Difetti Enzimatici
Archibald Garrod e l’alcaptonuria:
La nascita della Genetica biochimica
L’alcaptonuria....
33 anni prima della
formulazione della teoria
“un gene-un enzima” di
Beadle e Tatum
Al contrario degli individui normali,
l’acido omogentisico somministrato
somministrato ai pazienti
alcaptonurici veniva escreto
Conclusioni di Archibald Garrod
1. I singoli passaggi del metabolismo sono opera di enzimi individuali
2. In alcuni individui esiste un blocco di una tappa del metabolismo con
conseguente accumulo del substrato
3. Questa caratteristica è ereditaria e può essre trasmessa come
carattere recessivo
4. Riconobbe il principio dell’individualità chimica e genetica anche per le
idiosincrasie verso cibi e farmaci
Esempio di malattie
umane derivate da deficit
enzimatici causate da
difetti in geni singoli
Difetti enzimatici
Concetti generali
• Le enzimopatie sono quasi sempre recessive
(perdita di funzione)
• Accumulo del substrato e deficienza del
prodotto
• Substrati diffusibili versus substrati accumulati
in tessuti specifici (macromolecole)
• Deficit enzimatici multipli (cofattore comune)
• Omologie fenotipiche (comune via metabolica)
Relazioni tra Genotipo e Fenotipo
nelle malattie genetiche
DIFETTI ENZIMATICI:
Iperfenilalaninemia
Accumulo di
acido fenilpiruvico
danno cerebrale
BH4: tetraidrobiopterina
Test di Guthrie
Inibizione della crescita batterica da parte della beta-2-tienilalanina
è sbloccata dalla presenza di elevate concentrazioni di fenilalanina
(> 4mg/dl)
L’iperfenilalaninemia come prototipo di
malattia da sottoporre a screening neonatale
Mutazioni PKU (grande eterogeneità allelica)
Elevato numero di
eterozigoti composti
Correlazione genotipo-fenotipo
Diverse forme di iperfenilalaninemia
• Difetti della fenilalanina idrossilasi
• PKU classica
• PKU variante
• Iperfenilalaninemia non-PKU
• Difetti nel metabolismo della tetraidropterina
(terapia diversa dalla PKU)
• Fenilchetonuria materna
Eterogeneità delle frequenze alleliche
associate a malattie autosomiche recessive
nelle diverse popolazioni
Vantaggio dell’eterozigote, deriva genetica, effetto fondatore
Mucopolisaccaridosi
• Accumulo dei mucopolisaccaridi o
glicosamminoglicani (GAGs) nei lisosomi
per deficit enzima degradativo
• Eccesso GAGs nelle urine
• Interessamento multi-sistemico
• Diverse patologie
• Test di complementazione
Il test di complementazione
permette di scoprire se le mutazioni sono in
geni diversi
piastra
A
B
A
cellula
Co-coltivazione
B
Formazione di eterocarionte
mediante fusione cellulare
Iduronato-2-solfatasi
Eparan-sulfamidasi
Malattia I-Cell
(difetto enzima modificatore)
• Difetto dell’enzima N-acetylglucosamine-1phosphotransferase che trasferisce il gruppo
fosfato al mannosio del mannosio-6-fosfato
• Assenza del segnale per dirigere le proteine nei
lisosomi
• Presenza di inclusioni nel citoplasma
• Anomalie facciali, scheletriche, grave ritardo
della crescita e mentale
Deficit di alfa1-antitripsina
(inibitore serin-proteasi)
• Autosomica recessiva
• Riduzione della inibizione dell’elastasi
• Patologia ostruttiva del polmone
(enfisema) e cirrosi epatica
• Accumulo α1-AT nel RER del fegato
• Progressione dell’enfisema favorita dal
fumo
• Esempio di interazione con fattori
ambientali
Z
Influenza del fumo sul fenotipo del
deficit di α1-AT
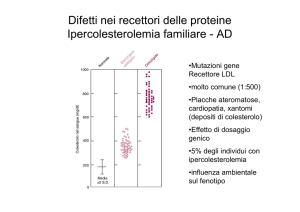
Ipercolesterolemia familiare
(autosomica dominante)
•Mutazioni gene
Recettore LDL
•Placche ateromatose,
cardiopatia, xantomi
(depositi di colesterolo)
•Effetto di dosaggio
genico
•5% degli individui con
ipercolesterolemia
5 Diverse classi di mutanti del
recettore per le LDL
Brown e Goldstein, 1979
Localizzazione delle mutazioni nel gene del
recettore delle LDL
Mutazioni del gene del recettore LDL
Crossing-over ineguale tra elementi
ripetuti di tipo Alu
Ruolo delle sequenze ripetitive nelle patologie e
nell’evoluzione
Mutazioni del dominio citoplasmatico
Deficit di
internalizzazione