MODULO 5 – IL METABOLISMO DEI COMPOSTI AZOTATI
RICHIAMI SULLA CHIMICA DI AMMINO ACIDI E PROTEINE
Introduzione
Gli amminoacidi sono composti organici caratterizzati dalla presenza nella molecola di un gruppo
carbossilico e di un gruppo amminico. Hanno un ruolo biologico fondamentale in quanto costituiscono i
blocchi da costruzione delle proteine e dei peptidi non proteici, che svolgono innumerevoli funzioni,
essenziali per la vita di tutti gli organismi. Possono inoltre essere utilizzati dalle cellule come fonte di
energia e alcuni di essi possiedono, come tali, anche importanti attività fisiologiche.
Obiettivi
Questa lezione presenta una rapida panoramica delle caratteristiche chimiche e fisiche principali di una
classe di amminoacidi, gli α-amminoacidi, che sono i costituenti delle proteine nell’uomo.
La conoscenza di queste nozioni di base è fondamentale per comprendere le correlazioni che esistono tra
le caratteristiche degli amminoacidi e la complessità di struttura e funzioni delle proteine.
Definizione
Gli amminoacidi sono composti che contengono sia un gruppo amminico (-NH2), basico, sia un gruppo
carbossilico (-COOH), acido. Negli organismi esistono molti di questi composti, ma quelli per noi più
interessanti e di cui ci occuperemo in questa lezione sono gli α-amminoacidi, i blocchi costituitivi delle
proteine nell’uomo.
Gli amino acidi come precursori delle proteine
Peptidi e proteine. Gli amminoacidi sono molecole di estrema importanza biologica proprio in quanto
costituiscono i peptidi e le proteine, macromolecole essenziali per la vita.
I peptidi sono polimeri formati da più amminoacidi, coniugati attraverso legami detti ammidici o peptidici.
Classe
Dipeptide
Tripeptide
Tetrapeptide
Pentapeptide
Esapeptide
Eptapeptide
Ottapeptide
Nonapeptide
Decapeptide
Oligopeptide
Polipeptide
Proteina
Un sistema di classificazione dei peptidi
Numero di amminoacidi
2
3
4
5
6
7
8
9
10
da 2 a 11
> 11
> 50 b
Il termine residuo amminoacidico indica un amminoacido nel contesto di un peptide. Si utilizzano i
termini oligopeptide e polipeptide per indicare rispettivamente macromolecole contenenti alcuni (in
genere da 2 a 10) o molti (più di 10) residui amminoacidici. Con il termine di proteina si indicano
polipeptidi con più di 50 residui che esercitano una funzione biologica solo se assumono una specifica
conformazione tridimensionale. Esistono quindi peptidi proteici e peptidi non proteici ed entrambi i tipi
di peptidi svolgono azioni biologiche importanti nel nostro organismo.
Proteine e funzioni. Ciascuna proteina svolge una o più funzioni nell’organismo, generalmente attraverso
l’interazione specifica con altre molecole. La conformazione (struttura tridimensionale) di ciascuna
proteina determina il tipo di interazioni possibili e quindi la specifica funzione; essa dipende dalla sequenza
dei residui amminoacidici contenuti nella proteina. L’abolizione (denaturazione) della conformazione
proteica determina la perdita della funzione.Tra le funzioni più importanti esercitate dalle proteine negli
organismi ricordiamo:
1. funzione strutturale (collagene)
2. funzione di trasporto (emoglobina, apolipoproteine, albumina)
3. funzione di difesa e protezione (immunoglobuline, fibrinogeno)
4. funzione di controllo e regolazione (ormoni, recettori di diversi ormoni, fattori di trascrizione)
5. funzione catalitica (tutti gli enzimi)
6. funzione di movimento (actina, miosina).
Peptidi non proteici
Anche numerosi peptidi non proteici esercitano diverse attività fisiologiche molto importanti. Alcuni
fungono da ormoni o sostanze regolatrici; per esempio, il glucagone, costituito da 29 residui
amminacidici, ha un ruolo essenziale nella regolazione del metabolismo glucidico e lipico; la vasopressina
(ormone antidiuretico), un nonapeptide, stimola il riassobimento dell’acqua a livello renale, mentre
l’ossitocina stimola la produzione di latte e la contrazione uterina durante la gravidanza. Altri peptici
agiscono da fattori di crescita e regolano divisione cellulare e crescita dei tessuti. Infine i peptidi oppiacei
(encefaline, endorfine) hanno un ruolo importante nella regolazione della sensazione del dolore:
interagiscono con recettori del sistema nervoso centrale che legano sostanze ad azione analgesica come la
morfina ed i derivati dell’oppio. Anche per i peptidi non proteici la funzione specifica correla direttamente
con la natura e la sequenza dei residui amminoacidici costitutivi, anche se non attraverso la
determinazione di una conformazione tridimensionale specifica.
Peptide
Numero di unità
amminoacidiche
Angiotensina II
Bradichinina
Gastrina
8
9
17
Glucagone
Endotelina 1
29
21
Metionina-encefalina
5
Ossitocina
9
Sostanza P
Fattore di rilascio della tirotropina
10
3
Vasopressina
(ormone antidiuretico [ADH])
9
Ruolo, funzione, attività
Regola la pressione del sangue
Un vasodilatatore e un diuretico
Stimola la secrezione di acido da parte dello
stomaco
Regola il metabolismo del glucosio
Regola la pressione del sangue; partecipa al
controllo del differenziamento durante lo
sviluppo embrionale
Un
peptide
oppiaceo
e
un
neurotrasmettitore; un analgesico naturale
Stimola la lattazione e la contrazione
dell'utero
Un neurotrasmettitore
Ormone ipotalamico che stimola il rilascio
della tirotropina da parte della ghiandola
pituitaria
Regola la pressione del sangue e il
riassorbimento dell'acqua da parte dei reni;
un neurotrasmettitore
Altre funzioni degli amino acidi
AA neurotrasmettitori. Anche se il ruolo fondamentale degli amminoacidi è quello di mattoni costitutivi di
proteine e peptici, essi possono svolgere altre funzioni importanti sotto forma di molecole singole.
Innanzitutto possono essere una fonte di energia: le cellule sono dotate di sistemi enzimatici capaci di
utilizzare lo scheletro carbonioso degli amminoacidi per formare glucosio o altri metaboliti, poi utilizzabili
per la produzione di ATP. Alcuni amminoacidi hanno un ruolo importante quali neurotrasmettitori, ovvero
quali messaggeri chimici nella comunicazione tra neuroni. Il glutammato (il sale dell’amminoacido acido
glutammico) è l’esempio più significativo, ma anche glicina e aspartato possono agire in modo simile. Altri,
come la tirosina, sono precursori necessari per la sintesi di ormoni o neurotrasmettitori.
α – amino acidi
Anche se nell’uomo sono stati individuati numerosi composti di natura amminoacidica, quelli più
importanti e abbondanti, in quanto contenuti nelle proteine, sono gli α-amminoacidi.
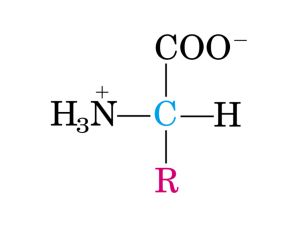
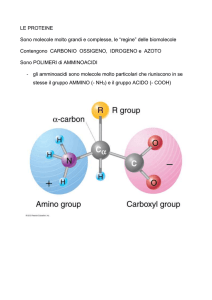
Essi sono caratterizzati dal fatto che entrambi i gruppi funzionali (amminico e carbossilico) sono legati allo
stesso carbonio, detto appunto carbonio α.
Esistono tuttavia anche amminoacidi diversi, che, in base allo spostamento del gruppo amminico rispetto a
quello carbossilico, sono definiti: β, γ o Δ amminoacidi.
Negli α-amminoacidi il carbonio centrale lega, oltre ai due gruppi funzionali, un atomo di idrogeno (-H) ed
una catena laterale o gruppo R (-R) di natura diversa nei diversi amminoacidi.
Gli aa delle proteine. Gli α-amminoacidi che si riscontrano nelle proteine sono 20 e la tabella ne indica
nome e abbreviazioni con cui vengono comunemente identificati.
Questi 20 amminoacidi si differenziano l’uno dall’altro esclusivamente in base alle caratteristiche chimiche
della catena laterale R. Peptidi e proteine sono costituiti quindi da questi 20 residui amminoacidici, che
possono essere legati in innumerevoli combinazioni di sequenza e numero. A questo livello di complessità,
si aggiunge il fatto che alcuni residui amminoacidici possono essere chimicamente modificati una volta
inseriti nel contesto della proteina (modifiche post-traduzionali). Un esempio tipico di questo fenomeno è
la fosforilazione (attacco di un gruppo fosfato) di residui di serina o treonina contenuti nella catena
amminoacidica di alcuni enzimi.
Amino acidi essenziali. I 20 α-ammioacidi che costituiscono le proteine sono distinti in essenziali e non
essenziali in base alla possibilità per le nostre cellule di sintetizzarli a partire da altri substrati. Otto
amminoacidi sono essenziali e devono necessariamente essere assunti con l’alimentazione. Per tutti gli
altri esistono sistemi enzimatici che ne rendono possibile la biosintesi endogena.
