ruolo dell’NPY nel comportamento alimentare. A conferma di ciò, dati tratti da un lavoro di
Hollopeter 1998 mostrarono che una dieta ad alto contenuto di grassi non sembrava influenzare, in questi
modelli, né il peso corporeo né il food intake. Comunque sebbene la dieta ad elevato contenuto di grassi
aumenti il peso dei loro cuscinetti adiposi, il peso corporeo reale di questi topi NPY -/-, sottoposti ad una
35
dieta ad alto contenuto di grassi, non è significativamente differente dai controlli nutriti con cibo normale.
Inoltre nei neonati dopo lesioni chimiche del nucleo arcuato dell’ipotalamo, indotte attraverso infusione di
mono-sodio-glutammato, non si riscontrano differenze a livello del food intake, del peso corporeo o del
peso dei cuscinetti adiposi, se si raffrontano topi knockout per NPY e topi wildtype. In maniera simile,
infusioni IP nei topi adulti di oro-tio-glucosio, che distrugge selettivamente i neuroni nell’ipotalamo ventromediale (VMH), conosciuto anche come il centro della sazietà, non mostrano differenze significative tra i
topi knockout e i controlli (197). Gli effetti della deficienza di NPY nella regolazione del food intake e
dell’omeostasi energetica, sono invece più ovvi quando i topi knockout per NPY vengono incrociati nel
background di topi ob/ob, deficienti per leptina. Questi topi knockout doppi mostrano una riduzione
significativa del fenotipo obeso dei topi ob/ob, accompagnato ad un ridotto food intake e un’aumentata
spesa energetica che influenzano lo sviluppo del diabete (195). Questo dimostra che l’NPY agisce
centralmente a valle della leptina.
Il modello knockout doppio per NPY e il topo obeso giallo (Ay), che ha un difetto nella pathway del
recettore 4 della melanocortina (MC4), non ha migliorato il fenotipo obeso. Studi sugli effetti della
deficienza di NPY in un modello di topo knockout per il peptide agouti related (AgRP), un peptide
oressigenico che agisce da antagonista MC4 endogeno, che è co-espresso nei neuroni NPY del nucleo
arcuato (198), come per l’altro modello su menzionato, non sembra influenzare il comportamento
nutrizionale o l’aumento di peso in condizioni normali, suggerendo che potrebbe essere stato attivato un
meccanismo compensatorio.
E’ anche interessante notare che i topi knockout per NPY e la galanina, rispetto ai wild-type, mangiano
molto di più e sono più pesanti (30%), rispondono ad una dieta ad alto apporto di grassi, aumentano di
peso e non sono in grado di regolare il loro peso normalmente dopo un cambio di dieta; i livelli di leptina,
insulina e glucosio sono elevati e un trattamento cronico con leptina causa perdita di peso (199).
2.17 Topi knockout per il recettore Y1
I recettori Y1, espressi principalmente nel sistema nervoso, compresi i nuclei nell’ipotalamo, si pensa
siano responsabili della regolazione del food intake e dell’omeostasi energetica. Nei roditori, infusioni ICV
del ligando [Leu31/Pro34]NPY, selettivo per i recettori Y1 e Y5, stimola fortemente il comportamento
nutrizionale. E’ interessante notare che, i modelli knockout per il recettore Y1 non mostrano alcuna
anomalia significativa riguardo il food intake e il peso corporeo, sebbene ci siano sottili cambiamenti in
36
tutte le linee di topo knockout analizzate. Ad esempio, i modelli descritti da Pedrazzini (200) mostrano
una lieve riduzione del food intake, sia nei topi che possono nutrirsi spontaneamente, che in quelli in cui il
feeding è NPY-indotto; il re-feeding indotto dal digiuno è fortemente diminuito. Nei topi Y1-/- più vecchi
sembra inoltre svilupparsi un fenotipo obeso, che è più accentuato nelle femmine rispetto ai maschi.
Questi topi mostrano anche iperinsulinemia associata ad una aumentata mobilizzazione del glucosio dal
tessuto adiposo e da glicogeno-sintesi. Questo fenotipo è il frutto quindi di una ridotta attività motoria e di
un diminuito ritmo metabolico (201).
I topi knockout generati da (202) e Kanatani (203) mostravano un fenotipo simile con food intake basale
inalterato, una iperinsulinemia lieve, un inizio di obesità ritardato e un aumento significativo della massa
grassa, particolarmente nei topi knockout femmine. Kushi et al. trovarono anche aumentati livelli di
uncoupling protein (UCP-1) nel tessuto adiposo bruno e ridotti livelli di UCP-2 nel tessuto adiposo bianco
suggerendo una diminuzione del dispendio energetico. Nel modello descritto da Kanatami (203), dopo
infusione ICV di NPY e di analoghi, si osserva una significativa riduzione del food intake a conferma,
ancora una volta, che il food intake stimolato da NPY è mediato attraverso questo recettore. Inoltre, i topi
Y1-/-, ob/ob mostrano una significativa riduzione del peso corporeo sia nei maschi che nelle femmine se
paragonati al fenotipo ob/ob (204). La principale causa del ridotto peso corporeo è la riduzione
dell’iperfagia. I topi doppio knockout (Y1-/-, ob/ob) generati da Lin (190) mostrano anch’essi una
significativa riduzione del fenotipo obeso, inclusa una significativa riduzione della massa grassa ed un
aumento dei parametri metabolici come l’insulina e il glucosio.
Il recettore Y1 è anche responsabile nel mediare il consumo volontario di alcool, poichè i topi che sono
privi del recettore Y1 mostrano un aumento nel consumo di alcool, se comparati ai topi wild-type, ed
inoltre, sono meno sensibili ai suoi effetti sedativi recuperando più rapidamente dal sonno alcool-indotto
(205). Hansel et al (2001) hanno anche evidenziato un nuovo ruolo dell’NPY nella neuro-rigenerazione,
poichè è stato mostrato in vitro e in vivo, in preparazioni di cervello di modelli Y1-/-, una riduzione
significativa nella proliferazione cellulare e nella generazione di nuovi neuroni (206).
2.18 Topi knockout per il recettore Y2
I recettori Y2 sono largamente espressi nel sistema nervoso centrale (SNC) con livelli particolarmente alti
trovati nel nucleo arcuato e nella barriera emato-encefalica; sono quindi dei candidati ideali nel mediare i
segnali periferici coinvolti nell’omeostasi energetica (207-209). Ciò è stato confermato dalla scoperta di
37
agonisti endogeni del Y2, come il PYY3-36 (rilasciato post-prandialmente dal tratto gastrointestinale) che
se iniettato perifericamente in dosi fisiologiche, inibisce il food intake e riduce l’aumento di peso nei topi
wild-type, ma non nei topi Y2 knockout (210).
2.19 Topi knockout per il recettore Y4
Topi knockout per il recettore Y4 hanno una significativa riduzione del peso corporeo e del food intake
(211), in particolare, i topi maschi hanno sia livelli plasmatici di PP, il ligando endogeno ad alta affinità per
questo recettore Y, elevati, che una massa ridotta di tessuto adiposo bianco. Gli aumenti dei livelli
plasmatici di PP in questi animali, che raggiungono valori simili a quelli riscontrati negli animali magri PP
transgenici (212), sono la spiegazione più verosimile per giustificare la riduzione sia del peso corporeo
che del food intake. I topi knockout doppi Y4-/-,ob/ob non hanno un fenotipo migliore, che rimane obeso e
diabetico. Comunque, la fertilità ritorna al 100% nei topi maschi ed è migliorata fino al 50% nelle femmine
(211). In questi modelli, il miglioramento significativo della fertilità conferma un ruolo prioritario del
recettore Y4 nella riproduzione.
I topi knockout doppi Y2-/-, Y4-/- mostrano un miglioramento del comportamento nutrizionale (213); infatti
possiedono un fenotipo magro, con un ridotto peso corporeo, una ridotta massa di tessuto adiposo
bianco, una ridotta leptinemia e insulinemia, suggerendo che gli aumenti nella termogenesi e nell’attività
locomotoria possano compensare gli elevati intake energetici. Inoltre il volume osseo è aumentato di tre
volte, il tutto associato ad una aumentata attività osteoblastica. Ciò suggerisce un’interazione sinergica
fra le due vie di segnale associate al Y2 e al Y4 nel regolare il volume osseo e l’adiposità.
2.20 Topi knockout per il recettore Y5
In ratti e topi, infusioni ICV di analoghi dell’NPY selettivi per il recettore Y5, hanno suggerito un ruolo
prioritario per questo recettore Y nel regolare il food intake, sebbene dati sui modelli knockout non
confermino questa osservazione. I topi privi del recettore Y5 si nutrono e crescono normalmente,
sviluppando un’obesità con inizio tardivo (> 30 settimane), il tutto accompagnato ad un aumento del food
intake e del peso corporeo (Marsh 1998). I topi Y5-/- non mostrano alcun cambiamento nel re-feeding
indotto dal digiuno. Infatti le risposte all’infusione ICV di NPY o analoghi dell’NPY sono minime o assenti.
Risultati simili sono stati ottenuti nei topi knockout generati da Kanatani (203).
38
Neanche i modelli Y5-/-;ob/ob mostrano miglioramenti del food intake e del peso corporeo (214). Sia nei
maschi che nelle femmine il food intake, il peso corporeo e l’adiposità non sono differenti da quelli
riscontrati nei topi ob/ob.
2.21 NPY: controllo dell’ espressione genica e proteica
In letteratura è noto che l’espressione genica dell’NPY e dei suoi recettori è sotto lo stretto controllo dei
peptidi e degli ormoni steroidei implicati nel controllo di importanti funzioni fisiologiche, come il
metabolismo energetico, il food intake, così come la riproduzione (147). Ad esempio, i glucocorticoidi, sia
in vitro (215, 216) che in vivo, come nell’ipotalamo di ratto (217-219), stimolano i livelli sia di mRNA che
del peptide NPY (216, 220); al contrario, l’insulina e la leptina (l’ormone del tessuto adiposo) li inibiscono
(221-224). L’espressione genica dell’NPY nell’ippocampo, una regione del cervello ricca di recettori
steroidei simili a quelli di tipo I del surrene, è selettivamente stimolata dall’aldosterone (225), così come a
livello ipotalamico, l’espressione genica del recettore Y1 è modulata dal corticosterone. Da notare che
tale effetto stimolatorio si riduce in seguito a surrenalectomia (226, 227).
L’insulina è un ormone che regola negativamente l’espressione dell’NPY, una scoperta che spiega
almeno in parte la riduzione del food intake e del peso corporeo osservata dopo somministrazione
cronica di insulina (228, 229). Nel ratto l’infusione di insulina, a livello dell’ARC e del PVN, determina una
marcata riduzione dell’espressione genica dell’NPY e del peptide, sia in animali normalmente nutriti
(224), che dopo digiuno; condizione conosciuta per aumentare questi parametri (223). L’azione inibente
dell’insulina è diretta e indipendente dai livelli plasmatici di glucosio, così come osservato da esperimenti
di infusione ICV, purché a dosi tali da non modificare né le concentrazioni plasmatiche di glucosio, né
quelle di insulina (223). Da notare che il trattamento sistemico simultaneo con insulina e glucorticoidi
previene, a livello ipotalamico, rispettivamente la stimolazione e l’espressione genica dell’NPY (219).
I neuroni ipotalamici dell’ARC che esprimono NPY, possedendo alti livelli di recettori OB-Rb, sono un
possibile target dell’ormone leptina, infatti nei topi ob/ob (230, 231), db/db e nel ratto fa/fa, la
somministrazione di leptina normalizza, nella regione dell’ARC, gli elevati livelli di mRNA dell’NPY (232).
Oltre la leptina, anche l’ormone della crescita (GH), un altro ormone correlato alle citochine, interagisce
direttamente con il sistema dell’NPY, stimolando sempre nel ratto, a livello dei neuroni dell’ARC,
attraverso i propri recettori, l’espressione genica dell’NPY (233). Tali effetti fanno parte del feedback
negativo che controlla la secrezione del GH (234).
39
I neuroni ipotalamici di ratto che producono il mediatore oressigenico NPY sembrano essere un target
anche per la ghrelina, un ormone peptidico acilato formato da 28 aminoacidi e secreto principalmente
dallo stomaco; una somministrazione centrale cronica del peptide ghrelina aumenta i livelli di mRNA
dell’NPY sino al 150% rispetto ai controlli (235).
NPY partecipa anche alla regolazione delle funzioni riproduttive influenzando il rilascio di LH; i recettori
estrogenici sono espressi in una subpopolazione di neuroni ipotalamici
secernenti NPY (236),
suggerendo quindi, che gli estrogeni possano modulare in modo diretto la produzione di NPY. Studi in
vivo condotti su ratti femmine, durante la fase del proestro, (estradiolo e progesterone circolante
aumentano) hanno evidenziato un aumento dell’immunoreattività NPY-simile nel PVN, nell’area preottica
mediana dell’ipotalamo (MPOA) e nell’ARC (237). Inoltre, stati di deficit da estrogeni, ad esempio dopo
ovariectomia, sono associati ad un’aumentata espressione genica dell’NPY nell’ARC, che torna a
decrescere dopo somministrazione esogena di estrogeni. Comunque, nei ratti alti livelli di estrogeni sono
associati ad anoressia cronica ed a livello ipotalamico, ad un’aumentata biosintesi di NPY, suggerendo
un possibile coinvolgimento di altri sistemi neuronali (238, 239). Anche se non confermato nella scimmia
rhesus (240), alcuni autori hanno riportato che il testosterone, in roditori castrati, è in grado di modulare
l’attività dell’NPY, determinandone un decremento dell’espressione genica; tale effetto viene abolito dopo
somministrazione di testosterone (241, 242).
In molti tessuti, i retinoidi sono implicati nella regolazione di funzioni quali lo sviluppo e la differenziazione,
incluso il sistema nervoso. L’acido retinoico è stato recentemente coinvolto nella complessa regolazione
della termogenesi e del dispendio energetico, poiché è in grado di modulare le proteine UCP-1 e
l’espressione della leptina (243, 244). Inoltre studi in vitro condotti sulla linea cellulare di neuroblastoma
umano, SH-SY5Y, (245) hanno dimostrato che l’acido retinoico è in grado di indurre un marcato
decremento dell’espressione genica dell’NPY già dopo 3 e 6 ore di incubazione fino ad arrivare ad una
completa soppressione dopo 12 e 24 ore.
Ad oggi è chiaro che anche alcuni fattori di crescita, espressi nel cervello, regolano l’espressione genica
dell’NPY come dimostrato in vitro, in differenti linee cellulari neuronali (147). I fattori crescita coinvolti
sono: il nerve growth factor (NGF; nelle cellule di feocromocitoma di ratto, PC12) (246, 247), il basic
fibroblast growth factor (in aggregati di cellule neuronali di ratto) (248), l’insulin-like growth factor-I (in
aggregati di cellule neuronali di ratti) (249), il brain-derived neurotrophic factor (neuroni corticali di ratti)
(250), l’epidermal growth factor (251), il neurotrophic-3, -4, -5 factor (252), il leukemia inhibitory factor
40
(LIF) (253). Nelle cellule PC12, è stato anche dimostrato che il trattamento con NGF attiva, in modo dose
dipendente, il promotore del gene del recettore Y1. Poiché l’NPY è stato coinvolto nel controllo delle
funzioni cognitive e forse nella genesi delle patologie correlate, queste scoperte potrebbero essere
importanti anche alla luce delle terapie neuroprotettive per le patologie neurodegenerative (147). Infine,
nel ratto il CNTF, un’altra citochina pleiotropica gp130-simile, con azione anoressante molto forte,
potrebbe agire, in parte, attraverso una down-regulation, a livello ipotalamico, dell’espressione genetica
dell’NPY (254).
2.22 Effetti sul comportamento alimentare e sul metabolismo
NPY è il prototipo degli ormoni che stimolano l’appetito; in particolar modo si è osservato che il peptide
favorisce l’intake di carboidrati (255, 256), che diminuiscono il dispendio energetico e che inducono, nel
tessuto adiposo bianco, l’attività dell’enzima lipasi (257). Nel ratto la somministrazione dell’NPY a livello
centrale, ha un effetto positivo sul bilancio energetico, stimolando selettivamente l'assunzione dei grassi
e dei carboidrati e favorendo il deposito dell'eccesso energetico sotto forma di tessuto adiposo;
un’infusione di NPY, nel quarto ventricolo cerebrale, aumenta l’assunzione di cibo, in modo dosedipendente e con un effetto prolungato nel tempo (tempi sperimentali di 60 e 120 minuti (258). Sempre
nel ratto, l’infusione di NPY nel PVN, oltre ad incrementare l’assunzione di cibo, provoca nel tessuto
adiposo bruno, una diminuzione dell’espressione dell’mRNA codificante per le proteine UCP-1, ma non
quella delle proteine UCP-2 e UCP-3, che sono espresse nel tessuto adiposo bianco e nel muscolo
scheletrico. Le proteine UCP sono proteine mediatrici della termogenesi, quindi coinvolte nella
regolazione del metabolismo energetico; l’NPY agisce in modo specifico e differente sull’espressione di
tali proteine regolando quindi il bilancio energetico (259).
Oltre agli effetti a livello centrale (controllo dell’assunzione di cibo, modulazione del rilascio di ACTH, GH
e ormone luteinizzante) è stato proposto anche un possibile coinvolgimento dell’NPY a livello periferico:
modulazione della secrezione di leptina (260)
isole di Langerhans, aumento della produzione di glucosio epatico, riduzione della termogenesi nel
tessuto adiposo bruno e aumento dell'attività della lipasi lipoproteica.
L'insulina rappresenta un segnale periferico delle riserve energetiche, costituisce un fattore fisiologico di
sazietà e, a differenza dell’NPY, inibisce l'assunzione di cibo e aumenta la termogenesi, favorendo la
perdita di peso. È noto che l'insulina è in grado di raggiungere il sistema nervoso centrale e quindi
41
l'ipotalamo, ma non si sa ancora come interagisca con i neuroni NPY secernenti. I glucocorticoidi
sarebbero, invece, essenziali per l'effetto dell’NPY sull'assunzione di cibo (261); infatti stimolano il
sistema NPY-ergico e sono permissivi rispetto ai suoi effetti adipogenici. Nei ratti un eccesso di
glucocorticoidi promuove l’aumento corporeo di grasso, l’iperinsulinemia (262, 263) ed aumenta, a livello
ipotalamico, sia l’espressione genica dell’NPY che quella dei suoi recettori (217, 220). A conferma di ciò,
nei ratti un intervento di surrenalectomia corregge la sindrome da obesità indotta da somministrazioni
croniche
di
NPY,
inibendo
l’aumento
di
peso,
l’iperfagia,
l’iperinsulinemia,
l’iperleptinemia,
l’ipertrigliceridemia; tutti questi effetti vengono ripristinati in seguito a somministrazione di glucocorticoidi
(264, 265). Resta ancora da chiarire il meccanismo con cui questi ultimi siano in grado di influenzare il
sistema dell’NPY, a livello sia centrale che degli adipociti (266).
2.23 Regolazione dei meccanismi neuroendocrini
Nell'ambito del sistema neuroendocrino, l’NPY esplica la sua attività di modulazione sia a livello
ipotalamico che ipofisarico, attraverso il sistema portale ipotalamo-ipofisi. Tra i diversi meccanismi che
risentono dell’azione del neuropeptide Y, ricordiamo l'asse ipotalamo-ipofisi-gonadi, l'asse ipotalamoipofisi-surrene e la secrezione di GH. In diverse specie animali, l’NPY ha un ruolo importante sia nella
stimolazione del rilascio basale, che nell'induzione del picco preovulatorio dell’LH. L'effetto stimolatorio
dell’NPY sulla secrezione di LH dipende però dalla presenza degli ormoni gonadici e comporta
l'amplificazione della risposta di altri segnali stimolatori. A livello dell'eminenza mediana, l’NPY
interagisce con i recettori Y1 e determina il rilascio di LHRH con un meccanismo mediato dall'aumento
dei livelli di calcio intracellulare. Quest’azione risulta notevolmente amplificata in quanto l’NPY, raggiunta
l'ipofisi anteriore, tramite il circolo portale ipotalamo-ipofisi, aumenta il rilascio di LH in risposta alla
presenza di LHRH. I recettori coinvolti agiscono mediante una proteina G accoppiata alla PLC, la cui
attivazione determina un aumento della concentrazione di Ca ++, dovuta sia alla liberazione dal pool
intracellulare, che ad un ingresso dall'ambiente extracellulare, attraverso i canali voltaggio dipendenti di
tipo L (267). Studi effettuati su diverse specie animali (268) hanno dimostrato che l’NPY ha un duplice
effetto sulla stimolazione del rilascio di LH e che tale effetto dipende dalla presenza degli steroidi gonadici
(269).
In particolare, si è osservato che il trattamento di ratti femmine ovariectomizzate con estrogeni determina
nell'eminenza mediana una riduzione dei livelli di NPY; questo effetto viene soppresso se, in
42
corrispondenza del picco di LH, vengono somministrati dei progestinici (238). Nei ratti castrati, invece, la
somministrazione di testosterone determina, nell'eminenza mediana, un effetto opposto, cioè un aumento
dei livelli dell’NPY. Si ritiene che questo diverso comportamento sia dovuto al nucleo dimorfico, in cui la
secrezione basale di NPY è indotta dall'androgenizzazione post-natale (270).
L’NPY ha una funzione modulatoria anche a livello dell'asse ipotalamo-ipofisi-surrene, stimolando il
rilascio di ACTH. Questo effetto è stato osservato microiniettando l’NPY nel nucleo paraventricolare, dove
sono localizzati, principalmente, i neuroni secernenti CRH (271, 272) e a livello ipofisarico, dove per
azione diretta dell’NPY si ha un aumento del rilascio di ACTH in risposta a basse concentrazioni di CRH.
Opposta è, invece, l'azione sulla secrezione del GH, infatti, la somministrazione nel terzo ventricolo,
dell’antisiero anti-NPY, determina un aumento della concentrazione plasmatica di GH. Nei ratti è stato
anche dimostrato, che la somministrazione ICV di NPY inibisce l’asse somatotropo, riducendo il
contenuto ipofisario di GH (273), abolendone il suo normale rilascio pulsatile nel plasma, e quindi
riducendone la sua concentrazione plasmatica (274).
2.24 NPY: cancro della mammella e della prostata
Nel tessuto prostatico normale sono presenti sia cellule neuroendocrine (NE) che fibre nervose
peptidergiche secernenti serotonina e alcuni neuropeptidi quali la cromogranina A, la bombesina, il
vasoactive intestinal peptide (VIP) e l’NPY. Tali prodotti secretori, che sono presenti nel 50% dei casi di
cancro alla prostata analizzati, agendo localmente attraverso specifici recettori presenti sulle cellule
stromali ed epiteliali, regolano, in modo paracrino, le diverse funzioni cellulari (275-277) e contribuiscono
alla fisiopatologia del cancro e alla sua progressione verso uno stadio di androgeno indipendenza, che
viene generalmente associato ad una prognosi peggiore (278, 279).
L’NPY, nello strato muscolare liscio della prostata dell’uomo, è localizzato principalmente nelle fibre
nervose e nelle cellule neuroendocrine (277, 280); infatti un’immunoreattività NPY-simile è risultata
positiva nel 75% dei casi analizzati (281), suggerendo un suo possibile ruolo nella crescita e nella
progressione del cancro alla prostata. Inoltre a livello dell’innervazione neuropeptidergica della prostata e
più in generale del tratto urogenitale maschile, le fibre nervose immunoreattive all’NPY e alla tirosina
idrossilasi sono le più abbondanti e sono localizzate soprattutto nello strato muscolare liscio della
prostata, nelle vescicole seminali e nei vasi deferenti, mentre è nell’epitelio di questi stessi organi che
sono più che altro presenti quelle positive al VIP (280).
43
Per quanto concerne il cancro alla mammella, uno studio condotto da Reubi ha evidenziato che l’NPY
potrebbe avere un ruolo importante nella progressione di questa patologia. Infatti l’85% dei pazienti con
cancro della mammella presentano un’elevata espressione dei recettori dell’NPY; in particolar modo il Y1
è espresso ad alti livelli, mentre il sottotipo Y2 è presente solo nel 24% dei casi analizzati. Al contrario,
nel tessuto normale, il sottotipo Y2 è predominante rispetto al Y1; la trasformazione neoplastica potrebbe,
quindi, indurre un cambiamento di espressione recettoriale da Y2 a Y1 (282).
La presenza dei recettori per NPY, nei due tumori analizzati, indica che l’NPY potrebbe avere un ruolo
importante nella progressione di questi tumori, suggerendo che analoghi radiomarcati dell’NPY,
potrebbero essere utilizzati sia a scopo diagnostico che terapeutico.
44
3. LA SOMATOSTATINA
3.1 Aspetti molecolari
Il peptide ipotalamico a cui venne attribuito il nome di Somatotropin Release Inhibitory Factor
(somatostatina), oggi conosciuto con il termine di somatostatina (283), è un peptide presente in quasi tutti
i tessuti dei mammiferi, in tutte le classi di vertebrati, persino nelle piante e nei procarioti ed è coinvolto
nella regolazione di numerose funzioni cellulari (284). Uno dei più importanti e meglio documentati effetti
della somatostatina è rappresentato dall’inibizione di alcuni processi di secrezione a livello dell’ipofisi
come la liberazione del GH e del Thyroid Stimulating Hormone (TSH). A livello del sistema
gastroenteropatico, la somatostatina regola la secrezione di insulina, di glucagone, di gastrina, del vaso
intestinal peptide (VIP), della secretina, e delle secrezioni esocrine a livello dello stomaco e del pancreas;
è coinvolta nella neurotrasmissione, così come nel controllo delle funzionalità renali, dell’angiogenesi e
del sistema immunitario. Da non dimenticare la sua attività antiproliferativa a livello delle cellule tumorali
(285).
Il peptide somatostatina è costituito da una sequenza di 14 aminoacidi (SS14) con un ponte disolfuro che
collega la cisteina in posizione 3 (Cys 3) con la cisteina in posizione 14 (Cys 14). Questo ponte, unitamente
alla presenza della sequenza aminoacidica costituita dagli aminoacidi in posizione 7-10 (Phe7, Trp8, Lys9
e Thr10), è ritenuto indispensabile allo svolgimento dell’attività biologica (286). In natura è stata
identificata anche un’altra forma di somatostatina, anch’essa biologicamente attiva, costituita da 28
aminoacidi (SS28) e localizzata per la prima volta nel Tetrahymena pyroformis (un protozoo ciliato) (287).
La differenza tra SS14 e SS28 risiede nell’estensione a livello dell’estremità carbossilica, di una
sequenza di ulteriori 14 aminoacidi (287). Nell’uomo, un solo gene localizzato a livello del braccio lungo
del cromosoma 3, codifica per entrambe le forme; tale gene, costituito da due esoni separati da un
introne, codifica per un trascritto, che tradotto da origine alla pre-prosomatostatina (pre-proSS), un
precursore di 116 aminoacidi, che subisce un rimaneggiamento mediante l’eliminazione di un peptide
segnale, costituito da 24 aminoacidi. Dal clivaggio tra gli aminoacidi glicina e alanina (Gly 24 e Ala25) (288)
si ottiene, quindi, un prodotto intermedio costituito da 92 aminoacidi, il proSS-1 (289). La complessa serie
di eventi che determina le modificazioni post-trascrizionali dell’ormone è caratterizzata dall’intervento di
una endopeptidasi che provvede ad eliminare un frammento aminoterminale operando un taglio a livello
dell’arginina in posizione 64 (Arg64) e dando così origine alla SS28 e a un peptide detto “non
somatostatina” sul cui ruolo biologico non sono disponibili informazioni precise. Un secondo enzima
45
isolato dalle cellule della corteccia cerebrale di ratto detto “somatostatina-28” convertasi, scinde la SS28
e conduce alla formazione di SS14. In quei tessuti dove l’enzima non è espresso (apparato intestinale), la
SS28 risulta essere la molecola biologicamente attiva come tale (290). Il catabolismo enzimatico della
SS14 coinvolge almeno cinque siti della molecola; tale processo ha inizio con una degradazione
proteolitica da parte dall’enzima encefalinasi, una endopeptidasi specifica (definita endopeptidasi neutra
o neprilisina) (291, 292), che scinde il legame tra gli aminoacidi 8-9 (Trp8-Lys9), dando origine a dei
frammenti che non sono in grado come tali di interagire con i recettori, in quanto privi della porzione attiva
della molecola. I frammenti ottenuti sono ulteriormente degradati da aminopeptidasi quali le esopeptidasi
specifiche che tagliano il legame in posizione 1-2 (Ala1-Gly2) e in posizione 2-3 (Gly2-Cys3) (286, 292), la
degradazione continua con l’intervento di due ulteriori endopeptidasi, che agiscono a livello del legame
fenilalanina-fenilalanina e treonina-fenilalanina rispettivamente in posizione 6-7 (Phe6-Phe7) e in
posizione 10-11 (Thr10-Phe11) (293). In particolare, per l’attività biologica è critica la perdita del legame
localizzato tra gli aminoacidi Trp8-Lys9, tanto che l’attacco delle endopeptidasi in questa sede dà origine a
frammenti privi di attività. Questa osservazione ha permesso di proteggere tale posizione con
arrangiamenti chimici, al fine di ottenere analoghi della somatostatina più duraturi (286).
3.2 Localizzazione anatomica e caratteristiche delle cellule che la producono
Nel 1968 Krulich et al. (283) identificarono, in estratti ipotalamici di ratto, un peptide in grado di inibire la
liberazione dell’ormone del GH e approssimativamente nello stesso periodo (1969) Hellman e Lernmark
(294) trovarono in estratti di pancreas, di ratto, una componente inibente la secrezione di insulina. Nel
1973 Brazeau (295) determinò la natura chimica di tale sostanza a livello ipotalamico e nel 1975,
mediante studi di immunofluorescenza, fu dimostrato che le cellule D degli isolotti del Langherans
secernevano la stessa sostanza, cioè la somatostatina (296-298). L’identificazione delle cellule che
producono somatostatina, la definizione delle loro caratteristiche strutturali e del loro significato
funzionale sono state il risultato di una lunga serie di ricerche e dell'impiego di tecniche istologiche
tradizionali, tecniche di microscopia elettronica, di immunoistochimica e di ibridazione in situ. Questi studi
hanno dimostrato che la somatostatina è prodotta dalle cellule D, degli isolotti del Langherans,
caratterizzate da un reticolo endoplasmatico rugoso relativamente sviluppato e da granuli osmiofili avvolti
da una membrana, di dimensioni comprese tra 200-400 nm. E’ risultato anche evidente che sono cellule
non segregate negli isolotti di Langherans, ma disseminate tra la componente esocrina del pancreas, sia
46
nell’ambito della porzione secernente acinosa, che nel contesto dei dotti (299). La collocazione periferica
si accorderebbe con una sorta di organizzazione funzionale dell’isolotto in due porzioni ben distinte: una
corticale, eterocellulare, che vede la coesistenza di cellule A (produttrici di glucagone), B (produttrici di
insulina), D (produttrici di somatostatina), ricca di vasi e di nervi, più suscettibile a rapide modificazioni
dell’attività secretoria, in rapporto a stimoli ambientali, ed una midollare, più centrale e omogenea dal
punto di vista citologico, in quanto in essa sono rappresentate, soprattutto, le cellule B. La posizione delle
cellule D potrebbe pertanto essere correlata alla funzione esercitata dalla somatostatina, a carico della
secrezione di insulina e glucagone (300, 301). Per quanto concerne l’apparato gastroenterico, la
somatostatina è stata trovata nelle cellule D dell’antro pilorico, del duodeno, dell’ileo e del grosso
intestino; quindi, le cellule D della mucosa ossintica mediano gli stimoli provenienti dai vasi o dai nervi,
senza avere alcun rapporto con il contenuto dello stomaco (299).
Ancora una volta l’immunofluorescenza ha permesso di individuare cellule che producono somatostatina
anche nella tiroide (298), a livello delle cellule parafollicolari e delle cellule D (302); tecniche di
immunoistochimica e di immunoelettronmicroscopia hanno inoltre evidenziato che nelle stesse cellule e
negli stessi granuli possono coesistere sia la calcitonina, che la somatostatina. A conferma di ciò, uno
stimolo funzionale, quale l’ipercalcemia, mobilizza la somatostatina dalle cellule parafollicolari insieme
alla calcitonina (303). La coesistenza di questi due ormoni suggerisce che la somatostatina funziona
come messaggero sia paracrino (influenza l’attività delle cellule vicine), che autocrino (regola la sua
stessa liberazione dalle cellule di origine) (302).
Nell’apparato respiratorio, la somatostatina è stata localizzata nel corso dello sviluppo, a livello dei
neuroni della tonaca muscolare e della sottomucosa dei bronchi, della muscolatura dei bronchioli
respiratori, dei dotti alveolari e dei tronchi nervosi o dei gangli dei bronchi principali, ma non nelle cellule
neuroendocrine (303). Nell’uomo adulto invece la somatostatina è assente nei tronchi nervosi dei bronchi
principali, nella sottomucosa bronchiolare e nei dotti alveolari (304). Un’immunoreattività SS14 e SS28
simile è stata anche riscontrata nelle popolazioni neuronali di tutto il sistema nervoso (305); in modo
specifico nella corteccia cerebrale (49%), nel peduncolo cerebrale (12%), nell’ipotalamo (7%), nel lobo
olfattorio (1%) (306, 307). I neuroni che contengono la somatostatina, a seconda della loro localizzazione
sono di due differenti tipi: interneuroni coinvolti in circuiti locali, o neuroni provvisti di lunghe proiezioni che
connettono regioni anatomiche distinte. Il corpo cellulare è ovale o rotondeggiante e le estensioni
citoplasmatiche sono spesso ramificate; i granuli presenti nei neuroni, sia a livello del soma che dei
47
prolungamenti cellulari, hanno un diametro di 80-110 nm, quindi più piccoli rispetto a quelli contenuti nelle
cellule D, ma più grandi rispetto alle vescicole che contengono altri tipi di peptidi (308). Da notare che a
livello neuronale la somatostatina coesiste con i classici neurotrasmettitori, quali la noradrenalina (nei
neuroni post-gangliari del ganglio mesenterico inferiore), il GABA (in una sottopopolazione di neuroni
corticali), l’NPY (nei neuroni corticali) e l’encefalina (nell’eminenza mediana) (309). Inoltre in neuroni
corticali cerebrali, in coltura, la somatostatina è stata anche colocalizzata con l’acetilcolinesterasi (310).
