Malattie genetiche
Il patrimonio genetico e’ contenuto nel DNA.
Il genoma umano e’ formato da 23 paia di cromosomi (1 paio di
eterocromosomi, 22 paia di autosomi).
La sequenza di tutto il genoma umano e’ ad oggi completa; il
sequenziamento consiste nell’individuare la sequenza di nucleotidi di tutto il
genoma umano (nucleotidi comprendono: adenina, timina, guanosina,
citosina).
Meno del 2% del genoma umano codifica per le proteine, la restante parte
ha funzioni non del tutto chiare (regolatorie, integrita’ strutturale).
Due individui qualunque hanno un patrimonio genetico uguale per il 99.5%,
le differenze sono codificate da meno dello 0.5% del DNA.
Le malattie genetiche sono causate da alterazioni del DNA e
sono molto frequenti.
Il 50% degli aborti spontanei e’ associato ad una anomalia
cromosomica.
Le malattie genetiche vengono classificate in tre grosse
categorie:
1) Malattie correlate a mutazione di singoli geni ad ampio
effetto (malattie mendeliane)
2) Malattie cromosomiche: dovute ad alterazioni nel numero o
nella struttura degli autosomi o dei cromosomi sessuali.
3) Malattie multigeniche complesse.
Le mutazioni possono essere:
1) Puntiformi nelle sequenze codificanti:
• nell’anemia falciforme viene ad essere modificata una base
della tripletta CTC che diventa CAC, l’aminoacido acido
glutammino viene sostituito dalla valina e la catena Beta
dell’Hb risulta essere modificata, gli eritrociti (drepanociti)
vanno piu’ facilmente incontro a lisi e tendono ad occludere
I piccoli vasi dando origine a fenomeni trombotici o
infartuali.
• Nella beta talassemia si ha la mancata o ridotta sintesi della
catena beta dell’Hb a seguito di mutazioni che modificano il
codone per l’acido glutammico in un codone di stop per cui
viene prodotta una catena beta prematuramente interrotta
e rapidamente degradata.
2) Mutazioni in sequenze non codificanti: riguardano sequenze
promotrici o intensificatrici, possono interferire con I processi di
trascrizione e traduzione.
3) Delezioni o inserzioni
4) Mutazioni da triplette ripetute (s.dell’X fragile).
In sintesi le mutazioni possono interferire con la sintesi proteica a
vari livelli:
-le delezioni e le mutazioni puntiformi che interessano il promotore
possono sopprimere la trascrizione
-le mutazioni che interessano gli introni (sequenze non codificanti)
possono determinare un’anomala maturazione dell’mRNA
-formazione di codoni di stop
Malattie Mendeliane
Le malattie mendeliane sono il risultato di mutazioni in singoli geni ad
ampio effetto.
Possono essere:
1) autosomiche dominanti: si manifestano in eterozigosi, almeno uno dei
due genitori e’ malato, possono essere colpiti sia maschi che femmine,
probabilita’ di 1:2 di generare figli malati, a volte frutto di mutazioni ex
novo a livello dell’oocita o dello spermatozoo, possono avere diversa
penetranza, diversa espressivita’
(esempi: ipercolesterolemia famigliare (alterazioni del recettore per le LDL),
s. di Marfan (conivolge un’alterata sintesi della fibrillina, proteina
extracellulare, s di Ehlers-Danlos, Malattia di Huntington, la
neurofibromatosi, la poliposi famigliare del colon)
2) autosomiche recessive: si manifestano quando entrambe gli alleli sono
mutati, i genitori sono portatori di un allele mutato, ma non manifestano la
malattia, probabilita’ di 1:4 di avere figli malati, se il gene mutato ha bassa
frequenza nella popolazione, spesso I figli malati sono frutto dell’unione di
consaguinei, penetranza spesso completa, esordio in eta’ precoce, spesso I
geni mutati codificano per proteine enzimatiche (difetti congeniti del
metabolismo)
(esempi: malattie da accumulo lisosomiale, glicogenosi, gangliosidosi,
malattia di Gaucher, deficit di alfa 1 antitripsina, anemia falciforme,
talassemie, s. di Elhers-Danlos, alcune atrofie muscolari neurogeniche).
3) Malattie legate al cromosma X: tutte le malattie legate al sesso sono
trasmesse dal cromosoma X (i maschi con mutazioni a carico dei geni sul
cromosoma Y di solito non sono fertili).
I maschi affetti non trasmettono la malattia ai figli, ma le femmine sono
tutte portatrici
Esempi: distrofia muscolare di Duchenne, Emofilia A e B, deficit di glucosio
6-fosfato deidrogenasi, s dell’X fragile, m. di Fabry.
Malattia di Fabry
ECG
La Malattia di Anderson-Fabry è un raro
disordine genetico provocato dalla
carenza dell’enzima lisosomiale αgalattosidasi A. La carenza di questo
enzima porta all’accumulo progressivo
di glicosfingolipidi, in particolare
globotriaosilceramide (GL-3), nei tessuti
viscerali e nell’endotelio vascolare di
tutto l’organismo.
4) Malattie legate alla trasmissione mitocondriale, matrilineare (miopatie
mitocondriali, cardiomiopatie mitocondriali, neuropatie mitocondriali).
Un’altra possible classificazione delle malattie mendeliane riguarda la
tipologia del difetto in termini di prodotto finale che si viene a creare in
conseguenza della mutazione genetica.