Proiezioni di fisher e enantiomeri L/D. La formula di proiezione di Fisher rappresenta il modo più semplice
per descrivere su un piano bidimensionale la struttura di un α-amminoacido. Ricordiamo che in questa
proiezione la catena carboniosa viene posta in una colonna verticale con il carbossile in alto.
Le linee verticali rappresentano i legami che, nello spazio tridimensionale, si allontanano
dall’osservatore, quelle orizzontali i legami che si avvicinano.
Da questa proiezione è facile osservare che il carbonio α di tutti gli α-amminoacidi (tranne glicina e prolina)
lega quattro residui diversi; è quindi un atomo chirale o asimmetrico, che da luogo a due configurazioni
assolute diverse, l’una l’immagine speculare dell’altra, definite enantiomeri.
Nella proiezione di Fisher, l’enantiomero L ha il gruppo amminico a sinistra, l’enantiomero Da destra. Gli
enantiomeri L e D possono essere distinti per la proprietà fisica di ruotare il piano della luce polarizzata in
senso opposto.
Gli α-amminoacidi presenti nelle proteine appartengono tutti alla serie L e molti organismi viventi, tra cui
l’uomo, non sono in grado di utilizzare gli amminoacidi della serie D.
Proprietà acido-base. Possedendo contemporaneamente almeno un gruppo carbossilico ed un gruppo
ammidico (ma altri gruppi protonabili possono essere presenti nelle catene laterali) gli amminoacidi
possono avere carica netta (la somma complessiva delle cariche dei diversi atomi della molecola) variabile.
Infatti questi gruppi funzionali si dissociano (perdita di uno ione idrogeno) o meno in base al pH
dell’ambiente in cui si trovano: in condizioni fisiologiche (pH 7.4), il gruppo carbossilico è deprotonato
(COO-) mentre quello amminico è protonato (NH3+); quindi la carica netta di amminoacidi con i soli due
gruppi funzionali principali è zero.
A pH molto acidi (< 2), anche il gruppo carbossilico è indissociato e la carica netta pari a +1. Infine a pH
fortemente basici (>10), il gruppo carbossilico è dissociato mentre quello amminico è indissociato: la
carica netta dell’amminoacido è –1.
Proprietà fisiche e Punto Isoelettrico. Questo comportamento dei gruppi protonabili principali e della
catena laterale determina alcune caratteristiche fisiche degli amminoacidi, quali la solubilità in acqua e
l’elevato punto di fusione. Gli stessi fenomeni a carico delle catene laterali si manifestano anche per i
residui amminoacidici nel contesto di peptidi e proteine. La presenza di cariche elettriche nei polimeri di
amminoacidi contribuisce alla formazione di interazioni elettrostatiche intra- ed intermolecolari che
contribuiscono sia al raggiungimento di una specifica conformazione sia alla specificità di interazione con
ligandi diversi. Alla presenza di gruppi protonati o meno si associa un’altra caratteristica fisica di
amminoacidi e peptidi: il punto isolelettrico, ovvero il valore di pH al quale la carica netta della molecola
è pari a zero. Questo valore può essere facilmente calcolato per gli amminoacidi di cui si conoscono
numero e caratteristiche di acidità dei gruppi protonabili; per i peptici il valore deve invece essere
determinato sperimentalmente. Caratteristiche degli α-amminoacidi. Gli α-amminoacidi possono essere
distinti in gruppi diversi in base alle caratteristiche chimio-fisiche delle catene laterali. Individuiamo:
amminoacidi idrofobici, amminoacidi neutri o polari ma non carichi, amminoacidi acidi, amminoacidi
basici.
Gli amminoacidi prevalentemente idrofobici possono avere catene laterali puramente alifatiche
(alanina, valina, leucina ed isoleucina) o aromatiche (enilalanina, inosina e triptofano); gli amminoacidi
neutri o polari presentano catene laterali eterogenee, ma non protonabili; comprendono serina e
treonina, che contengono un gruppo ossidrile, cisteina, che presenta un gruppo sulfidrile, glutammina e
asparagina, con gruppi ammidici non protonabili.
Gli amminoacidi acidi (acido glutammico ed aspartico) contengono gruppi carbossilici (possibili cariche
negative), mentre quelli basici(lisina, arginina ed istidina) hanno gruppi con atomi di azoto protonabili
(possibili cariche positive).
Residui importanti per la conformazione. Glicina e prolina si distinguono dagli altri amminoacidi: la catena
laterale del primo è rappresentata da un atomo di idrogeno.
E’ quindi l’amminoacido più piccolo, con flessibilità (rotazione degli angoli di legame) nettamente
superiore a quella di tutti gli altri. Questo spiega il fatto che, nel contesto di polipeptidi, si trovi in
corrispondenza di punti di ripiegamento brusco della catena. Nella prolina invece la catena laterale è un
anello che include l’azoto del gruppo ammidico principale (gruppo imminico); ciò rende l’amminoacido
estremamente rigido, con ovvie conseguenze conformazionali quando inserito nel contesto di catene
peptidiche.
Idrofilicità. La presenza di catene laterali con caratteristiche fisiche ben distinte influenza direttamente
l’idrofilicità di ciascun amminoacido, che sarà ovviamente elevata per quelli con gruppi protonabili oppure
polari, per diminuire progressivamente passando a quelli con catene neutre o idrofobiche. Questa
caratteristica fisica gioca un ruolo importante nel contesto dei peptici proteici, che normalmente svolgono
la propria funzione in ambiente acquoso: la conformazione più stabile dei peptici tenderà infatti a non
esporre all’ambiente circostante le catene laterali idrofobiche e, al tempo stesso, a porre sulla superficie di
contatto le catene laterali polari e cariche. Proprio questo fenomeno rappresenta una delle forze
determinanti per il raggiungimento della struttura tridimensionale finale delle proteine.
Il legame peptidico
Gli aminoacidi sono uniti dal legame peptidico. La formazione di peptidi deriva dalla concatenazione di
più α-amminoacidi attraverso legami ammidici (o peptidici) in cui il gruppo carbossilico di un
amminoacido reagisce con quello amminico di un altro amminoacido con eliminazione di una molecola
d’acqua. Più amminoacidi si possono unire a formare catene lineari, in cui entrambi i gruppi funzionali di
ciascun residuo amminoacidico sono coinvolti in legami peptidici.
Solo il primo amminoacido e l’ultimo presenteranno l’uno un gruppo amminico e l’altro un gruppo
carbossilico liberi. In una catena peptidica si può quindi individuare una direzionalità: il primo
amminoacido identifica l’estremità NH2 terminale (gruppo amminico libero) e l’ultimo l’estremità COOH
terminale (gruppo carbossilico libero).
Risonanza. Il legame peptidico presenta caratteristiche peculiari, che influenzano considerevolmente la
conformazione di catene polipeptidiche. E’ un legame forte, con una lunghezza di circa 1.32 Å, che è
maggiore di quella di un legame doppio ma inferiore a quella di un legame semplice. Il legame peptidico
può infatti essere descritto dalle due forme limite di risonanza rappresentate nella figura. Ne consegue
che è un legame rigido, ovvero che non consente rotazione lungo il proprio asse, e planare.
Configurazioni Cis e Trans. La rigidità del legame peptidico rende possibili isomeri geometrici,
caratterizzati da una configurazione cis o da una configurazione trans. Per ragioni di impedenza sterica, la
configurazione trans è molto più stabile e quindi assunta praticamente dalla totalità dei legami peptidici
negli organismi viventi.
I piani peptidici nella catena proteica
Angoli phi e psi. Il legame peptidico è anche planare: gli atomi coinvolti nel legame (C-N) e quelli ad essi
legati si trovano tutti sullo stesso piano, definito piano ammidico. Così una catena peptidica può essere
descritta, dal punto di vista strutturale, non solo come una sequenza di residui amminoacidici, ma anche
come una sequenza di piani peptidici. Ciascun carbonio α appartiene a due piani peptidici successivi e
rappresenta quindi il loro atomo di connessione.
Ramachandran Plots. Tale connessione avviene attraverso legami semplici tra Ca ed N amminico e tra Ca
e C carbossilico; sono legami non rigidi e quindi i piani peptidici sono liberi di ruotare l’uno rispetto
all’altro. Gli angoli di torsione (definiti phi e psi) possono avere valori diversi: per definizione sono possibili
tutti i valori compresi tra –180° e +180°; nella realtà, l’ingombro sterico dovuto alle catene laterali di
ciascun residuo amminoacidico limita notevolmente tali possibilità ed il fisico Ramachandran (grafico di
Ramachandran) ha stabilito come prevedere tali valori sulla base della natura degli amminoacidi coinvolti
nel legame peptidico.
Solo la glicina, avendo il solo idrogeno come catena laterale, rende possibile qualsiasi angolo di rotazione.
Questo spiega quanto accennato in precedenza in relazione alla corrispondenza tra glicina e punti di
maggiore flessibilità (torsione) nelle catene proteiche. Da quanto detto appare evidente che, alla
determinazione della conformazione strutturale di un polipeptide, concorrono forze e vincoli diversi,
quali:
i piani di rigidità del legame peptidico,
la possibilità di torsioni attorno agli angoli phi e psi,
l’ingombro sterico,
il grado di idrofilicità delle catene laterali dei vari residui.