Nell'ipotalamo la maggior parte dei corpi cellulari neuronali, positivi alla somatostatina, sono collocati in
vicinanza del terzo ventricolo, in tre o quattro strati che sono paralleli alla parete ventricolare; gli assoni
che derivano da queste cellule si portano caudalmente attraverso l’ipotalamo ed entrano nell'eminenza
mediana a livello del nucleo ventromediale (305, 311, 312). Parte delle fibre si estendono alla zona
neurovascolare del peduncolo ipofisario e sono, verosimilmente, coinvolte nella liberazione della
somatostatina nel sistema portale ipofisario.
Il peptide somatostatina è localizzato anche nelle fibre nervose, a livello di altri organi: nel fegato si trova
nelle fibre nervose che arrivano ai vasi sanguigni (313), nell’apparato genito-urinario, nel cuore e nella
cute è situato nelle fibre nervose associate alle fibre muscolari (314). La somatostatina è stata rilevata
anche nei fluidi biologici; infatti sia la forma SS14 che la SS28 sono presenti a livello ematico mentre il
tratto gastrointestinale rappresenta la maggior fonte di somatostatina circolante. L’ormone è secreto nel
liquido celebro-spinale e viene escreto nell’urina; anche il liquido seminale ne contiene alti livelli e
altrettanto ricco ne è il liquido amniotico (315, 316). La forma a 14 amminoacidi, ma non quella a 28,
viene attivamente trasportata nel latte materno dove la sua concentrazione è quattro volte più elevata
rispetto a quella del plasma (317, 318). Nella donna la massima concentrazione di somatostatina si
riscontra il primo giorno dopo il parto, mentre al quinto giorno il suo livello scende e si mantiene su di un
“plateau”. Nel neonato, la proteina presente nel latte resiste alla degradazione gastroenterica, dopodiché
raggiunge la porzione distale dell’intestino nel quale può svolgere delle funzioni di regolazione (319).
Le cellule che producono somatostatina, in particolare le cellule D, sono presenti durante la vita fetale. In
alcuni organi come il pancreas e il colon, il loro numero è più elevato rispetto alla vita adulta (320-322),
quindi si suppone che esse svolgano, durante la vita fetale, delle particolari funzioni (322), che
successivamente tendono a scomparire o vengono semplicemente ridimensionate. Nel feto umano le
cellule D sono individuabili a partire dall'ottava settimana, con una distribuzione non uniforme nei vari
organi; dalla quindicesima alla ventunesima settimana, il contenuto di somatostatina, a livello del
48
pancreas e del duodeno, è più elevato rispetto alle altre regioni del tratto gastrointestinale (323); dopo 5
mesi di gestazione, sulla sommità dei villi duodenali e del digiuno è possibile identificare estesi gruppi di
cellule contenenti granulazioni con elementi secernenti somatostatina (324); a livello ipotalamico
l’immunoreattività è invece scarsa (323).
3.3 Attività biologiche
L'ubiquitarietà della distribuzione anatomica della somatostatina si associa alla molteplicità dei suoi
organi bersaglio o, più precisamente, delle cellule la cui funzione specifica viene da essa regolata. La
possibilità di estrinsecare l'attività biologica è legata alla liberazione della somatostatina nello spazio
interstiziale o nel torrente circolatorio. L'influenza di questo peptide liberato dalle terminazioni nervose nel
sistema nervoso centrale e periferico o dalle cellule D del tratto gastroenterico si manifesta
sostanzialmente attraverso il liquido interstiziale (neurocrinia, paracrinia). Per quel che concerne invece la
liberazione da parte dei neuroni ipotalamici nel sistema portale ipofisario o la regolazione della
secrezione delle cellule A e B degli isolotti di Langerhans, viene chiamata in causa l'azione endocrina. Il
fatto che la somatostatina sia presente nella circolazione sistemica rende possibile una sua più ampia
influenza nell'ambito di uno stesso organo, un'azione simultanea su differenti organi e un'integrazione
degli effetti su uno o più tessuti bersaglio da parte dei differenti organi che la producono (325). A livello
cellulare la somatostatina interagisce con cinque differenti recettori (sst1, sst2, sst3, sst4 e sst5)
inducendo un segnale transmembrana che è alla base di effetti biologici quali la regolazione dei processi
di secrezione e la crescita cellulare (326, 327, 328).
3.4 Azioni a livello dei sistema nervoso centrale e periferico
Il profilo di espressione della somatostatina e dei suoi recettori, durante lo sviluppo, suggerisce il suo
coinvolgimento nella maturazione del sistema nervoso, cosi come, una sua diminuzione selettiva a livello
corticale, in età avanzata, fa ipotizzare un possibile ruolo trofico nel cervello (329). Alterazioni della
somatostatina sono associate a malattie del sistema nervoso centrale; infatti in soggetti affetti sia dal
morbo di Alzheimer, che dal morbo di Parkinson si riscontra una diminuzione di somatostatina nella
corteccia cerebrale e nel liquido cerebrospinale, così come nei soggetti depressi o epilettici (285). Nel
cervello, la somatostatina controlla la temperatura, il senso della fame e della sazietà, la percezione del
dolore, il sonno e la memoria. Studi in vivo hanno mostrato che nel ratto, iniezioni intracerebrali di
49
somatostatina producevano un aumento dell'attività motoria, disturbi del sonno e, in qualche caso,
voracità (330, 331); quelle intracerebroventricolari determinavano sia un effetto analgesico (interazione
con i recettori degli oppioidi o inibizione dei neuroni nocicettivi) (332), che una diminuzione, da un lato,
della secrezione di ACTH, del battito cardiaco, del livello plasmatico di adrenalina e di glucosio,
dell'attività motoria spontanea, mentre dall’altro un aumento dell'appetito, del consumo di ossigeno e
della secrezione gastrica (333). Da notare che gli esperimenti condotti sugli animali hanno evidenziato il
manifestarsi, accanto all’attività analgesica della somatostatina, anche degli effetti neurotossici legati alle
dosi del peptide impiegate ed alle modalità di somministrazione. A supporto di ciò, Gaumann e Yaksh
(334) evidenziarono che, nei ratti, le disfunzioni motorie successive all'iniezione intratecale di
somatostatina erano dose-dipendenti, mentre il blocco della percezione del dolore variava. L'esame
istologico indicava nucleolisi delle corna dorsali e ventrali, senza margini di sicurezza tra l'effetto
nocicettivo e quello tossico. Effetti simili sono stati osservati anche da Leblanc e Dirksen et al. (335, 336)
i quali riportarono che, nel ratto, gli effetti tossici, dovuti all'iniezione intratecale di somatostatina, erano
correlati alla dose e all'intervallo di somministrazione, laddove la capacità antinocicettiva risultava più
elevata se tale intervallo era breve. Per quel che riguarda studi sull'uomo, nel 1984 Chrubasik (337)
osservò in 8 pazienti terminali e in altri con dolore post-operatorio, ai quali era stata somministrata (via
intraventricolare, intratecale o epidurale) somatostatina (10-125 μg/ora), una riduzione significativa del
dolore; tale effetto non veniva annullato dagli antagonisti degli oppiodi (338).
La somatostatina, a livello del centro respiratorio situato nel midollo allungato, controlla la respirazione;
se iniettata nei ventricoli cerebrali dei mammiferi, induce apnea che può essere antagonizzata dalla somministrazione del naloxone, un antagonista degli oppioidi, suggerendo che ci sia una possibile interazione
tra la somatostatina e l’encefalina, entrambe liberate a livello delle terminazioni nervose (339). La
somatostatina regola anche la pressione arteriosa, agendo sul sistema simpatico e dando un effetto
ipotensivo (340), al quale può contribuire anche una diminuita secrezione di adrenalina da parte della
midollare del surrene (340, 341). La somatostatina non solo influenza l'attività elettrica spontanea dei
neuroni (329), ma interferisce, a livello del sistema nervoso centrale, con la liberazione di altri
neurotrasmettitori inclusi la dopamina, la 5-idrossitriptamina e la noradrenalina (342).
3.5 Regolazione delle funzioni ipotalamo-ipofisi
50
La somatostatina prodotta a livello ipotalamico, regola nell’ipofisi sia la secrezione del GH che quella del
TSH. L'effetto di tipo inibitorio è mediato dai neuroni somatostatinergici ipotalamici che indirizzano le loro
proiezioni verso l'eminenza mediana. Nel ratto, se questa via viene interrotta, si osserva un aumento
della secrezione del GH e del TSH; al contrario, se il sistema viene stimolato elettricamente, si assiste ad
un potenziamento dell'inibizione della secrezione del GH e del
TSH (343). Studi relativi
all'immunizzazione passiva verso l’ormone somatostatina hanno prodotto, come risultato, l'ipotesi che la
secrezione basale del GH sia sotto il controllo tonico della somatostatina, che blocca la liberazione del
GH in risposta al GHRH. Quest’ultimo è prodotto dai neuroni situati nel nucleo arcuato, i cui assoni si
proiettano anch'essi verso l'eminenza mediana, legandosi a specifici recettori di membrana. La
susseguente attivazione dell’enzima adenilato ciclasi determina un aumento di cAMP, che innesca una
catena di eventi che si traducono nella neosintesi di GH e nella sua liberazione da un pool preformato,
con il coinvolgimento di canali ionici di membrana (344). In particolar modo, la somatostatina inibisce la
liberazione del GH dal pool preformato, mentre è meno efficace nell’inibire la sintesi ex novo (345).
L'interazione tra GHRH e somatostatina si verifica non solo a livello ipofisario, ma anche attraverso
connessioni dirette nell'ipotalamo (346); pertanto, la somatostatina inibisce la liberazione del GH sia
mediante un'azione diretta sull'ipofisi, che attraverso una soppressione del GHRH, che a sua volta
modula la secrezione stessa della somatostatina. Quest’ultima subisce un "feedback" positivo dal GH e
dall’IGF-1 prodotto dal fegato (347, 348), così come dal glucagone e dalla bombesina, al contrario il
GABA e il glucosio inibiscono le cellule somatostatinergiche (349). Anche gli ormoni sessuali controllano i
livelli di somatostatina circolante, ad esempio, il testosterone li innalza, mentre gli estrogeni ne
determinano una diminuzione (350).
La secrezione del GH è pulsatile con picchi di secrezione durante le 24 ore; questo tipo di secrezione è
regolato proprio dall’effetto sinergico somatostatina/GHRH, che vengono liberati nella circolazione portale
in maniera ciclica. La somatostatina, di provenienza gastrointestinale, che raggiunge l'ipofisi, mediante la
circolazione sistemica, probabilmente non è determinante per la secrezione del GH, in quanto i livelli di
somatostatina, nel sangue periferico, sono molto più bassi rispetto a quelli del sangue portale ipofisario
(351). Nei soggetti normali, le altre funzioni ipofisarie come la secrezione di FSH, di LH, di ACTH e di
PRL non sembrano essere influenzate, anche se, in colture primarie di ipofisi fetale, gli analoghi selettivi
per il recettore sst2, inibiscono la secrezione di PRL (352). Inoltre, in soggetti affetti dal morbo di Addison
51
o con tumori ACTH-secernenti, la somatostatina riduce gli elevati livelli di ACTH, così come nei soggetti
acromegalici riduce quella di PRL (343).
La somatostatina modula anche la secrezione del TSH interagendo con il TRH a vari livelli: inibisce
l’aumento del calcio intracellulare, regola il numero di recettori per il TRH a livello ipofisario (353) e riduce
direttamente la liberazione del TRH a livello ipotalamico (354). Nei soggetti ipotiroidei, l’infusione di
somatostatina determina una riduzione significativa dei livelli di TSH, abolendo l’aumento notturno di TSH
(355).
3.6 Regolazione endocrina ed esocrina del pancreas
La somatostatina è in grado di controllare la funzione pancreatica sia a livello degli isolotti di Langherans,
sede della secrezione endocrina, che a livello degli acini pancreatici, dove vengono prodotti enzimi
digestivi come l’amilasi, la lipasi e la tripsina. La microarchitettura vascolare degli isolotti fa sì che il flusso
sanguigno dal centro dell’isolotto si porti alla periferia (356, 357) in modo che l’insulina prodotta dalle
cellule B regoli (in senso negativo) la secrezione del glucagone (cellule A) e della somatostatina (cellule
D), che a sua volta, in base alla direzione del flusso ematico, influenza sostanzialmente le funzioni del
tessuto esocrino perinsulare; al contrario le cellule A e B possono essere soggette al controllo paracrino
da parte del peptide e/o a controllo endocrino da parte della somatostatina presente a livello del torrente
circolatorio (357). Questo ormone svolge un’azione di tipo autocrino sulle cellule D, influenzando anche
l’attività delle cellule che producono il peptide PP (325). La secrezione di insulina e glucagone è ridotta
dall’aumento dei livelli di SS14 e SS28; in particolare la SS14 è dieci volte più potente nell’inibire la
secrezione del glucagone rispetto alla SS28, laddove quest’ultima è più efficace nell’inibire la secrezione
di insulina (358). Le cellule D pancreatiche dei mammiferi producono soprattutto SS14, mentre le cellule
D dell’intestino, dopo l’ingestione di cibo, producono SS28 che raggiunge nel plasma alte concentrazioni
e va a modulare la secrezione delle cellule B. Inoltre la SS28 inibisce la secrezione degli enzimi
pancreatici e del bicarbonato, riducendo il riempimento della cistifellea. L’azione a livello pancreatico
potrebbe quindi essere mediata dall’interazione con i gangli situati nel contesto dell’organo (325, 359).
3.7 Regolazione delle funzioni gastriche, intestinali ed epatiche
Studi condotti nel cane dimostrano che, a seguito dell'ingestione di un pasto, la somatostatina liberata dal
fondo dello stomaco, dall'antro pilorico e dall'intestino tenue è associata al suo aumento in circolo (360).
52
Anche in questo caso la modalità d'azione della somatostatina può essere duplice: un classico
meccanismo endocrino e la via paracrina (325, 360). La composizione chimica del cibo sembra ricoprire
un ruolo primario, rispetto alla distensione delle pareti gastriche, nella liberazione di somatostatina; a
livello dell’antro pilorico, lo stimolo principale è invece rappresentato dall’acidità della secrezione gastrica
(361, 362). La somatostatina nello stomaco stimola la secrezione mucosa, riduce quella di pepsinogeno,
del fattore intrinseco e dell’acido cloridrico, in modo sia diretto, che attraverso la soppressione della
secrezione di gastrina (363, 364). Infusioni di somatostatina riducono drasticamente la secrezione acida
dello stomaco sia nell'uomo che nel cane (365). La somatostatina inibisce altresì la secrezione
dell'istamina, dell'acetilcolina, potenti secretagoghi dell'acido cloridrico (366, 367), così come quella dei
peptidi correlati alla bombesina, che stimolano, a loro volta, la secrezione della gastrina (368). Quindi,
controllando la secrezione della gastrina, la somatostatina è in grado di regolare anche l'effetto trofico
esercitato sulle cellule enteroendocrine (369). La liberazione della gastrina è, a sua volta, inibita dall'acido
cloridrico, che stimola successivamente la produzione di somatostatina (370). Quest’ultima è coinvolta
non soltanto nel "feed-back" intragastrico tra acido cloridrico e gastrina, ma anche nel meccanismo denominato "bulbogastrone" e cioè il "feedback" duodeno-gastrico. Durante l'acidificazione del bulbo
duodenale si ha un'inibizione della funzione delle cellule parietali e una liberazione di somatostatina dalle
cellule D delle ghiandole fundiche, con un conseguente aumento della concentrazione plasmatica (371).
Alcuni ormoni, liberati a livello intestinale, quali la secretina e la motilina, mediano questo "feedback"
stimolando la secrezione di somatostatina sia dalla parete gastrica, che da quella intestinale; l’eccesso di
somatostatina inibisce la produzione di acido cloridrico (372). La somatostatina ritarda inoltre il ritmo di
riempimento dello stomaco (fase tardiva), inibisce la motilità intestinale (attraverso la modulazione della
liberazione di acetilcolina dipendente dal cAMP), regola il flusso sanguigno a livello enterico, la
proliferazione e la secrezione delle cellule della mucosa, inclusa quella neuroendocrina, nonché
l'assorbimento intestinale (325, 373). I livelli basali di somatostatina e la motilità intestinale fluttuano
durante il periodo tra due digestioni confermando il nesso esistente tra il peptide e la peristalsi intestinale
(374).
Nel fegato, la SS14 fa diminuire la secrezione di bile del 30%, deprime l'output di acidi biliari, colesterolo,
fosfolipidi, Na+, Cl- e K+ e riduce dal 15 al 30% il flusso ematico epatico (375).
3.8 Somatostatina e controllo dell’omeostasi nutrizionale
53
Un'assunzione incontrollata di nutrienti dall'intestino al sangue, potrebbe avere delle conseguenze
negative sull'omeostasi del “milieu interieur”. Infatti se ai vari nutrienti fosse consentito di entrare
liberamente nella circolazione, la loro concentrazione nel sangue si innalzerebbe in maniera abnorme e
prolungata causando una concentrazione intracellulare degli stessi non fisiologica, con un'attivazione di
vie metaboliche altrimenti inattive e l'accumulo di cataboliti con gravi disturbi metabolici. È pertanto
necessario che si realizzi un equilibrio tra l'assunzione delle sostanze a livello intestinale nella
circolazione ed il loro efflusso nei tessuti e nelle cellule (376). L'equilibrio si realizza, come sempre, per
un sottile gioco tra sostanze che stimolano e sostanze che inibiscono la digestione. Tra queste, la
somatostatina assume un ruolo di grande importanza, regolando le funzioni endocrine ed esocrine dello
stomaco, dell'intestino e del pancreas e la motilità del tubo gastroenterico, in parte mediante un
meccanismo di tipo endocrino, in parte mediante la via paracrina. Dopo l'ingestione di un pasto, la
somatostatina viene liberata dallo stomaco, dall'intestino tenue e dal pancreas, determinando una serie di
effetti, il cui risultato finale è dato da un'attenuazione dell'apporto di nutrienti dall'intestino alla
circolazione. Nel cane, uno stato di iposomatostatinemia, fa aumentare il livello post-prandiale dei
trigliceridi, laddove l’infusione intraportale del peptide riduce sia il livello dei carboidrati che dei trigliceridi
(325). Nell’uomo, infusioni endovenose di somatostatina fanno ridurre il livello basale e postprandiale di
insulina. Dal momento che l'insulina è un potente inibitore della somatostatina, l’iposomatostatinemia
potrebbe essere secondaria all’iperinsulinemia; d'altro canto essa potrebbe causare un aumento dei livelli
di insulina e quindi dell'assunzione di nutrienti nei soggetti obesi (325). I soggetti obesi presentano livelli
di somatostatina ridotti e virtualmente non hanno "somatostatin response" dopo il pasto, il che è
associato ad iperinsulinemia (377). Infine, la SS14 inibisce l'assorbimento del glucosio, dei grassi e degli
aminoacidi (325).
3.9 Somatostatina: modulazione delle funzioni renali, polmonari e del sistema immunitario
La somatostatina regola le funzioni renali sia agendo direttamente a livello del tubulo renale, sia
indirettamente influenzando le concentrazioni plasmatiche di ormoni vasoattivi coinvolti nella regolazione
del tono vascolare e nell'omeostasi del sodio e dell'acqua. È stato dimostrato, in esperimenti condotti in
volontari sani, che essa riduce il flusso plasmatico renale, il volume delle urine, l'escrezione urinaria di
sodio e potassio, il riassorbimento dei fosfati, ma aumenta l'osmolalità delle urine (378). La somatostatina
non modifica la concentrazione del fattore natriuretico atriale, della vasopressina e delle catecolamine,
54
ma induce una diminuzione dei livelli di renina mediante la soppressione della stimolazione della
corteccia surrenalica da parte del sistema renina-angiotensina (342). Effetti vasodinamici simili si
osservano nei diabetici e nei cirrotici, il che può rivestire un certo interesse nel trattamento di tali soggetti
(379).
Per quanto riguarda la corticale del surrene, esistono buone evidenze del fatto che la somatostatina
presente in circolo possa interagire con le cellule della glomerulare del surrene, modulando così la
produzione di aldosterone stimolata dall’angiotensina (343).
La somatostatina è stata estratta dal tessuto polmonare, per la prima volta, nel 1981 (380); si ritiene che
essa possa avere un ruolo nello sviluppo dell'organo, in quanto è stata localizzata, in feti di scimmie
rhesus, in alcuni corpi neuroepiteliali polmonari (381). In colture d'organo allestite in un mezzo di coltura
privo di siero e addizionato di fattori di crescita, la somatostatina determina l'allungamento e la
ramificazione delle porzioni distali delle strutture tubulari polmonari prelevate da feti di animali da
esperimento (382). A livello del polmone, la somatostatina viene degradata da almeno tre enzimi:
un'endopeptidasi, un'aminopeptidasi e un enzima tripsino-simile (383). Quest'attività potrebbe essere
relativa alla somatostatina prodotta localmente o rappresentare una sorta di meccanismo di riserva che
subentra allorché il metabolismo normale del peptide viene alterato da stati patologici (342).
La somatostatina influenza le funzioni immunitarie attraverso vari meccanismi che comprendono la
modulazione della produzione di immunoglobuline, la proliferazione dei linfociti, la citotossicità, la
produzione di citochine, la liberazione di mediatori dai basofili, il reclutamento degli eosinofili e la funzione
dei macrofagi. Inoltre essa inibisce la produzione di IgA e, in misura minore, di IgM da linfociti isolati dalle
placche di Peyer, dai linfonodi mesenterici e dalla milza (342). Per quel che concerne i linfociti e le cellule
linfoidi, la somatostatina ha un effetto bifasico sulla proliferazione spontanea e indotta: a basse
concentrazioni la inibisce e ad alte concentrazioni la stimola; questo tipo di risposta potrebbe essere
mediata da differenti recettori. La somatostatina riduce l'attività "natural killer" nei linfociti delle placche di
Peyer e in quelli splenici (384) e inibisce la produzione di linfotossina e del TNF dalle cellule periferiche
mononucleate, nonché, a dosi soprafisiologiche, di interferon γ (385, 386). La somatostatina sopprime la
liberazione di istamina indotta da IgE e di leucotrieni nei basofili e nei mastociti, ma a concentrazioni
elevate, stimola la liberazione di istamina dagli stessi mastociti, potrebbe, pertanto, avere un ruolo nelle
reazioni di ipersensibilità (387, 388). Per quel che concerne i macrofagi, la somatostatina abolisce la
risposta tumoricida dei macrofagi a dosi superiori a 1 nM, abolendo la chemiotassi e l’attività fagocitaria di
55
queste cellule (389, 390). I linfomi maligni ed i campioni tissutali dei pazienti affetti da tubercolosi e
sarcoidosi esprimono recettori per la somatostatina, mentre nelle malattie granulomatose i recettori
scompaiono dopo trattamento con corticosteroidi (391).
3.10 I recettori per la somatostatina
Le diverse azioni fisiologiche della somatostatina sono mediate dall'interazione con specifici recettori in
grado di legare l'ormone con elevata affinità e selettività. Gli studi di caratterizzazione molecolare hanno
permesso di identificare almeno 5 sottotipi di recettori per la somatostatina (sst), numerati da 1 a 5 in
base all'ordine nel quale essi sono stati identificati; strutturalmente sono simili pur avendo differenti
caratteristiche farmacologiche. Sia nell'uomo che in altre specie animali, tali recettori sono codificati da
una famiglia di 5 geni non allelici localizzati su cromosomi differenti (392, 393). I geni che codificano per
le varie classi di sst mancano in genere di introni, ad eccezione del gene sst2 che contiene un introne
criptico, che dà origine a due isoforme recettoriali, sst2A e sst2B, le quali differiscono tra loro per la
lunghezza della coda carbossiterminale intracellulare, composta rispettivamente da 369 e 346 aminoacidi
(394). Nella specie umana, i recettori sst pur variando nella composizione aminoacidica da 359 (sst2B) a
418 amminoacidi (sst3), hanno 105 aminoacidi che risultano conservati filogeneticamente. Dal punto di
vista strutturale, essi appartengono, come per l’NPY, alla famiglia dei recettori accoppiati alle proteine
eterotrimeriche ad attività GTPasica (proteine G). Tali recettori di natura glicoproteica mostrano la
porzione N-terminale in sede extracellulare e presentano una struttura a "serpentina" attraverso la
membrana plasmatica, così da formare sette domini transmembrana, con tre anse extracellulari, tre anse
intracellulari e una coda C-terminale intracellulare (395). Sia nel dominio N-terminale che nella seconda
ansa extracellulare, sono presenti dei residui di asparagina (Asn) soggetti ad ammino glicosilazione; la
presenza di tali siti giustifica il ruolo svolto dagli oligosaccaridi nella funzionalità dei recettori. È risaputo
infatti, che la componente in carboidrati è importante nel determinare il legame ad alta affinità della
somatostatina naturale e dei suoi analoghi (396). La natura e l'estensione della componente in carboidrati
è variabile tra le 5 sottoclassi di recettori ed è il recettore sst2 a presentarsi come quello maggiormente
glicosilato.
I recettori sst possiedono, inoltre, diversi siti di fosforilazione tra la seconda e la terza ansa citoplasmatica
e nella regione carbossiterminale intracellulare. I sottotipi sst1, sst2, sst4 e sst5, ma non il sottotipo sst3,
contengono dei residui conservati di cisteina, localizzati 12 aminoacidi a valle del settimo dominio
56
transmembrana, che fungono da siti di palmitoilazione. La presenza di residui di acido palmitoilico
permettono l'aggancio della catena carbossiterminale del recettore alla membrana; ciò induce una
particolare conformazione della molecola rendendo inaccessibili quei siti che, una volta fosforilati, portano
a desensitizzazione del recettore stesso (397). I recettori sst mostrano alcune proprietà strutturali tipiche;
infatti possiedono delle sequenze caratteristiche che potremmo definire "un marcatore specifico"
dell'intera famiglia; tali sequenze comprendono un sito di fosforilazione Arg-Ser-Glu a livello della terza
ansa intracellulare e la sequenza Leu-Tyr-Ala-Asn-Ser-Cys-Ala-Asn-Pro-Ile-Leu-Tyr (LYANSCANPILY)
presente nel settimo dominio transmembrana dei recettori sst1, sst2, sst3 e sst4. Il recettore sst5 invece,
presenta un residuo di valina al posto dell’isoleucina (393). I domini transmembrana rappresentano le
zone recettoriali più conservate, con un grado di identità tra le sequenze aminoacidiche dei vari sottotipi
compreso tra il 46 e il 61% (398, 399); la maggiore identità di sequenza è presente fra i recettori sst1 e
sst4 (61%), così come tra sst3 e sst5 (59%). In particolar modo, basandosi anche sul profilo
farmacologico dell’interazione degli analoghi sintetici della somatostatina, è possibile suddividere gli sst in
due sottoclassi, la prima costituita da sst1 e sst4 (bassa affinità per gli analoghi) e la seconda da sst2,
sst3, sst5 (alta affinità per gli analoghi) (400).
Il raffronto tra le sequenze aminoacidiche degli sst ha mostrato una buona similarità (82-96%) tra quelli
presenti nell'uomo, nel ratto, nel topo e nel maiale. La sequenza di sst1 risulta essere quella
filogeneticamente più conservata, con il 97% d’identità tra la proteina umana e quella di ratto (398, 399).
Tra i recettori a sette domini transmembrana finora identificati, i recettori per gli oppioidi risultano essere i
più simili a quelli sst, con un'analogia strutturale attorno al 37%; infatti alcuni analoghi della somatostatina
sono in grado di legare i recettori degli oppioidi (401) e nella linea cellulare T47D, di carcinoma
mammario, la morfina è in grado di interagire con i recettori sst2, determinando una riduzione della
crescita cellulare. In genere, il dominio di legame dei recettori a sette domini transmembrana comprende
una serie di aminoacidi non contigui, presenti nei domini transmembrana e nel dominio N-terminale, che
formano una specie di “tasca” all’interno della struttura del recettore. Da qui scaturisce l’ipotesi che gli
analoghi sintetici della somatostatina simili all’octreotide interagiscano con i recettori sst in una cavità di
legame formata da residui idrofobici, con una forte carica elettrica (Asn 276 e Phe294 in sst2), localizzati
esclusivamente tra i domini transmembrana III e IV. La presenza dei residui Gln 291 e Ser305 sulla parte
esterna dei domini transmembrana VI e VII di sst1 potrebbe quindi prevenire l’ingresso dell’analogo in
questa cavità. I ligandi naturali SS14 e SS28, essendo più lunghi e flessibili, rispetto agli analoghi
57
sintetici, potrebbero assumere una conformazione che permetterebbe loro di entrare nella cavità di
legame di tutti e cinque i recettori sst, oppure potrebbe interagire con un altro epitopo dei recettori stessi
(402, 403). Greenwood (404) mediante esperimenti di mutagenesi e di delezione genica, ha dimostrato il
coinvolgimento della seconda ansa extracellulare (ma non della prima e della terza, così come del
dominio N-terminale) nell’interazione della somatostatina e dell’octreotide, con il recettore sst5 umano,
quindi, il dominio di legame ai recettori sst coinvolge i domini transmembrana da III a VII con il contributo
della seconda ansa extracellulare. Sia SS14 che SS28 legano tutti i sottotipi recettoriali, anche se SS14
mostra una selettività inferiore per i recettori sst1 e sst4 e SS28 appare più selettiva per il recettore sst5.
Non sembrano invece esistere differenze nelle proprietà di legame alle sottoclassi sst2A e sst2B (405).
3.11 Distribuzione
I dati sulla distribuzione dei recettori della somatostatina nel SNC sono stati ottenuti principalmente nei
roditori, dimostrando come essi siano espressi in modo distinto, ma talvolta sovrapposto. A livello del
SNC del ratto sono in genere espresse tutte e cinque le sottoclassi sst. In particolare studi di
localizzazione cellulare hanno evidenziato la loro presenza a livello del corpo cellulare e dei terminali
assonici e dendritici (406). Le maggiori concentrazioni sono rilevabili a livello della corteccia, del nucleo
striato, dell’ippocampo, dell’amigdala, del bulbo olfattivo e dell’area preottica (398). Analizzando i livelli di
espressione dei singoli sottotipi, è possibile osservare una maggiore espressione di sst1 e sst2 a livello
della corteccia, di sst1 e sst3 a livello dell’amigdala e di sst3 e sst5 a livello dell’ipotalamo-area preottica;
in genere il recettore sst4 è poco espresso nel SNC (407). Paradossalmente, nel cervelletto si trovano
elevati livelli di mRNA per il recettore sst3 associati però ad un ridotto contenuto di proteina recettoriale
(408). La presenza di mRNA per i recettori sst1, sst2, sst3 e sst4 in aree quali la neocorteccia,
l’ippocampo e l’amigdala giustifica il ruolo della somatostatina nella regolazione di attività complesse
come l’attività motoria, l’apprendimento e la memoria; inoltre la loro identificazione a livello della corteccia
piriforme suggerisce un ruolo primario della somatostatina nella modulazione delle informazioni
sensoriali. I recettori sst2 e sst4 sono anche espressi ad alti livelli nell’abenula, area di interconnessione
tra i gangli della base, le strutture mesolimbiche e i nuclei del rafe (406).
Nel ratto a livello ipotalamico sono presenti gli mRNA di tutti e cinque i sottotipi recettoriali (408); tuttavia
l’osservazione, in omogenati di ipotalamo, che il legame della somatostatina non viene inibito
dall’octreotide, suggerisce la preponderanza del sottotipo sst1, recettore al quale l’octreotide si lega con
58
bassa affinità. A livello ipofisario dell’uomo adulto sono espressi i sottotipi sst1, sst2, sst3 e sst5, mentre
sst4 è espresso transitoriamente solo durante lo sviluppo della ghiandola, risultando assente nell’adulto
(409). Al contrario di quanto avviene in omogenati di ipotalamo, in preparati di membrane di ipofisi, il
legame della somatostatina viene inibito dall’octreotide, facendo quindi supporre la presenza
preponderante dei recettori per i quali questo analogo ha una elevata affinità (sst2, sst3, sst5). Sempre
nel ratto, a livello ipofisario l’espressione del recettore sst5 appare nel 70% delle cellule somatotropo e
solo nel 21% delle cellule gonadotrope, mentre il recettore sst2 appare più espresso nelle cellule
tireotrope (409).
Nelle isole pancreatiche è stata dimostrata la presenza di tutti e cinque i sottotipi recettoriali (410), così
come nei linfociti circolanti (411) e nello stomaco (412). Un’espressione moderata dei recettori sst1 e sst5
è osservabile a livello dell’intestino tenue, dove peraltro sono presenti bassi livelli di espressione dei
recettori sst3 e sst4; questa struttura si caratterizza anche per l’assenza di espressione del recettore sst2
(412). Elevati livelli di sst2 sono invece presenti a livello della ghiandola surrenale dove è stata osservata
anche una moderata presenza dei recettori sst1 e sst3. Fegato e milza presentano un’elevata
espressione di sst3 e una moderata o ridotta espressione degli altri sottotipi; al contrario, il recettore sst4
risulta altamente espresso nel cuore e nei polmoni, mentre nel rene sst3 e sst4 sono parimenti espressi
(284). In particolare nel tessuto prostatico umano, sia livello dell’epitelio che del compartimento stromale
sono stati trovati i recettori sst1, sst2, sst3 (413), suggerendo un ruolo paracrino nella regolazione
dell’attività di questa ghiandola. I recettori sst4 e sst5, meno abbondanti, sono confinati nelle cellule
epiteliali (326, 414). È stato ampiamente dimostrato che, nell’uomo, l'espressione dei recettori sst è
presente in una varietà di tessuti tumorali (391, 415, 416) e il recettore sst2 appare essere il sottotipo più
diffuso, mentre gli altri sottotipi sono presenti in percentuale più variabile. Per i tumori prostatici benigni e
maligni, invece, si è osservata una mancanza dell’isoforma sst2 e poiché questo sottotipo è responsabile
dell’effetto antiproliferativo della somatostatina si verifica una condizione di vantaggio di crescita e di
peggioramento della prognosi (414). La presenza dei recettori per la somatostatina, nei tessuti tumorali,
ha portato ad ipotizzare che la perdita del controllo della proliferazione da parte delle cellule cancerose,
possa anche essere la conseguenza di possibili mutazioni, e quindi alterazioni funzionali, di tali recettori
(416).