In relazione ai meccanismi implicati nelle mutazioni, sono state definite
cinque categorie di difetti:
1. Difetti enzimatici e loro conseguenze
2. Difetti dei sistemi recettoriali di membrana e dei sistemi di
trasporto
3. Alterazioni di struttura, funzione e quantita’ di proteine non
enzimatiche
4. Mutazioni responsabili di alterate reazioni ai farmaci
5. Difetti delle proteine che regolano la crescita cellulare.
Difetti enzimatici
Le conseguenze biochimiche di un difetto enzimatico possono portare a:
1) accumulo del substrato ---> m. da accumulo lisosomiale
(autosomiche recessive) ---> le glicogenosi, le mucopolisaccaridosi, le
sfingolipidosi
1. M. di Tay-Sachs, una gangliosidosi dovuta al deficit della esosaminidasi A
che causa accumulo di gangliosidi soprattutto nel SNC e autonomo e nella
retina
2. M. Di Niemann-Pick, dovuta ad accumulo di sfingomielina, con
conseguente coinvolgimento neurologico ed epatosplenomegalia
3. M. Di Gaucher, il difetto riguarda la glucocerebrosidasi, vi e’ accumulo dei
glucocerebrosidi nei fagociti soptattutto nella milza, nel fegato, nel midollo
osseo, nei linfonodi
2) Blocco metabolico con ridotta quantita’ di prodotto finale (albinismo)
3) incapacita’ di inattivare un substrato dannoso per i tessuti (deficit di alfa
1 – antitripsina).
Albinismo
La figura rappresenta l'albero genealogico di una famiglia, seguita per 3
generazioni.
I quadrati e i cerchi pieni indicano rispettivamente maschi e femmine affetti
da una malattia ereditaria.
Di quale tipo di ereditarietà si tratta?
Difetti del sistema recettoriale di
membrana e di trasporto
Difetti dei sistemi recettoriali:
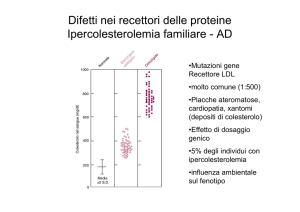
L’ipercolesterolemia famigliare e’ una “malattia recettoriale”
dovuta ad una mutazione del gene che codifica per il recettore
per le LDL.
E’ una malattia autosomica dominante, tra le piu’ frequenti, gli
eterozigoti hanno un livello plasmatico di colesterolo molto
elevato, gli omozigoti hanno livelli di 5-6 volte superiori alla
norma, l’aterosclerosi compare molto precocemente.
La genetica dell’ipercolesterolemia famigliare
complessa, vi sono centinaia di mutazioni
e’
molto
Ipercolesterolemia famigliare
Difetti delle proteine di trasporto:
- dell’ossigeno: Talassemie, Anemia falciforme
- degli ioni: Fibrosi cistica
Anemia Falciforme
Talassemia Maggiore
Alterazioni di struttura, funzione e
quantita’ di proteine strutturali
1. extracellulari: s. di Ehlers-Danlos dovuta ad
alterazioni nella struttra del collagene fibrillare,
osteogenesi imperfecta, s. di Marfan
2. della membrana cellulare: distrofia muscolare
Sindrome di Marfan
Mutazioni resposnsabili
di alterate reazioni ai farmaci
Deficit della glucosio 6 fosfato deidrogenasi: possono
prodursi gravi crisi emolitiche
Difetti delle proteine
che regolano la crescita cellulare
Difetti a carico dei proto-oncogeni e dei geni
oncosoppressori: un ruolo importante nella patogenesi
dei tumori
Malattie Cromosomiche
Il cariotipo umano e’ formato da 46 cromosomi (22 coppie omologhe di
autosomi e una coppia (XX o XY) di cromosomi sessuali
La cariotipizzazione e’ lo strumento fondamentale del citogenetista.
Le malattie cromosomiche possono derivare da un alterato numero di
cromosomi trisomie, monosomie) oppure da alterazioni nella struttura di
uno o piu’ cromosomi.
Esempi di malattie che interessano gli autosomi: trisomia 21 o s. di Down,
trisomia 18 o s. di Edwards, s. di DiGeorge da delezione del cromosoma
22q11.2 (1:4000, cardiopatie congenite, anomalie dle palato, dismorfismi
facciali,ritardo nello sviluppo, immunodeficienza)
Sindrome di Down
Esempi di malattie che interessano i cromosomi
sessuali: s. di Turner o ipogonadismo femminile
(monosomia X), s. di Klinefelter o ipogonadismo
maschile (XXY).
XO = Sindrome di Turner
XXY, XXXY, XXYY = Sindrome di Klinefelter
Malattie multigeniche
Sono causate da interazioni tra varianti genetiche e
fattori ambientali
Esempi: diabete tipo 1 e tipo 2, l’aterosclerosi,
l’ipertensione arteriosa.
Tecniche per lo studio
delle alterazioni del DNA
PCR: amplificazione esponenziale del DNA
Sequenziamento del DNA
Marcatori polimorfici: polimorfismi
Analisi molecolari con tecniche di ibridazione: southern blot
(ibridazione di sonde marcate)
Analisi dell’RNA (applicazione pratica per l’identificazione dei
virus a RNA es. HIV e HCV o per la stratificazione molecolare
dei tumori)