Forze che determinano la struttura tridimensionale delle proteine
Forze per la struttura 3D. Abbiamo visto come la definizione di proteina implica l’assunzione da parte della
catena amminoacidica di una conformazione tridimensionale specifica che determina la funzione del
polimero stesso. In termini generali le proteine possono essere distinte in globulari (simili a gomitoli) o
fibrose (simili a bastoncini). Tuttavia ciascuna proteina ha una sua forma specifica e unica. Diversi fattori e
forze concorrono alla definizione di questa struttura. Si è precedentemente discusso come una delle forze
trainanti è quella che porta le catene laterali idrofobiche verso l’interno della struttura, al riparo da
interazioni con l’ambiente acquoso, esponendo contemporaneamente quelle idrofiliche sulla superficie di
contatto. Al raggiungimento di questo obiettivo concorre anche la formazione di ponti idrogeno che
coinvolgono i gruppi amminici e carbossilici dello scheletro peptidico degli amminoacidi idrofobici:
quando impegnate in tali legami, queste parti potenzialmente idrofiliche possono accomodarsi lontane
dall’ambiente acquoso, insieme alle catene laterali apolari. Il mantenimento della forma tridimensionale è
inoltre raggiunto grazie all’istaurarsi di interazioni elettrostatiche tra catene laterali cariche e di
interazioni idrofobiche tra catene laterali apolari.
Ponti disolfuro. Anche alcuni legami covalenti possono dare un contributo importante; è il caso dei ponti
disolfuro tra i gruppi sulfidrile (-SH) di cisteine localizzate in punti distanti della stessa catena o di catene
diverse (nel caso di proteine multimeriche). La forza di questi legami comporta una notevole
stabilizzazione della forma tridimensionale definitiva.
Ordini di struttura delle proteine
Anche se ciascuna proteina ha una forma specifica e diversa da quella di ogni altro polipeptide, quando si
confrontano e studiano conformazioni di proteine diverse si possono trovare elementi in comune, che
aiutano a capire come tale livello di complessità possa essere raggiunto partendo da catene di 20 elementi
costitutivi diversi. Per semplificare lo studio della forma delle proteine si distinguono quattro ordini di
struttura diversi, identificati con i termini di struttura primaria, secondaria, terziaria e quaternaria.
La struttura primaria indica la sequenza dei residui amminoacidici che costituiscono la catena. Tale
sequenza è determinata dalla sequenza dei nucleotidi presenti nel gene codificante per la specifica
proteina.
Ogni proteina ha una sequenza specifica che è un determinante importante per la struttura finale.Questa
catena di amminoacidi non rimane in uno stato completamente esteso, ma tende a ripiegarsi e
arrotolarsi. A livello locale, si osserva spesso la tendenza a formare strutture ordinate, che mantengono
caratteristiche simili in tutte le proteine e costituiscono appunto la struttura secondaria. Essa include α
eliche e foglietti β. L’unione di tutte le porzioni della catena con una precisa struttura secondaria e non in
una organizzazione tridimensionale complessiva definisce la struttura terziaria, che corrisponde quindi alla
forma definitiva di una proteina costituita da una singola subunità. In molti casi, la funzione biologica può
essere svolta solo quando più subunità uguali o diverse si assemblano in complessi proteici multimerici; in
questo caso la forma e l’organizzazione dell’intero complesso viene definita struttura quaternaria.
Un esempio di proteina: l’emoglobina
Struttura dell’emoglobina e l’EME. L’emoglobina è un esempio di proteina con struttura quaternaria;
nell’adulto (emoglobina A, HbA) è infatti costituita da 2 catene α e 2 catene β, unite in un’unica struttura
globulare attraverso interazioni non covalenti. Ciascuna sub-unità adatta in un particolare ripiegamento
idrofobico una molecola non proteica, l’eme, che, grazie all’atomo di ferro in esso contenuto, può legare
reversibilmente composti quali l’ossigeno, l’anidide carbonica, il monossido di carbonio. L’emoglobina
contenuta nei globuli rossi è la molecola che consente l’efficace trasporto dell’ossigeno nel nostro sangue,
catturando 4 molecole di ossigeno/molecola di emoglobina a livello dei polmoni e cedendole nei distretti
periferici. Curve di saturazione per O2 e effetti dell’ossigenazione sulla struttura 4°. La struttura
quaternaria dell’emoglobina è fondamentale per consentire la funzione dell’emoglobina, che si basa sulla
reversibilità del legame con l’ossigeno. Il grafico confronta, per diversi valori di pressione parziale di
ossigeno (asse X, PO2), la saturazione in ossigeno (asse Y, YO2) dell’emoglobina e della mioglobina, una
proteina dei muscoli. Quest’ultima è una proteina monomerica, formata da un’unica sub-unità molto
simile alle catene presenti nell’emoglobina. Osserviamo come la mioglobina raggiunge la saturazione in
ossigeno anche in presenza di poco ossigeno nell’ambiente, mentre l’emoglobina raggiunge gli stessi livelli
di saturazione solo in presenza di molto ossigeno. Questo significa che nei polmoni, dove la PO2 è elevata,
l’emoglobina può caricarsi di ossigeno, ma, quando raggiunge distretti periferici, in cui la PO2 è inferiore,
rilascia l’ossigeno, che può così raggiungere le cellule.
Questo fenomeno è reso possibile dalla presenza delle 4 sub-unità; infatti, quando una subunità
dell’emoglobina lega una molecola di ossigeno, va incontro a lievi modifiche conformazionali che vengono
trasmesse alle sub-unità vicine, aumentandone la capacità di legare a loro volta l’ossigeno.
Tale fenomeno è tipico di proteine multimeriche e viene definito allosterismo. La caratteristica principale
delle proteine allosteriche è quella di poter modificare la propria conformazione e, di conseguenza, la
propria funzione, in seguito ad interazioni con molecole diverse, dette effettori allosterici.
Riepilogo
Gli amminoacidi sono composti organici caratterizzati dalla presenza nella molecola di un gruppo
carbossilico e di un gruppo amminico. I più importanti per le cellule sono gli α amminoacidi, in cui
entrambi i gruppi chimici sono legati allo stesso atomo di carbonio, detto appunto carbonio α. Essi sono
infatti gli elementi costitutivi delle proteine e dei peptidi, che svolgono innumerevoli funzioni essenziali
per la vita di tutti gli organismi.
Gli α amminoacidi presenti nelle proteine di cellule eucariotiche sono 20 e si distinguono per la natura
chimica della catena laterale R, che determina le caratteristiche chimico-fisiche (punto isoelettrico,
idrofobicità e idrofilia) di ciascun amminoacido. Alcuni amminoacidi sono essenziali per il nostro
organismo e devono essere apportati con la dieta.
L’unione degli amminoacidi a formare catene polipeptidiche avviene attraverso un legame chimico di tipo
ammidico tra il gruppo amminico di un amminoacido e quello carbossilico di un altro. Il legame peptidico è
un legame rigido, che non consente rotazione attorno al proprio asse. Determina quindi punti di rigidità
nel contesto di una catena peptidica, influenzando significativamente la conformazione tridimensionale
assunta dalla stessa.
La capacità funzionale di ciascuna proteina dipende strettamente dal mantenimento di una struttura
tridimensionale specifica (struttura terziaria o quaternaria per proteine multimeriche), correlata alla
sequenza degli amminoacidi nella catena polipeptidica (struttura primaria), ma caratterizzata da motivi
tridimensionali riscontrabili in molte proteine diverse (struttura secondaria: α elica e foglietto β).
DIGESTIONE E ASSORBIMENTO DEI COMPOSTI AZOTATI
Introduzione
L’organismo umano non è in grado di sintetizzare tutti i diversi tipi di L-α-amminoacidi necessari per la
sintesi proteica; alcuni di essi sono essenziali e devono essere apportati con l’alimentazione. Le proteine
che si assumono con la dieta non sono importanti in quanto tali, bensì quale fonte principale di
amminoacidi per l’organismo. Esse sono molecole di considerevoli dimensioni, contenenti sia legami
covalenti forti che altri numerosi legami deboli, che ne determinano la conformazione. Per poter essere
assorbite vanno incontro ad un processo digestivo che prevede due tappe:
la denaturazione proteica,
l’idrolisi dei legami peptidici.
Queste fasi avvengono nello stomaco e nell’intestino, per un’azione coordinata del succo acido gastrico e
degli enzimi contenuti nelle secrezioni enteriche. Una volta semplificate a tripeptidi, dipeptidi e singoli
amminoacidi, possono essere assorbite dagli enterociti e veicolate nel sangue per confluire nel pool degli
aminoacidi liberi.
Obiettivi
Questa unità descrive i processi biochimici che consentono la digestione delle proteine assunte con la
dieta ed i meccanismi di assorbimento intestinale degli aminoacidi.
Metabolismo degli Amino Acidi: aspetti generali
Diversi processi biochimici vitali dell’organismo umano utilizzano composti azotati quali gli amminoacidi,
le porfirine e le ammine biogene. Le cellule umane tuttavia non posseggono sistemi enzimatici che
consentono di incorporare l’azoto inorganico presente nell’atmosfera (N2) in composti di natura organica.