3.12 Attivazione e meccanismi intracellulari
59
Il legame della somatostatina con i propri recettori induce l'attivazione o l'inibizione di effettori
intracellulari, mediante il coinvolgimento delle proteine a struttura eterotrimerica (formate dalle tre
subunità: α, β, γ) in grado di legare e idrolizzare il GTP, comunemente note come proteine G. Uno degli
effettori intracellulari più studiato è l’adenilato ciclasi (AC), che attiva la via di segnale AC-cAMP-protein
chinasi A (PKA) (417). Studi condotti in cellule isolate che esprimono i diversi recettori sst hanno
dimostrato che, l'accoppiamento funzionale di tali recettori con l'enzima adenilato ciclasi coinvolge le
proteine G αi1 e G αi2 (418), mentre le G αi3 e la G αo1 sembrano essere coinvolte, rispettivamente,
nella regolazione da parte della somatostatina dei canali del potassio e del calcio (419). Non sono
ancora del tutto noti i siti di interazione del recettore con le proteine G, anche se una sequenza
consenso per l'accoppiamento, è stata identificata a livello della terza ansa citoplasmatica dei
recettori sst2, sst3, sst4 e sst5 (420). Comunque, è stato osservato che alcune azioni mediate
dalla somatostatina attraverso il recettore sst1 non sono sensibili alla tossina della pertosse,
portando ad ipotizzare che la particolare sequenza della terza ansa citoplasmatica di questo recettore
non permetta l'accoppiamento con la proteina G. La costruzione di una chimera molecolare del
recettore sst1 con la sequenza della terza ansa citoplasmatica, proveniente da un altro recettore
(come ad esempio il recettore δ per gli oppioidi), solitamente accoppiato alle proteine G, ha permesso
di ottenere un recettore sst1 ibrido capace di interagire con le proteine G, confermando l'importanza,
in tale meccanismo, del dominio intracellulare corrispondente alla terza ansa citoplasmatica (420). In
contrasto con tali evidenze è stato descritto che, in fibroblasti di topo, l’effetto di stimolo della
somatostatina sulla pompa Na+/H+, mediato dal recettore sst1, non viene bloccato dalla tossina della
pertosse, ma soprattutto non coinvolge la terza ansa citoplasmatica (421).
Il recettore sst2 purificato è in grado di associarsi con le proteine Gαi2, Gαi3 e Gαo2 (418, 419). Come noto, il
recettore sst2 esiste in due isoforme (sst2A e sst2B) e dagli studi condotti è stato possibile osservare che
l'isoforma sst2A mostra un'efficienza di accoppiamento con le proteine G minore rispetto all'isoforma
sst2B (420). La causa di tale differenza sembra essere dovuta ad un effetto di ingombro sterico della
parte carbossi-terminale, che nell’isoforma sst2A è più lunga.
3.13 Effetti mediati dal sistema adenilato ciclasi-protein chinasi A
60
In senso generale, si ritiene che tutti i recettori sst clonati, e in particolare le isoforme umane dei
recettori sst2, sst3 e sst5, siano quelle per cui l'accoppiamento all'AC, mediante proteine G sensibili
alla tossina della pertosse, è stato stabilito con maggiore certezza (392, 398, 419, 422).
Per quanto concerne il recettore sst1, sebbene un limitato numero di studi riportava che era accoppiato
all’AC attraverso una proteina G, sensibile alla tossina della pertosse (PTX-sensibile) (423), ad oggi è
ben dimostrato che gli agonisti che legano il sottotipo recettoriale sst1 possono inibire la via dell’AC in
maniera PTX-sensibile (424). Esperimenti condotti su cellule ovariche di criceto cinese (CHO), che
esprimevano rispettivamente il sottotipo sst1 umano (425) e di ratto (426), hanno confermato che,
l’inibizione dell’attività dell’AC è mediata dalle proteine G i3 e Gi1/2.
Analizzando il sottotipo sst2, solo l’isoforma sst2B sembrava essere accoppiata all'AC. Allo scopo
di chiarire le basi molecolari di tali differenze, Reisine (420) ha generato una forma di recettore sst2B
troncata al punto in cui la sequenza aminoacidica diverge rispetto a quella dell'sst2A. Questo
recettore troncato media l'inibizione della formazione di cAMP indotta dalla somatostatina, suggerendo
che il terminale -COOH dei recettori sst2 non è importante ai fini dell'accoppiamento con l'AC, ma è la
porzione C-terminale del sottotipo sst2A ad inibire questo accoppiamento. Il recettore sst2A è
associato con le proteine Gαi2, Gαi3 e Gαo2 (418, 419), ma non con la proteina G αi1, necessaria per
l'accoppiamento con l'AC. Questa differenza nella modalità di associazione con la proteina Gαi1
potrebbe spiegare l'effetto opposto dei due sottotipi recettoriali sst2 sull'attività di AC, anche se
esperimenti di co-espressione, condotti nella linea cellulare CHO, hanno dimostrato che il sottotipo sst2A
può inibire l’AC attraverso le proteine G i1 (427).
Anche il recettore sst3 è accoppiato, mediante la proteina G αi1, all'AC e alla successiva riduzione dei
livelli intracellulari di cAMP (392, 428). Studi di mutagenesi hanno mostrato come, per tale interrelazione, sia necessaria l'estremità C-terminale e l'accoppiamento con la proteina Gαi1 (420). In termini
fisiologici, i meccanismi sopra descritti sono alla base dell'effetto inibitorio della somatostatina sulla
sintesi e sulla secrezione del GH, da parte delle cellule somatotrope ipofisarie. Infatti, l'espressione
genica e il rilascio del GH sono direttamente controllate dal GHRH e dalla somatostatina in modo
antagonistico, rispettivamente, tramite l'aumento e la diminuzione dei livelli intracellulari di cAMP
(429). Infine i sottotipi sst4 (424) e sst5, in modo simile a tutti gli altri sottotipi sst, inibiscono l’AC
attraverso le proteine G, sensibili alla tossina della pertosse (422). Comunque, se l’agonista è ad alte
concentrazioni, sst5 può anche mediare l’attivazione dell’AC, come dimostrato nelle cellule COS (cellule
61
renali di scimmia) e CHO, attraverso un meccanismo apparentemente coinvolgente l’attivazione delle
proteine Gs (430).
3.14 Effetti mediati dal sistema PLC/IP3
Il possibile utilizzo da parte dei recettori sst della via intracellulare mediata dall’attivazione della PLC, con
formazione di IP3 è controversa e quindi non viene ritenuta una via di segnale normalmente utilizzata
(431).
Il recettore sst1, così come dimostrato, in vitro, sui modelli cellulari CHO (425) e COS (432), ma non nella
linea cellulare F4C1, tumore ipofisario di ratto (433), attiva la via della PLC, con susseguente produzione
dell’IP3.
Anche sst2A stimola la PLC, nelle linee cellulari COS (432), F4C1 (433) e GM4C1 (434). Tale effetto è
solo parzialmente sensibile alla tossina della pertosse, suggerendo che le proteine G, sia sensibili che
insensibili a questa tossina, possano essere coinvolte nel collegamento di sst2A a questa via. Sempre
nelle cellule COS, transfettate per sst3, sst4 e sst5, è stato dimostrato che, l’attivazione della PLC è solo
parzialmente sensibile alla tossina della pertosse, ed, il sottotipo sst5 è il più efficace nell’attivare la PLC
e la produzione di IP3 (432).
3.15 Effetti mediati da variazioni delle concentrazioni ioniche intracellulari
I recettori sst possono risultare accoppiati a diversi sottotipi di canali del potassio (rettificatori ritardati,
rettificatori in entrata, canali sensibili all'ATP e canali a grande conduttanza calcio-dipendenti) (420, 435,
437). Esperimenti di ricombinazione molecolare hanno dimostrato che, l'accoppiamento funzionale tra i
recettori sst e i canali del potassio è mediato dall'attivazione della proteina Gαi3 (438). L'attivazione dei
canali del potassio, da parte dei recettori della somatostatina, induce una iperpolarizzazione reversibile
della membrana cellulare, con conseguente cessazione dei potenziali d'azione spontanei e una riduzione
delle concentrazioni intracellulari di Ca+2 (327). Oltre a questo effetto indiretto, sull'ingresso di calcio nella
cellula, i recettori sst2, in particolare, possono anche esercitare un effetto diretto di blocco dei canali del
Ca+2 voltaggio-dipendenti, attraverso la proteina Gαo2 (439). A livello di alcune popolazioni di neuroni del
cervello di ratto, tale modulazione sembra coinvolgere i canali del calcio di tipo N e di tipo P/Q (440). D'altro
canto, è stata anche dimostrata un'azione inibitoria della somatostatina sui canali del Ca+2, mediata
dall'attivazione della guanilato ciclasi con formazione del cGMP, attivazione di una chinasi cGMP-
62
dipendente e conseguente fosforilazione dei canali del Ca+2 (441). In apparente contrasto con questo
effetto inibitorio, esercitato dalla somatostatina, è stato osservato che essa è in grado di indurre anche
un'attivazione sia dei canali del Ca+2, che del potassio, attraverso una defosforilazione dei canali stessi,
dovuta all'attivazione di una serina-treonina fosfatasi (437). Infine, il recettore sst1 può essere accoppiato
all'attivazione della pompa Na+/H+ attraverso una via di segnale insensibile alla tossina della pertosse,
mentre il recettore sst2 non risulta accoppiato con questo trasportatore ionico (421).
3.16 Effetti mediati da modificazioni dell’attività fosfatasica
Le fosfatasi fanno parte di un’ampia famiglia di enzimi che comprendono forme transmembranali,
recettoriali e non recettoriali. Esse hanno due diversi siti preferenziali di azione; si dividono infatti in
fosfatasi con prevalente attività verso gli aminoacidi serina/treonina oppure tirosina. Inoltre esistono
fosfatasi in serina/treonina di tipo I e II; quest’ultime defosforilano anche i residui tirosinici.
La somatostatina può svolgere la sua azione inibitoria sulla proliferazione cellulare principalmente
attraverso l'attività dì proteine tirosina-fosfatasi, benché siano state descritte anche interazioni con
fosfatasi in serina e treonina (442, 443). A conferma dell'importanza del ruolo svolto dalle fosfatasi, è
stato postulato che la mancanza degli effetti inibitori della somatostatina sia da imputare non ad un'alterata
espressione del recettore per l'ormone, ma ad un mancato accoppiamento fra il recettore e la fosfatasi
stessa. Esperimenti condotti sulla linea cellulare trasformata PCC13, derivante dalla tiroide, la cui
proliferazione è stimolata dall’insulina e dal TSH ed inibita dalla somatostatina, hanno permesso di
osservare che il blocco dell’attività della tirosina-fosfatasi, con vanadato, produce un aumento della
velocità di crescita di queste cellule. Inoltre, dal momento che il trattamento con somatostatina è
accompagnato da un aumento dell’attività tirosina-fosfatasica, l’incremento di tale attività enzimatica è un
segnale citostatico per questa linea cellulare (444). Si è inoltre osservato che, nella linea cellulare CHO
(445), gli effetti della somatostatina mediati dal recettore sst2 sono trasdotti da un sistema intracellulare ad
attività tirosina-fosfatasica. L'attivazione di sst2 da parte dell'ormone, comporta la dissociazione della
fosfatasi dal recettore stesso e un aumento della sua attività. Nelle cellule di controllo la fosfatasi è fosforilata
in tirosina, mentre in quelle trattate con somatostatina, si osserva una defosforilazione di questi
residui con attivazione dell’enzima fosfatasi, a dimostrazione del ruolo chiave della fosfatasi come
trasduttore dei segnali antiproliferativi della somatostatina a livello del sottotipo sst2 (445).
Complessivamente, l'interazione della somatostatina con le diverse classi di fosfatasi, fornisce un
63
esempio di come l'attività biologica possa venire regolata da fini meccanismi che coinvolgono fosforilazioni
reversibili che hanno un ruolo fondamentale nella fisiologia della cellula (446).
Infine un’altra via di trasduzione del segnale intracellulare è quella d ella MAPK. Nelle cellule CHO che
esprimono sst1, la somatostatina antagonizza l’effetto proliferativo indotto dal fibroblast growth factor
(FGF) attraverso la stimolazione di ERK1, attraverso la subunità della proteina G, sensibile alla tossina
della pertosse, Gβ (447). L’induzione di ERK1 dipende dall’attivazione della MAPK chinasi attraverso la
cascata MEK1/2, Raf-1, Ros, PI3K e della tirosin chinasi citoplasmatica, appartenente alla famiglia c-src.
L’attivazione della tirosin fosfatasi SHP-2 é necessaria per avere la massima attivazione di ERK1 e Raf-1,
dimostrando un cross-talk tra le vie fosfotirosin-fosfatasi e MAPK. Poiché SHP-2 lega costitutivamente csrc, si é ipotizzato che c-src, per il quale la completa attività dipende dalla defosforilazione di un singolo
residuo fosfotirosinico, sia un diretto substrato di SHP-2 (447). Le due varianti del recettore sst2A e
sst2B, mediano una prolungata attivazione di entrambe le forme ERK1 e ERK2, attraverso la subunità G
(448). Comunque, le due isoforme differiscono nella loro capacità di attivare la MAPK p38 o l’PI3K,
esercitando così effetti opposti sulla proliferazione. Questi dati suggeriscono che le sequenze C-terminali
delle due isoforme giocano un ruolo importante nel reclutare le distinte vie chinasiche (448).
Nelle cellule CHO stabilmente transfettate per il recettore sst4, la somatostatina induce l’attivazione di
entrambe MAPK e/o MAPK chinasi, via proteina G sensibile alla tossina della pertosse (449). Da notare
che un’attivazione acuta di ERK1/2, al contrario della long lasting activation, è PKC-indipendente.
L’effetto proliferativo indotto dalla somatostatina è associato ad una prolungata attivazione della MAPK
(450).
Quando espresso nelle cellule CHO, sst5 inibisce l’attività della MAPK attraverso un meccanismo
coinvolgente l’inibizione del guanilato ciclasi e la formazione di cGMP (451).
3.17 Dimerizzazione dei recettori della somatostatina
Sebbene un ampio numero di recettori di membrana mediano la loro azione come dimeri (Heldin 1995),
inizialmente si credeva che i recettori accoppiati alle proteine G (GPCRs) funzionassero generalmente
come monomeri. Studi di farmacologia, biochimica e fisica, basati sulla BRET (bioluminescence
resonance energy transfer) e pbFRET (photobleaching fluorescence resonance energy transfer), hanno
evidenziato che i GPCRs, includendo i membri della famiglia dei recettori della somatostatina, possano
funzionare sia come dimeri, che oligomeri (452). Tra i recettori sst, la omo-etero dimerizzazione è stata
64
dimostrata per la prima volta per il sottotipo umano sst5 (hsst5) (453). Studi in vitro, sulla linea cellulare
CHO, hanno evidenziato che i monomeri sono predominanti nelle cellule che esprimono bassi livelli di
recettori transfettati, dimostrando quindi, che i recettori sst5 non formano dimeri in assenza dell’agonista
e che i trattamenti con somatostatina determinano un aumento dose dipendente della dimerizzazione del
recettore. Il sottotipo hsst5 può formare associazioni eterodimeriche con hsst1, ma non con hsst4,
suggerendo che la eterodimerizzazione è ristretta a specifiche combinazioni dei sottotipi recettoriali (453).
Associazioni tra il recettore hsst5 e la forma lunga del recettore umano D2 (D2R), un sottotipo che forma
omodimeri a livello della membrana plasmatica, sono state evidenziate nelle cellule CHO co-esprimenti i
due recettori (454). Mentre gli eterodimeri non sono localizzabili in assenza di stimolazione agonista, il
trattamento di cellule con dopamina, SS14 o entrambe gli agonisti, determina un aumento dell’efficienza
della pbFRET, suggerendo che, l’eterodimerizzazione è indotta da ciascun agonista. L’analisi
farmacologica dell’interazione hsst-D2R mostra che le cellule co-esprimenti i due recettori hanno una
attività funzionale maggiore se stimolati da entrambe somatostatina e ligandi dopaminergici (453). Anche
sst2A e sst3 possono formare omodimeri quando co-espressi in cellule HEK293. Mentre l’eterodimero
sst2A–sst3 si comporta come l’omodimero sst2A, esso non riproduce le caratteristiche farmacologiche
dell’omodimero suggerendo che, l’interazione fisica di sst3 con sst2A potrebbe indurre l’inattivazione
funzionale del sottotipo sst3 (455).
3.18 Analoghi della somatostatina
La somatostatina, benché proposta per il trattamento di numerose patologie umane, non può essere
considerata un farmaco ideale, perché presenta una breve emivita plasmatica (2-3 minuti) e necessita,
quindi, di essere somministrata per infusione endovenosa continua, con conseguente effetto “rebound”
sulla secrezione dell’ormone stesso (456, 457). Nel tentativo di superare queste limitazioni si è pensato di
sviluppare degli analoghi sintetici che, pur conservando le stesse caratteristiche biologiche e
farmacologiche dell’ormone ipotalamico, presentassero un’emivita più lunga e fossero meno aggredibili
dagli enzimi proteolitici. Negli anni successivi l’identificazione chimica della somatostatina furono
sintetizzati peptidi detti “full size”, che conservavano, seppur modificata, la struttura di 14 amminoacidi
dell’ormone nativo. Questi composti però non presentavano caratteristiche biologiche superiori alla
somatostatina (445). Negli anni ‘70 però, venne osservato che anche piccoli peptidi di 6-8 aminoacidi
mantenevano l’attività biologica dell’ormone naturale e l’attenzione si concentrò su questi composti (458).
65
I più potenti analoghi della somatostatina oggi disponibili conservano la conformazione ciclica
responsabile dell’attività biologica e mancano del legame aminoacidico 8-9, sito di attacco delle peptidasi;
alla costituzione del gruppo farmacoforo concorrono inoltre la sostituzione degli aminoacidi in posizione 1
e 4 con epimeri destrogiri (459), che danno una maggiore selettività recettoriale rispetto alla
somatostatina. Di tutti gli agonisti di sintesi della somatostatina oggi disponibili, solo alcuni sono entrati
nella pratica clinica: in particolare sono attualmente impiegati in terapia l’octreotide (460), il lanreotide e il
vapreotide. Dal punto di vista strutturale, l’octreotide mantiene il ponte disolfuro tra le due cisteine, mentre
la struttura ciclica è ridotta a soli sei aminoacidi rispetto ai dodici della somatostatina nativa. La presenza
del triptofano destrogiro, in posizione 4, nell’octreotide ha permesso di aumentare considerevolmente
l’affinità della molecola per il recettore sst2 (461); inoltre la presenza di un altro aminoacido destrogiro (DPhe¹), sull’estremità C-terminale e un aminoacido idrossilato (Thr-olo) in posizione 8, all’estremità Nterminale, rallentano la velocità di degradazione del peptide (462). L’octreotide è stato il primo analogo a
essere disponibile per uso clinico sperimentale a metà degli anni ‘80, utilizzato soprattutto per l’effetto
inibitorio sulla secrezione del GH (286). Il lanreotide rappresenta un altro octapeptide ciclico inizialmente
noto come BIM 23014 o somatuline, caratterizzato dalla presenza del gruppo D-βNal nella porzione
carbossiterminale; come gli altri agonisti della somatostatina, inoltre, presenta un ponte disolfuro e una
struttura ciclica esaminoacidica. Analogamente a quanto osservato per l’octreotide, questo insieme di
sostituzioni chimiche rende la molecola più resistente alla degradazione enzimatica, più attiva
biologicamente e più affine ai recettori cellulari di tipo 2. Il lanreotide, inoltre, si lega al recettore sst5 con
una affinità maggiore di quella esibita dall’octreotide (409, 463). Tale composto mostra anche una
potente azione inibitoria sulla proliferazione cellulare, conseguente a danno vascolare sistemico (464) ed
evoca un effetto soppressivo sulle risposte immunitarie, mediate dalle interazioni leucociti-endotelio
(465).
Il vapreotide è anch’esso un analogo octapeptidico ciclico, caratterizzato da un ponte disolfuro e da una
struttura ciclica dove gli aminoacidi tiroxina e valina sono rispettivamente in posizione 3 e 6, come nel
lanreotide. Il composto eptaciclico definito TT-232 nasce da modifiche dell’octreotide: rimozione del
residuo tirosinico in posizione 6 e sostituzione della fenilalanina in posizione 3, con una residuo tirosinico;
tale composto non inibisce il rilascio di GH, ma ha un effetto antiproliferativo in una grande varietà di
modelli in vitro e in vivo (466, 467). Un composto correlato al TT-232, in cui il ponte disolfuro tra le due
cisteine risulta sostituito da un ponte solfuro, tra due alanine, è stato chiamato Lan-7; questo analogo
66
mostra citotossicità selettiva nei confronti delle cellule tumorali, evocando meccanismi di tipo apoptotico
senza interferire sulla crescita delle cellule normali (468). Gli analoghi octapeptidici lineari ovviamente
non possiedono il ponte disolfuro, ma nelle posizioni 1 e 4 presentano l’epimero destrogiro degli stessi
aminoacidi presenti nella molecola degli agonisti ciclici. In aggiunta, nel composto BIM 23056,
l’aminoacido in posizione 8 è rappresentato dal gruppo D-βNal, mentre nella molecola del BIM 23052
l’aminoacido in posizione 6 è costituito dalla treonina (Thr). Questi analoghi risultano legarsi
preferenzialmente al sottotipo sst5 (BIM 23056), nonché a quello di tipo 3 (BIM 23052) (469, 470).
3.19 Somatostatina e suoi analoghi: controllo della crescita cellulare
La capacità da parte della somatostatina e dei suoi analoghi di controllare la proliferazione di cellule normali e
trasformate, è stata esplorata in modelli animali, in linee cellulari di derivazione murina o umana e in tumori
umani (linee cellulari) impiantati in topi atimici (471, 472). Esistono anche dati provenienti da studi clinici, i
quali indicano che, la somatostatina e i suoi analoghi hanno degli effetti nel trattamento di differenti
patologie neoplastiche (473, 474). L'inibizione della crescita cellulare è sostenuta da due tipi di meccanismo:
indiretti e diretti. I primi sfruttano gli effetti sistemici della somatostatina e dei suoi analoghi, giustificando
così l'effetto antiproliferativo riscontrato in tumori nei quali sono assenti i recettori per la somatostatina.
Tali meccanismi sono rappresentati da: a) inibizione della liberazione o dell'azione degli ormoni coinvolti
nella regolazione della crescita cellulare (quali l’insulina, il GH, la PRL, la gastrina, la secretina e la
colecistochinina). In particolare, la PRL interviene nello sviluppo del cancro della prostata (475) e il GH
riveste un ruolo fondamentale nella crescita dei tumori mammari (476); gli ormoni gastrointestinali,
insieme ai fattori di crescita, controllano lo sviluppo dei tumori del pancreas e del colon-retto (471, 477);
b) inibizione della liberazione o dell'azione, di fattori di crescita quali l’IGF, l'EGF, il PDGF, l'FGF, il TGF-α.
L'inibizione dell'IGF-1 è sia diretta, che mediata dall'inibizione del GH. L'IGF-1 è un importante mitogeno per
numerosi tipi di cellule trasformate, inibisce l'apoptosi, sollecita la mobilità cellulare e facilita la crescita delle
cellule endoteliali (478, 479). In particolare è stato dimostrato che la crescita dei tumori mammari umani
impiantati nel topo è ridotta, rispetto ai controlli, negli animali che geneticamente manifestano una
deficienza di IGF-1, anche se ambedue i gruppi ricevono lo stesso supporto estrogenico (480). Inoltre, il
lanreotide stimola selettivamente la formazione della proteina "IGF binding protein 1", interferendo così,
con l'azione dell'IGF-1 a livello recettoriale (481). La somatostatina può ridurre anche la secrezione di
bombesina e di Gastrin-Releasing Peptide, fattori autocrini nel carcinoma a piccole cellule del polmone
(482, 483); c) inibizione dell’angiogenesi legata, in parte, all’inibizione della liberazione di IGF-1 (481). Tale
67
azione è particolarmente importante nelle prime fasi dello sviluppo neoplastico, per spiegare il marcato effetto
di riduzione della crescita che gli analoghi sintetici producono in differenti modelli animali, dopo l’impianto di un
tumore (472); d) modulazione delle funzioni del sistema immunitario. Sono state osservate infatti,
modifiche nell’attività delle cellule “natural killer” murine, dopo trattamento con somatostatina e in pazienti
affetti da carcinomi metastatici trattati con l’octreotide (342, 484).
I meccanismi d’azione diretti sono, invece, rappresentati dagli effetti prodotti dal legame della
somatostatina o dei suoi analoghi di sintesi ai recettori specif ici, determinando l’attivazione di vari
secondi messaggeri, come descritto in precedenza. Tra gli eventi post-recettoriali, particolare
importanza viene attribuita alla modulazione degli enzimi fosforilanti, che integrano e amplificano il
messaggio di vari fattori di crescita e sono considerati cruciali per la trasmissione del segnale
mitogenico dalla membrana al nucleo. L'aumento della fosfotirosin-fosfatasi induce la defosforilazione
dell'EGF-R, provvisto di attività tirosina-chinasica, e ciò si traduce in una incapacità del recettore di
mediare il segnale proliferativo del fattore di crescita (485, 486). Ormoni quali la dopamina e l'LHRH
sfruttano lo stesso meccanismo per produrre un arresto della crescita cellulare e in cellule di carcinoma
della mammella è stato dimostrato che il trattamento con antiestrogeni fa aumentare una
fosfotirosinfosfatasi di membrana, strettamente correlata all'effetto antitumorale di queste sostanze
(487, 488). Un'ulteriore via implicata nell'estrinsecazione dell'effetto antiproliferativo, a seguito
dell'interazione degli analoghi della somatostatina con il proprio recettore, è l'inibizione dell'attività della
MAPK stimolata dall'IGF-1, il cui segnale è mediato da un recettore ad attività tirosina-chinasica (489). La
somatostatina può inibire direttamente l'espressione dei geni c-fos e c-jun, impedendo la formazione di
un fattore di trascrizione denominato AP1, così come dimostrato in una linea cellulare di adenoma
ipofisario di ratto e in cellule parietali dello stomaco, dove l'octreotide, mediante l'azione di alcune
fosfatasi, è in grado di inibire la formazione di AP1 (442, 490).
L'apoptosi è un evento dovuto alla deliberata attivazione di un programma genetico in cui la cellula è
coinvolta attivamente; essa consiste in una serie di modificazioni morfologiche e biochimiche che
provocano una degradazione irreversibile del DNA genomico, alterazioni nucleari e frammentazione
della cellula. L'induzione dell'apoptosi è mediata da sst3 e da modificazioni conformazionali di p53
"wild-type", defosforilazione-dipendenti, e dall'induzione del gene bax. L'azione su p53 si verifica
rapidamente e precede il verificarsi dell'apoptosi (491).
68
3.20 Effetto antiproliferativo in cellule non trasformate
Per quel che concerne le cellule normali, la somatostatina svolge un ruolo nei processi fisiologici di
rinnovamento delle cellule epiteliali e nella risposta dell'epitelio a vari tipi di danno. A questo proposito,
risultati interessanti sono stati ottenuti in enterociti normali ottenuti da cripte dell'intestino tenue di ratto,
dove la somatostatina (492) ha la capacità di inibire completamente sia la sintesi del DNA (493), che
l'espressione dei protooncogeni regolati dall'EGF (494). Nelle cellule follicolari tiroidee non trasformate e ben
differenziate di ratto, FRTL5, la somatostatina inibisce sia la sintesi del DNA, stimolata dal TSH e dall'IGF-1, che
la proliferazione cellulare (495). Anche nelle linea cellulare PCC13, la somatostatina, interagendo con il
recettore sst4, induce l'inibizione della crescita cellulare stimolata dall'insulina e dal TSH, attraverso un
blocco della progressione del ciclo cellulare, dalla fase G1 alla fase S; tale legame determina, inoltre, un
aumento del 100% dell'attività fosfotirosin-fosfatasica (496). Il controllo sulla crescita si esercita non solo
sulle cellule epiteliali normali, ma anche su quelle muscolari e stromali. Infatti, il lanreotide determina una
riduzione della proliferazione di elementi stromali ottenuti da iperplasia prostatica benigna umana (497).
Inoltre nei conigli, in cui era stata provocata un'arteriosclerosi accelerata delle coronarie, mediante
trapianto cardiaco eterotopico, il lanreotide attenua l'iperplasia dell'intima e dello strato muscolare. Lo
stesso analogo inibisce l'incorporazione di timidina triziata nelle arterie coronarie del maiale. Ciò assume
particolare importanza se si pensa che, in pazienti che hanno subito un trapianto cardiaco o
un'angioplastica, la proliferazione delle cellule muscolari lisce rappresenta la causa principale
dell’ostruzione del lume delle arterie coronarie (498).
Un modello interessante per lo studio della regolazione della crescita delle cellule normali, da parte della
somatostatina è rappresentato dalle colture primarie di colangiociti di ratto polarizzati, in cui le superfici
apicale e basolaterale sono facilmente accessibili. Le cellule sviluppano giunzioni intercellulari serrate
mantenendo il loro fenotipo e rispondendo all'esposizione alla somatostatina. Se la superficie basolaterale
viene trattata con il peptide, essa manifesta una riduzione del 60% dei livelli di cAMP, indotti da forskolina,
suggerendo che su di essa vengono preferenzialmente espressi recettori specifici dal momento che, il
fenomeno non si verifica se al trattamento con somatostatina viene esposta la superficie apicale (499).
Esperimenti condotti in vivo hanno dimostrato che, in ratti in cui era stato effettuato sopra al muscolo
gastrocnemio un impianto sottocutaneo di matrice ossea demineralizzata, allo scopo di indurre
ossificazione, la somatostatina determina una diminuzione della proliferazione cellulare valutata
mediante l’incorporazione di timidina triziata e una riduzione dell'osteogenesi (500).
69
3.21 Effetto antiproliferativo in linee cellulari neoplastiche
Per quel che riguarda le linee cellulari coltivate in vitro esiste, in letteratura, una certa variabilità delle
condizioni di coltura impiegate, laddove nelle sperimentazioni relative all'impianto delle stesse linee,
nei topi atimici, si riscontrano notevoli discrepanze nel dosaggio degli analoghi utilizzati. Tuttavia, i
risultati depongono a favore della capacità della somatostatina e dei suoi analoghi nel controllare la
crescita della maggior parte dei tumori studiati, interagendo per lo più con recettori specifici. In
qualche caso appare evidente che l'azione della somatostatina e degli analoghi consiste nella
soppressione degli effetti proliferativi di ormoni o fattori di crescita che, a loro volta, interagiscono con
recettori specifici spesso provvisti di attività tirosina-chinasica. Risultano interessanti, le informazioni
relative al potenziamento da parte degli analoghi della somatostatina dell'effetto di altri farmaci, quali
gli antiestrogeni, gli analoghi dell'LHRH e i chemioterapici. In cellule derivate da tumori degli acini
pancreatici del ratto (ARA-2J), la somatostatina e l’octreotide inibiscono la crescita cellulare con la
mediazione di specifici recettori (501, 502), mentre le linee di carcinoma pancreatico di ratto, BxPC-3 e
SOJ-6, vengono solo debolmente inibite dalla somatostatina (503).
Nella linea di adenocarcinoma gastrico MKN45 sia in vitro, che dopo impianto in topi atimici, l’octreotide
induce una riduzione sia della crescita, che della secrezione di gastrina (501, 504).
Nelle linee cellulari di cancro del colon umano (CX1, X56, C170, LIM 1215, LIM 1863, LIM 2405, LIM
2412, SW 480, SW 620, RIP, DRUM) e di ratto (DHD/K12) è emersa l’efficacia della somatostatina e dei
suoi analoghi sintetici, nell’inibire la crescita cellulare (505, 506), dove l’attivazione della fosfatasi sembra
essere coinvolta nella realizzazione di tale effetto (507). Nelle linee cellulari di cancro della tiroide,
derivate da tumori umani primitivi o da metastasi linfonodali, si è osservato un effetto bifasico
dell’octreotide: a basse dosi stimola la proliferazione, mentre a dosi più alte inibisce la crescita cellulare e
l’invasione neoplastica (508).
Nel carcinoma polmonare a piccole cellule, in particolare nella linea cellulare NCI H69, la somatostatina è
incapace di influenzare la proliferazione cellulare in vitro (509), a differenza del lanreotide e
dell’octreotide, che risultano invece essere efficaci sia in vitro, che dopo impianto in topi atimici (510).
Nelle cellule MMQ, derivate da un tumore ipofisario PRL-secernente di ratto, si osserva una evidente
risposta alla somatostatina (511) e all’octreotide, caratterizzata da un blocco del ciclo cellulare tra la fase
G0 e G1 (512).
70
La somatostatina e i suoi analoghi inibiscono, in vitro, anche la crescita di cellule di leucemia linfoblastica
umana (513), di osteosarcoma umano (SK-ES1), di melanoma B16 e del sarcoma S180 (514), impiantate
in topi atimici (515).
Anche nelle linee di carcinoma mammario umano MCF-7 e CG-5, la somatostatina, l’octreotide (516), il
TT-232 e il lanreotide (514) sono provvisti di una marcata attività antiproliferativa. In particolare, le linee
cellulari estrogeno-sensibili esprimono livelli variabili di sst1, sst2, sst5, poiché è stato dimostrato che gli
estrogeni regolano in senso positivo l’espressione di sst2, interagendo con il proprio recettore (517).
3.22 Effetto antiproliferativo nelle linee cellulari neoplastiche di prostata umana
La somatostatina e i suoi analoghi sono efficaci nell’inibire, in vitro, la crescita delle cellule cancerose
prostatiche umane sia androgeno-dipendenti, che androgeno-indipendenti. Nelle cellule LNCaP, derivate
da adenocarcinoma umano androgeno-dipendente, e in cui gli EGF-R sono indotti dagli androgeni, la
SS14 controlla la proliferazione e inibisce la sintesi proteica, mediante l’attivazione di fosfotirosin-fosfatasi
(518). L’analogo RC-160 inibisce la crescita delle cellule di adenocarcinoma umano PC3 e DU145,
androgeno-indipendenti, impiantate in topi atimici, determinando una diminuzione significativa degli EGFR, nonché dei livelli sia di GH, che di gastrina (519, 520). L’analogo TT-232, nelle cellule PC3, induce una
marcata apoptosi e qualora le cellule siano impiantate in topi atimici, determina una riduzione della
massa tumorale pari al 60% del volume dei tumori (nelle stesse condizioni RC-160 determina solo il 40%
di riduzione) (514, 520).
71
4. SCOPO DEL LAVORO
Questo lavoro è stato svolto con l’obiettivo di valutare il possibile ruolo del neuropeptide Y sulla
proliferazione delle linee cellulari di PCa umano LNCaP (androgeno dipendente) e DU145 e PC3
(androgeno indipendenti) e, in particolare, di comprendere i possibili meccanismi molecolari alla base di
tale evento. Le scarse conoscenze fisiopatologiche e l’assenza di terapie specifiche per il carcinoma
prostatico androgeno-indipendente suggeriscono la necessità di approfondire e studiare i meccanismi
molecolari alla base dell’evoluzione fisio-patologica del carcinoma.
Il presupposto molecolare del nostro studio è stato valutare, nelle tre linee cellulari studiate, l’espressione
del recettore Y1 sia a livello genico (RT-PCR) che proteico (analisi Western blot). Inoltre, si è valutata,
mediante analisi immunoistochimica, la presenza del Y1 anche a livello di campioni bioptici di tessuto
tumorale. Per chiarire se l’NPY potesse agire anche a livello del PCa in modo autocrino-paracrino, è stata
valutata, sempre tramite analisi RT-PCR, l’espressione del suo mRNA nelle stesse linee cellulari.
Dopo aver verificato il presupposto molecolare per una possibile azione biologica dell’NPY sulle cellule,
sono stati condotti esperimenti di proliferazione cellulare a dosi diverse di NPY per 96 ore, per valutare
l’effetto sulla proliferazione. Successivi esperimenti di proliferazione, mediante l’utilizzo di un antagonista
specifico per il recettore Y1, BIBP3226 (521), hanno evidenziato il ruolo chiave di questo recettore nel
mediare sia gli effetti proliferativi che antiproliferativi. Per chiarire i diversi effetti esercitati dall’NPY sulla
proliferazione cellulare si è ritenuto fondamentale, in ciascuna linea cellulare, stabilire quali vie di segnale
intracellulari fossero attivate dal sistema NPY/Y1.
Poiché dalla letteratura è noto che i recettori Y1 sono generalmente accoppiati alla cascata della MAPK
(149), mediante l’analisi Western Blot è stata valutata, dopo trattamento con NPY, la variazione del grado
di fosforilazione della via di segnale MAPK/ERK1/2. Essendo noto il coinvolgimento della MAPK negli
eventi di proliferazione sia in senso stimolatorio che inibitorio, sono stati condotti esperimenti di
incorporazione di timidina triziata dopo pretrattamento con un inibitore selettivo di MEK (U0126). Poiché
in alcuni sistemi cellulari è stato osservato che l’attivazione della PKC (522) è un evento a monte della
fosforilazione di ERK1/2, mediante l’utilizzo del GF109203X, un inibitore selettivo della PKC, abbiamo
inoltre studiato la relazione esistente tra PKC, ERK1/2 e proliferazione cellulare. Per essere sicuri che
l’attivazione della via MAPK/ERK/1/2 fosse effettivamente mediata dal Y1, abbiamo valutato, mediante
l’analisi Western blot, la capacità dell’NPY di attivare ERK1/2 dopo pretrattamento con il BIBP3226. Sono
72
stati inoltre valutati anche gli altri due segnali intracellulari notoriamente associati al sistema NPY/Y1:
l’inibizione dei livelli di cAMP e l’incremento dei livelli intracellulari di Ca ++ (149). Anche in questo caso,
nelle linee cellulari in cui uno dei due sistemi sopradescritti è risultato stimolato dall’NPY, sono stati
condotti esperimenti con il BIBP3226.
Poiché in letteratura sono presenti dati esigui concernenti il PCa ed eventuali neuropeptidi coinvolti nel
ciclo cellulare, abbiamo caratterizzato la linea cellulare di PCa umano androgeno dipendente LNCaP,
anche per il sistema somatostatina, un peptide ipotalamico ad azione antiproliferativa (523). Si è ritenuto
utile iniziare questo studio proprio da questa linea cellulare poiché, anche a livello clinico, l’androgeno
dipendenza rappresenta il primo stadio del PCa. Come per il sistema dell’NPY, anche in questo è stata
valutata inizialmente sia l’espressione genica (analisi RT-PCR) della somatostatina che dei suoi cinque
sottotipi recettoriali (sst1, sst2A, sst3, sst4, sst5), cosi come quella proteica (analisi Western blot). Si è
voluto inoltre verificare anche la presenza sia genica (analisi RT-PCR) che proteica (analisi
immunocitochimica) del tumor suppressor gene hZAC e del prostatic specific antigen (PSA) (analisi RTPCR). Dopo aver verificato il presupposto molecolare per una possibile azione biologica della
somatostatina, abbiamo quindi valutato l’effetto sulla proliferazione cellulare mediante un trattamento
dose dipendente di somatostatina e di alcuni analoghi sintetici recettore-specifici, per 24, 48, 72 ore.
73
5. MATERIALI E METODI
Colture cellulari: Le linee cellulari di PCa umano androgeno dipendenti LNCaP, androgeno indipendenti
DU145 e PC3, quelle di neuroblastoma umano SH-SY5Y e SK-N-MC e quelle di carcinoma polmonare
non a piccole cellule Calu-6 (American Type Culture Collection, Rockville, MD, USA) sono state coltivate
in monostrato, in un incubatore a 37°C, al 5% di CO 2. Per le linee cellulari LNCaP, DU145 e PC3, il
medium di coltura è stato l’RPMI 1640, con 10 mg/L di rosso fenolo (Biochrom, Berlin, Germany), 5/10%
di siero fetale bovino (FBS; Gibco, Grand Island, NY, USA); per le cellule SH-SY5Y e SK-N-MC, è stato
usato come medium il MEM contenente aminoacidi non essenziali, sodio piruvato 1 nM, 10 mg/L di rosso
fenolo e 10% di FBS; per le cellule Calu-6 è stato usato come medium il MEM contenente il 10% di FBS,
1% di aminoacidi non essenziali, 20 mg/dl gentamicina, 200 mM glutamina e 1 mM di sodio piruvato.
Estrazione dell’RNA e analisi RT-PCR: Le cellule, ad una confluenza del 70-80%, sono state lavate con
PBS con Ca++/Mg++ freddo (+4°C), raccolte e l’RNA totale è stato estratto usando il Tri Reagent (SigmaAldrich, Milano, Italia), secondo il classico metodo fenolo-cloroformio (524). L’analisi dell’espressione
genica, mediante la metodica RT-PCR, è stata condotta su 1 g di RNA totale, quantificato dopo una
digestione con DNAase, utilizzando il kit Deossiribonucleasi I (Sigma-Aldrich, Milano, Italia). La reazione
di RT è stata condotta in un volume finale di 50 μL, utilizzando il kit commerciale GeneAmp (Applied
Biosystems, Milano, Italia). I primer specifici utilizzati per l’identificazione dell’NPY, del sottotipo
recettoriale Y1, della β actina (gene costitutivo), della somatostatina, di sst1, sst2, sst3, sst4, sst5, di
ZAC,
del
PSA,
e
del
18S
(gene
costitutivo)
sono
rispettivamente:
e
(CCAgATACTACTCggCgCTgCgACACTACA-3’)
5’gAATgCATgATACTTTATTTAAACACACATATATACAA3’)
NPY
NPY
Y1
(525);
forward
reverse
forward
(5’-
TATACCACTCTTCTCTTggTgCTg-3’) e Y1 reverse (5’-CTggAAgTTTTTgTTCAggAACCCA-3’) (92); β
actina
forward
(5’-TgACggggTCACCCACACTgTgCCCATCTA-3’)
(CTAgAAGCATTTgCggTggACgATggAgg-3’)
(526);
e
β
somatostatina
actina
reverse
forward
(5’(5’-
gATgCTgTCCTgCCgCCTCCAg-3’) e somatostatina reverse (5’-ACAggATgTgAAAgTCTTCCA-3’); sst1
forward (5’-ATggTggCCCTCAAggCCgg-3’) e sst1 reverse (5’-CgCggTggCgTAATAgTCAA-3’); sst2A
forward (5’-GCCAAGATGAAGACCATCAC-3’) e sst2A reverse (5’-GATGAACCCTGTGTACCAAGC-3’);
sst3 forward (5'-TCATCTGCCTCTGCTACC-3’) e sst3 reverse (5'-GAGCCCAAAGAAGGCAGG-3’); sst4
forward (5’-ATCTTCGCAGACACCAGACC-3’) e sst4 reverse (5’-ATCAAGGCTGGTCACGACGA-3’); sst5
74
forward (5'-CCGTCTTCATCATCTACACGG-3’) e sst5 reverse (5’-GGCCAGGTTGACGATGTTGA-3’);
PSA forward (5’TggACAgggggCAAAAgCAC3’) e PSA reverse (5’AggACACAgAgAggACAAAA3’) (527);
ZAC
forward
(5’AACCggAAAgACCACCTgAAAAACCAC3’)
e
ZAC
reverse
(5’gTCgCACATCCTTCCgggTgTAgA3’) (528); 18S forward (5’-CTCGCTCCTCTCCTACTTGG-3’) e 18S
reverse (5’-CCATCGAAAGTTGATAGGGC-3’). I prodotti di amplificazione sono stati separati per
elettroforesi, in un gel di agarosio al 2%, contenente etidio bromuro.
Analisi immunoistochimica: Per studiare l’espressione del sottotipo recettoriale Y1, a livello tissutale, è
stata eseguita un’indagine immunoistochimica impiegando dei preparati istologici di carcinoma prostatico
umano. Sezioni di tessuto inclusi in paraffina, sono state fatte aderire su vetrini precedentemente trattati
con poly-L-lisina. I campioni sono stati liberati dalla paraffina attraverso due lavaggi, di 10 minuti
ciascuno, con Clearene. Dopo incubazione per 10 minuti con acqua ossigenata al 3%, per inibire le
perossidasi endogene, è stata allestita l’analisi immunoistochimica secondo la procedura dell’avidinabiotina-perossidasi. Le sezioni sono state incubate per 18 ore a +4°C con l’anticorpo anti-Y1 (antisiero
purificato, contro questo recettore; gentilmente donato dal Dr. B. Bunnemann, GSK, Verona), diluito
1:100 in un tampone Tris salino (TBS: Tris-HCl 0.05 M contenente 0.85% di NaCl 0.5M) a pH 7.4,
addizionato dell’1% di albumina sierica bovina, 0,1% di sodio azide, 1% di siero di capra e 5% di siero
normale umano. Il giorno seguente, dopo tre lavaggi con TBS-Triton a pH 7.4, i preparati sono stati
incubati con antisiero secondario biotinato, diluito 1:200 in TBS e addizionato con siero normale umano al
5%, per un’ora a temperatura ambiente. Si è proceduto quindi all’amplificazione della reazione mediante
incubazione delle sezioni per 30 minuti, a temperatura ambiente, con il complesso avidina-biotinaperossidasi (Vector Laboratories, California, USA). Lo sviluppo della reazione è stato eseguito mediante
immersione dei preparati in una soluzione acquosa di 3.3 diaminobenzidina allo 0,03% in 90 mL di
tampone TBS, addizionata con 100 µL di perossido di idrogeno al 3%. L’arresto dello sviluppo della
reazione è stato ottenuto per immersione del vetrino in acqua distillata in modo da poter controllare la
colorazione al microscopio ottico.
La stessa analisi utilizzando uno specifico anticorpo primario anti-hZAC (prodotto in coniglio da Pagotto e
coll. (529)) diluito 1:800 in un buffer di PBS w/o Ca2+/Mg2+ 0.01 M pH 7.4 contenente lo 0.1% di Triton e lo
0.5% di BSA è stata condotta sulle cellule androgeno dipendenti LNCaP.
Analisi Western Blot: Le tre linee cellulari prostatiche sono state raccolte in RIPA buffer (0.05 M di 1M
tris HCl Stock pH 7.7, 0.15 M NaCl, 0.8% di Triton 10%, 0.08% di SDS, 10 mM di 0.5 M EDTA, 100 µM di
75
Sodio vanadato, 0.8% di Sodio deossicolico, 50 mM di NaF, 5 mM di acido iodoacetico, acqua distillata)
(530) contenente l’1% di inibitore delle proteasi (Sigma-Aldrich, Milano, Italia) allo scopo di prevenire la
degradazione enzimatica. La concentrazione proteica dei campioni è stata quantificata con il kit BCA
protein assay (Pierce, Rockford, IL), utilizzando una curva standard di albumina di siero bovino (BSA,
Sigma in RIPA buffer (2 mg/mL). La densità ottica (OD) degli standard e dei campioni è stata letta ad una
lunghezza d’onda () di 550 nm. I punti sono poi stati interpolati secondo un’equazione quadratica del tipo
y=a+bx+cx2. Sia i campioni (75 μg), risospesi in 2X Laemmli buffer, che il Raimbow marker (10 μL,
Amersham Bioscience, Milano, Italia) sono stati separati mediante SDS-PAGE (Ready-Gel, 10%, Bio-Rad
Laboratories, Milano, Italia). Le proteine sono state trasferite a 30 volt, per 18 ore, su una membrana di
nitrocellulosa (Bio-Rad Laboratories, Milano, Italia). Dopo i passaggi di lavaggio e bloccaggio, il blot è
stato incubato (+4°C), tutta la notte, con una specifica diluizione dell’anticorpo primario (anti-Y1: 1:700,
descritto precedentemente; anti-ERK1/2 1:900 e anti-pERK 1:100 (entrambi Santa Cruz Biotechnology,
Santa Cruz, CA)). La membrana viene infine incubata con l’anticorpo secondario, coniugato con
perossidasi di rafano (Santa Cruz Biotechnology, Santa Cruz, CA) per 1 ora e 30 minuti a temperatura
ambiente. L’immunoreattività specifica è stata evidenziata utilizzando la soluzione SuperSignal West Pico
Substrate (Pierce, Rockford, IL, USA). Blottando un controllo negativo sulla membrana ed incubandolo
soltanto con anticorpo secondario, nessuna banda è stata rilevata.
Per quanto riguarda l’analisi Western blot dei cinque recettori ssts, le cellule LNCaP e Calu-6 (controllo
positivo) (531), sono state solubilizzate in un buffer di lisi (20 mM HEPES, 5 mM EDTA, 3 mM EGTA, 150
mM NaCl, 1 mM fenilmetilsulfonil fluoruro (PMSF), 10 µg/ml Soybean trypsin inhibitor, 10 µg/ml
leupeptina, 50 µg/ml bacitracina, 4 mg/ml dodecil-β-D-maltoside) per 1 ora a +4°C e susseguentemente
centrifugate per 1 ora a +4°C a 100.000 x g. Le proteine glicosilate sono state immobilizzate su colonne,
precedentemente equilibrate con buffer di lisi, utilizzando 0.5 ml di wheat germ agglutinin (WGA, Vector
Laboratories, Inc., Burlingame, CA). La colonna è stata quindi lavata ed eluita con il buffer di lisi
contenente N,N,N-triacetyl-chitotriose 3 mM (Sigma-Aldrich, Milano, Italia). Il contenuto proteico
dell’eluato è stata valutato mediante il saggio di Bradford. Dieci µg di proteine glicosilate sono stati
denaturati e frazionati attraverso SDS-PAGE (Ready-Gel, 12,5%, Bio-Rad Laboratories, Milano, Italia) e
trasferiti elettroforeticamente su membrane di nitrocellulosa (Hybond C-extra). Dopo il transfer, i siti di
binding aspecifici sono stati bloccati incubando le membrane, per un’ora a +22°C, con un buffer salino di
Tris Base-Tween (TBS-T: 0,02 M Tris Base, 0,137 M NaCl, 0,5% Tween 20; pH 7,6 aggiustato con HCl
76
1M) contenente il 5% di latte non grasso. Dopo 5 lavaggi con TBS-T, le membrane sono state incubate
per 16 ore a +4°C con una soluzione (TBS-T contenenti 1% di BSA) 1:500 di anticorpi policlonali specifici
per sst1, sst2A, sst3, sst5 (Santa Cruz Biotechnology, Santa Cruz, CA). Le membrane sono state lavate
5 volte con TBS-T, incubate per 1 ora a 22°C con una soluzione 1:2000 di un anticorpo secondario (antirabbit) coniugato con perossidasi di rafano (Santa Cruz Biotechnology, Santa Cruz, CA), lavate ed
immerse per 1 minuto nella soluzione di chemiluminescenza. Successivamente le membrane sono state
esposte per circa 30 secondi per generare gli immunoblots.
Studi di proliferazione cellulare: Per gli esperimenti di conta cellulare, le cellule LNCaP, DU145 e PC3
sono state seminate in Petri dal diametro di 60 mm. Dopo 72 ore dalla semina, il medium è stato
sostituito con il medium sperimentale (RPMI 1640/0,1% BSA o 2% di FBS) e le cellule trattate sia con
NPY umano (Bachem, Bubendorf, Svizzera), a concentrazioni comprese tra 10-7 e 10-10M, che con un
antagonista Y1-specifico, BIBP3226 (Bachem, Bubendorf, Svizzera). Dopo 96 ore di incubazione, le
cellule sono state raccolte con tripsina 0,05%/EDTA 0.02% e risospese in RPMI 1640. La conta cellulare
e la vitalità delle stesse è stata valutata attraverso la conta con trypan-blue. Lo stesso protocollo è stato
utilizzato per valutare l’efficacia della SS14 (10-7-10-11M; Sigma-Aldrich, Milano, Italia) sulla proliferazione
della linea cellulare androgeno dipendente LNCaP.
Per quanto riguarda gli studi condotti utilizzando l’incorporazione di timidina triziata, le cellule LNCaP,
DU145 e PC3 sono state seminate in multiwell da 24, rispettivamente ad una densità di 3,5 x 10 4, 1,5 x
104 e 2 x 104 cellule per pozzetto. Dopo 24 ore dalla semina, il medium è stato sostituito con il medium
sperimentale (RPMI 1640/0,1% BSA o 2% di FBS) contenente un inibitore specifico della MAP chinasi
(MEK), U0126 (10-6 M; Biomol, Plymouth Meeting, PA, USA) o della PKC, GF109203X (10-7 M; SigmaAldrich, Milano, Italia). Trenta minuti dopo, è stato aggiunto NPY umano (10 -8 M; Bachem, Bubendorf,
Svizzera) per ulteriori 48 ore. Sei ore prima della fine dell’incubazione, sono stati aggiunti 2 μCi/pozzetto.
Dopo un’incubazione di 30 minuti con TCA 5%, le cellule sono state risospese in 300 μL di NaOH 0,1 N e
5 ml di liquido di scintillazione. I cpm sono stati misurati utilizzando un β-counter. Per valutare gli effetti
sull’incorporazione di timidina triziata in cellule LNCaP dopo trattamento con SS14 e alcuni analoghi
sintetici specifici (10-7-10-11M; tabella 1), si è seguito lo stesso protocollo, ad eccezione della durata del
trattamento, che è stato di 24 ore anziché 48 ore.
Effetto dell’NPY sulle concentrazioni di cAMP: Le cellule LNCaP, DU145 e PC3 sono state seminate
in multiwell da 24 pozzetti. Una volta a confluenza, sono state lavate con RPMI 1640 ed incubate a 37°C,
77
per 15 minuti, con 200 μl di RPMI 1640 contenente isobutilmetilxantina 0,5 mM (Sigma-Aldrich, Milano,
Italia). Successivamente, sono stati aggiunti, ad ogni pozzetto, per ulteriori 15 minuti, 200 μl di forskolina
(Sigma-Aldrich, Milano, Italia) e di NPY umano 10-8 M. Le cellule sono state raccolte utilizzando 500 μl di
etanolo freddo al 75% e le membrane precipitate, centrifugando a 10000 rpm, per 3 minuti. Il surnatante è
stato risospeso in un tampone RIA (radioimmunoassay) ed il contenuto di cAMP quantificato con un kit
RIA commerciale (Amersham Bioscience Europe, Milano, Italia).
Effetto del trattamento di NPY sulle [Ca+2]i: L’effetto dell’NPY sui livelli intracellulari di Ca++ è stato
determinato attraverso l’utilizzo della sonda fluorescente fura-2 acetossimetil estere (fura-2; Molecular
Probes, Eugene, OR, USA), secondo il protocollo di Grynkiewicz (532). Le cellule sono state seminate su
vetrini aventi un diametro di 12 mm e, una volta raggiunta la confluenza del 70%, sono state incubate con
fura-2 (1 M) a 37°C per 45/60 minuti e successivamente lavate due volte con una soluzione salina (NaCl
1 M, KCl 1 M, MgCl2 1 M, HEPES pH 7.4 1 M e glucosio 1 M). I vetrini sono stati quindi inseriti, uno per
volta, in una cuvetta del fluorometro (Fast Filter Polariser, mod. LS 50B, Perkin Elmer, Norwalk, CT,
USA), a cui è stata aggiunta la soluzione salina contenente CaCl 2 1 mM. Una volta stabilizzato il sistema,
è stato aggiunto NPY 10-8 M ed è stata misurata la fluorescenza. La corrispondente concentrazione di
Ca++ intracellulare è stata quantificata con l’equazione descritta da Grynkiewicz (532). Per ogni vetrino è
stata effettuata la calibrazione del fluorimetro mediante l’aggiunta di ionomicina 10 -3 M (concentrazione
finale di 2.7 M) e di digitonina 50 mM (concentrazione finale di 100 M) che permettono di valutare la
massima quantità di Ca++ legata al fura-2, e EGTA 0.5 M che consente invece di valutare i minimi livelli di
Ca++ legati al fura-2 perché, avendo una maggior affinità per il Ca++ rispetto al fura-2, lo sottrae ad esso.
Analisi statistica dei dati: L'analisi statistica è stata realizzata usando il programma Prism 4.0. I dati
sono presentati come media dei valori ottenuti ± errore standard. La lettera N indica il numero di replicati
in un esperimento. La significatività delle differenze ottenute tra i gruppi dei trattamenti è stata valutata
con l'analisi della varianza (ANOVA), seguita dal Tukey test, considerata significativa per valori di p<0,05.
78
6. RISULTATI
Espressione dell’NPY e del Y1. L’espressione dell’NPY e del Y1 sono state inizialmente valutate a
livello genico attraverso l’analisi RT-PCR. Il gene dell’NPY presente nella linea cellulare di neuroblastoma
umano SH-SY5Y usata come controllo positivo, non è espresso in nessuna delle tre linee di carcinoma
prostatico (PCa) studiate (figura 1, A). Un prodotto di 354 bp, corrispondente al Y1, insieme ad un’altra
banda più grande (+ 97 bp), corrispondente a 451 bp, è presente in tutte le linee cellulari di PCa ed
anche nella linea cellulare di neuroblastoma umano SK-N-MC, utilizzata come controllo positivo (533).
L’analisi RT-PCR per la actina (660 bp), gene ad espressione costitutiva, è stata effettuata per
confermare la presenza di cDNA nei campioni utilizzati (Figura 1, B). L’analisi Western blot, utilizzando un
anticorpo anti-Y1 specifico, ha mostrato in tutte le linee cellulari di PCa, una banda specifica
immunoreattiva all’altezza di 70 kDa (figura 2, A); l’analisi immunoistochimica, utilizzando lo stesso
anticorpo, su campioni post-operatori di carcinoma prostatico ha evidenziato che il recettore Y1 è
presente anche a livello della membrana plasmatica delle cellule epiteliali tumorali (figura 2, B).
Effetto dell’NPY sulla proliferazione cellulare delle PCa. Un trattamento di 96 ore con NPY, a
concentrazioni comprese tra 10-10 e 10-7 M, determina una riduzione significativa della proliferazione nelle
linee cellulari LNCaP e DU145, con un effetto massimo di inibizione, rispettivamente del -45/63% (alla
dose di NPY 10-8 M) e -33/35% (alla dose di NPY 5X10-8 M); al contrario, un marcato effetto proliferativo è
stato osservato per l’altra linea cellulare androgeno indipendente PC3, con un massimo effetto alla dose
di NPY 10-8 M (+36/60%) (figura 3). Successivamente, abbiamo valutato se l’effetto sulla proliferazione
cellulare fosse mediato dal recettore Y1, presente in tutte e tre le linee di carcinoma prostatico analizzate.
Il trattamento con BIBP3226, un antagonista specifico per il recettore Y1 (521), alla concentrazione di 1
M abolisce entrambi gli effetti proliferativo (PC3) e antiproliferativo (LNCaP e DU145) dell’NPY (figura
4). Il solo trattamento con NPY e BIBP3226 non modifica la sopravvivenza cellulare.
Effetto del trattamento con NPY sulle vie intracellulari di segnale. Per comprendere i meccanismi
intracellulari alla base degli effetti dell’NPY sulla proliferazione cellulare, abbiamo valutato nello specifico
il coinvolgimento della via MAPK/ERK1/2 dopo trattamento delle linee cellulari di PCa con NPY 10 -8 M. Le
cellule LNCaP mostrano un’attivazione costitutiva della via ERK1/2 senza subire alcuna modificazione da
79
parte dell’NPY. Al contrario, nelle cellule DU145 e PC3, il trattamento con l’NPY determina un marcato
aumento della fosforilazione di ERK1/2 entro 5-15 minuti, mentre nelle PC3 tale attivazione è transitoria,
con un ritorno ai valori basali già a 30 minuti; nelle DU145 i livelli rimangono elevati per almeno 6 ore
(figura 5). Inoltre in ambedue le linee cellulari androgeno indipendenti, DU145 e PC3, un pretrattamento
(45 minuti) con BIBP3226 (10-6 M) riduce marcatamente la fosforilazione NPY-dipendente di ERK1/2;
nella linea cellulare DU145, il BIBP3226 da solo attiva la fosforilazione di ERK1/2 (figura 7). Per valutare
se l’attivazione di ERK1/2, da parte dell’NPY, fosse realmente collegata alla proliferazione cellulare,
abbiamo condotto esperimenti di proliferazione, mediante valutazione dell’incorporazione di timidina
triziata, dopo trattamento con l’inibitore di MEK (U0126) e NPY 10 -8 M (figura 8). Nelle cellule LNCaP, il
pretrattamento con UO126 10-6 M, seguito dal trattamento con NPY 10-8 M, determina un effetto inibitorio
maggiore rispetto a quello mediato dal solo NPY, suggerendo che l’effetto dell’NPY sulla proliferazione
cellulare è almeno in parte indipendente dall’attivazione di ERK1/2. Nelle cellule DU145, la riduzione della
proliferazione indotta dall’NPY non è influenzata dal trattamento con UO126; al contrario nelle cellule
PC3 l’efficacia dell’NPY nello stimolare la proliferazione cellulare è prevenuta dal trattamento con UO126
(NPY vs NPY + UO126: p < 0,001), indicando che l’attivazione di ERK1/2 è coinvolta in questo
meccanismo. Nelle cellule LNCaP e DU145, i trattamenti con l’NPY e il GF109203X (inibitore di PKC)
riducono la proliferazione cellulare allo stesso modo di quanto avviene con il solo NPY, mentre nelle
cellule PC3, l’aggiunta di GF109203X annulla l’effetto proliferativo indotto dall’NPY. In alcuni sistemi
cellulari è stato dimostrato che l’attivazione della PKC è un evento a monte della fosforilazione di ERK1/2
(522); questo sembra il caso delle cellule PC3, poiché il pretrattamento con GF109203X blocca la
fosforilazione di ERK1/2 indotta dall’NPY (figura 6). Al contrario, nelle cellule DU145 il pretrattamento con
GF109203X non influenza la fosforilazione di ERK1/2 indotta dall’NPY, suggerendo che quest’ultimo
evento è indipendente dalla PKC (figura 6).
Dato che il recettore Y1 è generalmente accoppiato all’inibizione dell’adenilato ciclasi e all’incremento dei
livelli intracellulari di Ca++ (93), abbiamo preso in considerazione anche queste due vie. Per gli studi sul
cAMP, le cellule LNCaP, DU145 e PC3 sono state trattate per 15 minuti con NPY 10 -8 M in presenza o in
assenza di forskolina (FSK) 5 M. In tutte le tre linee cellulari, il trattamento con l’NPY da solo non
modifica i livelli basali di cAMP, mentre, come atteso, l’esposizione alla FSK induce un marcato
incremento delle concentrazioni intracellulari di cAMP. Il trattamento con l’NPY riduce l’aumento di cAMP
indotto da FSK solo nelle PC3, mentre risulta inefficace nelle LNCaP e nelle DU145 (figura 9),
80
suggerendo che i recettori dell’NPY modulano negativamente l’adenilato ciclasi solo nelle PC3. Per
valutare l’effetto del trattamento dell’NPY sui livelli intracellulari di calcio, le cellule prostatiche sono state
incubate con la sonda fluorescente fura-2 per 45/60 minuti e quindi esposte a NPY 10-8 M. La
concentrazione di Ca++ intracellulare corrispondente è stata calcolata mediante l’equazione di
Grynkiewicz (532). Il trattamento con NPY non produce alcun cambiamento della concentrazione di Ca ++
nelle tre linee considerate. L’integrità e la corretta responsività dei sistemi cellulari usati sono state
successivamente valutate mediante l’esposizione all’adenosina trifosfato (ATP) 100 µM che, come
riportato da Janssens (534), induce un marcato incremento di Ca++ intracellulare nelle linee cellulari
androgeno indipendenti, DU145 e PC3, ma non in quella androgeno dipendente, LNCaP, per la quale è
stato utilizzato come controllo positivo lo inoforo A23187 10-6 M (535) (figura 10).
Espressione della SS14, dei cinque ssts, di ZAC e del PSA. Nella linea cellulare di PCa umano
androgeno dipendente LNCaP, l’espressione genica della SS14, dei suoi cinque sottotipi recettoriali
(sst1, sst2A, sst3, sst4, sst5), del tumor suppressor gene ZAC e del PSA è stata valutata mediante analisi
RT-PCR. Come controllo positivo è stato utilizzato un campione di NFPA (non functional pituitary
adenoma). Le cellule LNCaP esprimono sia la SS14 (349 bp), che i recettori sst1, sst2A, sst3 e sst5,
corrispondenti a specifiche bande rispettivamente di 318 bp, 414 bp, 95 bp e 226 bp. Nessuna banda è
stata invece evidenziata, all’altezza aspettata (321 bp), per il recettore sst4 (figura 11). Le cellule LNCaP
esprimono anche il gene ad azione antiproliferativa ZAC (308 bp) e il PSA (376 bp). L’analisi RT-PCR per
il 18S (217 bp), gene ad espressione costitutiva è stata effettuata per confermare la presenza di cDNA
nei campioni utilizzati (figura 12). Mediante analisi immunocitochimica, utilizzando un anticorpo anti-ZAC,
è stata evidenziata a livello nucleare la proteina ZAC (figura 13), mentre l’analisi Western blot, utilizzando
anticorpi ssts specifici, ha confermato i precedenti risultati dell’espressione genica, evidenziando bande
immunoreattive specifiche, sia nelle cellule LNCaP che nelle Calu-6 (controllo positivo) del peso di 62
kDa per sst1 e sst2 (figura 14), di 47 kDa per sst3 e 80 kDa per sst5 (figura 15).
Effetto del trattamento con SS14 e analoghi sulla proliferazione delle cellule LNCaP.
Per valutare gli effetti della SS14 e di alcuni analoghi sintetici specifici sull’incorporazione di timidina
triziata, per ogni composto sono stati valutati tre tempi di trattamento: 24, 48 e 72 ore. Considerando per
la maggior parte di questi composti significativi i dati ottenuti dopo 24 ore di trattamento, è emerso che la
81
SS14 esercita un effetto antiproliferativo con un picco di inibizione massima alla concentrazione di 10-10
M, corrispondente ad una riduzione del 43.5% (p<0.01 vs. controllo) (figura 16, A). A supporto di ciò,
trattamenti per 96 ore con SS14 hanno evidenziato una riduzione massima del numero di cellule (conta
cellulare) del - 36%, ad una concentrazione di 10-9 M (Figura 16, B). Il lanreotide, un analogo ad alta
affinità per il recettore sst2 (IC50 0,75 nM) e in minor misura per sst5 (IC50 12,7 nM), ha mostrato un
effetto antiproliferativo con inibizione massima alla dose di 10 -11 M. In questo caso l’incorporazione di
timidina è stata ridotta del -55.3% (p<0.001 vs. controllo) (figura 17, A). Il composto A, affine ad sst2
(IC50 0,29 nM) e sst5 (IC50 0,67 nM), è risultato efficace a concentrazioni comprese tra 10 -8 e 10-11 M,
riducendo l’incorporazione di timidina triziata del -62% (p0.001 vs. controllo) (Figura 17, B). Il composto
B, specifico per sst5 (IC50 2,4 nM), ha evidenziato dopo 24 ore di trattamento un effetto antiproliferativo
massimo di -81% alla concentrazione di 10-11 M (p<0.001 vs. controllo) (Figura 18).