Questo processo, denominato fissazione dell’azoto, è realizzato solo da alcuni microrganismi e vegetali, e
l’uomo può semplicemente apportare composti azotati dall’esterno, attraverso l’alimentazione, e
utilizzarli quali blocchi di partenza per le biosintesi endogene.
Le proteine, essendo formate da amminoacidi, rappresentano la maggiore fonte di azoto organico.
L’assunzione di proteine garantisce da un lato la disponibilità degli amminoacidi essenziali, e quindi la
sintesi proteica endogena, dall’altro il mantenimento di un pool di amminoacidi liberi necessario per la
biosintesi di tutti gli altri composti azotati.
Circa il 2% delle proteine endogene viene rinnovato quotidianamente. Anche se una buona percentuale
degli amminoacidi liberati da questo turnover vengono riutilizzati per i processi biosintetici, una parte
viene invece eliminata e deve essere quindi rimpiazzata attraverso l’apporto esogeno. Gli amminoacidi
non possono infatti essere accumulati sotto forma di riserve come i lipidi o i carboidrati. Mentre eventuali
eccessi vengono trasformati in altri metaboliti, eventuali deficit non possono essere colmati
semplicemente da trasformazioni metaboliche endogene, in quanto non esistono sistemi enzimatici in
grado di produrre gli otto amminoacidi essenziali.
L’apporto di amminoacidi con la dieta è quindi necessario per la vita. Anche se il loro compito principale è
di natura strutturale, dobbiamo ricordare che gli amminoacidi possono svolgere, in condizioni particolari,
anche un ruolo energetico nell’organismo, ovvero essere utilizzati per produrre glucosio.
Digestione e assorbimento degli amino acidi
Digestione ed assorbimento. Le proteine sono macromolecole complesse, formate da aminoacidi uniti da
forti legami peptidici in catene che generalmente assumono conformazioni tridimensionali complesse.
Per poter essere assorbite, le proteine devono subire un processo di semplificazione notevole, che porta
alla liberazione dei singoli amminoacidi o dei dipeptidi e tripeptidi presenti nella catena polipeptidica.
Il processo inizia nello stomaco, grazie al succo fortemente acido secreto dalle cellule parietali, che
consente la denaturazione iniziale della massa proteica.
In questa sede inizia anche la vera lisi dei legami peptidici ad opera della proteasi pepsina; il processo
prosegue e viene portato a termine nell’intestino, dove altre proteasi presenti nel secreto pancreatico e
negli enterociti completano l’idrolisi dei legami covalenti.
Denaturazione Proteica. Molto spesso le proteine alimentari subiscono un’iniziale processo di
denaturazione nel corso dei processi di cottura. L’esposizione ad alte temperature distrugge i legami
deboli (ponti idrogeno, interazioni elettrostatiche, interazioni idrofobiche) che contribuiscono alla
definizione della conformazione proteica.
Lo stesso effetto viene raggiunto nello stomaco, ad opera dell’acido cloridrico presente nel succo gastrico.
Il venir meno di tutte le interazioni intra- ed intercatenarie determina la trasformazione delle proteine in
semplici strutture lineari, in cui i singoli legami peptidici sono facilmente accessibili all’attacco di enzimi
proteolitici.
Pepsina. Nel lume gastrico inizia anche l’idrolisi delle proteine esogene. Le cellule gastriche secernono
uno zimogeno, il pepsinogeno, che viene attivato ad enzima attivo, la pepsina, ad opera del pH acido.
L’attivazione comporta il distacco di una porzione della catena amminoacidica, con conseguente
esposizione del sito attivo. Le molecole di pepsina attivate dall’acidità ambientale velocizzano l’attivazione
del resto del pepsinogeno con un processo autocatalitico.
La pepsina è un endopeptidasi che scinde specificamente legami peptidici, in cui sono coinvolti
amminoacidi aromatici o bicarbossilici; in questo modo degrada le proteine denaturate in grossi
frammenti polipetidici.
Endopepeptidasi. Le proteine grossolanamente frammentate giungono nel duodeno, dove le secrezioni
pancreatiche e biliari neutralizzano l’acidità del secreto gastrico, rendendo l’ambiente modicamente
basico. Il secreto pancreatico contiene endopeptidasi ed esopeptidasi che continuano la degradazione
delle proteine; si tratta di:
tripsina,
chimotripsina ed elastasi (endopeptidasi)
procarbossipeptidasi A e B (esopeptidasi).
Questi enzimi sono rilasciati nel lume intestinale come zimogeni (tripsinogeno, chimotripsinogeno,
proelastasi e procarbossipeptidasi). Un enzima prodotto dalle cellule della mucosa intestinale,
l’enterochinasi, attiva per proteolisi il tripsinogeno liberando la tripsina, che, a sua volta attiva gli altri
zimogeni pancreatici.
Le tre endopeptidasi hanno specificità diverse e, idrolizzando i legami peptidici interni delle catene
polipeptidiche, riducono le catene amminoacidiche a piccoli frammenti. Questi sono attaccati dalle
cabossipeptidasi che, idrolizzando i legami peptidici posti all’estremità carbossi-teminale dei peptidi,
liberano singoli amminoacidi e accorciano progressivamente le catene amminoacidiche.
Le cellule della parete intestinale producono o espongono sulla loro superficie enzimi quali
l’amminopeptidasi o specifiche dipeptidasi, che scindendo rispettivamente i legami peptidici posti
all’estremità amino-terminale dei peptidi, oppure quelli contenuti in specifici dipeptidi, contribuiscono
alla liberazione dei singoli amminoacidi costitutivi delle proteine assunte con l’alimentazione.
Assorbimento AA. I prodotti finali della digestione proteica sono amminoacidi liberi, dipeptidi e
tripeptidi; e tutti vengono assorbiti dalla mucosa enterica. Dipeptidi e tripeptidi possono essere
efficacemente semplificati all’interno degli enterociti, ad opera di aminopeptidasi citoplasmatiche. Gli
amminoacidi a pH fisiologico presentano i gruppi amminico e carbossilico in forma protonata (-NH3+ e –
COO-), e quindi, in quanto ioni, non possono superare la membrana plasmatica passivamente. Esistono
trasportatori attivi o facilitati che veicolano questi composti attraverso le membrane; sono dotati di
specificità diverse per i vari tipi di amminoacidi e molti di essi utilizzano il gradiente della concentrazione
di sodio, esistente ai due lati delle membrane, quale fonte di energia per il trasporto.
Riepilogo
La digestione delle proteine è un processo fondamentale per garantire l’apporto di amminoacidi
all’organismo. Il processo digestivo inizia con la denaturazione della complessa struttura tridimensionale
delle molecole ad opera del pH gastrico. La linearizzazione della catena amminoacidica rende attaccabili i
legami peptidici dalle endopeptidasi e carbossipeptidasi. La pepsina presente nel secreto gastrico inizia
l’idrolisi dei legami, che viene completata nel lume intestinale dalle peptidasi secrete dal pancreas. Questi
enzimi, secreti in forma inattiva, vengono attivati per proteolisi parziale e determinano la semplificazione
delle proteine ad amminoacidi liberi, dipeptidi e tripeptidi. Dipeptidasi ed amminopeptidasi presenti
sulla membrana e nel citoplasma degli enterociti completano il processo digestivo. L’assorbimento degli
amminoacidi attraverso la mucosa intestinale avviene attraverso l’intervento di trasportatori specifici.
CARATTERISTICHE GENERALI DEL CATABOLISMO AMMINOACIDICO
Introduzione
Gli animali non partecipano al processo di fissazione dell’azoto, ovvero non sono in grado di ridurre
l’azoto atmosferico ad azoto amminico, nitrati e nitriti. Essi assumono ed eliminano azoto allo stato di
ossidazione dell’ammoniaca. Si utilizza il termine di bilancio dell’azoto per indicare l’equilibrio tra azoto
apportato con l’alimentazione e quello escreto con urina e feci. L’azoto eliminato deriva principalmente
dal catabolismo delle proteine e quindi degli amminoacidi. In condizioni normali, gli amminoacidi non
sono una fonte energetica importante per il nostro organismo, tuttavia, in alcune situazioni, non potendo
essere accumulati, essi vengono attivamente ossidati. Il loro catabolismo può essere distinto in due fasi
separate:
1. l’eliminazione del gruppo amminico,
2. l’utilizzo dello scheletro carbonioso.
La prima fase è molto delicata in quanto genera ammoniaca (NH3), un composto tossico per gli animali;
una serie di reazioni spendono energia per trasformare l’ammoniaca in un composto meno tossico e
facilmente eliminabile. Quando questi meccanismi biochimici non funzionano correttamente possono
insorgere sintomatologie cliniche gravi, dovute appunto all’accumulo di ammoniaca nel sangue.
Obiettivi
In questa lezione sono descritti i processi biochimici che determinano e regolano l’ossidazione degli
amminoacidi. In particolar modo vengono descritte le reazioni che portano all’eliminazione del gruppo
amminico degli amminoacidi sotto forma di urea e quelle che consentono l’utilizzo dello scheletro
carbonioso di tali composti. La lezione include anche alcuni cenni alle patologie che insorgono quando
questi processi sono difettosi.