82
7. DISCUSSIONE
Dal presente studio è emerso che l’attivazione del sottotipo recettoriale Y1, presente nelle linee cellulari
di PCa umano studiate, ha un effetto sia stimolatorio (PC3) che inibitorio (LNCaP e DU145) sulla
proliferazione cellulare. I sistemi di trasduzione del segnale associati a questi effetti sembrano differire a
seconda della linea cellulare: nelle cellule LNCaP, non c’è alcun coinvolgimento di ERK1/2, mentre nelle
cellule DU145 l’NPY induce una rapida e sostenuta fosforilazione di ERK1/2. Al contrario, nelle cellule
PC3, l’effetto proliferativo dell’NPY sembra essere mediato dalla cascata PKC-ERK1/2, così come
dall’inibizione della produzione di cAMP indotto da FSK. L’analisi dell’espressione genica dell’mRNA del
Y1, in aggiunta al prodotto principale di 354 bp mostra un prodotto della grandezza di 451 bp.
Quest’ultimo contiene un piccolo introne (97 bp) con un codone di stop, che potrebbe codificare per una
variante del recettore Y1 con solo cinque regioni transmembrana (93, 536). Riguardo il possibile ruolo
funzionale svolto da questo introne, in letteratura è stato proposto un suo coinvolgimento soltanto nella
produzione della proteina Y1, attraverso un meccanismo post-trascrizionale (537).
L’analisi Western blot del Y1 ha mostrato una banda di 70 kDa, così come riportato da precedenti lavori
condotti sia in vitro che ex vivo (538, 539), che mostrano come il Y1 è espresso a livello del tessuto
prostatico (111). Trattamenti con NPY riducono la proliferazione cellulare nella linea cellulare LNCaP e
DU145, mentre stimolano quella delle PC3. La concentrazione alla quale si ottengono i massimi effetti
(NPY 10-8 M) per ogni linea cellulare è in accordo con il range d’azione dei peptidi (282), così come con
l’affinità dell’NPY ai siti di legame specifici presenti nelle cellule PC3 (Kd: 30 nM) (245). L’attivazione del
recettore Y1 sembra mediare tali effetti, così come dimostrato dagli esperimenti condotti utilizzando
l’inibitore specifico BIBP3226 per il Y1. Questi dati suggeriscono che le altre isoforme recettoriali
dell’NPY, il Y2 nelle PC3 e Y4 nelle DU145 (osservazioni personali), non dovrebbero essere coinvolte
nella modulazione della proliferazione cellulare. Anche in letteratura è riportato che il Y1 sembra mediare
sia l’effetto proliferativo che antiproliferativo in differenti tessuti e sistemi cellulari, incluse la muscolatura
liscia vascolare, le cellule endoteliali (540), le cellule gliali danneggiate e le cellule dell’epitelio olfattorio
dei roditori (541). Gli effetti mediati dall’NPY sulle cellule di PCa umano sembrano abbastanza specifici
per la proliferazione cellulare, poiché questo peptide non ha effetti né sull’invasione né sulla migrazione
delle cellule PC3 (542), LNCaP e DU145 (543). Le due linee cellulari androgeno indipendenti hanno un
comportamento opposto, poiché il Y1 media rispettivamente un effetto proliferativo (PC3) o
83
antiproliferativo (DU145). I nostri risultati sulla via MAPK/ERK1/2 hanno evidenziato delle differenze
sostanziali tra le tre linee cellulari. Le cellule LNCaP mostrano un’attivazione costitutiva di questa via,
così come altre linee cellulari (544), con nessuna modulazione da parte dell’NPY che quindi riduce la
proliferazione cellulare delle LNCaP attraverso meccanismi che potrebbero essere, almeno in parte,
indipendenti dall’attivazione di ERK1/2, come ad esempio
la via della IP3K (545, 546). La perdita
apparente del coinvolgimento di ERK1/2 nel controllo della crescita cellulare delle LNCaP non sembra
esclusiva per l’NPY, ma è stata anche dimostrata per il diidrotestosterone, il principale fattore trofico
prostatico (547). In entrambe le linee cellulari androgeno indipendenti DU145 e PC3, il trattamento con
NPY stimola rapidamente la fosforilazione di ERK1/2, che velocemente ritorna a livelli basali nelle cellule
PC3, ma mostra livelli persistenti ed elevati (almeno 6 ore) nelle cellule DU145. Questo differente profilo
di attivazione della MAPK/ERK/1/2 è associato con un opposto effetto antiproliferativo esercitato dal
sistema NPY/Y1, anche perché in ambedue queste linee cellulari abbiamo dimostrato che l’attivazione di
ERK1/2 NPY-mediata è Y1-dipendente. I nostri dati si allineano a studi precedenti, condotti su cellule
tumorali o immortalizzate, che associavano una persistente attivazione di ERK1/2 ad un effetto
antiproliferativo (548), come osservato nelle cellule DU145, mentre una rapida e transitoria attivazione di
ERK1/2 ad un incremento della proliferazione (PC3). Ciò supporta il concetto che, almeno per queste due
linee cellulari, la classificazione di androgeno indipendente non è fondamentale per spiegare le differenze
viste nella modulazione della crescita cellulare. Il coinvolgimento della MAPK nella modulazione della
crescita cellulare da parte dell’NPY, via recettore Y1, come già descritto in altri sistemi cellulari può
essere via PKC (549), o PKC-indipendente (96). I nostri dati mostrano che l’attivazione della PKC è
necessaria per la fosforilazione di ERK1/2 nelle cellule PC3, mentre nelle cellule DU145, questo processo
è chiaramente PKC-indipendente, suggerendo la dicotomia nella risposta all’NPY delle due linee cellulari
androgeno indipendenti. Inoltre, i dati qui riportati indicano che il trattamento con NPY inibisce l’accumulo
di cAMP indotto dalla FSK solo nelle cellule PC3, ma non nelle cellule LNCaP e DU145. Al contrario di
quanto avviene per le cellule neuronali che esprimono i recettori per l’NPY, in nessuna delle cellule di
PCa studiate l’NPY modifica i livelli di Ca+2 intracellulare. Sebbene i nostri dati evidenzino l’assenza del
gene dell’NPY nelle tre linee di PCa studiate, escludendo quindi possibili effetti autocrini, esso è presente
nella prostata dell’uomo (281), quindi l’NPY potrebbe essere rilasciato dalle cellule neuroendocrine o
dalle terminazioni nervose per raggiungere i recettori specifici vicino alle cellule epiteliali di origine
tumorale; infatti i meccanismi neuroendocrini potrebbero giocare un ruolo cruciale nella regolazione
84
locale della crescita e della differenziazione delle cellule di PCa, in modo particolare nella fase di
androgeno indipendenza (550).
Poiché il PCa è un tumore eterogeneo che nelle fasi più avanzate perde la responsività alle manipolazioni
ormonali convenzionali, si stanno sperimentando dei trattamenti endocrini alternativi quali l’utilizzo di
antagonisti per LHRH, analoghi per LHRH con radicali citotossici e più recentemente la somatostatina e i
suoi analoghi sintetici. L’effetto antiproliferativo della somatostatina è stato documentato su diversi tipi di
tumori benigni e maligni (551); tuttavia a causa dei suoi effettivi non recettore specifici e della breve
emivita plasmatica, sono stati disegnati e sintetizzati degli analoghi più resistenti alla degradazione
metabolica e quindi con una maggiore durata d’azione. Partendo dal presupposto che studi precedenti in
vitro hanno evidenziato differenze nell’espressione dei diversi sottotipi recettoriali tra tessuto prostatico
normale e tumorale (414), e poiché l’esatta funzione mediata dai diversi sottotipi recettoriali in cellule di
PCa non è ad oggi ben definita, abbiamo voluto analizzare nello specifico gli effetti mediati dalla
somatostatina, come già avviene per numerose altre linee cellulari (395), e di alcuni analoghi recettorespecifici sulla proliferazione della linea di PCa umano androgeno dipendente LNCaP. In questo studio,
l’analisi RT-PCR e Western blot hanno evidenziato, nelle cellule LNCaP, la presenza di tutti i recettori
ssts ad eccezione del sottotipo sst4. Basandoci sul fatto che in letteratura c’è una crescente evidenza che
la somatostatina regola la proliferazione cellulare, sia arrestando la crescita attraverso i recettori sst1,
sst2A, sst4 e sst5, sia inducendo apoptosi attraverso il sottotipo sst3 (552), abbiamo valutato l’efficacia di
analoghi sintetici della somatostatina, nell’influenzare la crescita delle cellule LNCaP. I nostri dati sulla
proliferazione
cellulare
mostrano
che
sia
la
somatostatina,
che
i
due
analoghi
specifici
contemporaneamente per sst2 e sst5 (lanreotide e composto A) e quello monospecifico per sst5
(composto B) sono in grado di inibire, rispetto al controllo, l’incorporazione di timidina in un range di dosi
compreso tra 10-9 M e 10-11 M, così come riportato da altri studi che hanno valutato le stesse sostanze
(518, 553). Poiché la ricerca nel campo dei tumor suppressing genes (TSG) è stata ampiamente estesa
negli anni passati proprio per la loro rilevanza nell’iniziazione e nella progressione delle patologie
tumorali, un possibile coinvolgimento in questi processi è stato anche suggerito per uno dei TSG più
recentemente identificato, hZAC, una zinc-finger protein in grado di controllare contemporaneamente
l’apoptosi e la progressione del ciclo cellulare arrestando le cellule in G1. L’espressione di hZAC è stata
studiata in diverse linee cellulari di tumore della mammella (554) dove si è osservata una diminuita
espressione del gene o addirittura una perdita della funzione del gene rispetto al controllo normale. La
85
proteina hZAC è stata da noi identificata, mediante analisi ICC, nel nucleo dellle linee cellulari di
carcinoma prostatico LNCaP. Essendo stata recentemente dimostrata una correlazione tra la
somatostatina e l’espressione di hZAC a livello della ghiandola ipofisaria (555), sarebbe interessante
studiare gli effetti degli analoghi della somatostatina sull’espressione di hZAC; questo tipo di studio
potrebbe chiarire infatti le modalità di azione di questi analoghi più specifici e potenti, e suggerire nuove
applicazioni terapeutiche di questi composti.
Il dosaggio plasmatico del PSA viene utilizzato normalmente in clinica in associazione ad altri esami
specifici come test diagnostico di patologie prostatiche, in particolare è un test di conferma utile nel caso
di tumori alla prostata e nel follow-up post operatorio e terapeutico con un indiscutibile valore prognostico.
Nell’ambito del nostro studio la valutazione dei livelli sia di mRNA del PSA che della proteina stessa
rilasciata nel medium di coltura, in seguito a trattamento con quegli analoghi della somatostatina in grado
di bloccare la proliferazione cellulare, potrebbero suggerire utili informazioni sull’efficacia di questi
composti riproducendo in vitro ciò che ci si aspetterebbe in vivo (556) in presenza di molecole in grado di
bloccare la proliferazione tumorale.
86
8. TABELLE
Composti
sst1
sst2
sst3
sst4
sst5
SS14
1,95
0,25
1,2
1,77
1,41
Lanreotide
2129
0,75
98
18,26
12,7
A
1020
0,29
133
>1000
0,67
B
1152
166
1000
1618
2,4
Tabella 1
Specificità recettoriale della SS14 umana e di alcuni suoi analoghi agonisti. I valori riportati si riferiscono
alle IC50 (nM).
87
9. FIGURE
9. BIBLIOGRAFIA
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
Kirby RS 2001 Prostate Cancer
Wilson JD 1984 Sexual Differentiation: Basic and Clinical Aspects. Raven Press, New York
Wilson JD 1992 Syndromes of androgen resistance. Biol Reprod 46:168-73
Donahoe PK, Budzik GP, Trelstad R, Mudgett-Hunter M, Fuller A, Jr., Hutson JM, Ikawa H,
Hayashi A, MacLaughlin D 1982 Mullerian-inhibiting substance: an update. Recent Prog Horm
Res 38:279-330
Jost A 1965 In: Organogenesis
Brading AF, Turner WH 1994 The unstable bladder: towards a common mechanism. Br J Urol
73:3-8
Garde SV, Sheth AR, Shah MG, Kulkarni SA 1991 Prostate--an extrapituitary source of folliclestimulating hormone (FSH): occurrence, localization, and de novo biosynthesis and its hormonal
modulation in primates and rodents. Prostate 18:271-87
Hodge KK, McNeal JE, Terris MK, Stamey TA 1989 Random systematic versus directed
ultrasound guided transrectal core biopsies of the prostate. J Urol 142:71-4; discussion 74-5
Ayala AG, Ro JY, Babaian R, Troncoso P, Grignon DJ 1989 The prostatic capsule: does it
exist? Its importance in the staging and treatment of prostatic carcinoma. Am J Surg Pathol
13:21-7
Brawer MK, Peehl DM, Stamey TA, Bostwick DG 1985 Keratin immunoreactivity in the benign
and neoplastic human prostate. Cancer Res 45:3663-7
Esposti PL 1971 Cytologic malignancy grading of prostatic carcinoma by transrectal aspiration
biopsy. A five-year follow-up study of 469 hormone-treated patients. Scand J Urol Nephrol 5:199209
Gleason DF 1977 Histologic grading and clinical staging of prostatic cancer
Serio M, Salerno R 2004 ORMONI E TUMORI ENDOCRINO-DIPENDENTI
Crawford ED, Eisenberger MA, McLeod DG, Spaulding JT, Benson R, Dorr FA,
Blumenstein BA, Davis MA, Goodman PJ 1989 A controlled trial of leuprolide with and without
flutamide in prostatic carcinoma. N Engl J Med 321:419-24
Ben-Josef E, Yang SY, Ji TH, Bidart JM, Garde SV, Chopra DP, Porter AT, Tang DG 1999
Hormone-refractory prostate cancer cells express functional follicle-stimulating hormone receptor
(FSHR). J Urol 161:970-6
Smith JR, Freije D, Carpten JD, Gronberg H, Xu J, Isaacs SD, Brownstein MJ, Bova GS,
Guo H, Bujnovszky P, Nusskern DR, Damber JE, Bergh A, Emanuelsson M, Kallioniemi
OP, Walker-Daniels J, Bailey-Wilson JE, Beaty TH, Meyers DA, Walsh PC, Collins FS, Trent
JM, Isaacs WB 1996 Major susceptibility locus for prostate cancer on chromosome 1 suggested
by a genome-wide search. Science 274:1371-4
Bruchovsky N, Lesser B, Van Doorn E, Craven S 1975 Hormonal effects on cell proliferation in
rat prostate. Vitam Horm 33:61-102
Isaacs JT 1984 Antagonistic effect of androgen on prostatic cell death. Prostate 5:545-57
Oberhammer F, Wilson JW, Dive C, Morris ID, Hickman JA, Wakeling AE, Walker PR,
Sikorska M 1993 Apoptotic death in epithelial cells: cleavage of DNA to 300 and/or 50 kb
fragments prior to or in the absence of internucleosomal fragmentation. Embo J 12:3679-84
Wright SC, Zheng H, Zhong J, Torti FM, Larrick JW 1993 Role of protein phosphorylation in
TNF-induced apoptosis: phosphatase inhibitors synergize with TNF to activate DNA
fragmentation in normal as well as TNF-resistant U937 variants. J Cell Biochem 53:222-33
Pienta KJ, Esper PS 1993 Risk factors for prostate cancer. Ann Intern Med 118:793-803
Ghanadian R, Puah CM, O'Donoghue EP 1979 Serum testosterone and dihydrotestosterone in
carcinoma of the prostate. Br J Cancer 39:696-9
Bruchovsky N, Wilson JD 1968 The conversion of testosterone to 5-alpha-androstan-17-betaol-3-one by rat prostate in vivo and in vitro. J Biol Chem 243:2012-21
Glantz GM 1964 Cirrhosis And Carcinoma Of The Prostate Gland. J Urol 91:291-3
Blair A, Fraumeni JF 1978 Geographic patterns of prostate cancer in the United States. J Natl
Cancer Inst 61:1379-84
Giovannucci E, Rimm EB, Colditz GA, Stampfer MJ, Ascherio A, Chute CC, Willett WC 1993
A prospective study of dietary fat and risk of prostate cancer. J Natl Cancer Inst 85:1571-9
88
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
Le Marchand L, Kolonel LN, Wilkens LR, Myers BC, Hirohata T 1994 Animal fat consumption
and prostate cancer: a prospective study in Hawaii. Epidemiology 5:276-82
Whittemore AS, Wu AH, Kolonel LN, John EM, Gallagher RP, Howe GR, West DW, Teh CZ,
Stamey T 1995 Family history and prostate cancer risk in black, white, and Asian men in the
United States and Canada. Am J Epidemiol 141:732-40
Pollard M, Luckert PH 1985 Promotional effects of testosterone and dietary fat on prostate
carcinogenesis in genetically susceptible rats. Prostate 6:1-5
Peto R, Doll R, Buckley JD, Sporn MB 1981 Can dietary beta-carotene materially reduce
human cancer rates? Nature 290:201-8
Hill PB, Wynder EL 1979 Effect of a vegetarian diet and dexamethasone on plasma prolactin,
testosterone and dehydroepiandrosterone in men and women. Cancer Lett 7:273-82
Gu FL, Xia TL, Kong XT 1994 Preliminary study of the frequency of benign prostatic hyperplasia
and prostatic cancer in China. Urology 44:688-91
Yoshizawa K, Willett WC, Morris SJ, Stampfer MJ, Spiegelman D, Rimm EB, Giovannucci E
1998 Study of prediagnostic selenium level in toenails and the risk of advanced prostate cancer.
J Natl Cancer Inst 90:1219-24
Heinonen OP, Albanes D, Virtamo J, Taylor PR, Huttunen JK, Hartman AM, Haapakoski J,
Malila N, Rautalahti M, Ripatti S, Maenpaa H, Teerenhovi L, Koss L, Virolainen M, Edwards
BK 1998 Prostate cancer and supplementation with alpha-tocopherol and beta-carotene:
incidence and mortality in a controlled trial. J Natl Cancer Inst 90:440-6
Tretli S, Engeland A, Haldorsen T, Hakulinen T, Horte LG, Luostarinen T, Schou G,
Sigvaldason H, Storm HH, Tulinius H, Vaittinen P 1996 Prostate cancer--look to Denmark? J
Natl Cancer Inst 88:128
Schwartz GG, Hulka BS 1990 Is vitamin D deficiency a risk factor for prostate cancer?
(Hypothesis). Anticancer Res 10:1307-11
Rooney C, Beral V, Maconochie N, Fraser P, Davies G 1993 Case-control study of prostatic
cancer in employees of the United Kingdom Atomic Energy Authority. Bmj 307:1391-7
Wynder EL, Mabuchi K, Whitmore WF, Jr. 1971 Epidemiology of cancer of the prostate.
Cancer 28:344-60
Herbert JT, Birkhoff JD, Feorino PM, Caldwell GG 1976 Herpes simplex virus type 2 and
cancer of the prostate. J Urol 116:611-2
McCombs RM 1977 Role of oncornaviruses in carcinoma of the prostate. Cancer Treat Rep
61:131-2
Feminella J, Lattimer J 1974 An apparent increase in genital carcinomas among wives of men
with prostatic carcinoma: an epidemiologic survey. Pirquet Bulletin of Clinical Medicine 20:3-9
Viola MV, Fromowitz F, Oravez S, Deb S, Finkel G, Lundy J, Hand P, Thor A, Schlom J 1986
Expression of ras oncogene p21 in prostate cancer. N Engl J Med 314:133-7
Kim HG, Kassis J, Souto JC, Turner T, Wells A 1999 EGF receptor signaling in prostate
morphogenesis and tumorigenesis. Histol Histopathol 14:1175-82
Schuurmans AL, Bolt J, Veldscholte J, Mulder E 1991 Regulation of growth of LNCaP human
prostate tumor cells by growth factors and steroid hormones. J Steroid Biochem Mol Biol 40:1937
Kim JH, Sherwood ER, Sutkowski DM, Lee C, Kozlowski JM 1991 Inhibition of prostatic tumor
cell proliferation by suramin: alterations in TGF alpha-mediated autocrine growth regulation and
cell cycle distribution. J Urol 146:171-6
Chodak GW, Thisted RA, Gerber GS, Johansson JE, Adolfsson J, Jones GW, Chisholm
GD, Moskovitz B, Livne PM, Warner J 1994 Results of conservative management of clinically
localized prostate cancer. N Engl J Med 330:242-8
Varrault A, Ciani E, Apiou F, Bilanges B, Hoffmann A, Pantaloni C, Bockaert J, Spengler D,
Journot L 1998 hZAC encodes a zinc finger protein with antiproliferative properties and maps to
a chromosomal region frequently lost in cancer. Proc Natl Acad Sci U S A 95:8835-40.
Bilanges B, Varrault A, Mazumdar A, Pantaloni C, Hoffmann A, Bockaert J, Spengler D,
Journot L 2001 Alternative splicing of the imprinted candidate tumor suppressor gene ZAC
regulates its antiproliferative and DNA binding activities. Oncogene 20:1246-53.
Varrault A, Bilanges B, Mackay DJ, Basyuk E, Ahr B, Fernandez C, Robinson DO, Bockaert
J, Journot L 2001 Characterization of the methylation-sensitive promoter of the imprinted ZAC
gene supports its role in transient neonatal diabetes mellitus. J Biol Chem 276:18653-6.
Huang SM, Schonthal AH, Stallcup MR 2001 Enhancement of p53-dependent gene activation
by the transcriptional coactivator Zac1. Oncogene 20:2134-43.
89
51.
52.
53.
54.
55.
56.
57.
58.
59.
60.
61.
62.
63.
64.
65.
66.
67.
68.
69.
70.
71.
72.
73.
74.
75.
76.
77.
78.
Abdollahi A, Roberts D, Godwin AK, Schultz DC, Sonoda G, Testa JR, Hamilton TC 1997
Identification of a zinc-finger gene at 6q25: a chromosomal region implicated in development of
many solid tumors. Oncogene 14:1973-9.
Smith RJ, Arnaud P, Konfortova G, Dean WL, Beechey CV, Kelsey G 2002 The mouse Zac1
locus: basis for imprinting and comparison with human ZAC. Gene 292:101-12
Tatemoto K, Carlquist M, Mutt V 1982 Neuropeptide Y-a novel brain peptide with structural
similarities to peptide YY and pancreatic polypeptide. Nature 313:404-406
Tatemoto K 1982 Isolation and characterization of peptide YY (PYY), a candidate gut hormone
that inhibits pancreatic exocrine secretion. Proc Natl Acad Sci U S A 79:2514-8
Kimmel JR, Hayden LJ, Pollock HG 1975 Isolation and characterization of a new pancreatic
polypeptide hormone. J Biol Chem 250:9369-76
Gehlert DR 2004 Introduction to the reviews on neuropeptide Y. Neuropeptides 38:135-40
Larhammar D 1996 Evolution of neuropeptide Y, peptide YY and pancreatic polypeptide.
Regulatory Peptides 62:1-11
Minth CD, Bloom SR, Polak JM, Dixon JE 1984 Cloning, characterization, and DNA sequence
of a human cDNA encoding neuropeptide tyrosine. Proceedings of the National Academy of
Sciences of the United States of America 81:4577-4581
Schwartz TW, Fuhlendorff J, Kjems LL, Kristensen MS, Vervelde M, O'Hare M, Krstenansky
JL, Bjornholm B 1990 Signal epitopes in the three-dimensional structure of neuropeptide Y.
Interaction with Y1, Y2, and pancreatic polypeptide receptors. Ann N Y Acad Sci 611:35-47
Cerda-Reverter JM, Larhammar D 2000 Neuropeptide Y family of peptides: structure,
anatomical expression, function, and molecular evolution. Biochem Cell Biol 78:371-92
Minth CD, Dixon JE 1990 Expression of the human neuropeptide Y gene. The Journal of
Biological Chemistry 256:12933-12939
Sperk G, Marksteiner J, Gruber B, Bellmann R, Mahata M, Ortler M 1992 Functional changes
in neuropeptide Y- and somatostatin-containing neurons induced by limbic seizures in the rat.
Neuroscience 50:831-846
Sperk G 1994 Kainic acid seizures in the rat. Prog Neurobiol 42:1-32
Sperk G, Lassmann H, Baran H, Kish SJ, Seitelberger F, Hornykiewicz O 1983 Kainic acid
induced seizures: neurochemical and histopathological changes. Neuroscience 10:1301-15
Marksteiner J, Sperk G, Maas D 1989 Differential increases in brain levels of neuropeptide Y
and vasoactive intestinal polypeptide after kainic acid-induced seizures in the rat. Naunyn
Schmiedebergs Arch Pharmacol 339:173-7
Kelly RB, Grote E 1993 Protein targeting in the neuron. Annu Rev Neurosci 16:95-127
Higuchi H, Costa E, Yang HY 1988 Neuropeptide Y inhibits the nicotine-mediated release of
catecholamines from bovine adrenal chromaffin cells. J Pharmacol Exp Ther 244:468-74.
Brakch N, Rist B, Beck-Sickinger AG, Goenaga J, Wittek R, Burger E, Brunner HR,
Grouzmann E 1997 Role of prohormone convertases in pro-neuropeptide Y processing:
coexpression and in vitro kinetic investigations. Biochemistry 36:16309-20
Hopsu-Havu VK, Glenner GG 1966 A new dipeptide naphthylamidase hydrolyzing glycyl-prolylbeta-naphthylamide. Histochemie 7:197-201
Grandt D 1993 Proteolytic processing by dipeptidyl aminopeptidase IV generates receptor
selectivity for peptide YY. Med. Klin. 88:143-145
Stange T, Kettmann U, Holzhausen HJ 1996 Immunoelectron microscopic single and double
labelling of aminopeptidase N (CD 13) and dipeptidyl peptidase IV (CD 26). Acta Histochem
98:323-31
Shibuya-Saruta H, Kasahara Y, Hashimoto Y 1996 Human serum dipeptidyl peptidase IV
(DPPIV) and its unique properties. J Clin Lab Anal 10:435-40
Yaron A, Mlynar D 1968 Aminopeptidase-P. Biochem Biophys Res Commun 32:658-63
Ryan JW, Denslow ND, Greenwald JA, Rogoff MA 1994 Immunoaffinity purifications of
aminopeptidase P from guinea pig lungs, kidney and serum. Biochem Biophys Res Commun
205:1796-802
Mentlein R, von Kolszynski M, Sprang R, Lucius R 1990 Proline-specific proteases in
cultivated neuronal and glial cells. Brain Res 527:159-62
Medeiros Mdos S, Turner AJ 1996 Metabolism and functions of neuropeptide Y. Neurochem
Res 21:1125-32
Allen LG, Kalra PS, Crowley WR, Kalra SP 1985 Comparison of the effects of neuropeptide Y
and adrenergic transmitters on LH release and food intake in male rats. Life Sci 37:617-23.
Grouzmann E, Comoy E, Bohuon C 1989 Plasma neuropeptide Y concentrations in patients
with neuroendocrine tumors. J Clin Endocrinol Metab 68:808-13
90
79.
80.
81.
82.
83.
84.
85.
86.
87.
88.
89.
90.
91.
92.
93.
94.
95.
96.
97.
98.
99.
100.
Grouzmann E, Aubert JF, Waeber B, Brunner HR 1992 A sensitive and specific two-site,
sandwich-amplified enzyme immunoassay for neuropeptide Y. Peptides 13:1049-54
Nagaki S, Fukamauchi F, Sakamoto Y, Higuchi H, Miki N, Miki N, Ono M, Sadamatsu M,
Kato N, Osawa M 2000 Upregulation of brain somatostatin and neuropeptide Y following
lidocaine-induced kindling in the rat. Brain Research 852:470-474
Allen Y, Adrian T, Allen J, Tatemoto K, Crow T, Bloom S, Polak J 1983 Neuropeptide Y
distribution in the rat brain. Science 221:877-879
Heilig M, Widerlov E 1990 Neuropeptide Y: an overview of central distribuition, functional
aspects, and possible involvement in neuropsychiatric illnesses. Acta Psychiatrica Scandinavica
82:95-114
Adrian TE, Gu J, Allen JM, Tatemoto K, Polak JM, Bloom SR 1984 Neuropeptide Y in the
human male genital tract. Life Sciences 35:2643-2648
Dawbarn D, Hunt S, Emson P 1984 Neuropeptide Y: regional distribution chromatographic
characterization and immunohistochemical demonstration in post-mortem human brain. Brain
Research 296:168-173
Allen JM, Gibson SJ, Adrian TE, Polak JM, Bloom SR 1984 Neuropeptide Y in human spinal
cord. Brain Research 308:145-148
Rowan S, Todd AJ, Spike RC 1993 Evidence that neuropeptide Y is present in gabaergic
neurons in the superficial dorsal horn of the rat spinal cord. Neuroscience 53:537-545
Ekblad E, Edvinsson L, Wahlestedt C, Uddman R, Hakanson R, Sundler F 1984
Neuropeptide Y co-exists and co-operates with noradrenaline in perivascular nerve fibers. Regul
Pept 8:225-35
Lundberg JM, Terenius SL, Hokfelt T, Martling CR, Tatemoto K, Mutt V, Polak J, Bloom S,
Goldstein M 1982 Neuropeptide Y (NPY)-like immunoreactivity in peripheral noradrenergic
neurons and effects of NPY on sympathetic function. Acta Physiologica Scandinavica 116:477480
Samuelson U, Dallsgard C 1985 Action and localization of neuropeptide Y in the human
fallopian tube. Neurosciences Letters:49-58
Bagnoli P, Dal Monte M, Casini G 2003 Expression of neuropeptides and their receptors in the
developing retina of mammals. Histol Histopathol 18:1219-42
Michel MC, Beck-Sickinger A, Cox H, Doods HN, Herzog H, Larhammar D, Quirion R,
Schwartz T, Westfall T 1998 XVI. International Union of Pharmacology recommendations for the
nomenclature of neuropeptide Y, peptide YY, and pancreatic polypeptide receptors. Pharmacol
Rev 50:143-50
Larhammar D, Blomqvist AG, Yee F, Jazin E, Yoo H, Wahlestedt C 1992 Cloning and
functional expression of a human neuropeptide Y/peptide YY receptor of the Y1 type. Journal of
Biological Chemistry 267:10935-10938
Herzog H, Hort YJ, Ball HJ, Hayes G, Shine J, Selbie LA 1992 Cloned human neuropeptide Y
receptor couples to two different second messenger systems. Proceedings of the National
Academy of Sciences of the United States of America 89:5794-5798
Lundell I, Berglund MM, Starback P, Salaneck E, Gehlert DR, Larhammar D 1997 Cloning
and characterization of a novel neuropeptide Y receptor subtype in the zebrafish. DNA Cell Biol
16:1357-63
Mullins DE, Guzzi M, Xia L, Parker EM 2000 Pharmacological characterization of the cloned
neuropeptide Y y(6) receptor. Eur J Pharmacol 395:87-93
Mullins DE, Zhang X, Hawes BE 2002 Activation of extracellular signal regulated protein kinase
by neuropeptide Y and pancreatic polypeptide in CHO cells expressing the NPY Y(1), Y(2), Y(4)
and Y(5) receptor subtypes. Regul Pept 105:65-73
Gerald C, Walker MW, Vaysse PJ, He C, Branchek TA, Weinshank RL 1995 Expression
cloning and pharmacological characterization of a human hippocampal neuropeptide Y/peptide
YY Y2 receptor subtype. J Biol Chem 270:26758-61
Bard JA, Walker MW, Branchek TA, Weinshank RL 1995 Cloning and functional expression of
a human Y4 subtype receptor for pancreatic polypeptide, neuropeptide Y, and peptide YY.
Journal of Biological Chemistry 270:26762-26765
Stenkamp RE, Filipek S, Driessen CA, Teller DC, Palczewski K 2002 Crystal structure of
rhodopsin: a template for cone visual pigments and other G protein-coupled receptors. Biochim
Biophys Acta 1565:168-82
Larhammar D, Wraith A, Berglund MM, Holmberg SK, Lundell I 2001 Origins of the many
NPY-family receptors in mammals. Peptides 22:295-307
91
101.
102.
103.
104.
105.
106.
107.
108.
109.
110.
111.
112.
113.
114.
115.
116.
117.
118.
119.
120.
121.
Salaneck E, Fredriksson R, Larson ET, Conlon JM, Larhammar D 2001 A neuropeptide Y
receptor Y1-subfamily gene from an agnathan, the European river lamprey. A potential ancestral
gene. Eur J Biochem 268:6146-54
Larhammar D, Salaneck E 2004 Molecular evolution of NPY receptor subtypes. Neuropeptides
38:141-51
Starback P, Lundell I, Fredriksson R, Berglund MM, Yan YL, Wraith A, Soderberg C,
Postlethwait JH, Larhammar D 1999 Neuropeptide Y receptor subtype with unique properties
cloned in the zebrafish: the zYa receptor. Brain Res Mol Brain Res 70:242-52
Fredriksson R, Larson ET, Yan YL, Postlethwait JH, Larhammar D 2004 Novel neuropeptide
Y Y2-like receptor subtype in zebrafish and frogs supports early vertebrate chromosome
duplications. J Mol Evol 58:106-14
Eva C, Keinanen K, Monyer H, Seeburg P, Sprengel R 1990 Molecular cloning of a novel G
protein-coupled receptor that may belong to the neuropeptide receptor family. FEBS Lett 271:814
Krause J, Eva C, Seeburg PH, Sprengel R 1992 Neuropeptide Y1 subtype pharmacology of a
recombinantly expressed neuropeptide receptor. Mol Pharmacol 41:817-21
Berglund MM, Holmberg SK, Eriksson H, Gedda K, Maffrand JP, Serradeil-Le Gal C,
Chhajlani V, Grundemar L, Larhammar D 1999 The cloned guinea pig neuropeptide Y receptor
Y1 conforms to other mammalian Y1 receptors. Peptides 20:1043-53
Holmberg SK, Mikko S, Boswell T, Zoorob R, Larhammar D 2002 Pharmacological
characterization of cloned chicken neuropeptide Y receptors Y1 and Y5. J Neurochem 81:462-71
Blomqvist AG, Roubos EW, Larhammar D, Martens GJ 1995 Cloning and sequence analysis
of a neuropeptide Y/peptide YY receptor Y1 cDNA from Xenopus laevis. Biochim Biophys Acta
1261:439-41
Salaneck E, Ardell DH, Larson ET, Larhammar D 2003 Three neuropeptide Y receptor genes
in the spiny dogfish, Squalus acanthias, support en bloc duplications in early vertebrate evolution.
Mol Biol Evol 20:1271-80
Gregor P, Millham ML, Feng Y, DeCarr LB, McCaleb ML, Cornfield LJ 1996 Cloning and
characterization of a novel receptor to pancreatic polypeptide, a member of the neuropeptide Y
receptor family. FEBS Letters 381:58-62
Berglund MM, Lundell I, Eriksson H, Soll R, Beck-Sickinger AG, Larhammar D 2001 Studies
of the human, rat, and guinea pig Y4 receptors using neuropeptide Y analogues and two distinct
radioligands. Peptides 22:351-6
Lundell I, Boswell T, Larhammar D 2002 Chicken neuropeptide Y-family receptor Y4: a
receptor with equal affinity for pancreatic polypeptide, neuropeptide Y and peptide YY. J Mol
Endocrinol 28:225-35
Batterham RL, Le Roux CW, Cohen MA, Park AJ, Ellis SM, Patterson M, Frost GS, Ghatei
MA, Bloom SR 2003 Pancreatic polypeptide reduces appetite and food intake in humans. J Clin
Endocrinol Metab 88:3989-92
Borowsky B, Walker MW, Bard J, Weinshank RL, Laz TM, Vaysse P, Branchek TA, Gerald C
1998 Molecular biology and pharmacology of multiple NPY Y5 receptor species homologs. Regul
Pept 75-76:45-53
Matsumoto M, Nomura T, Momoses K, Ikeda Y, Kondou Y, Akiho H, Togami J, Kimura Y,
Okada M, Yamaguchi T 1996 Inactivation of a novel neuropeptide Y/peptide YY receptor gene in
primate species. Journal of Biological Chemistry 271:27217-27220
Rose PM, Lynch JS, Frazier ST, Fisher SM, Chung W, Battaglino P, Fathi Z, Leibel R,
Prabhavathi F 1997 Molecular genetics analysis of a human neuropeptide Y receptor. The
human homolog of the murine "Y5" receptor may be a pseudogene. Journal of Biological
Chemistry 272:3622-3627
Burkhoff A, Linemeyer DL, Salon JA 1998 Distribution of a novel hypothalamic neuropeptide Y
receptor gene and its absence in rat. Molecular Brain Research 53:311-316
Starback P, Wraith A, Eriksson H, Larhammar D 2000 Neuropeptide Y receptor gene y6:
multiple deaths or resurrections? Biochem Biophys Res Commun 277:264-9.
Wraith A, Tornsten A, Chardon P, Harbitz I, Chowdhary BP, Andersson L, Lundin LG,
Larhammar D 2000 Evolution of the neuropeptide Y receptor family: gene and chromosome
duplications deduced from the cloning and mapping of the five receptor subtype genes in pig.
Genome Res 10:302-10
Gehlert DR, Schober DA, Beavers L, Gadski R, Hoffman JA, Smiley DL, Chance RE,
Lundell I, Larhammar D 1996 Characterization of the peptide binding requirements for the
cloned human pancreatic polypeptide-preferring receptor. Mol Pharmacol 50:112-8
92
122.
123.
124.
125.
126.
127.
128.
129.
130.
131.
132.
133.
134.
135.
136.
137.
138.
139.
140.
141.
142.
Rose PM, Fernandes P, Lynch JS, Frazier ST, Fisher SM, Kodukula K, Kienzle B, Seethala
R 1995 Cloning and functional expression of a cDNA encoding a human type 2 neuropeptide Y
receptor. J Biol Chem 270:29038
Sharma P, Holmberg SK, Eriksson H, Beck-Sickinger AG, Grundemar L, Larhammar D
1998 Cloning and functional expression of the guinea pig neuropeptide Y Y2 receptor. Regul
Pept 75-76:23-8
Salaneck E, Holmberg SK, Berglund MM, Boswell T, Larhammar D 2000 Chicken
neuropeptide Y receptor Y2: structural and pharmacological differences to mammalian Y2(1).
FEBS Lett 484:229-34
Gerald C, Walker MW, Criscione L, Gustafson EL, Batzl-Hartmann C, Smith KE, Vaysse P,
Durkin MM, Laz TM, Linemeyer DL, Schaffhauser AO, Whitebread S, Hofbauer KG, Taber
RI, Branchek TA, Weinshank RL 1996 A receptor subtype involved in neuropeptide-Y-induced
food intake. Nature 382:168-71.
Hu Y, Bloomquist BT, Cornfield LJ, DeCarr LB, Flores-Riveros JR, Friedman L, Jiang P,
Lewis-Higgins L, Sadlowski Y, Schaefer J, Velazquez N, McCaleb ML 1996 Identification of a
novel hypothalamic neuropeptide Y receptor associated with feeding behavior. J Biol Chem
271:26315-9
Herzog H, Darby K, Ball H, Hort Y, Beck-Sickinger A, Shine J 1997 Overlapping gene
structure of the human neuropeptide Y receptor subtypes Y1 and Y5 suggests coordinate
transcriptional regulation. Genomics 41:315-319
Nei M, Gojobori T 1986 Simple methods for estimating the numbers of synonymous and
nonsynonymous nucleotide substitutions. Mol Biol Evol 3:418-26
Felsenstein KM, Lewis-Higgins L 1993 Processing of the beta-amyloid precursor protein
carrying the familial, Dutch-type, and a novel recombinant C-terminal mutation. Neurosci Lett
152:185-9
Lundell I, Eriksson H, Marklund U, Larhammar D 2001 Cloning and characterization of the
guinea pig neuropeptide Y receptor Y5. Peptides 22:357-63
Eva C, Oberto A, Sprengel R, Genazzani E 1992 The murine NPY-1 receptor gene. Structure
and delineation of tissue-specific expression. FEBS Lett 314:285-8
Nakamura M, Sakanaka C, Aoki Y, Ogasawara H, Tsuji T, Kodama H, Matsumoto T, Shimizu
T, Noma M 1995 Identification of two isoforms of mouse neuropeptide Y-Y1 receptor generated
by alternative splicing. Isolation, genomic structure, and functional expression of the receptors. J
Biol Chem 270:30102-10
Parker HY, Kwon HM, Lim HJ, Hong BK, Lee JY, Park BE, Jang Y, Cho SY, Kim HS 2001
Potential role of leptin in angiogenesis: leptin induces endothelial cell proliferation and expression
of matrix metalloproteinases in vivo and in vitro. Exp Mol Med 33:95-102
Fabry M, Langer M, Rothen-Rutishauser B, Wunderli-Allenspach H, Hocker H, BeckSickinger AG 2000 Monitoring of the internalization of neuropeptide Y on neuroblastoma cell line
SK-N-MC. Eur J Biochem 267:5631-7
Ball H, Shine J, Herzog H 1995 Multiple promoters regulate tissue-specific expression of the
human NPY-Y1 receptor gene. The Journal of Biological Chemistry 270:27272-27276
Aakerlund L, Gether U, Fuhlendorff J, Schwartz TW, Thastrup O 1990 Y1 receptors for
neuropeptide Y are coupled to mobilization of intracellular calcium and inhibition of adenylate
cyclase. FEBS Lett 260:73-8
Motulsky HJ, Michel MC 1988 Neuropeptide Y mobilizes Ca2+ and inhibits adenylate cyclase in
human erythroleukemia cells. Am J Physiol 255:E880-5
Mullins D, Kirby D, Hwa J, Guzzi M, Rivier J, Parker E 2001 Identification of potent and
selective neuropeptide Y Y(1) receptor agonists with orexigenic activity in vivo. Mol Pharmacol
60:534-40
Lundberg JM, Tatemoto K 1982 Pancreatic polypeptide family (APP, BPP, NPY and PYY) in
relation to sympathetic vasoconstriction resistant to alpha-adrenoceptor blockade. Acta Physiol
Scand 116:393-402.
Rudolf K, Eberlein W, Engel W, Wieland HA, Willim KD, Entzeroth M, Wienen W, BeckSickinger AG, Doods HN 1994 The first highly potent and selective non-peptide neuropeptide Y
Y1 receptor antagonist: BIBP3226. Eur J Pharmacol 271:R11-3
Cervin A, Lindberg S, Mercke U, Uddman R 1992 Neuropeptide Y in the rabbit maxillary sinus
modulates cholinergic acceleration of mucociliary activity. Acta Otolaryngol 112:872-81
Lundberg JM, Modin A 1995 Inhibition of sympathetic vasoconstriction in pigs in vivo by the
neuropeptide Y-Y1 receptor antagonist BIBP 3226. Br J Pharmacol 116:2971-82
93
143.
144.
145.
146.
147.
148.
149.
150.
151.
152.
153.
154.
155.
156.
157.
158.
159.
160.
161.
162.
163.
164.
165.
Lutz CM, Frankel WN, Richards JE, Thompson DA 1997 Neuropeptide Y receptor genes on
human chromosome 4q31-q32 map to conserved linkage groups on mouse chromosomes 3 and
8. Genomics 41:498-500
Gehlert DR, Beavers LS, Johnson D, Gackenheimer SL, Schober DA, Gadski RA 1996
Expression cloning of a human brain neuropeptide Y Y2 receptor. Mol Pharmacol 49:224-8
Beck-Sickinger AG, Grouzmann E, Hoffmann E, Gaida W, van Meir EG, Waeber B, Jung G
1992 A novel cyclic analog of neuropeptide Y specific for the Y2 receptor. Eur J Biochem
206:957-64
Martel JC, Fournier A, St-Pierre S, Dumont Y, Forest M, Quirion R 1990 Comparative
structural requirements of brain neuropeptide Y binding sites and vas deferens neuropeptide Y
receptors. Mol Pharmacol 38:494-502
Magni P 2003 Hormonal control of the neuropeptide Y system. Current Protein & Peptide
Science 4:45-57
Lacroix JS, Ulman LG, Potter EK 1994 Modulation by neuropeptide Y of parasympathetic
nerve-evoked nasal vasodilatation via Y2 prejunctional receptor. Br J Pharmacol 113:479-84
Pedrazzini T, Pralong F, Grouzmann E 2003 Neuropeptide Y: the universal soldier. Cellular and
Molecular Life Sciences 60:350-377
Wahlestedt C, Regunathan S, Reis DJ 1992 Identification of cultured cells selectively
expressing Y1-, Y2-, or Y3- type receptors for neuropeptide Y/peptide YY. Life Sci 50:L7-12
Foucart S, Bleakman D, Bindokas VP, Miller RJ 1993 Neuropeptide Y and pancreatic
polypeptide reduce calcium currents in acutely dissociated neurons from adult rat superior
cervical ganglia. J Pharmacol Exp Ther 265:903-9
Grundemar L, Wahlestedt C, Reis DJ 1991 Neuropeptide Y acts at an atypical receptor to
evoke cardiovascular depression and to inhibit glutamate responsiveness in the brainstem. J
Pharmacol Exp Ther 258:633-8
Balasubramaniam A, Sheriff S, Rigel DF, Fischer JE 1990 Characterization of neuropeptide Y
binding sites in rat cardiac ventricular membranes. Peptides 11:545-50
Dumont Y, Satoh H, Cadieux A, Taoudi-Benchekroun M, Pheng LH, St-Pierre S, Fournier A,
Quirion R 1993 Evaluation of truncated neuropeptide Y analogues with modifications of the
tyrosine residue in position 1 on Y1, Y2 and Y3 receptor sub-types. Eur J Pharmacol 238:37-45
Jacques D, Cadieux A, Dumont Y, Quirion R 1995 Apparent affinity and potency of BIBP3226,
a non-peptide neuropeptide Y receptor antagonist, on purported neuropeptide Y Y1, Y2 and Y3
receptors. Eur J Pharmacol 278:R3-5
McCullough LA, Westfall TC 1996 Mechanism of catecholamine synthesis inhibition by
neuropeptide Y: role of Ca2+ channels and protein kinases. J Neurochem 67:1090-9
Cavadas C, Silva AP, Mosimann F, Cotrim MD, Ribeiro CA, Brunner HR, Grouzmann E 2001
NPY regulates catecholamine secretion from human adrenal chromaffin cells. J Clin Endocrinol
Metab 86:5956-63
Lundell I, Blonquist AG, Berglund MM, Schober DA, Johnson D, Sta MA, Gadski RA,
Gehlert DR, Larhammar D 1995 Cloning of human receptor of the NPY receptor family with high
affinity for pancreatic polypeptide and peptide YY. Journal of Biological Chemistry 270:2912329128
Darby K, Eyre HJ, Lapsys N, Copeland NG, Gilbert DJ, Couzens M, Antonova O,
Sutherland GR, Jenkins NA, Herzog H 1997 Assignment of the Y4 receptor gene (PPYR1) to
human chromosome 10q11.2 and mouse chromosome 14. Genomics 46:513-5
Lundell I, Statnick MA, Johnson D, Schober DA, Starback P, Gehlert DR, Larhammar D
1996 The cloned rat pancreatic polypeptide receptor exhibits profound differences to the
orthologous receptor. Proc Natl Acad Sci U S A 93:5111-5
Voisin T, Goumain M, Lorinet AM, Maoret JJ, Laburthe M 2000 Functional and molecular
properties of the human recombinant Y4 receptor: resistance to agonist-promoted
desensitization. J Pharmacol Exp Ther 292:638-46
Berglund MM, Schober DA, Esterman MA, Gehlert DR 2003 Neuropeptide Y Y4 receptor
homodimers dissociate upon agonist stimulation. J Pharmacol Exp Ther 307:1120-6
Yan H, Yang J, Marasco J, Yamaguchi K, Brenner S, Collins F, Karbon W 1996 Cloning and
functional expression of cDNAs encoding human and rat pancreatic polypeptide receptors. Proc
Natl Acad Sci U S A 93:4661-5
Flood JF, Morley JE 1989 Dissociation of the effects of neuropeptide Y on feeding and memory:
evidence for pre- and postsynaptic mediation. Peptides 10:963-6
Stanley BG, III VLW, Donias HW, Ha LH, Spears LC 1993 The lateral hypothalamus: a primary
site mediating excitatory amino acid-elicited eating. Brain Res 630:41-49
94
166.
167.
168.
169.
170.
171.
172.
173.
174.
175.
176.
177.
178.
179.
180.
181.
182.
183.
184.
185.
186.
187.
188.
Balasubramaniam A, Sheriff S, Johnson ME, Prabhakaran M, Huang Y, Fischer JE, Chance
WT 1994 [D-TRP32]neuropeptide Y: a competitive antagonist of NPY in rat hypothalamus. J Med
Chem 37:811-5
Haynes AC, Arch JR, Wilson S, McClue S, Buckingham RE 1998 Characterisation of the
neuropeptide Y receptor that mediates feeding in the rat: a role for the Y5 receptor? Regul Pept
75-76:355-61
Hwa JJ, Witten MB, Williams P, Ghibaudi L, Gao J, Salisbury BG, Mullins D, Hamud F,
Strader CD, Parker EM 1999 Activation of the NPY Y5 receptor regulates both feeding and
energy expenditure. Am J Physiol 277:R1428-34.
Gregor P, Feng Y, DeCarr LB, Cornfield LJ, McCaleb ML 1996 Molecular characterization of a
second mouse pancreatic polypeptide receptor and its inactivated human homologue. J Biol
Chem 271:27776-81
Kassis S, Olasmaa M, Terenius L, Fishman PH 1987 Neuropeptide Y inhibits cardiac
adenylate cyclase through a pertussis toxin-sensitive G protein. J Biol Chem 262:3429-31
Westlind-Danielsson A, Unden A, Abens J, Andell S, Bartfai T 1987 Neuropeptide Y
receptors and the inhibition of adenylate cyclase in the human frontal and temporal cortex.
Neurosci Lett 74:237-42
Daniels AJ, Lazarowski ER, Matthews JE, Lapetina EG 1989 Neuropeptide Y mobilizes
intracellular Ca2+ and increases inositol phosphate production in human erythroleukemia cells.
Biochem Biophys Res Commun 165:1138-44
Reynolds EE, Yokota S 1988 Neuropeptide Y receptor-effector coupling mechanisms in cultured
vascular smooth muscle cells. Biochem Biophys Res Commun 151:919-25
Shigeri Y, Nakajima S, Fujimoto M 1995 Neuropeptide YY1 receptors-mediated increase in
intracellular Ca2+ concentration via phospholipase C-dependent pathway in porcine aortic
smooth muscle cells. J Biochem (Tokyo) 118:515-20
Lobaugh LA, Blackshear PJ 1990 Neuropeptide Y stimulation of myosin light chain
phosphorylation in cultured aortic smooth muscle cells. J Biol Chem 265:18393-9
Nie M, Selbie LA 1998 Neuropeptide Y Y1 and Y2 receptor-mediated stimulation of mitogenactivated protein kinase activity. Regul Pept 75-76:207-13
Mannon PJ, Mele JM 2000 Peptide YY Y1 receptor activates mitogen-activated protein kinase
and proliferation in gut epithelial cells via the epidermal growth factor receptor. Biochemical
Journal 350:655-661
Bleakman D, Colmers WF, Fournier A, Miller RJ 1991 Neuropeptide Y inhibits Ca2+ influx into
cultured dorsal root ganglion neurones of the rat via a Y2 receptor. Br J Pharmacol 103:1781-9
Lynch JW, Lemos VS, Bucher B, Stoclet JC, Takeda K 1994 A pertussis toxin-insensitive
calcium influx mediated by neuropeptide Y2 receptors in a human neuroblastoma cell line. J Biol
Chem 269:8226-33
Freitag C, Svendsen AB, Feldthus N, Lossl K, Sheikh SP 1995 Coupling of the human Y2
receptor for neuropeptide Y and peptide YY to guanine nucleotide inhibitory proteins in
permeabilized SMS-KAN cells. J Neurochem 64:643-50
Shigeri Y, Fujimoto M 1994 Y2 receptors for neuropeptide Y are coupled to three intracellular
signal transduction pathways in a human neuroblastoma cell line. J Biol Chem 269:8842-8
Nakamura M, Aoki Y, Hirano D 1996 Cloning and functional expression of a cDNA encoding a
mouse type 2 neuropeptide Y receptor. Biochim Biophys Acta 1284:134-7
Grouzmann E, Meyer C, Burki E, Brunner H 2001 Neuropeptide Y Y2 receptor signalling
mechanisms in the human glioblastoma cell line LN319. Peptides 22:379-86
Rettori V, Milenkovic L, Aguila M, Cann SM 1990 Physiologically significant effect of
neuropeptide Y to suppress growth hormone release by stimulating somatostatin discharge.
Endocrinology 126:2296-2301
Wong LB, Park CL, Yeates DB 1998 Neuropeptide Y inhibits ciliary beat frequency in human
ciliated cells via nPKC, independently of PKA. Am J Physiol 275:C440-8
Walker MW, Smith KE, Bard J, Vaysse PJ, Gerald C, Daouti S, Weinshank RL, Branchek TA
1997 A structure-activity analysis of the cloned rat and human Y4 receptors for pancreatic
polypeptide. Peptides 18:609-12
Sun L, Miller RJ 1999 Multiple neuropeptide Y receptors regulate K+ and Ca2+ channels in
acutely isolated neurons from the rat arcuate nucleus. J Neurophysiol 81:1391-403
Sun L, Philipson LH, Miller RJ 1998 Regulation of K+ and Ca++ channels by a family of
neuropeptide Y receptors. J Pharmacol Exp Ther 284:625-32
95
189.
190.
191.
192.
193.
194.
195.
196.
197.
198.
199.
200.
201.
202.
203.
204.
205.
206.
207.
208.
209.
Bischoff A, Puttmann K, Kotting A, Moser C, Buschauer A, Michel MC 2001 Limited signal
transduction repertoire of human Y(5) neuropeptide Y receptors expressed in HEC-1B cells.
Peptides 22:387-94
Lin S, Boey D, Herzog H 2004 NPY and Y receptors: lessons from transgenic and knockout
models. Neuropeptides 38:189-200
Inui A, Okita M, Nakajima M, Momose K, Ueno N, Teranishi A, Miura M, Hirosue Y, Sano K,
Sato M, Watanabe M, Sakai T, Watanabe T, Ishida K, Silver J, Baba S, Kasuga M 1998
Anxiety-like behavior in transgenic mice with brain expression of neuropeptide Y. Proc Assoc Am
Physicians 110:171-82
Thiele TE, Marsh DJ, Ste Marie L, Bernstein IL, Palmiter RD 1998 Ethanol consumption and
resistance are inversely related to neuropeptide Y levels. Nature 396:366-9
Michalkiewicz M, Michalkiewicz T 2000 Developing transgenic neuropeptide Y rats. Methods
Mol Biol 153:73-89
Kaga T, Inui A, Okita M, Asakawa A, Ueno N, Kasuga M, Fujimiya M, Nishimura N, Dobashi
R, Morimoto Y, Liu IM, Cheng JT 2001 Modest overexpression of neuropeptide Y in the brain
leads to obesity after high-sucrose feeding. Diabetes 50:1206-10
Erickson JC, Hollopeter G, Palmiter RD 1996 Attenuation of the obesity syndrome of ob/ob
mice by the loss of neuropeptide Y. Science 274:1704-1707
Bannon AW, Seda J, Carmouche M, Francis JM, Norman MH, Karbon B, McCaleb ML 2000
Behavioral characterization of neuropeptide Y knockout mice. Brain Res 868:79-87
Hollopeter G, Erickson JC, Palmiter RD 1998 Role of neuropeptide Y in diet-, chemical- and
genetic-induced obesity of mice. Int J Obes Relat Metab Disord 22:506-12
Qian S, Chen H, Weingarth D, Trumbauer ME, Novi DE, Guan X, Yu H, Shen Z, Feng Y,
Frazier E, Chen A, Camacho RE, Shearman LP, Gopal-Truter S, MacNeil DJ, Van der Ploeg
LH, Marsh DJ 2002 Neither agouti-related protein nor neuropeptide Y is critically required for the
regulation of energy homeostasis in mice. Mol Cell Biol 22:5027-35
Hohmann JG, Teklemichael DN, Weinshenker D, Wynick D, Clifton DK, Steiner RA 2004
Obesity and endocrine dysfunction in mice with deletions of both neuropeptide Y and galanin. Mol
Cell Biol 24:2978-85
Pedrazzini T, Seydoux J, Kunstner P, Aubert JF, Grouzmann E, Beermann F, Brunner HR
1998 Cardiovascular response, feeding behavior and locomotor activity in mice lacking the NPY
Y1 receptor. Nat Med 4:722-6
Burcelin R, Brunner H, Seydoux J, Thorensa B, Pedrazzini T 2001 Increased insulin
concentrations and glucose storage in neuropeptide Y Y1 receptor-deficient mice. Peptides
22:421-7
Kushi A, Sasai H, Koizumi H, Takeda N, Yokoyama M, Nakamura M 1998 Obesity and mild
hyperinsulinemia found in neuropeptide Y-Y1 receptor-deficient mice. Proc Natl Acad Sci U S A
95:15659-64
Kanatani A, Mashiko S, Murai N, Sugimoto N, Ito J, Fukuroda T, Fukami T, Morin N,
MacNeil DJ, Van der Ploeg LH, Saga Y, Nishimura S, Ihara M 2000 Role of the Y1 receptor in
the regulation of neuropeptide Y-mediated feeding: comparison of wild-type, Y1 receptordeficient, and Y5 receptor-deficient mice. Endocrinology 141:1011-6
Pralong FP, Gonzales C, Voirol MJ, Palmiter RD, Brunner HR, Gaillard RC, Seydoux J,
Pedrazzini T 2002 The neuropeptide Y Y1 receptor regulates leptin-mediated control of energy
homeostasis and reproductive functions. Faseb J 16:712-4
Thiele TE, Koh MT, Pedrazzini T 2002 Voluntary alcohol consumption is controlled via the
neuropeptide Y Y1 receptor. J Neurosci 22:RC208
Howell OW, Scharfman HE, Herzog H, Sundstrom LE, Beck-Sickinger A, Gray WP 2003
Neuropeptide Y is neuroproliferative for post-natal hippocampal precursor cells. J Neurochem
86:646-59
Broberger C, Landry M, Wong H, Walsh JN, Hokfelt T 1997 Subtypes Y1 and Y2 of the
neuropeptide Y receptor are respectively expressed in pro-opiomelanocortin- and neuropeptideY-containing neurons of the rat hypothalamic arcuate nucleus. Neuroendocrinology 66:393-408
Baskin DG, Breininger JF, Schwartz MW 1999 Leptin receptor mRNA identifies a
subpopulation of neuropeptide Y neurons activated by fasting in rat hypothalamus. Diabetes
48:828-33
King PJ, Williams G, Doods H, Widdowson PS 2000 Effect of a selective neuropeptide Y Y(2)
receptor antagonist, BIIE0246 on neuropeptide Y release. Eur J Pharmacol 396:R1-3
96
210.
211.
212.
213.
214.
215.
216.
217.
218.
219.
220.
221.
222.
223.
224.
225.
226.
227.
228.
229.
230.
231.
Batterham RL, Cowley MA, Small CJ, Herzog H, Cohen MA, Dakin CL, Wren AM, Brynes
AE, Low MJ, Ghatei MA, Cone RD, Bloom SR 2002 Gut hormone PYY(3-36) physiologically
inhibits food intake. Nature 418:650-4
Sainsbury A, Schwarzer C, Couzens M, Jenkins A, Oakes SR, Ormandy CJ, Herzog H 2002
Y4 receptor knockout rescues fertility in ob/ob mice. Genes Dev 16:1077-88
Ueno N, Inui A, Iwamoto M, Kaga T, Asakawa A, Okita M, Fujimiya M, Nakajima Y, Ohmoto
Y, Ohnaka M, Nakaya Y, Miyazaki JI, Kasuga M 1999 Decreased food intake and body weight
in pancreatic polypeptide-overexpressing mice. Gastroenterology 117:1427-32
Sainsbury A, Baldock PA, Schwarzer C, Ueno N, Enriquez RF, Couzens M, Inui A, Herzog
H, Gardiner EM 2003 Synergistic effects of Y2 and Y4 receptors on adiposity and bone mass
revealed in double knockout mice. Mol Cell Biol 23:5225-33
Marsh DJ, Hollopeter G, Kafer KE, Palmiter RD 1998 Role of the Y5 neuropeptide Y receptor
in feeding and obesity. Nat Med 4:718-21.
Allen L, Tischler A, Lee Y, Bloom S 1984 Neuropeptide Y (NPY) in PC 12 phaeocromocytoma
cultures: responses to dexamethasone and nerve growth factor. Neuroscience 116:291-296
Barnea A, Cho G, Hajibeigi A, Aguila MC, Magni P 1991 Dexamethasone-induced
accumulation of neuropeptide-Y by aggregating fetal brain cells in culture: a process dependent
on the developmental age of the aggregates. Endocrinology 129:931-938
Corder R, Pralong F, Turnill D, Saudan D, Miller A, Gaillard R 1988 Dexamethasone
treatment increases neuropeptide Y levels in rat hypotalamic neurones. Life Sciences 43:18791886
McKibbin P, Cotton S, Carthy HM, Williams G 1992 The effect of dexamethasone on
neuropeptide Y concentrations in specific hypothalamic regions. Life Science 51:1301-1307
Wilding JPH, Gilbey SG, Lambert PD, Gathei MA, Bloom SR 1993 Increases in neuropeptide
Y content and gene expression in the hypothalamus of rats treated with dexamethasone are
prevented by insulin. Neuroendocrinology 57:581-587
Akabayashi A, Watanabe Y, Wahlestedt C, McEwen BS, Paez X, Leibowitz SF 1994
Hypothalamic neuropeptide Y, its gene expression and receptor activity: relation to circulating
corticosterone in adrenalectomized rats. Brain Research 665:201-212
Sahu A, Sninsky CA, Kalra PS, Kalra SP 1990 Neuropeptide Y concentration in microdissected
hypothalamic regions and in vitro release from the medial basal hypotalamus-preoptic area of
streptozotocin-diabetic rats with and without insulin substitution therapy. Endocrinology 126:192198
Abe M, Saito M, Ikeda H, Shimazu T 1991 Increased neuropeptide content in the arcuatoparaventricular hypothalamic neuronal system in both insulin-dependent and non-insulindependent diabetic rats. Brain Research 539:223-227
Schwartz MW, Sipols A, Marks J, Sanagora G, White JD, Scheurink A, Kahn SE, Baskin
DG, Woods SC, Figlewicz DP, Jr DP 1992 Inhibition of hypothalamic neuropeptide Y gene
expression by insulin. Endocrinology 130:3608-3616
Wang J, Leibowitz KL 1997 Central insulin inhibits hypothalamic galanin and neuropeptide Y
gene expression and peptide release in intact rats. Brain Research 777:231-236
Watanabe Y, Akabayashi A, McEwen BS 1995 Adrenal steroid regulation of neuropeptide Y
(NPY) mRNA: differences between dentate hilus and locus coeruleus and arcuate nucleus.
Molecular Brain Research 28:135-140
Larsen PJ, Jessop DS, Chowdrey HS, Lightman SL, Mikkelsen JD 1994 Chronic
administration of glucocorticoids directly upregulates prepro-neuropeptide Y and Y1-receptor
mRNA levels in the arcuate nucleus of the rat. Journal of Neuroendocrinology 6:153-159
Wisialowski T, Parker R, Preston E, Sainsbury A, Kraegen E, Herzog H, Cooney G 2000
Adrenalectomy reduces neuropeptide Y-induceed insulin release and NPY receptor expression in
the rat ventromedial hypothalamus. Journal of Clinical Investigation 105:1253-1259
Woods SC, Lotter EC, McKay LD, Jr DP 1979 Chronic intracerebroventricular infusion of insulin
reduces food intake and body weight of baboons. Nature 282:503-505
Vanderweele DA, Haraczkiewicz E, Itallie TBV 1982 Elevated insulin and satiety in obese and
normal weight rats. Appetite 3:99-109
Schwartz MW, Baskin DG, Bukowski TR, Kuijper JL, Foster D, Lasser G, Prunkard DE,
Porte DJ, Woods SC, Seeley RJ, Weigle DS 1996 Specificity of leptin action on elevated blood
glucose levels and hypothalamic neuropeptide Y gene expression in ob/ob mice. Diabetes
45:531-535
Wilding JPH 1996 Metabolic action of neuropeptide Y and their relevance to obesity.
Biochemical Society Transactions 24:576-581
97
232.
233.
234.
235.
236.
237.
238.
239.
240.
241.
242.
243.
244.
245.
246.
247.
248.
249.
250.
251.
252.
Sanacora G, Kershaw M, Finkelstein JA, White JD 1990 Increased hypothalamic content of
preproneuropeptide Y messenger ribonucleic acid in genetically obese Zucker rats and its
regulation by food deprivation. Endocrinology 127:730-737
Chan YY, Steiner RA, Clifton DK 1996 Regulation of hypothalamic neuropeptide Y neurons by
growth hormone in the rat. Endocrinology 137:1319-1325
Minami S, Kamegai J, Sugihara H, Suzuki N, Wakabayashi I 1998 Growth hormone inhibits its
own secretion by acting on the hypothalamus through its receptors on neuropeptide Y neurons in
the arcuate nucleus and somatostatin neurons in the periventricular nucleus. Endocrine Journal
45:S19-26
Shintani M, Ogawa Y, Ebihara K, Aizawa-Abe M, Miyanaga F, Takaya K, Hayashi T, Inoue G,
Hosoda K, Kojima M, Kangawa K, Nakao K 2001 Ghrelin, an endogenous growth hormone
secretagogue, is a novel orexigenic peptide that antagonizes leptin action through the activation
of hypothalamic neuropeptide Y/Y1 receptor pathway. Diabetes Feb;50(2):227-32.
Chen P, Li C, Haskell-Luevano C, Cone RD, Smith MS 1999 Altered expression of agoutirelated protein and its colocalization with neuropeptide Y in the arcuate nucleus of the
hypothalamus during lactation. Endocrinology 140:2645
Leibowitz SF, Akabayashi A, Alexander JT, Wang J 1998 Gonadal steroids and hypothalamic
galanin and neuropeptide Y: role in eating behavior and body weight control in female rats.