Catabolismo degli amino acidi: aspetti generali
Gli amminoacidi sono la principale forma di azoto nell’organismo. Essi sono utilizzati principalmente per la
sintesi proteica e, a differenza di quanto accade per acidi grassi e glucosio, non possono essere accumulati
in quanto tali. Quando la loro assunzione con la dieta è in eccesso rispetto alle esigenze biosintetiche,
oppure quando la disponibilità di altri nutrienti è insufficiente per mancato apporto dall’esterno (digiuno)
o stati dismetabolici (p.es.: diabete), gli amminoacidi possono essere attivamente ossidati, e il loro
scheletro carbonioso utilizzato quale fonte di energia.
Poiché gli amminoacidi contengono un gruppo amminico, la loro ossidazione prevede una tappa
fondamentale, in cui tale gruppo viene separato dal resto della struttura molecolare ed indirizzato verso
processi biochimici che ne consentono l’eliminazione. La restante parte della molecola può poi essere
trasformata in intermedi metabolici importanti (glucidi, lipidi e corpi chetonici) ed essere utilizzata da
circuiti biochimici ossidativi che portano alla produzione di energia chimica.
Le transamminasi
Biosintesi dell’urea. Gli animali eliminano l’azoto proveniente dai composti azotati sotto forma di tre
diversi prodotti finali:
ammoniaca,
acido urico,
urea.
L’uomo è un organismo ureotelico, che elimina l’azoto sotto forma di urea. La biosintesi dell’urea avviene
attraverso una serie di tappe diverse:
1. la transamminazione,
2. la deamminazione ossidativa del glutammato,
3. il trasporto dell’ammoniaca,
4. il ciclo dell’urea.
Le reazioni di transamminazione consentono il trasferimento del gruppo amminico α da un amminoacido
donatore (nella figura amminoacido 1) ad un chetoacido accettore (chetoacido 1), creando in questo
modo il chetoacido corrispondente all’amminoacido di partenza (chetoacido 2) e un nuovo amminoacido
(amminoacido 2). Si tratta di reazioni reversibili, che non comportano la perdita netta di un gruppo
amminico, bensì semplicemente il suo trasferimento. Sono quindi reazioni importanti anche dal punto di
vista anabolico, in quanto consentono di creare nuovi amminoacidi (non essenziali) a partire da altri.
Specificità e coenzima PLP. Il piridossal fosfato (PLP), derivato dalla vitamina B6, è il cofattore presente
nel sito attivo di tutte le transaminasi, che sono accomunate dallo stesso meccanismo di reazione. Il
coenzima agisce da trasportatore intermedio del gruppo amminico ceduto da un amminoacido. Quando
quest’ultimo lascia il sito attivo sotto forma di chetoacido, un secondo chetoacido si lega al sito attivo e
riceve il gruppo amminico dal PLP, trasformandosi in un amminoacido. Ciascuna transferasi è specifica per
una coppia di substrati, ma non per l’altra coppia.
Così l’alanina piruvato transaminasi trasforma alanina in piruvato (e viceversa) interagendo con coppie
diverse di α -chetoacido/ α -amminoacido, mentre la glutammato α -chetoglutaratotransaminasi
trasforma specificamente α -chetoglutarato in glutammato e viceversa.
Transamminasi e medicina. Le trasamminasi sono enzimi presenti in molti tessuti, tuttavia la
concentrazione maggiore si riscontra nel fegato, organo in cui può avvenire la produzione di urea, e nel
miocardio. Come molti altri enzimi sono riscontrabili anche nei liquidi biologici (soprattutto nel sangue) in
conseguenza del normale processo di ricambio cellulare.
La quantificazione della concentrazione sierica di due tipi di transamminasi l’alanina-amminotransferasi
(ALT) e l’aspartato-amminotransferasi (AST), ha una valenza importante nella diagnostica medica. Il primo
enzima (ALT) è un marcatore biochimico utile per valutare patologie epatiche. Le maggiori elevazioni
(valori normali sono 35-40 Unità/L) si riscontrano in corso di danno epatocellulare (epatiti). Incrementi
della concentrazione della AST (valori normali = 35-40 Unità/L) si manifestano invece in caso di infarto
miocardio, ma anche in seguito a patologie epatiche (epatiti, carcinomi epatici) e muscolari (traumi,
interventi chirurgici, distrofie).
La deamminazione del glutammato
Il glutammato. Dal punto di vista catabolico, l’importanza delle reazioni di transamminazione deriva dal
fatto che l’accettore più frequente o finale è l’ α -chetoglutarato. In questi casi si ha la formazione di
glutammato per trasferimento del gruppo amminico di vari amminoacidi. Reazioni quali quelle catalizzate
dagli enzimi Aspartato Aminotransferasi (AST) e Alanina Aminotransferasi (ALT), trasferendo il gruppo
amminico sull’ α -chetoglutarato non determinano l’eliminazione del gruppo azotato potenzialmente
tossico, ma lo convogliano su un unico metabolita, il glutammato. Questo fenomeno è importante in
quanto il glutammato è l’amminoacido che con maggiore facilità va incontro a processi di deamminazione.
Deamminazione. La deamminazione ossidativa del glutammato è catalizzata dalla glutammato
deidrogenasi, un enzima localizzato nella matrice mitocondriale, che richiede NAD+ o NADP+quale
coenzima ossidante. Il gluatammato formatosi nel citosol ad opera delle transaminasi entra nei
mitocondri e subisce il distacco del gruppo amminico, liberato sotto forma di ammoniaca, trasformandosi
nuovamente in α -chetoglutarato.
Metaboliti. La liberazione di ammoniaca dagli amminoacidi è quindi un processo che richiede l’azione
coordinata di transaminasi e glutammato deidrogenasi. Questo processo è particolarmente efficace a
livello epatico, dove l’attività della glutammato deidrogenasi è sottoposta a regolazione allosterica da
parte di diversi metaboliti:
GTP,
ATP,
NADH,
ADP.
I primi tre hanno un effetto inibitore, l’ultimo ha un effetto attivante.
Sempre a livello epatico avviene la trasformazione dell’ammoniaca in urea. Anche la deamminazione del
glutammato è una reazione completamente reversibile e quindi ha un ruolo anche nei processi anabolici,
potendo catalizzare l’attacco di un gruppo amminico all’ α -chetoglutarato, con conseguente formazione di
glutammato.
Produzione di glutammina. L’ammoniaca prodotta nei tessuti extraepatici a partire dagli amminoacidi e
da altri composti azotati deve essere trasportata al fegato per essere trasformata in urea. Essendo un
prodotto altamente tossico, non viene liberata come tale, bensì rapidamente incorporata in composti
organici. La via principale di questo processo è catalizzata dall’enzima glutamminasintetasi, che aggiunge
ammoniaca al carbossile terminale del glutammato, trasformandolo appunto in glutammina.
La reazione consuma un legame altamente energetico dell’ATP. La glutammina liberata nel sangue viene
attivamente captata dal fegato, ove va incontro a deamminazione (glutamminasi) formando glutammato,
che può essere ulteriormente trasformato in α -chetoglutarato.
Ciclo Alanina-Glucosio. Un circuito metabolico particolare ed importante si instaura tra muscolo e fegato
in condizioni in cui la degradazione proteica ai fini energetici è abbastanza elevata. I gruppi amminici
derivati dagli amminoacidi vengono trasferiti normalmente al glutammato; questo può essere trasportato
al fegato come glutammina, oppure essere convertito per transamminazione in alanina utilizzando come
chetoacido accettore il piruvato proveniente dalla glicolisi. L’alanina viene trasportata al fegato e
ritrasformata in glutammato, che entra nel processo di deamminazione ossidativa, e piruvato.
Quest’ultimo viene incanalato verso la neosintesi di glucosio che, rilasciato nel sangue sarà captato dai
muscoli. Questo ciclo glucosio-alanina rappresenta un meccanismo di economia metabolica, in quanto fa
sì che il fegato spenda energia per sintetizzare il glucosio, mentre il muscolo utilizza l’ATP solo ai fini della
contrazione.
L’eliminazione dell’azoto: il ciclo dell’urea
L'eliminazione dell'azoto. L’eliminazione dell’ammoniaca procede attraverso un percorso biochimico
ciclico denominato ciclo dell’urea. Esso prevede più tappe enzimatiche, di cui alcune avvengono nei
mitocondri ed altre nel citosol. Tappa preliminare del ciclo iniziale è l’incorporazione dell’ammoniaca
presente all’interno dei mitocondri nel composto carbammil-fosfato. La reazione avviene nei mitocondri,
ad opera dall’enzima carbammilfosfato sintetasi I, che utilizza ammoniaca, anidride carbonica (sotto
forma di HCO3-) e 2 molecole di ATP. La reazione è fortemente irreversibile ed è attivata allostericamente
da N-acetilglutammato.
Esiste anche un’isoforma citosolica di questo enzima (carbammilfosfato sintetasi II), che interviene nella
biosintesi delle pirimidine.
Ciclo dell’UREA. Il carbammilfosfato è un composto attivato che entra nel ciclo dell’urea reagendo con
ornitina per formare citrullina. La citrullina esce quindi dai mitocondri e il ciclo continua nel citsol. Un
secondo gruppo amminico viene apportato al ciclo dell’urea tramite una reazione di transamminazione
che trasferisce il gruppo azotato dall’amminoacido aspartato alla citrullina. La reazione, catalizzata
dall’argininosuccinato sintetasi consuma una molecola di ATP.
La successiva scissione dell’argininosuccinato produce fumarato e arginina. Il fumarato può entrare nel
ciclo di Krebs, mentre l’arginina viene idrolizzata ad opera dell’enzima citosolico arginasi, che produce
urea ed ornitina, il substrato iniziale del ciclo. L’urea così prodotta passa nel sangue per essere escreta dai
reni con l’urina; l’ornitina invece viene veicolata nei mitocondri per iniziare un nuovo ciclo.
La stechiometria del ciclo dell’urea può essere riassunta nella reazione:
CO2 + 2NH4+ + 3 ATP + Aspartato + H2O -> UREA + 2ADP + AMP + PPi + Fumarato.
Dato che il pirofosfato prodotto nella formazione di carbammilfosfato viene ulteriormente idrolizzato a
fosfato inorganico, il ciclo prevede il consumo di quattro legami altamente energetici. Tuttavia il fumarato
generato nel ciclo viene trasformato in malato e questo in ossalacetato, con produzione di una molecola di
NADH.
Poiché la riossidazione del coenzima ridotto ad opera della catena respiratoria produce circa 3 molecole di
ATP, si può affermare che il costo energetico del ciclo dell’urea non così elevato. La velocità con cui
procede il ciclo dell’urea è strettamente legata alla dieta. Se sono assunte molte proteine oppure se si è in
una fase di digiuno, si ha un’intensa attività di degradazione proteica, con conseguente incremento del
flusso produttivo di urea. La regolazione della velocità del ciclo avviene soprattutto attraverso
meccanismi a lungo termine che determinano maggiore o minore biosintesi degli enzimi coinvolti nel ciclo
e della carbammilfosfato sintetasi.
Destino dello scheletro carbonioso
Se l’organismo investe energia per eliminare il gruppo amminico degli amminoacidi sotto forma di urea,
può tuttavia ricavare energia dall’ossidazione dello scheletro carbonioso delle stesse molecole. In
condizioni normali il catabolismo degli amminoacidi può contribuire per una percentuale del 10-15% alla
produzione totale di energia chimica. Il catabolismo dei diversi amminoacidi presenti nelle proteine
converge verso la formazione di prodotti che sono in grado di entrare nel ciclo di Krebs per essere quindi
completamente ossidati ad anidride carbonica ed acqua.
Alcuni di essi possono tuttavia essere utilizzati per la gluconeogenesi o la chetogenesi.
In particolar modo sei amminoacidi vengono trasformati in piruvato (e successivamente acetil-CoA),
cinque in α -chetoglutarato, quattro in succinil-CoA, due in fumarato, due in ossalacetato: nel loro
insieme, potendo dar luogo a molecole utilizzabili per la gluconeogenesi, questi amminoacidi vengono
definiti glucogenici. Altri amminoacidi producono acetoacetil-CoA ed acetil-CoA, che possono essere
utilizzati per produrre corpi chetonici; vengono così definiti chetogenici. Per alcuni amminoacidi, una
porzione dello scheletro carbonioso è chetogenica e l’altra porzione è glucogenica: rientrano quindi in
entrambi i gruppi. Solo lisina e leucina sono esclusivamente chetogenici.
Amminoacidi glucogenici e cheto genetici. Lo scheletro carbonioso (o parte di esso) di: alanina, glicina,
cisteina, serina, treonina, triptofano, viene degradato a piruvato. Quest’ultimo è generalmente convertito
in acetil-CoA ed entra nel ciclo dell’acido citrico. Ovviamente può essere trasformato anche in
ossalacetato e rifornire la gluconeogenesi. Una parte della struttura di: triptofano, fenilalanina, tirosina,
leucina, isoleucina, produce acetil-CoA direttamente oppure attraverso la formazione di acetoacetil-CoA.
Fenilalanina e Tirosina. Fenilchetonuria (PKU). Il catabolismo della fenilalanina inizia con una reazione di
idrossilazione dell’anello benzenico generando tirosina. Questa tappa biochimica è catalizzata dall’enzima
fenilalanina idrossilasi. La mancanza dell’enzima è associata ad una grave patologia, nota come
fenichetonuria.
La fenilalanina, non potendo essere adeguatamente catabolizzata, si accumula e da luogo a metaboliti
alternativi quali:
fenilpiruvato,
fenilacetato,
fenillattato,
che si possono facilmente riscontrare nelle urine. Se presenti nei primi giorni di vita, questi metaboliti
alterano i normali processi di sviluppo cerebrale portando a ritardo mentale. Per questo è obbligatorio
eseguire screening sui neonati (vengono quantificati i metaboliti sopraccitati), in modo da attuare, in caso
di test positivo, adeguate misure preventive (eliminare la fenilalanina dalla dieta).
Amminoacidi glucogenici. Lo scheletro carbonioso di prolina glutammato, glutammina, arginina ed
istidina è convertito ad α -chetoglutarato. Quello di metionina, treonina, valina ed isoleucina genera
invece succinil-CoA. Aspartato ed asparagina sono trasformati in ossalacetato, mentre fenilalanina e
tirosina producono anche fumarato.
Riepilogo
Gli amminoacidi derivati dalla degradazione delle proteine endogene o apportate dall’esterno possono
essere utilizzati nell’organismo per produrre energia chimica. Se limitato in condizioni normali, questo
fenomeno catabolico può divenire importante in condizioni di digiuno o in stati patologici (diabete) che
precludono il normale utilizzo di altri nutrienti. In questi stati la demolizione della massa muscolare e il
catabolismo amminoacidico risultano sensibilmente aumentati.
Il catabolismo degli amminoacidi può essere distinto in due fasi: l’eliminazione del gruppo amminico
sotto forma di urea, e la degradazione ossidativa dello scheletro carbonioso. La prima parte viene
espletata tramite processi di transamminazione che convogliano la maggior parte dei gruppi azotati dei
diversi amminoacidi sul glutammato. Quest’ultimo può essere efficacemente deamminato ad opera della
glutammato deidrogenasi. L’ammoniaca staccata dal glutammato viene convogliata verso la produzione di
urea, un composto organico meno tossico dell’ammoniaca stessa.
Il processo richiede il consumo di energia chimica e avviene esclusivamente nel fegato. L’urea viene poi
rilasciata nel sangue ed eliminata dal rene con l’urina.
Lo scheletro carbonioso degli amminoacidi è ovviamente la parte che può essere ossidata per produrre
energia. Complessivamente gli amminoacidi sono distinti in:
glucogenici, se danno luogo a metaboliti che possono essere trasformati in glucosio attraverso la
gluconeogenesi,
chetogenici, se generano acetoacetil-CoA e corpi chetonici.
La distinzione tra i due gruppi non è netta in quanto lo scheletro carbonioso di molti amminoacidi può dar
luogo a entrambi i tipi di metaboliti. L’assenza o l’errato funzionamento di enzimi che partecipano al
catabolismo amminoacidico può dar luogo a patologie. In particolar modo la fenilchetonuria deriva
dall’impossibilità di degradare correttamente la fenilalanina.
CARATTERISTICHE GENERALI DELL’ANABOLISMO AMMINOACIDICO
Introduzione
Tutti i 20 amminoacidi sono biologicamente importanti e necessari ai fini della sintesi proteica nelle cellule
umane, tuttavia, dal punto di vista nutrizionale, possono essere distinti in essenziali e non essenziali. Le
cellule sono dotate di vie enzimatiche capaci di produrre il secondo tipo di amminoacidi, ma non quelli
essenziali, che devono essere assunti con gli alimenti. Le vie biosintetiche degli amminoacidi non
essenziali sono relativamente più semplici rispetto a quelle che potrebbero generare gli amminoacidi
essenziali; si può quindi ipotizzare che la perdita della possibilità di sintesi degli amminoacidi essenziali
abbia rappresentato un vantaggio evolutivo per la sopravvivenza.
Obiettivi
La lezione presenta una rapida panoramica delle vie enzimatiche che garantiscono la sintesi degli
amminoacidi non essenziali. Contiene inoltre rapidi cenni ad importanti metaboliti derivati dagli
amminoacidi stessi.
Amino Acidi Non-Essenziali ed Essenziali
La maggior parte dei batteri e delle piante è in grado di sintetizzare tutti e 20 gli amminoacidi necessari
per la sintesi proteica. Le cellule umane hanno invece perso la capacità di produrre una decina di
amminoacidi, e mantenuto quella di produrre gli altri dieci. Gli amminoacidi vengono quindi distinti in due
categorie: quelli essenziali e quelli non essenziali. I primi devono essere recuperati con il cibo, i secondi
possono essere prodotti a partire da altri amminoacidi e da metaboliti intermedi della glicolisi e del ciclo
di Krebs.
Cenni sulla sintesi degli amino acidi non essenziali
Incorporazione di NH4+ in composti organici. Oltre all’assunzione di proteine con la dieta, l’organismo
umano può incorporare azoto in molecole organiche a partire dall’ammoniaca (NH3) attraverso le reazioni
catalizzate dagli enzimi glutammato deidrogenasi e glutammina sintasi. Il primo enzima catalizza
l’amminazione riduttiva dell’ α -chetoglutarato a produrre glutammato.