Endocrinology 139:1771-1780
Crowley W, Tessel R, O'Donohue T, Adler B, Kalra S 1985 Effects of ovarian hormones on the
concentrations of immunoreactive neuropeptide Y in discrete brain regions of the female rat:
correlation with serum LH and median eminence LHRH. Endocrinology 117:1151-1155
Diano S, Kalra SP, Sakamoto H, Horvath TL 1998 Leptin receptors in estrogen receptorcontaining neurons of the female rat hypothalamus. Brain Research 812:256
Majdoubi ME, Ramaswamy S, Sahu A, Plant TM 2000 Effects of orchidectomy on levels of the
mRNAs encoding gonadotropin-releasing hormone and other hypothalamic peptides in the adult
male rhesus monkey (Macaca mulatta). Journal of Neuroendocrinology 12:167
Sahu A, Kalra SP, Crowley WR, Kalra PS 1989 Testosterone rises neuropeptide-Y
concentration in selected hypothalamic sites and in vitro release from the medial basal
hypothalamus of castrated male rats. Endocrinology 124:410
Urban JH, Bauer-Dantoin AC, Levine JE 1993 Neuropeptide Y gene expression in the arcuate
nucleus: sexual dimorphism and modulation by testosterone. Endocrinology 132:139
Alvarez R, Andres Jd, Yubero P, Vinas O, Mampel T, Iglesias R, Giralt M, Villarroya F 1995
A novel regulatory pathway of brown fat thermogenesis. Retinoic acid is a transcriptional activator
of the mitochondrial uncoupling protein gene. Journal of Biological Chemistry 270:5666-5673
Kumar MV, Scarpace PJ 1998 Differential effects of retinoic acid on uncoupling protein-1 and
leptin gene expression. Journal of Endocrinology 157:237-243
Magni P, Bernasconi M, Pagano C, Calcagno A, Vettor R, Motta M 2001 Biological actions of
leptin in a human neuroblastoma cell line: effects on cell proliferation and neuropeptide Y
expression. Endocrine Society Meeting, Denver, CO
Allen JM, Martin JB, Heinrich G 1987 Neuropeptide Y gene expression in PC 12 cells and its
regulation by nerve growth factor: a model for developmental regulation. Brain Research 427:3943
Sabol SL, Higuchi H 1990 Transcriptional regulation of the neuropeptide Y gene by nerve
growth factor: antagonism by glucocorticoids and potentiation by adenosine 3',5'-monophosphate
and phorbol ester. Molecular Endocrinology 4:384-392
Barnea A, Cho G 1993 Basic fibroblast growth factor selectively amplifies the functional state of
neurons producing neuropeptide Y but not somatostatin in cultures of fetal brain cells: evidence
for a cooperative interaction with insulin-like growth factor-1. Endocrinology 133:1895-1898
Barnea A, Cramer O, Cho G 1993 Truncated insulin-like growth factor I: a potent inducer of
neuropeptide Y production by aggregate cultures of fetal brain cells. Endocrine Journal 1:11-17
Takei N, Sasaoka K, Higuchi H, Endo Y, Hatanaka H 1996 BDNF increases the expression of
neuropeptide Y mRNA and promotes differentiation/maturation of neuropeptide Y-positive
cultered cortical neurons from embrionic and postnatal rats. Molecular Brain Research 37:283289
Colmers WF, Wahlestedt C (eds) 1993 The biology of neuropeptide Y and related peptides.
Humana Press, Totawa, NJ
Barnea A, Aguila-Mansilla N, Chute HT, Welcher AA 1996 Comparison of neurotrophin
regulation of human and rat neuropeptide Y (NPY) neurons: induction of NPY production in
aggregate cultures derived from rat but not from human fetal brains. Brain Research 732:52-60
98
253.
254.
255.
256.
257.
258.
259.
260.
261.
262.
263.
264.
265.
266.
267.
268.
269.
270.
271.
272.
273.
274.
275.
276.
Freidin M, Dougherty M, Kessler JA 1993 Cell density regulates neuropeptide Y expression in
cultured sympathetic neurons. Brain Research 615:135-140
Pu S, Dhillon H, Moldawer LL, Kalra PS, Kalra SP 2000 Neuropeptide Y counteracts the
anorectic and weight reducing effects of ciliary neurotropic factor. J Neuroendocrinol 12:827-32.
Stanley BG, Daniel DR, Chin AS, Leibowitz SF 1985 Paraventricular nucleus injections of
peptide YY and neuropeptide Y preferentially enhance carbohydrate ingestion. Peptides 6:12051211
Welch CC, Grace MK, Billington CJ, Levine AS 1994 Preference and diet type affect
macronutrient selection after morphine, NPY, norepinephrine, and deprivation. Am J Physiol
266:R426-33
Pesonen U, Huupponen R, Rouru J, Koulu M 1992 Hypothalamic neuropeptide expression
after food restriction in Zucker rats: evidence of persistent neuropeptide Y gene activation.
Molecular Brain Research 16:255-260
Corp ES, McQuade J, Krasnicki S, Conze DB 2001 Feeding after fourth ventricular
administration af neuropeptide Y receptor agonists in rats. Peptides 22:493-499
DiBona GF 2002 Neuropeptide Y. Am J Physiol Regul Integr Comp Physiol 282:R635-6.
Serradeil-LeGal C, Lafontan M, Raufaste D, Marchand J, Pouzet B, Casellas P, Pascal M,
Maffrand JP, Fur GL 2000 Characterization of NPY receptors controlling lipolysis and leptin
secretion in human adipocytes. FEBS Letters 475:150-156
Bing C, wang Q, Pickavance L, Willials G 1996 The central regulation of energy homoeostasis:
roles of neuropeptide Y and other brain peptides. Regulatory peptides in the control of food intake
et metabolism 21:559-565
Rebuffe-Scrive M, Walsh UA, McEwen B, Rodin J 1992 Effect of chronic stress and
exogenous glucocorticoids on regional fat distribution and metabolism. Physiol Behav 52:583-590
Dallman MF, Strack AM, Akana SF, Bradbury MJ, Hanson ES, Scribner KA, Smith M 1993
Feast and famine: critical role of glucocorticoids with insulin in daily energy flow. Front
Neuroendocrinol 14:303-347
Zakrzewska KE, Sainsbury A, Cusin I, Rouru R, Jeanrenaud B, Rohner-Jeanrenaud EF
1999 Selective dependence of intracerebroventricular neuropeptide Y-elicited effects on central
glucocorticoids. Endocrinology 140:3183-3187
Sainsbury A, Wilks D, Cooney GJ 2000 Interaction between adrenal glucocorticoids and
parasympathetic activation in mediating hyperinsulinemia during chronic central neuropeptide Y
infusion in rats. Diabetologia 43:859-865
Sainsbury A, Herzog H 2001 Inhibitory effects of central neuropeptide Y on the somatotropic
and gonadotropic axis in male rats are independent of adrenal hormones. Peptides 22:467-471
Kalra SP, Crowley WR 1992 Neuropeptide Y: a novel neuroendocrine peptide in the control of
pituitary hormone secretion, and its relation to luteinizing hormone. Frontiers in
Neuroendocrinology 13:1-46
Kalra S, Crowley W 1984 Differential effects of pancreatic polypeptide on luteinizing hormone
release in female rats. Neuroendocrinology 38:511-513
Kalra SP, Crowley WR, Kalra PS 1986 Different modes of NPY receptor activation may regulate
LH secretion and feeding behaviour. Society for Neuroscience meeting(Abst) 12:1494
Gorsky R 1984 Sexual differentiation of brain structure in rodents. New York: Raven Press
Walhestedt C, Skagerberg G, Ekman R, Heilig M, Sundler D, Hakanson R 1987 Neuropeptide
Y (NPY) in the area of the hypothalamic paraventricular nucleus activates the pituitary adrenal
axis in the rat. Brain Research 41:33-38
Leibowitz SF, Sladeck C, Spencer L, Tempel D 1988 Neuropeptide Y, epinephrine and
norepinephrine in the paraventricular nucleus: stimulation of feeding and the release of
corticosterone, vasopressin and glucose. Brain Research Bulletin 21:905-912
Catzeflis C, Pierroz DD, Rohner-Jeanrenaud F, Rivier JE, Sizonenko PC, Aubert ML 1993
Neuropeptide Y administered chronically in to the lateral ventricle profoundly inhibits both the
gonadotropic and the somatotropic axis in intact adult female rats. Endocrinology 132:224-234
Pierroz DD, Catzeflis C, Aebi AC, Rivier JE, Aubert ML 1996 Chronic administration of
neuropeptide Y into the lateral ventricle inhibits both both the pituitary-testicular axis and growth
hormone and insulin-like growth factor I secretion in intact adult male rats. Endocrinology 137:312
Gkonos PJ, Krongrad A, Roos BA 1995 Neuroendocrine peptides in the prostate. Urological
Research 23:81-87
di Sant'Agnese PA 2001 Neuroendocrine differentiation in prostatic carcinoma: an update on
recent developments. Ann Oncol 12:S135-40.
99
277.
278.
279.
280.
281.
282.
283.
284.
285.
286.
287.
288.
289.
290.
291.
292.
293.
294.
295.
296.
297.
298.
299.
300.
301.
302.
Ventura S, Pennefather JN, Mitchelson F 2002 Cholinergic innervation and function in the
prostate gland. Pharmacology & Therapeutics 94:93-112
Cohen RJ, Glezerson G, Haffejee Z, Afrika D 1990 Prostatic carcinoma: histological and
immunohistological factors affecting prognosis. British Journal of Urology 66:405-410
Abrahamsson PA 1999 Neuroendocrine cells in tumor growth of the prostate. EndocrineRelated Cancer 6:503-519
Tainio H 1995 Peptidergic innervation of the human prostate, seminal vesicle and vas deferens.
Acta Histochemica 97:113-119
Mack D, Hacker GW, Hauser-Kronberger C, Frick J, Dietze O 1997 Vasoactive intestinal
polypeptide (VIP) and neuropeptide tyrosine (NPY) in prostate carcinoma. European Journal of
Cancer 33:317-318
Reubi JC, Gugger M, Waser B, Schaer JC 2001 Y1-Mediated Effect of Neuropeptide Y in
Cancer: Breast Carcinomas as Targets. Cancer Research 61:4636-4641
Krulich L, Dhariwal AP, McCann SM 1968 Stimulatory and inhibitory effects of purified
hypothalamic extracts on growth hormone release from rat pituitary in vitro. Endocrinology
83:783-90
Corleto VD, Nasoni S, Panzuto F, Cassetta S, Delle Fave G 2004 Somatostatin receptor
subtypes: basic pharmacology and tissue distribution. Digestive and Liver Disease 36:S8-S16
Sica G 1999 Attività biologiche della somatostatina. In: La somatostatina e i suoi analoghi. Lo
stato dell'arte; pp 53-67
Brevini TAL 1999 Somatostatina e suoi analoghi di sintesi. In: La somatostatina e i suoi
analoghi. Lo stato dell'arte.; pp 19-31
Berelowitz M, LeRoith D, von Schenk H, Newgard C, Szabo M, Frohman LA, Shiloach J,
Roth J 1982 Somatostatin-like immunoactivity and biological activity is present in Tetrahymena
pyriformis, a ciliated protozoan. Endocrinology 110:1939-44
Goodman RH, Aron DC, Roos BA 1983 Rat pre-prosomatostatin. Structure and processing by
microsomal membranes. J Biol Chem 258:5570-3
Benoit R, Esch F, Bennett HP, Ling N, Ravazzola M, Orci L, Mufson EJ 1990 Processing of
prosomatostatin. Metabolism 39:22-5
Patel YC, Galanopoulou A 1995 Processing and intracellular targeting of prosomatostatinderived peptides: the role of mammalian endoproteases. Ciba Found Symp 190:26-40;
discussion 40-50
Sakurada C, Yokosawa H, Ishii S 1990 The degradation of somatostatin by synaptic membrane
of rat hippocampus is initiated by endopeptidase-24.11. Peptides 11:287-92
Feindt J, Krisch B, Lucius R, Mentlein R 1997 Meningeal cells are targets and inactivation
sites for the neuropeptide somatostatin. Brain Res Mol Brain Res 44:293-300
Mentlein R, Dahms P 1994 Endopeptidases 24.16 and 24.15 are responsible for the degradation
of somatostatin, neurotensin, and other neuropeptides by cultivated rat cortical astrocytes. J
Neurochem 62:27-36
Hellman B, Lernmark A 1969 Inhibition of the in vitro secretion of insulin by an extract of
pancreatic alpha-1 cells. Endocrinology 84:1484-8
Brazeau P, Vale W, Burgus R, Ling N, Butcher M, Rivier J, Guillemin R 1973 Hypothalamic
polypeptide that inhibits the secretion of immunoreactive pituitary growth hormone. Science
179:77-9
Orci L, Baetens D, Dubois MP, Rufener C 1975 Evidence for the D-cell of the pancreas
secreting somatostatin. Horm Metab Res 7:400-2
Polak JM, Pearse AG, Grimelius L, Bloom SR 1975 Growth-hormone release-inhibiting
hormone in gastrointestinal and pancreatic D cells. Lancet 1:1220-2
Hokfelt T, Efendic S, Hellerstrom C, Johansson O, Luft R, Arimura A 1975 Cellular
localization of somatostatin in endocrine-like cells and neurons of the rat with special references
to the A1-cells of the pancreatic islets and to the hypothalamus. Acta Endocrinol Suppl (Copenh)
200:5-41
Bordi C, D'Adda T, Baggi MT, Pilato FP 1989 Structure and function of endocrine cells in the
oxyntic (acid-secreting) mucosa of human stomach. Scand J Gastroenterol Suppl 166:115-21;
discussion 138-7
Orci L, Unger RH 1975 Functional subdivision of islets of Langerhans and possible role of D
cells. Lancet 2:1243-4
Orci L 1982 Macro- and micro-domains in the endocrine pancreas. Diabetes 31:538-65
DeLellis RA, May L, Tashjian AH, Jr., Wolfe HJ 1978 C-cell granule heterogeneity in man. An
ultrastructural immunocytochemical study. Lab Invest 38:263-9
100
303.
304.
305.
306.
307.
308.
309.
310.
311.
312.
313.
314.
315.
316.
317.
318.
319.
320.
321.
322.
323.
324.
325.
326.
327.
328.
329.
330.
331.
Scopsi L 1990 Non-calcitonin genes-derived neurohormonal polypeptides in normal and
pathologic thyroid C cells. In: Progress in surgical pathology; pp 185-229
Hislop AA, Wharton J, Allen KM, Polak JM, Haworth SG 1990 Immunohistochemical
localization of peptide-containing nerves in human airways: age-related changes. Am J Respir
Cell Mol Biol 3:191-8
Johansson O, Hokfelt T, Elde RP 1984 Immunohistochemical distribution of somatostatin-like
immunoreactivity in the central nervous system of the adult rat. Neuroscience 13:265-339
Patel YC, Reichlin S 1978 Somatostatin in hypothalamus, extrahypothalamic brain, and
peripheral tissues of the rat. Endocrinology 102:523-30
Mengod G, Rigo M, Savasta M, Probst A, Palacios JM 1992 Regional distribution of
neuropeptide somatostatin gene expression in the human brain. Synapse 12:62-74
Pelletier G 1991 The brain as an endocrine organ. Blackwell Scientific, Boston
Patel YC 1992 General apsects of the biology and function of somatostatin, Berlin
Delfs JR, Zhu CH, Dichter MA 1984 Coexistence of acetylcholinesterase and somatostatinimmunoreactivity in neurons cultured from rat cerebrum. Science 223:61-3
Krisch B 1979 Immunohistochemical results on the distribution of somatostatin in the
hypothalamus and in limbic structures of the rat. J Histochem Cytochem 27:1389-90
Finley JC, Maderdrut JL, Roger LJ, Petrusz P 1981 The immunocytochemical localization of
somatostatin-containing neurons in the rat central nervous system. Neuroscience 6:2173-92
Feher E, Fodor M, Feher J 1992 Ultrastructural localization of somatostatin- and substance Pimmunoreactive nerve fibers in the feline liver. Gastroenterology 102:287-94
Polak JM, Bloom SR 1986 Somatostatin localization in tissues. Scand J Gastroenterol Suppl
119:11-21
Patel Y, Rao K, Reichlin S 1977 Somatostatin in human cerebrospinal fluid. N Engl J Med
296:529-33
Sasaki A, Yoshinaga K 1989 Immunoreactive somatostatin in male reproductive system in
humans. J Clin Endocrinol Metab 68:996-9
Werner H, Amarant T, Millar RP, Fridkin M, Koch Y 1985 Immunoreactive and biologically
active somatostatin in human and sheep milk. Eur J Biochem 148:353-7
Holst N, Jenssen TG, Burhol PG 1990 A characterization of immunoreactive somatostatin in
human milk. J Pediatr Gastroenterol Nutr 10:47-52
Sica G 1999 Localizzazione della somatostatina e caratteristiche delle cellule che la producono.
In: La somatostatina e i suoi analoghi, Lo stato dell'arte; pp 1-17
Alumets J, Sundler F, Hakanson R 1977 Distribution, ontogeny and ultrastructure of
somatostatin immunoreactive cells in the pancreas and gut. Cell Tissue Res 185:465-79
Lehy T, Peranzi G, Cristina ML 1981 Correlative immunocytochemical and electron microscopic
studies: identification of (entero)glucagon- somatostatin- and pancreatic polypeptide-likecontaining cells in the human colon. Histochemistry 71:67-80
Stefan Y, Grasso S, Perrelet A, Orci L 1983 A quantitative immunofluorescent study of the
endocrine cell populations in the developing human pancreas. Diabetes 32:293-301
Chayvialle JA, Paulin C, Dubois PM, Descos F, Dubois MP 1980 Ontogeny of somatostatin in
the human gastro-intestinal tract, endocrine pancreas and hypothalamus. Acta Endocrinol
(Copenh) 94:1-10
Kobayashi S, Iino S 1993 Segi's cap: group formation of gut endocrine cells at the tip of the villi
in human embryonal intestine. Nagoya J Med Sci 56:43-52
Schusdziarra V 1996 Somatostatin: Biological actions and pathophysiology. In: Octreotide: From
basic scienze to clinical medicine.; pp 35-53
Reubi JC, Waser B, Schaer JC, Markwalder R 1995 Somatostatin receptors in human prostate
and prostate cancer. J Clin Endocrinol Metab 80:2806-14
Csaba Z, Dournaud P 2001 Cellular biology of somatostatin receptors. Neuropeptides 35:1-23
Ferone D, Kwekkeboom DJ, Pivonello R, Bogers ADJJ, Colao A, Lamberts SWJ, Van
Hagen PM, Hofland LJ 2001 In vivo and in vitro expression of somatostatin receptors in two
human thymomas with similar clinical presentation and different histological features.
Epelbaum J 1986 Somatostatin in the central nervous system: physiology and pathological
modifications. Prog Neurobiol 27:63-100
Aponte G, Leung P, Gross D, Yamada T 1984 Effects of somatostatin on food intake in rats.
Life Sci 35:741-6
Vecsei L, Kiraly C, Bollok I, Nagy A, Varga J, Penke B, Telegdy G 1984 Comparative studies
with somatostatin and cysteamine in different behavioral tests on rats. Pharmacol Biochem
Behav 21:833-7
101
332.
333.
334.
335.
336.
337.
338.
339.
340.
341.
342.
343.
344.
345.
346.
347.
348.
349.
350.
351.
352.
353.
354.
355.
356.
357.
Taddese A, Nah SY, McCleskey EW 1995 Selective opioid inhibition of small nociceptive
neurons. Science 270:1366-9
Moore MR, Black PM 1991 Neuropeptides. Neurosurg Rev 14:97-110
Gaumann DM, Yaksh TL 1988 Intrathecal somatostatin in rats: antinociception only in the
presence of toxic effects. Anesthesiology 68:733-42
Leblanc R, Gauthier S, Gauvin M, Quirion R, Palmour R, Masson H 1988 Neurobehavioral
effects of intrathecal somatostatinergic treatment in subhuman primates. Neurology 38:1887-90
Dirksen R, Lerou J, Nijhuis GM, Booij LH, Jurna I 1990 Intrathecal somatostatin produces
effects dependent on the interval between catheter implantation and drug injection. Life Sci
47:1347-54
Chrubasik J, Meynadier J, Blond S, Scherpereel P, Ackerman E, Weinstock M, Bonath K,
Cramer H, Wunsch E 1984 Somatostatin, a potent analgesic. Lancet 2:1208-9
Penn RD, Paice JA, Kroin JS 1990 Intrathecal octreotide for cancer pain. Lancet 335:738
Harfstrand A, Fuxe K, Kalia M, Agnati LF 1985 Somatostatin induced apnoea: prevention by
central and peripheral administration of the opiate receptor blocking agent naloxone. Acta Physiol
Scand 125:91-5
Frisinghelli A, Crema C, Bevilacqua M, Chebat E, Turiel M 1991 [Hemodynamic response to
somatostatin at rest and during sympathetic activation in idiopathic orthostatic hypotension].
Cardiologia 36:199-206
Van Dijk G, Balkan B, Lindfeldt J, Bouws G, Scheurink AJ, Ahren B, Steffens AB 1994
Contribution of liver nerves, glucagon, and adrenaline to the glycaemic response to exercise in
rats. Acta Physiol Scand 150:305-13
Holt R.I.G., McGregor AM 1997 Somatostatin - from Basic Science to Clinical Application.
Excerpta Medica, Hong Kong
Patel Y 1992 General aspects of the biology and function of somatostatin. In: Somatostatin:
Basic and clinical aspects of neuroscience.; pp 1-28
Herrington J, Hille B 1994 Growth hormone-releasing hexapeptide elevates intracellular calcium
in rat somatotropes by two mechanisms. Endocrinology 135:1100-8
Cuttler L 1996 The regulation of growth hormone secretion. Endocrinol Metab Clin North Am
25:541-71
Horvath S, Palkovits M, Gorcs T, Arimura A 1989 Electron microscopic immunocytochemical
evidence for the existence of bidirectional synaptic connections between growth hormonereleasing hormone- and somatostatin-containing neurons in the hypothalamus of the rat. Brain
Res 481:8-15
Berelowitz M, Szabo M, Frohman LA, Firestone S, Chu L, Hintz RL 1981 Somatomedin-C
mediates growth hormone negative feedback by effects on both the hypothalamus and the
pituitary. Science 212:1279-81
Katakami H, Downs TR, Frohman LA 1988 Inhibitory effect of hypothalamic medial preoptic
area somatostatin on growth hormone-releasing factor in the rat. Endocrinology 123:1103-9
Argente J, Garcia-Segura LM, Pozo J, Chowen JA 1996 Growth hormone-releasing peptides:
clinical and basic aspects. Horm Res 46:155-9
Chowen JA, Garcia-Segura LM, Gonzalez-Parra S, Argente J 1996 Sex steroid effects on the
development and functioning of the growth hormone axis. Cell Mol Neurobiol 16:297-310
Chihara K, Minamitani N, Kaji H, Arimura A, Fujita T 1981 Intraventricularly injected growth
hormone stimulates somatostatin release into rat hypophysial portal blood. Endocrinology
109:2279-81
Shimon I, Yan X, Taylor JE, Weiss MH, Culler MD, Melmed S 1997 Somatostatin receptor
(SSTR) subtype-selective analogues differentially suppress in vitro growth hormone and prolactin
in human pituitary adenomas: Novel potential therapy for functional pituitary tumors. Journal of
Clinical Investigation 100:2386-2392
Schlegel W, Winiger BP, Mollard P, Vacher P, Wuarin F, Zahnd GR, Wollheim CB, Dufy B
1987 Oscillations of cytosolic Ca2+ in pituitary cells due to action potentials. Nature 329:719-21
Lucke C, Hoffken B, von zur Muhlen A 1975 The effect of somatostatin on TSH levels in
patients with primary hypothyroidism. J Clin Endocrinol Metab 41:1082-4
Weeke J, Christensen SE, Hansen AP, Laurberg P, Lundbaek K 1980 Somatostatin and the
24 h levels of serum TSH, T3, T4, and reverse T3 in normals, diabetics and patients treated for
myxoedema. Acta Endocrinol (Copenh) 94:30-7
Fujita T 1973 Insulo-acinar portal system in the horse pancreas. Arch Histol Jpn 35:161-71
Taborsky GJ, Jr. 1983 Evidence of a paracrine role for pancreatic somatostatin in vivo. Am J
Physiol 245:E598-603
102
358.
359.
360.
361.
362.
363.
364.
365.
366.
367.
368.
369.
370.
371.
372.
373.
374.
375.
376.
377.
378.
379.
380.
381.
Amherdt M, Patel YC, Orci L 1987 Selective binding of somatostatin-14 and somatostatin-28 to
islet cells revealed by quantitative electron microscopic autoradiography. J Clin Invest 80:1455-8
Gyr K, Beglinger C, Kohler E, Trautzl U, Keller U, Bloom SR 1987 Circulating somatostatin.
Physiological regulator of pancreatic function? J Clin Invest 79:1595-600
Chayvialle JA, Miyata M, Rayford PL, Thompson JC 1980 Effects of test meal, intragastric
nutrients, and intraduodenal bile on plasma concentrations of immunoreactive somatostatin and
vasoactive intestinal peptide in dogs. Gastroenterology 79:844-52
Schusdziarra V, Harris V, Conlon JM, Arimura A, Unger R 1978 Pancreatic and gastric
somatostatin release in response to intragastric and intraduodenal nutrients and HCl in the dog. J
Clin Invest 62:509-18
Lucey MR, Wass JA, Rees LH, Dawson AM, Fairclough PD 1989 Relationship between gastric
acid and elevated plasma somatostatinlike immunoreactivity after a mixed meal.
Gastroenterology 97:867-72
Saffouri B, Weir G, Bitar K, Makhlouf G 1979 Stimulation of gastrin secretion from the perfused
rat stomach by somatostatin antiserum. Life Sci 25:1749-53
Park J, Chiba T, Yamada T 1987 Mechanisms for direct inhibition of canine gastric parietal cells
by somatostatin. J Biol Chem 262:14190-6
Colturi TJ, Unger RH, Feldman M 1984 Role of circulating somatostatin in regulation of gastric
acid secretion, gastrin release, and islet cell function. Studies in healthy subjects and duodenal
ulcer patients. J Clin Invest 74:417-23
Guillemin R 1976 Somatostatin inhibits the release of acetylcholine induced electrically in the
myenteric plexus. Endocrinology 99:1653-4
Sandvik AK, Waldum HL 1988 The effect of somatostatin on baseline and stimulated acid
secretion and vascular histamine release from the totally isolated vascularly perfused rat
stomach. Regul Pept 20:233-9
Schusdziarra V, Bender H, Pfeffer A, Pfeiffer EF 1984 Modulation of acetylcholine-induced
secretion of gastric bombesin-like immunoreactivity by cholinergic and histamine H2-receptors,
somatostatin and intragastric pH. Regul Pept 8:189-98
Ryberg B, Tielemans Y, Axelson J, Carlsson E, Hakanson R, Mattson H, Sundler F, Willems
G 1990 Gastrin stimulates the self-replication rate of enterochromaffinlike cells in the rat stomach.
Effects of omeprazole, ranitidine, and gastrin-17 in intact and antrectomized rats.
Gastroenterology 99:935-42
Holst JJ, Orskov C, Seier-Poulsen S 1992 Somatostatin is an essential paracrine link in acid
inhibition of gastrin secretion. Digestion 51:95-102
Schusdziarra V, Rouiller D, Harris V, Unger RH 1979 Release of gastric somatostatin-like
immunoreactivity during acidification of the duodenal bulb. Gastroenterology 76:950-3
Lucey MR, Fairclough PD, Wass JA, Kwasowski P, Medbak S, Webb J, Rees LH 1984
Response of circulating somatostatin, insulin, gastrin and GIP, to intraduodenal infusion of
nutrients in normal man. Clin Endocrinol (Oxf) 21:209-17
Zyznar ES, Pietri AO, Harris V, Unger RH 1981 Evidence for the hormonal status of
somatostatin in man. Diabetes 30:883-6
Aizawa I, Itoh Z, Harris V, Unger RH 1981 Plasma somatostatin-like immunoreactivity during
the interdigestive period in the dog. J Clin Invest 68:206-13
Magnusson I, Einarsson K, Angelin B, Nyberg B, Bergstrom K, Thulin L 1989 Effects of
somatostatin on hepatic bile formation. Gastroenterology 96:206-12
Schusdziarra V 1992 The physiological role of somatostatin in the regulation of nutrient
homeostasis. In: Somatostatin: Basic and clinical aspects of neuroscience.; pp 43-54
Schusdziarra V, Lawecki J, Ditschuneit HH, Lukas B, Maier V, Pfeiffer EF 1985 Effect of lowdose somatostatin infusion on pancreatic and gastric endocrine function in lean and obese
nondiabetic human subjects. Diabetes 34:595-601
Walker BJ, Nelstrop GA, Joekes AM 1983 Somatostatin and water excretion. Lancet 1:1101-2
Gines A, Salmeron JM, Gines P, Jimenez W, Salo J, Piera C, Claria J, Rivera F, Arroyo V,
Rodes J 1992 Effects of somatostatin on renal function in cirrhosis. Gastroenterology 103:186874
Wood SM, Wood JR, Ghatei MA, Lee YC, O'Shaughnessy D, Bloom SR 1981 Bombesin,
somatostatin and neurotensin-like immunoreactivity in bronchial carcinoma. J Clin Endocrinol
Metab 53:1310-2
Dayer AM, De Mey J, Will JA 1985 Localization of somatostatin-, bombesin-, and serotonin-like
immunoreactivity in the lung of the fetal rhesus monkey. Cell Tissue Res 239:621-5
103
382.
383.
384.
385.
386.
387.
388.
389.
390.
391.
392.
393.
394.
395.
396.
397.
398.
399.
400.
401.
402.
403.
404.
405.
406.
Hilfer SR, Schneck SL, Brown JW 1986 The effect of culture conditions on cytodifferentiation of
fetal mouse lung respiratory passageways. Exp Lung Res 10:115-36
Ruggere MD, Patel YC 1985 Somatostatin metabolism by the isolated rat lung. Endocrinology
117:1870-3
Agro A, Padol I, Stanisz AM 1991 Immunomodulatory activities of the somatostatin analogue
BIM 23014c: effects on murine lymphocyte proliferation and natural killer activity. Regul Pept
32:129-39
Yousefi S, Ghazinouri A, Vaziri N, Tilles J, Carandang G, Cesario T 1990 The effect of
somatostatin on the production of human interferons by mononuclear cells. Proc Soc Exp Biol
Med 194:114-8
Yousefi S, Vaziri N, Carandang G, Le W, Yamamoto R, Granger G, Ocariz J, Cesario T 1991
The paradoxical effects of somatostatin on the bioactivity and production of cytotoxins derived
from human peripheral blood mononuclear cells. Br J Cancer 64:243-6
Theoharides TC, Douglas WW 1981 Mast cell histamine secretion in response to somatostatin
analogues: structural considerations. Eur J Pharmacol 73:131-6
Goetzl EJ, Payan DG 1984 Inhibition by somatostatin of the release of mediators from human
basophils and rat leukemic basophils. J Immunol 133:3255-9
Jenkins SA, Baxter JN, Day DW, Al-Sumidaie AM, Leinster SJ, Shields R 1986 The effects of
somatostatin and SMS 201-995 on experimentally-induced pancreatitis and endotoxaemia in rats
and on monocyte activity in patients with cirrhosis and portal hypertension. Klin Wochenschr 64
Suppl 7:100-6
Peck R 1987 Neuropeptides modulating macrophage function. Ann N Y Acad Sci 496:264-70
van Hagen PM, Krenning EP, Kwekkeboom DJ, Reubi JC, Anker-Lugtenburg PJ,
Lowenberg B, Lamberts SW 1994 Somatostatin and the immune and haematopoetic system; a
review. Eur J Clin Invest 24:91-9
Yamada Y, Post SR, Wang K, Tager HS, Bell GI, Seino S 1992 Cloning and functional
characterization of a family of human and mouse somatostatin receptors expressed in brain,
gastrointestinal tract, and kidney. Biochemistry 89:251-255
Rohrer L, Raulf F, Bruns C, Buettner R, Hofstaedter F, Schule R 1993 Cloning and
characterization of a fourth human somatostatin receptor. Proc Natl Acad Sci U S A 90:4196-200
Bell GI, Yasuda K, Kong H, Law SF, Raynor K, Reisine T 1995 Molecular biology of
somatostatin receptors. Ciba Found Symp 190:65-79; discussion 80-8
Patel YC 1999 Somatostatin and its receptor family. Front Neuroendocrinol 20:157-98
Rens-Domiano S, Reisine T 1991 Structural analysis and functional role of the carbohydrate
component of somatostatin receptors. J Biol Chem 266:20094-102
O'Dowd BF, Hnatowich M, Caron MG, Lefkowitz RJ, Bouvier M 1989 Palmitoylation of the
human beta 2-adrenergic receptor. Mutation of Cys341 in the carboxyl tail leads to an uncoupled
nonpalmitoylated form of the receptor. J Biol Chem 264:7564-9
Patel YC, Greenwood MT, Panetta R, Demchyshyn L, Niznik H, Srikant CB 1995 The
somatostatin receptor family. Life Sci 57:1249-65
Reisine T, Bell GI 1995 Molecular biology of somatostatin receptors. Endocr Rev 16:427-42
Bass RT, Buckwalter BL, Patel BP, Pausch MH, Price LA, Strnad J, Hadcock JR 1996
Identification and characterization of novel somatostatin antagonists. Mol Pharmacol 50:709-15
Evans CJ, Keith DE, Jr., Morrison H, Magendzo K, Edwards RH 1992 Cloning of a delta
opioid receptor by functional expression. Science 258:1952-5
Nehring RB, Meyerhof W, Richter D 1995 Aspartic acid residue 124 in the third transmembrane
domain of the somatostatin receptor subtype 3 is essential for somatostatin-14 binding. DNA Cell
Biol 14:939-44
Strnad J, Hadcock JR 1995 Identification of a critical aspartate residue in transmembrane
domain three necessary for the binding of somatostatin to the somatostatin receptor SSTR2.
Biochem Biophys Res Commun 216:913-21
Greenwood MT, Hukovic N, Kumar U, Panetta R, Hjorth SA, Srikant CB, Patel YC 1997
Ligand binding pocket of the human somatostatin receptor 5: mutational analysis of the
extracellular domains. Mol Pharmacol 52:807-14
Patel YC, Srikant CB 1994 Subtype selectivity of peptide analogs for all five cloned human
somatostatin receptors (hsstr 1-5). Endocrinology 135:2814-7
Dournaud P, Gu YZ, Schonbrunn A, Mazella J, Tannenbaum GS, Beaudet A 1996
Localization of the somatostatin receptor SST2A in rat brain using a specific anti-peptide
antibody. J Neurosci 16:4468-78
104
407.
408.
409.
410.
411.
412.
413.
414.
415.
416.
417.
418.
419.
420.
421.
422.
423.
424.
425.
426.
427.
428.
429.
430.
431.