Questa reazione rappresenta una tappa fondamentale dei processi catabolici (l’azoto di diversi
amminoacidi viene convogliato per transamminazione sul glutammato, che poi va incontro a
deamminazione), ma anche la reazione iniziale per la sintesi di molti amminoacidi, sempre attraverso i
processi di transamminazione.
La reazione catalizzata dall’enzima glutammina sintasi consente l’incorporazione di azoto inorganico nella
glutammina a partire da glutammato. La reazione è accoppiata all’idrolisi di un legame altamente
energetico dell’ATP.
L’azoto ammidico della glutammina è la fonte di azoto per la sintesi di molti altri amminoacidi, di purine e
pirimidine e di altri composti azotati. Questo spiega perché l’attività della glutammina sintasi,
rappresentando un crocevia importane del metabolismo azotato, è sottoposta a fine regolazione,
prevalentemente di tipo allosterico: l’attività viene inibita quando la concentrazione di glutammina è
elevata, mentre viene stimolata se i livelli di glutammina sono bassi, ma alti quelli di ATP ed α chetoglutarato, substrati di partenza della reazione.
Reazioni di transamminazione. A patire dal glutammato, attraverso reazioni di transamminazione che
trasferiscono il gruppo azotato su vari chetoacidi, vengono sintetizzati molti amminoacidi.
In particolar modo alanina ed aspartato vengono prodotti per trasferimento dell’ammino-gruppo
rispettivamente su piruvato ed ossalacetato. Tutte le reazioni richiedono come coenzima il piridossalfosfato (vitamina B6).
ASPARAGINA. La sintesi dell’asparagina a partire dall’aspartato ad opera dell’asparagina sintasi è simile a
quella della glutammina.
La differenza fondamentale è che nel caso dell’asparagina, il gruppo amminico proviene dalla glutammina.
In questa reazione non si ha quindi incorporazione netta di azoto inorganico in molecole organiche.
TIROSINA, SERINA, GLICINA, PROLINA E ARGININA. La tirosina è sintetizzata dalla fenilalanina ad opera
della fenilalanina idrossilasi.
La reazione prevede l’incorporazione di ossigeno molecolare nell’anello aromatico (posizione para) della
fenilalanina e richiede la presenza del cofattore tetraidrobiopterina.
L’assenza di fenilalanina idrossilasi è causa della malattia fenilchetonuria.
La serina è invece sintetizzata a partire dal 3-fosfo-glicerato attraverso tre tappe successive. A sua volta
rappresenta il precursore per la sintesi di glicina e di cisteina.
Infine prolina e arginina sono sintetizzate a partire dal glutammato; la prima attraverso una reazione di
ciclizzazione, la seconda nell’ambito del ciclo dell’urea attraverso l’intermedio ornitina.
Amminoacidi come precursori di composti azotati
Amminoacidi come precursori. Gli amminoacidi sono i precursori fondamentali per la sintesi proteica e
nel nostro organismo circa 400g di amminoacidi al giorno sono utilizzati per il turnover proteico. Una
quota inferiore viene destinata alla sintesi di un numero notevole di composti contenenti azoto e coinvolti
in numerosi processi fisiologici di importanza rilevante.
Tra questi:
purine e pirimidine (basi azotate),
porfirine,
ossido nitrico,
catecolamine,
melanina,
istamina,
serotonina,
glutatione.
Sintesi di Ossido Nitrico.L’ossido nitrico (NO) è un gas prodotto dall’enzima ossido nitrico sintasi a partire
dall’arginina.
Tale gas si diffonde rapidamente nelle cellule e determina un incremento del GMP ciclico, (composta
analogo all’AMP ciclico, in cui l’adenina dell’AMP è sostituita dalla guanina), con successiva dilatazione dei
vasi sanguigni e riduzione della pressione ematica.
Farmaci quali la nitroglicerina, utilizzati per contrastare l’angina pectoris (ischemia miocardia), si basano
proprio sulla loro capacità di essere rapidamente trasformati in NO ed indurre una rapida dilatazione delle
coronarie.
Biosintesi delle Catecolamine. Le cellule di origine neuronale e quelle della midollare del surrene
convertono la tirosina ad adrenalina e noradrenalina, composti che esplicano funzione di
neurotrasmettitore (circuiti nervosi adrenergici) ed anche di ormoni, secreti in condizioni di stress e
capaci di modulare il metabolismo degli zuccheri e dei lipidi.
La sintesi passa attraverso la formazione di dopa (ad opera dell’enzima tirosina idrossilasi) e dopamina
(per decarbossilazione della dopa).
La L-dopa è un farmaco importante nella terapia del morbo di Parkinson, caratterizzato da una marcata
riduzione di dopamina associata alla morte di neuroni in regioni specifiche del cervello.
Nei melanociti la tirosina è convertita a melanina attraverso un percorso enzimatico che inizia con la
formazione di dopa ad opera della tirosina idrossilasi.
La melanina è il pigmento che determina il colore di pelle, occhi e capelli.
Produzione di GABA. La sintesi di γ-amminobutirrato avviene principalmente nel tessuto cerebrale, ove
agisce da neurotrasmettitore inibitorio.
Il composto si forma per decarbossilazione del glutammato ad opera della glutammato decarbossilasi.
Istamina. L’istamina è una molecola prodotta nei mastociti per decarbossilazione dell’istidina. Quando
liberata da queste cellule, esercita un potente effetto vasodilatatorio.
È coinvolta inoltre nelle risposte allergiche ed immunitarie e nella modulazione della secrezione acida a
livello gastrico.
Metabolismo del Triptofano: Formazione di Serotonina. L’idrossilazione del triptofano a 5idrossitriptofano è catalizzata dalla tirosina idrossilasi. La successiva decarbossilazione porta alla sintesi di
serotonina (5-idrossitriptamina).
A livello cerebrale la serotonina è coinvolta nella regolazione del sonno, dell’umore e dell’appetito. È
prodotta anche dalle piastrine (aggregazione) e dalla muscolatura liscia (vasocostrizione).
Farmaci che modulano l’azione della serotonina sono utilizzati nella terapia della depressione (inibitori del
riassorbimento o della degradazione), dell’emicrania e della schizofrenia.
Creatina e Creatinina. Tre amminoacidi, glicina, arginina e metionina, partecipano alla sintesi di creatina.
La prima tappa avviene nel rene e comporta il trasferimento di un gruppo guanidinico dall’arginina alla
glicina con formazione di guanidinacetato. La successiva metilazione di questo composto (nel fegato) per
mezzo della S-adenosilmetionina, porta alla creatina.
La forma fosforilata della creatina (fosfocreatina) è un’importante riserva di energia chimica a pronta
disponibilità per il muscolo.La deidratazione non enzimatica (spontanea) della creatina porta alla
formazione di cratinina, che viene liberata nel sangue ed eliminata per via urinaria.
La quantità di creatina escreta con l’urina è generalmente costante e proporzionale alla massa muscolare
dell’individuo. La concentrazione della creatinina urinaria è quindi un parametro utile per la valutazione
della funzionalità renale.
Sintesi delle Porfirine. Le porfirine sono composti ciclici formati da quattro anelli pirrolici. Hanno la
capacità di formare complessi con ioni metallici, che si legano agli atomi di azoto dei pirroli.
Esempi importanti sono l’eme dell’emoglobina, che contiene ferro e la clorofilla che contiene magnesio.
Nell’uomo sono numerose le proteine che contengono eme quale fattore proteico; tra queste
l’emoglobina, la mioglobina ed i citocromi.
La sintesi delle porfirine inizia dall’unione del succinil-CoA (metabolita del ciclo di Krebs) con
l’amminoacido glicina, con formazione di acido Δ -amminolevulinico (ALA) ad opera dell’enzima ALAsintasi(enzima chiave regolatore dell’intero processo).
La condensazione di due molecole di ALA porta alla formazione del porfobilinogeno, molecola che
contiene l’anello pirrolico. Quattro molecole di porfobilinogeno sono unite a formare l’anello
tetrapirrolico.
Anomalie genetiche o acquisite nella sintesi delle porfirine si associano a stati patologici denominati
porifirie, con sintomatologia variabile, ma spesso caratterizzata da anemia, dolore addominale e
fotosensibilità. Dal catabolismo dell’eme deriva la bilirubina.
Riepilogo
Gli amminoacidi essenziali devono essere apportati con l’alimentazione perché l’organismo è privo delle
vie enzimatiche per la loro biosintesi. Quelli non essenziali sono invece prodotti dalle cellule utilizzando
percorsi metabolici brevi. Fondamentali sono l’incorporazione di azoto (dall’ammoniaca) nell’acido α chetoglutarico, a formare glutammato, e nel glutammato stesso, a formare glutammina. Dal glutammato,
per transamminazione, si formano altri amminoacidi. Vie specifiche consentono la sintesi di:
asparagina,
serina,
glicina,
prolina,
arginino,
tirosina.