Raulf F, Perez J, Hoyer D, Bruns C 1994 Differential expression of five somatostatin receptor
subtypes, SSTR1-5, in the CNS and peripheral tissue. Digestion 55 Suppl 3:46-53
Bruno JF, Xu Y, Song J, Berelowitz M 1993 Tissue distribution of somatostatin receptor
subtype messenger ribonucleic acid in the rat. Endocrinology 133:2561-7
O'Carroll AM, Krempels K 1995 Widespread distribution of somatostatin receptor messenger
ribonucleic acids in rat pituitary. Endocrinology 136:5224-7
Patel YC 1997 Molecular pharmacology of somatostatin receptor subtypes. J Endocrinol Invest
20:348-67
van Hagen PM 1996 Somatostatin receptor expression in clinical immunology. Metabolism
45:86-7
Le Romancer M, Cherifi Y, Levasseur S, Laigneau JP, Peranzi G, Jais P, Lewin MJ, ReylDesmars F 1996 Messenger RNA expression of somatostatin receptor subtypes in human and
rat gastric mucosae. Life Sci 58:1091-8
Dizeyi N, Konrad LK, Bjartell A, Wu H, Gadaleanu V, Hansson J, Helboe L, Abrahamsson
PA 2002 Localization and mRNA expression of somatostatin receptor subtypes in human
prostatic tissue and prostate cancer cell lines. Urologic Oncology 7:91-98
Sinisi AA, Bellastella A, Prezioso D, Nicchio MR, Lotti T, Salvatore M, Pasquali D 1997
Different expression patterns of somatostatin receptor subtypes in cultured epithelial cells from
human normal prostate and prostate cancer. J Clin Endocrinol Metab 82:2566-9
Reubi JC, Schaer JC, Laissue JA, Waser B 1996 Somatostatin receptors and their subtypes in
human tumors and in peritumoral vessels. Metabolism 45:39-41
Robbins RJ 1996 Somatostatin and cancer. Metabolism 45:98-100
Moller LN, Stidsen CE, Hartmann B, Holst JJ 2003 Somatostatin receptors. Biochimica et
Biophysica Acta 1616:1-84
Luthin DR, Eppler CM, Linden J 1993 Identification and quantification of Gi-type GTP-binding
proteins that copurify with a pituitary somatostatin receptor. J Biol Chem 268:5990-6
Law SF, Yasuda K, Bell GI, Reisine T 1993 Gi alpha 3 and G(o) alpha selectively associate with
the cloned somatostatin receptor subtype SSTR2. J Biol Chem 268:10721-7
Reisine T, Woulfe D, Raynor K, Kong H, Heerding J, Hines J, Tallent M, Law S 1995
Interaction of somatostatin receptors with G proteins and cellular effector systems. Ciba Found
Symp 190:160-7; discussion 167-70
Hou C, Gilbert RL, Barber DL 1994 Subtype-specific signaling mechanisms of somatostatin
receptors SSTR1 and SSTR2. J Biol Chem 269:10357-62
O'Carroll AM, Lolait SJ, Konig M, Mahan LC 1992 Molecular cloning and expression of a
pituitary somatostatin receptor with preferential affinity for somatostatin-28. Mol Pharmacol
42:939-46
Rens-Domiano S, Law SF, Yamada Y, Seino S, Bell GI, Reisine T 1992 Pharmacological
properties of two cloned somatostatin receptors. Mol Pharmacol 42:28-34
Kaupmann K, Bruns C, Hoyer D, Seuwen K, Lubbert H 1993 Distribution and second
messenger coupling of four somatostatin receptor subtypes expressed in brain. FEBS Lett
331:53-9
Kubota A, Yamada Y, Kagimoto S, Shimatsu A, Imamura M, Tsuda K, Imura H, Seino S,
Seino Y 1994 Identification of somatostatin receptor subtypes and an implication for the efficacy
of somatostatin analogue SMS 201-995 in treatment of human endocrine tumors. Journal of
Clinical Investigation 93:1321-1325
Hadcock JR, Strnad J, Eppler CM 1994 Rat somatostatin receptor type 1 couples to G proteins
and inhibition of cyclic AMP accumulation. Mol Pharmacol 45:410-6
Kagimoto S, Yamada Y, Kubota A, Someya Y, Ihara Y, Yasuda K, Kozasa T, Imura H, Seino
S, Seino Y 1994 Human somatostatin receptor, SSTR2, is coupled to adenylyl cyclase in the
presence of Gi alpha 1 protein. Biochem Biophys Res Commun 202:1188-95
Yasuda K, Rens-Domiano S, Breder CD, Law SF, Saper CB, Reisine T, Bell GI 1992 Cloning
of a novel somatostatin receptor, SSTR3, coupled to adenylylcyclase. J Biol Chem 267:20422-8
Bertherat J, Bluet-Pajot MT, Epelbaum J 1995 Neuroendocrine regulation of growth hormone.
Eur J Endocrinol 132:12-24
Carruthers AM, Warner AJ, Michel AD, Feniuk W, Humphrey PP 1999 Activation of adenylate
cyclase by human recombinant sst5 receptors expressed in CHO-K1 cells and involvement of
Galphas proteins. Br J Pharmacol 126:1221-9
Schonbrunn A 1990 Somatostatin action in pituitary cells involves two independent transduction
mechanism. Metabolism 39:96-100
105
432.
433.
434.
435.
436.
437.
438.
439.
440.
441.
442.
443.
444.
445.
446.
447.
448.
449.
450.
451.
452.
453.
Akbar M, Okajima F, Tomura H, Majid MA, Yamada Y, Seino S, Kondo Y 1994 Phospholipase
C activation and Ca2+ mobilization by cloned human somatostatin receptor subtypes 1-5, in
transfected COS-7 cells. FEBS Lett 348:192-6
Chen L, Fitzpatrick VD, Vandlen RL, Tashjian AH, Jr. 1997 Both overlapping and distinct
signaling pathways for somatostatin receptor subtypes SSTR1 and SSTR2 in pituitary cells. J Biol
Chem 272:18666-72
Hipkin RW, Friedman J, Clark RB, Eppler CM, Schonbrunn A 1997 Agonist-induced
desensitization, internalization, and phosphorylation of the sst2A somatostatin receptor. J Biol
Chem 272:13869-76
Wang HL, Bogen C, Reisine T, Dichter M 1989 Somatostatin-14 and somatostatin-28 induce
opposite effects on potassium currents in rat neocortical neurons. Proc Natl Acad Sci U S A
86:9616-20
Sims SM, Lussier BT, Kraicer J 1991 Somatostatin activates an inwardly rectifying K+
conductance in freshly dispersed rat somatotrophs. J Physiol 441:615-37
White RE, Schonbrunn A, Armstrong DL 1991 Somatostatin stimulates Ca(2+)-activated K+
channels through protein dephosphorylation. Nature 351:570-3
Yatani A, Codina J, Sekura RD, Birnbaumer L, Brown AM 1987 Reconstitution of somatostatin
and muscarinic receptor mediated stimulation of K+ channels by isolated GK protein in clonal rat
anterior pituitary cell membranes. Mol Endocrinol 1:283-9
Ikeda SR, Schofield GG 1989 Somatostatin blocks a calcium current in rat sympathetic ganglion
neurones. J Physiol 409:221-40
Viana F, Hille B 1996 Modulation of high voltage-activated calcium channels by somatostatin in
acutely isolated rat amygdaloid neurons. J Neurosci 16:6000-11
Meriney SD, Gray DB, Pilar GR 1994 Somatostatin-induced inhibition of neuronal Ca2+ current
modulated by cGMP-dependent protein kinase. Nature 369:336-9
Todisco A, Seva C, Takeuchi Y, Dickinson CJ, Yamada T 1995 Somatostatin inhibits AP-1
function via multiple protein phosphatases. Am J Physiol 269:G160-6
Srikant CB, Shen SH 1996 Octapeptide somatostatin analog SMS 201-995 induces
translocation of intracellular PTP1C to membranes in MCF-7 human breast adenocarcinoma
cells. Endocrinology 137:3461-8
Florio T, Scorziello A, Thellung S, Salzano S, Berlingieri MT, Fusco A, Schettini G 1997
Oncogene transformation of PC Cl3 clonal thyroid cell line induces an autonomous pattern of
proliferation that correlates with a loss of basal and stimulated phosphotyrosine phosphatase
activity. Endocrinology 138:3756-63
Lopez F, Esteve JP, Buscail L, Delesque N, Saint-Laurent N, Theveniau M, Nahmias C,
Vaysse N, Susini C 1997 The tyrosine phosphatase SHP-1 associates with the sst2
somatostatin receptor and is an essential component of sst2-mediated inhibitory growth signaling.
J Biol Chem 272:24448-54
Fischer N 1989 Anorexia nervosa and unresolved rapprochement conflicts. A case study. Int J
Psychoanal 70:41-54.
Florio T, Yao H, Carey KD, Dillon TJ, Stork PJ 1999 Somatostatin activation of mitogenactivated protein kinase via somatostatin receptor 1 (SSTR1). Mol Endocrinol 13:24-37
Sellers LA, Alderton F, Carruthers AM, Schindler M, Humphrey PP 2000 Receptor isoforms
mediate opposing proliferative effects through gbetagamma-activated p38 or Akt pathways. Mol
Cell Biol 20:5974-85
Bito H, Mori M, Sakanaka C, Takano T, Honda Z, Gotoh Y, Nishida E, Shimizu T 1994
Functional coupling of SSTR4, a major hippocampal somatostatin receptor, to adenylate cyclase
inhibition, arachidonate release and activation of the mitogen-activated protein kinase cascade. J
Biol Chem 269:12722-30
Sellers LA 1999 Prolonged activation of extracellular signal-regulated kinase by a protein kinase
C-dependent and N17Ras-insensitive mechanism mediates the proliferative response of G(i/o)coupled somatostatin sst(4) receptors. J Biol Chem 274:24280-8
Cordelier P, Esteve JP, Bousquet C, Delesque N, O'Carroll AM, Schally AV, Vaysse N,
Susini C, Buscail L 1997 Characterization of the antiproliferative signal mediated by the
somatostatin receptor subtype sst5. Proc Natl Acad Sci U S A 94:9343-8
Devi LA 2000 G-protein-coupled receptor dimers in the lime light. Trends Pharmacol Sci 21:3246
Rocheville M, Lange DC, Kumar U, Patel SC, Patel RC, Patel YC 2000 Receptors for
dopamine and somatostatin: formation of hetero-oligomers with enhanced functional activity.
Science 288:154-7
106
454.
455.
456.
457.
458.
459.
460.
461.
462.
463.
464.
465.
466.
467.
468.
469.
470.
471.
472.
473.
474.
475.
476.
477.
478.
Ng GY, O'Dowd BF, Lee SP, Chung HT, Brann MR, Seeman P, George SR 1996 Dopamine
D2 receptor dimers and receptor-blocking peptides. Biochem Biophys Res Commun 227:200-4
Pfeiffer M, Koch T, Schroder H, Klutzny M, Kirscht S, Kreienkamp HJ, Hollt V, Schulz S
2001 Homo- and heterodimerization of somatostatin receptor subtypes. Inactivation of sst(3)
receptor function by heterodimerization with sst(2A). J Biol Chem 276:14027-36
Di Carlo F 1999 Somatostatina e suoi analoghi: azioni farmacologiche. In: La somatostatina e i
suoi analoghi. Lo stato dell'arte.; pp 87-95
Di Carlo F 1999 Analoghi della somatostatina: forme farmaceutiche e cinetica. In: la
somatostatina e i suoi analoghi. Lo stato dell'arte; pp 97-102
Barnes AJ, Long RG, Adrian TE, Vale W, Brown MR, Rivier JE, Hanley J, Ghatei MA,
Sarson DL, Bloom SR 1981 Effect of a long-acting octapeptide analogue of somatostatin on
growth hormone and pancreatic and gastrointestinal hormones in man. Clin Sci (Lond) 61:653-6
Morgan BA, Coy D, Taylor J, Murphy W 1997 The influence of structure on subtype selectivity
and efficacy at somatostatin receptors. Journal of Endocrinology Investigation 20 (Suppl.7):5-7
Bauer W, Briner U, Doepfner W, Haller R, Huguenin R, Marbach P, Petcher TJ, Pless 1982
SMS 201-995: a very potent and selective octapeptide analogue of somatostatin with prolonged
action. Life Sci 31:1133-40
Coy DH, Taylor JE 1996 Receptor-specific somatostatin analogs: correlations with biological
activity. Metabolism 45:21-3
Battershill PE, Clissold SP 1989 Octreotide. A review of its pharmacodynamic and
pharmacokinetic properties, and therapeutic potential in conditions associated with excessive
peptide secretion. Drugs 38:658-702
Srikant CB, Patel YC 1981 Receptor binding of somatostatin-28 is tissue specific. Nature
294:259-60
Sidney EJ, Hampl V, Nelson DP, Archer SL, Foegh ML, Cathapermal SS, Weir EK 1996 The
somatostatin analog angiopeptin does not reduce chronic hypoxic pulmonary hypertension in
rats. Proc Soc Exp Biol Med 213:43-9
Leszczynski D, Dunsky K, Josephs MD, Zhao Y, Foegh ML 1995 Angiopeptin, a somatostatin14 analogue, decreases adhesiveness of rat leukocytes to unstimulated and IL-1 beta-activated
rat heart endothelial cells. Life Sci 57:PL217-23
Keri G, Mezo I, Vadasz Z, Horvath A, Idei M, Vantus T, Balogh A, Bokonyi G, Bajor T,
Teplan I, et al. 1993 Structure-activity relationship studies of novel somatostatin analogs with
antitumor activity. Pept Res 6:281-8
Keri G, Mezo I, Horvath A, Vadasz Z, Balogh A, Idei M, Vantus T, Teplan I, Mak M, Horvath
J, et al. 1993 Novel somatostatin analogs with tyrosine kinase inhibitory and antitumor activity.
Biochem Biophys Res Commun 191:681-7
Zheng H, Fink D, Li H, Jiang X, Aebi S, Law P, Goodman M, Howell SB 1997 In vitro
antineoplastic activity of a novel lanthionine-containing peptide. Clin Cancer Res 3:1323-30
Raynor K, Murphy WA, Coy DH, Taylor JE, Moreau JP, Yasuda K, Bell GI, Reisine T 1993
Cloned somatostatin receptors: identification of subtype-selective peptides and demonstration of
high affinity binding of linear peptides. Mol Pharmacol 43:838-44
Raynor K, O'Carroll AM, Kong H, Yasuda K, Mahan LC, Bell GI, Reisine T 1993
Characterization of cloned somatostatin receptors SSTR4 and SSTR5. Mol Pharmacol 44:385-92
Redding TW, Schally AV 1984 Inhibition of growth of pancreatic carcinomas in animal models
by analogs of hypothalamic hormones. Proc Natl Acad Sci U S A 81:248-52
Sica G, Iacopino F 1999 Somatostatina e suoi analoghi: controllo della crescita cellulare. In: La
somatostatina e i suoi analoghi. Lo stato dell'arte; pp 69-85
Sica G, Di Carlo F 1999 Somatostatina e suoi analoghi: tumori neuroendocrini. In: La
somatostatina e i suoi analoghi. Lo stato dell'arte; pp 117-126
Sica G, Angelucci C, Iacopino F 1999 Somatostatina e suoi analoghi: impiego terapeutico in
tumori non endocrini. In: La somatostatina e i suoi analoghi. Lo stato dell'arte
Schally AV, Redding TW, Paz-Bouza JI, Comaru-Schally AM, Mathe G 1987 Current concept
for improving treatment of prostate cancer based on combination of LH-RH agonists with other
agents. Prog Clin Biol Res 243A:173-97
Malarkey WB, Kennedy M, Allred LE, Milo G 1983 Physiological concentrations of prolactin can
promote the growth of human breast tumor cells in culture. J Clin Endocrinol Metab 56:673-7
Schally AV, Redding TW 1987 Somatostatin analogs as adjuncts to agonists of luteinizing
hormone-releasing hormone in the treatment of experimental prostate cancer. Proc Natl Acad Sci
U S A 84:7275-9
Macaulay VM 1992 Insulin-like growth factors and cancer. Br J Cancer 65:311-20
107
479.
480.
481.
482.
483.
484.
485.
486.
487.
488.
489.
490.
491.
492.
493.
494.
495.
496.
497.
498.
499.
500.
501.
Nakao-Hayashi J, Ito H, Kanayasu T, Morita I, Murota S 1992 Stimulatory effects of insulin and
insulin-like growth factor I on migration and tube formation by vascular endothelial cells.
Atherosclerosis 92:141-9
Yang XF, Beamer WG, Huynh H, Pollak M 1996 Reduced growth of human breast cancer
xenografts in hosts homozygous for the lit mutation. Cancer Res 56:1509-11
Lamberts SW, Krenning EP, Reubi JC 1991 The role of somatostatin and its analogs in the
diagnosis and treatment of tumors. Endocr Rev 12:450-82
Moody TW, Russell EK, O'Donohue TL, Linden CD, Gazdar AF 1983 Bombesin-like peptides
in small cell lung cancer: biochemical characterization and secretion from a cell line. Life Sci
32:487-93
Cuttitta F, Carney DN, Mulshine J, Moody TW, Fedorko J, Fischler A, Minna JD 1985
Bombesin-like peptides can function as autocrine growth factors in human small-cell lung cancer.
Nature 316:823-6
Ritts RE, Kvols LK, Strehlo B, Jacobsen D 1989 Immunologic studies of patients with
malignant neuroendocrine carcinomas and responses to somatostatin analogue (SMS 201-995,
Sandostatin). American Association of Cancer Research
Hierowski MT, Liebow C, du Sapin K, Schally AV 1985 Stimulation by somatostatin of
dephosphorylation of membrane proteins in pancreatic cancer MIA PaCa-2 cell line. FEBS Lett
179:252-6
Pan MG, Florio T, Stork PJ 1992 G protein activation of a hormone-stimulated phosphatase in
human tumor cells. Science 256:1215-7
Florio T, Pan MG, Newman B, Hershberger RE, Civelli O, Stork PJ 1992 Dopaminergic
inhibition of DNA synthesis in pituitary tumor cells is associated with phosphotyrosine
phosphatase activity. J Biol Chem 267:24169-72
Freiss G, Vignon F 1994 Antiestrogens increase protein tyrosine phosphatase activity in human
breast cancer cells. Mol Endocrinol 8:1389-96
Cattaneo MG, Amoroso D, Gussoni G, Sanguini AM, Vicentini LM 1996 A somatostatin
analogue inhibits MAP kinase activation and cell proliferation in human neuroblastoma and in
human small cell lung carcinoma cell lines. FEBS Lett 397:164-8
Todisco A, Campbell V, Dickinson CJ, DelValle J, Yamada T 1994 Molecular basis for
somatostatin action: inhibition of c-fos expression and AP-1 binding. Am J Physiol 267:G245-53
Sharma K, Patel YC, Srikant CB 1996 Subtype-selective induction of wild-type p53 and
apoptosis, but not cell cycle arrest, by human somatostatin receptor 3. Mol Endocrinol 10:168896
Baliga BS, Borowitz SM 1988 Effects of growth and differentiation inducing factors on protein
kinase-C of cultured intestinal crypt cells. Biochem Biophys Res Commun 154:278-84
Conteas CN, Majumdar AP 1987 The effects of gastrin, epidermal growth factor, and
somatostatin on DNA synthesis in a small intestinal crypt cell line (IEC-6). Proc Soc Exp Biol Med
184:307-11
Hodin RA, Saldinger P, Meng S 1995 Small bowel adaptation: counterregulatory effects of
epidermal growth factor and somatostatin on the program of early gene expression. Surgery
118:206-10; discussion 210-1
Tsuzaki S, Moses AC 1990 Somatostatin inhibits deoxyribonucleic acid synthesis induced by
both thyrotropin and insulin-like growth factor-I in FRTL5 cells. Endocrinology 126:3131-8
Florio T, Scorizello A, Fattore M, D'Alto V, Salzano S, Rossi G, Berlingieri MT, Fusco A,
Schettini G 1996 Somatostatin inhibits PC Cl3 thyroid cell proliferation through the modulation of
phosphotyrosine activity. Impairment of the somatostatinergic effects by stable expression of E1A
viral oncogene. J Biol Chem 271:6129-36
Tatoud R, Degeorges A, Prevost G, Hoepffner JL, Gauville C, Millot G, Thomas F, Calvo F
1995 Somatostatin receptors in prostate tissues and derived cell cultures, and the in vitro growth
inhibitory effect of BIM-23014 analog. Mol Cell Endocrinol 113:195-204
Foegh ML 1992 Angiopeptin: a treatment for accelerated myointimal hyperplasia? J Heart Lung
Transplant 11:S28-31
Vroman B, LaRusso NF 1996 Development and characterization of polarized primary cultures of
rat intrahepatic bile duct epithelial cells. Lab Invest 74:303-13
Weiss RE, Reddi AH, Nimni ME 1981 Somatostatin can locally inhibit proliferation and
differentiation of cartilage and bone precursor cells. Calcif Tissue Int 33:425-30
Watson SA, Morris DL, Durrant LG, Robertson JF, Hardcastle JD 1992 Inhibition of gastrinstimulated growth of gastrointestinal tumour cells by octreotide and the gastrin/cholecystokinin
receptor antagonists, proglumide and lorglumide. Eur J Cancer 28A:1462-7
108
502.
503.
504.
505.
506.
507.
508.
509.
510.
511.
512.
513.
514.
515.
516.
517.
518.
519.
520.
521.
522.
523.
Christophe J 1994 Pancreatic tumoral cell line AR42J: an amphicrine model. Am J Physiol
266:G963-71
Takeda Y, Escribano MJ 1991 Effects of insulin and somatostatin on the growth and the colony
formation of two human pancreatic cancer cell lines. J Cancer Res Clin Oncol 117:416-20
Watson SA, Durrant LG, Morris DL 1990 The effect of the E2 prostaglandin enprostil, and the
somatostatin analogue SMS 201 995, on the growth of a human gastric cell line, MKN45G. Int J
Cancer 45:90-4
Qin Y, Schally AV, Willems G 1991 Somatostatin analogue RC-160 inhibits the growth of
transplanted colon cancer in rats. Int J Cancer 47:765-70
Radulovic S, Miller G, Schally AV 1991 Inhibition of growth of HT-29 human colon cancer
xenografts in nude mice by treatment with bombesin/gastrin releasing peptide antagonist (RC3095). Cancer Res 51:6006-9
Smith JP, Solomon TE 1988 Effects of gastrin, proglumide, and somatostatin on growth of
human colon cancer. Gastroenterology 95:1541-8
Hoelting T, Duh QY, Clark OH, Herfarth C 1996 Somatostatin analog octreotide inhibits the
growth of differentiated thyroid cancer cells in vitro, but not in vivo. J Clin Endocrinol Metab
81:2638-41
Bepler G, Carney DN, Gazdar AF, Minna JD 1987 In vitro growth inhibition of human small cell
lung cancer by physalaemin. Cancer Res 47:2371-5
Taylor JE, Bogden AE, Moreau JP, Coy DH 1988 In vitro and in vivo inhibition of human small
cell lung carcinoma (NCI-H69) growth by a somatostatin analogue. Biochem Biophys Res
Commun 153:81-6
Judd AM, Login IS, Kovacs K, Ross PC, Spangelo BL, Jarvis WD, MacLeod RM 1988
Characterization of the MMQ cell, a prolactin-secreting clonal cell line that is responsive to
dopamine. Endocrinology 123:2341-50
Cheung NW, Boyages SC 1995 Somatostatin-14 and its analog octreotide exert a cytostatic
effect on GH3 rat pituitary tumor cell proliferation via a transient G0/G1 cell cycle block.
Endocrinology 136:4174-81
Santini V, Lamberts SW, Krenning EP, Backx B, Lowenberg B 1995 Somatostatin and its
cyclic octapeptide analog SMS 201-995 as inhibitors of proliferation of human acute
lymphoblastic and acute myeloid leukemia. Leuk Res 19:707-12
Keri G, Erchegyi J, Horvath A, Mezo I, Idei M, Vantus T, Balogh A, Vadasz Z, Bokonyi G,
Seprodi J, Teplan I, Csuka O, Tejeda M, Gaal D, Szegedi Z, Szende B, Roze C, Kalthoff H,
Ullrich A 1996 A tumor-selective somatostatin analog (TT-232) with strong in vitro and in vivo
antitumor activity. Proc Natl Acad Sci U S A 93:12513-8
Pinski J, Schally AV, Halmos G, Szepeshazi K, Groot K 1996 Somatostatin analog RC-160
inhibits the growth of human osteosarcomas in nude mice. Int J Cancer 65:870-4
Setyono-Han B, Henkelman MS, Foekens JA, Klijn GM 1987 Direct inhibitory effects of
somatostatin (analogues) on the growth of human breast cancer cells. Cancer Res 47:1566-70
Xu Y, Berelowitz M, Bruno JF 1998 Characterization of the promoter region of the human
somatostatin receptor subtype 2 gene and localization of sequences required for estrogenresponsiveness. Molecular and Cellular Endocrinology 139:71-77
Brevini TAL, Bianchi R, Motta M 1993 Direct inhibitory effect of somatostatin on the growth of
the human prostatic cancer cell line LNCaP: possible mechanism of action. Journal of Clinical
Endocrinology & Metabolism 77:626-631
Pinski J, Halmos G, Schally AV 1993 Somatostatin analog RC-160 and bombesin/gastrinreleasing peptide antagonist RC-3095 inhibit the growth of androgen-independent DU-145 human
prostate cancer line in nude mice. Cancer Lett 71:189-96.
Pinski J, Schally AV, Halmos G, Szepeshazi K 1993 Effect of somatostatin analog RC-160 and
bombesin/gastrin releasing peptide antagonist RC-3095 on growth of PC-3 human prostatecancer xenografts in nude mice. Int J Cancer 55:963-7
Cho YR, Kim CW 2004 Neuropeptide Y promotes beta-cell replication via extracellular signalregulated kinase activation. Biochemical and Biophysical Research Communications 314:773780
Nanzer AM, Khalaf S, Mozid AM, Fowkes RC, Patel MV, Burrin JM, Grossman AB,
Korbonits M 2004 Ghrelin exerts a proliferative effect on a rat pituitary somatotroph cell line via
the mitogen-activated protein kinase pathway. Eur J Endocrinol 151:233-40
Bousquet C, Guillermet J, Vernejoul F, Lahlou H, Buscail L, Susini C 2004 Somatostatin
receptors and regulation of cell proliferation. Dig Liver Dis 36 Suppl 1:S2-7
109
524.
525.
526.
527.
528.
529.
530.
531.
532.
533.
534.
535.
536.
537.
538.
539.
540.
541.
542.
543.
544.
Chomczinski P, Sacchi N 1987 Single-step method of RNA isolation by acid guanidinium
thiocyanate-phenol- chloroform extraction. Analytical Biochemistry 162:156-159
Kennedy AL, Harakall SA, Lynch SW, Brass KM, Hardwick JC, Mawe GM, Parsons RL 1998
Expression and physiological actions of neuropeptide Y in guinea pig parasympathetic cardiac
ganglia. Journal of the Autonomic Nervous System 71:190-195
Ponte P, Ng S-Y, Engel J, Gunning P, Kedes L 1984 Evolutionary conservation in the
untranslated regions of actin mRNAs: DNA sequence of a human beta-actin cDNA. Nucleic Acids
Research 12:1687-1696
Franco OE, Onishi T, Yamakawa K, Arima K, Yanagawa M, Sugimura Y, Kawamura J 2003
Mitogen-activated protein kinase pathway is involved in androgen-independent PSA gene
expression in LNCaP cells. Prostate 56:319-25
Pagotto U, Arzberger T, Theodoropoulou M, Grubler Y, Pantaloni C, Saeger W, Losa M,
Journot L, Stalla GK, Spengler D 2000 The expression of the antiproliferative gene ZAC is lost
or highly reduced in nonfunctioning pituitary adenomas. Cancer Res 60:6794-9
Pagotto U, Arzberger T, Ciani E, Lezoualc'h F, Pilon C, Journot L, Spengler D, Stalla GK
1999 Inhibition of Zac1, a new gene differentially expressed in the anterior pituitary, increases cell
proliferation. Endocrinology 140:987-96.
Gilbert C, Rollet-Labelle E, Caon AC, Naccache PH 2002 Immunoblotting and sequential lysis
protocols for the analysis of tyrosine phosphorylation-dependent signaling. Journal of
Immunological Methods 271:185-201
Ferone D, Arvigo M, Semino C, Jaquet P, Saveanu A, Taylor JE, Moreau JP, Culler MD,
Albertelli M, Minuto F, Barreca A 2005 Somatostatin and dopamine receptor expression in lung
carcinoma cells and effects of chimeric somatostatin-dopamine molecules on cell proliferation.
Am J Physiol Endocrinol Metab
Grynkiewicz G, Poenie M, Tsien RY 1985 A new generation of Ca2+ indicators with greatly
improved fluorecence properties. Journal of Biological Chemistry 260:3440-3450
Li AJ, Ritter S 2005 Functional expression of neuropeptide Y receptors in human neuroblastoma
cells. Regul Pept 129:119-24
Janssens R, Boeynaems JM 2001 Effects of extracellular nucleotides and nucleosides on
prostate carcinoma cells. British Journal of Pharmacology 132:536-546
Letari O, Malgaroli A, Morgan DW, Welton AF, Nicosia S 1991 Cytosolic calcium ion and
arachidonic acid release and metabolism in macrophages. European Journal of Pharmacology
206:211-219
Nilsson T, Erlinge D, Cantera L, Edvinsson L 1996 Contractile effects of neuropeptide Y in
human subcutaneous resistance arteries are mediated by Y1 receptors. Journal of
Cardiovascular Pharmacology 28:764-768
Marklund U, Bystrom M, Gedda K, Larefalk A, Juneblad K, Nystrom S, Ekstrand AJ 2002
Intron-mediated expression of the human neuropeptide Y Y1 receptor. Mol Cell Endocrinol
188:85-97
Sheikh SP, Williams JA 1990 Structural characterization of Y1 and Y2 receptors for
neuropeptide Y and peptide YY by affinity cross-linking. Journal of Biological Chemistry
265:8304-8310
Wieland HA, Eckard CP, Doods HN, Beck-Sickinger AG 1998 Probing of the neuropeptide YY1-receptors interaction with anti-receptor antibodies. European Journal of Biochemistry
255:595-603
Erlinge D, Brunkwall J, Edvinsson L 1994 Neuropeptide Y stimulates proliferation of human
vascular smooth muscle cells: cooperation with noradrenaline and ATP. Regulatory Peptides
50:259-265
Hansel DE, Eipper BA, Ronnett GV 2001 Neuropeptide Y functions as a neuroproliferative
factor. Nature 410:940-4
Nagakawa O, Ogasawara M, Fujii H, Murakami K, Murata J, fuse H, Saiki I 1998 Effect of
prostatic neuropeptides on invasion and migration of PC-3 prostate cancer cells. Cancer Letters
133:27-33
Nagakawa O, Ogasawara M, Murata J, Fuse H, Saiki I 2001 Effect of prostatic neuropeptides
on migration of prostate cancer cell lines. International Journal of Urology 8:65-70
Gallego C, Gupta SK, Heasley LE, Qian NX, Johnson GL 1992 Mitogen-activated protein
kinase activation resulting from selective oncogene expression in NIH 3T3 and rat 1a cells. Proc
Natl Acad Sci U S A 89:7355-9
110
545.
546.
547.
548.
549.
550.
551.
552.
553.
554.
555.
556.
Robidoux J, Simoneau L, St-Pierre S, Ech-Chadli H, Lafond J 1998 Human
syncytiotrophoblast NPY receptors are located on BBM and activate PLC-to-PKC axis. Am J
Physiol 274:E502-9
Milenkovic I, Weick M, Wiedemann P, Reichenbach A, Bringmann A 2004 Neuropeptide Yevoked proliferation of retinal glial (Muller) cells. Graefes Arch Clin Exp Ophthalmol 242:944-50
Guo C, Luttrell LM, Price DT 2000 Mitogenic signaling in androgen sensitive and insensitive
prostate cancer cell lines. J Urol 163:1027-32.
Mabuchi S, Ohmichi M, Kimura A, Ikebuchi Y, Hisamoto K, Arimoto-Ishida E, Nishio Y,
Takahashi K, Tasaka K, Murata Y 2004 Tamoxifen inhibits cell proliferation via mitogenactivated protein kinase cascades in human ovarian cancer cell lines in a manner not dependent
on the expression of estrogen receptor or the sensitivity to cisplatin. Endocrinology 145:1302-13
Mannon PJ, Kaiser LM 1997 Retinoic acid is a negative regulator of the neuropeptide Y/peptide
YY Y1 receptor gene in SK-N-MC cells. Journal of Neurochemistry 68:20-25
di Sant'Agnese PA, Cockett ATK 1996 Neuroendocrine differentiation in prostatic malignancy.
Cancer 78:357-361
Vainas IG 2001 Octreotide in the management of hormone-refractory prostate cancer.
Chemotherapy 47 Suppl 2:109-26
Sharma K, Patel YC, Srikant CB 1999 C-terminal region of human somatostatin receptor 5 is
required for induction of Rb and G1 cell cycle arrest. Mol Endocrinol 13:82-90
Zatelli MC, Tagliati F, Taylor JE, Rossi R, Culler MD, Degli Uberti EC 2001 Somatostatin
receptor subtypes 2 and 5 differentially affect proliferation in vitro of the human medullary thyroid
carcinoma cell line TT. Journal of Clinical Endocrinology & Metabolism 86:2161-2169
Bilanges B, Varrault A, Basyuk E, Rodriguez C, Mazumdar A, Pantaloni C, Bockaert J,
Theillet C, Spengler D, Journot L 1999 Loss of expression of the candidate tumor suppressor
gene ZAC in breast cancer cell lines and primary tumors. Oncogene 18:3979-88.
Theodoropoulou M, Pagotto U, Arzberger T 2002 The tumor suppressor gene ZAC1 is a novel
mediator of the somatostatin antiproliferative action in the rat pituitary tumor cell line GH3. In:
ABST OC2.6 ENAC, Munich, Germany (ed)
Berruti A, Dogliotti L, Mosca A, Tarabuzzi R, Torta M, Mari M, Gorzegno G, Fontana D,
Angeli A 2001 Effects of the somatostatin analog lanreotide on the circulating levels of
chromogranin-A, prostate-specific antigen, and insulin-like growth factor-1 in advanced prostate
cancer patients. Prostate 47:205-11
111