Gli amminoacidi sono i blocchi costitutivi fondamentali delle proteine, ma hanno un ruolo importante per
la sintesi di vari metaboliti, con funzioni diverse e fondamentali per l’organismo. Tra questi alcuni
neurotrasmettitori (catecolamine, serotonina, acido γ -amminobutirrico), le porfirine e la creatina.
IL METABOLISMO DELLE BASI AZOTATE
Introduzione
Le basi azotate sono composti eterociclici (contengono azoto) con un ruolo biochimico importante: sono
componenti fondamentali dei nucleotidi, che, a loro volta, rappresentano i blocchi costitutivi degli acidi
nucleici quali il DNA e l’RNA, e possono inoltre fungere da composti donatori di energia chimica (ATP e
GTP) o essere utilizzati per la sintesi di coenzimi quali NAD+, FAD+ e coenzima A. Le basi azotate sono
distinte in due classi: le pirimidine (citosina e timina) e le purine (adenina e guanina). L’uomo sintetizza
tali composti a partire da intermedi anfibolici di altri metabolismi. La degradazione delle purine dà luogo
ad acido urico, mentre quella delle pirimidine esita in più composti diversi. Anomalie che coinvolgono i
percorsi metabolici delle basi azotate si manifestano nell’uomo sotto forma di varie patologie, tra cui le
principali sono:
la gotta,
la sindrome di Lesh-Nyhan,
la carenza di adenosina deaminasi,
l’aciduria orotica.
Obiettivi
La lezione inizia con una rapida panoramica delle caratteristiche chimiche fondamentali delle basi azotate
e dei nucleotidi. Tratta poi i punti salienti dell’anabolismo e del catabolismo delle basi azotate, includendo
alcuni semplici riferimenti alle principali patologie umane associate ad anomalie nell’ambito di tali processi
metabolici.
Basi eterocicliche
Le basi azotate presenti negli acidi nucleici appartengono a due classi: purine e pirimidine. Sono molecole
planari, aromatiche ed eterocicliche (ovvero contengono un anello con uno o più vertici occupati da atomi
diversi dal carbonio; in questo caso dall’azoto). Le purine presentano due anelli e sono rappresentate da
adenina e guanina. Le pirimidine contengono un solo anello e includono citosina, timina e uracile.
Nucleosidi
Le basi azotate sono componenti fondamentali dei nucleosidi e dei nucleotidi. Per nucleosidi si intendono
composti costituiti da una base azotata legata ad una molecola di ribosio o deossiribosio. Il legame è di
tipo N-glicosidico e coinvolge il carbonio anomerico dello zucchero e l’atomo di azoto numero 9 delle
purine e numero 1 delle pirimidine.
Il nome dei nucleosidi deriva da quello della base azotata contenuta; avremo quindi adenosina,
guanosina, citidina e timina quando contengono ribosio o gli stessi nomi preceduti dal prefisso deossi-,
quando contengono deossiribosio.
Nucleotidi
I nucleotidi si formano dall’unione dei nucleosidi con una o più molecole di acido fosforico, che si
coniugano tramite legame estere al carbonio 5’ (più spesso) o 3’ del ribosio. In base al numero di fosfati
legati allo zucchero si definiscono:
i nucleotidi monofosfato (AMP adenosina monofosfato, GMP guanosina monofosfato, CMP
citidina monofosfato e TMP timidina monofosfato),
i nucleotidi difosfato (ADP, GDP, CDP e TDP, come sopra con difosfato invece di monofosfato),
i nucleotidi trifosfato (ATP, GTP, CTP e TTP, come sopra con trifosfato invece di monofosfato).
Quando in numero maggiore di uno, i fosfati sono coniugati tra loro tramite legame anidridico altamente
energetico. Per questo i nucleotidi trifosfato sono molecole che possono agire da donatori immediati di
energia libera.
Funzione dei nucleotidi
Oltre al ruolo importante di molecole deposito di energia chimica, i nucleotidi sono mattoni costitutivi
degli acidi nucleici.
In particolar modo i ribonucleotidi, che contengono ribosio, formano l’acido nucleico RNA, mentre i
desossiribonucleotidi, contenenti deossi-ribosio, formano il DNA. I nucleotidi sono poi utilizzati per la
sintesi di alcuni coenzimi, quali il coenzima A, il NAD+ ed il FAD+.
Ed ancora alcuni nucleotidi monofosfato (AMP o GMP) sono importanti molecole segnale, utilizzate quali
mediatori in diversi processi cellulari.
Digestione dei nucleotidi
Gli acidi nucleici ingeriti con gli alimenti vengono degradati a mononucleotidi dalle ribonucleasi e
desossiribonucleasi presenti nel secreto pancreatico e dalle polinucleotidasi presenti sulle membrane
dell’intestino tenue. Fosfatasi non specifiche idrolizzano i nucleotidi a nucleosidi.
Questi possono essere assorbiti come tali, oppure essere ulteriormente semplificati grazie all’intervento di
nucleosidasi, che liberano basi azotate e pentoso, oppure di nucleoside fosforilasi, che producono
pentoso-1-fosfato e base azotata.
La maggior parte delle purine e pirimidine apportate con la dieta non vengono utilizzate per la sintesi di
nucleotidi, ma sono in realtà degradate a livello intestinale (adenina e guanina sono convertite ad acido
urico) ed escrete con l’urina.
Biosintesi dei nucleotidi
Nell’uomo e nella maggior parte dei vertebrati la sintesi dei nucleotidi avviene generalmente de novo,
ovvero da precursori semplici quali amminoacidi, zuccheri e CO2.
Esiste parallelamente anche una seconda possibilità, definita via di salvataggio, che ricicla le basi libere
prodotte dal turn-over intracellulare e dall’assorbimento dei nucleotidi assunti con l’alimentazione.
I processi fondamentali della sintesi de novo sono:
1. la sintesi delle basi azotate,
2. la fosforilazione delle purine,
3. la fosforilazione delle pirimidine.
Le purine sono sintetizzate direttamente come nucleotidi, a partire dal fosforibosilpirofosfato (PRPP), cui
vengono aggiunti progressivamente gli atomi che costituiscono l’anello purinico. Il PRPP deriva dal ribosio5-fosfato, a sua volta prodotto dalla via dei pentoso fosfati. Inoltre è un intermedio nella via di recupero,
nella sintesi di NAD+ e NADP+ e dei nucleotidi pirimidinici.
Biosintesi dei nucleotidi Purinici
Gli atomi necessari per la sintesi dell’inosina monofosfato (IMP), il primo intermedio biosintetico con un
sistema ad anello purinico completo e precursore di AMP e GMP, sono forniti da glicina, glutammina,
aspartato, anidride carbonica ed il cofattore folato.
Dieci reazioni successive portano dal PRPP all’IMP. La prima, catalizzata dall’enzima PRPPammidotransferasi, prevede la donazione di un gruppo amminico da parte della glutammina, a formare
fosforibosilammina.
La successiva addizione di atomi donati da glicina (atomi di carbonio 4 e 5 e azoto 7 dell’anello), da
glutammina (azoto 3), da molecole di tetraidrofolato (carbonio 8 e 2), da aspartato (azoto 1) e dalla
CO2(carbonio 6), associata alla chiusura dell’anello, portano alla progressiva formazione del nucleoside
IMP.
Sei molecole di ATP sono consumate in queste tappe, per fornire l’energia chimica necessaria per fare
tappe biosintetiche endoergoniche (accoppiamento delle reazioni).
Un’ulteriore molecola di ATP è consumata per la produzione di PRPP: in questo caso avviene un vero e
proprio trasferimento del gruppo pirofosfato dall’ATP al carbonio 1 del ribosio-5-fosfato.
Formazione di AMP e GMP dall’ IMP. Dopo la sintesi di IMP, il processo biosintetico si dirama e tramite
una breve serie di tappe enzimatiche porta alla produzione di AMP e GMP.
Successivamente i nucleotidi monofosfato possono essere convertiti in nucleotide difosfato attraverso
processi di fosforilazione catalizzati da chinasi specifiche che, consumando ATP, aggiungono successivi
gruppi fosforilici.
Un analogo percorso porta alla formazione di GTP dal GDP. L’ATP viene invece formato a partire dall’ADP
nel contesto della fosforilazione ossidativa, della glicolisi e del ciclo di Krebs.
Regolazione della biosintesi delle purine: un classico esempio di feedback negativo. Poiché la sintesi di
IMP e nucleotidi purinici consuma ATP ed intermedi anfibolici quali glicina, gluatammina e
tetraidrofolato è importante che essa sia strettamente regolata, in modo da essere commisurata alle
esigenze della cellula. Il meccanismo di regolazione fondamentale è un classico esempio di regolazione a
feedback negativo e si estrinseca soprattutto controllando la velocità d’azione dell’enzima PRPP sintetasi
e quindi determinando la concentrazione di PRPP, metabolita di partenza della biosintesi. IMP, ma
soprattutto AMP e GMP agiscono da regolatori allosterici negativi sull’enzima. Essendo il PRPP utilizzato
anche da altre vie metaboliche, il controllo si esercita anche più specificamente sull’enzima PRPP-ammido
transferasi, enzima peculiare della sintesi delle purine, inibito allostericamente da AMP e GMP.
Questi meccanismi influiscono sul flusso complessivo dei metaboliti nel percorso biosintetico, ma AMP e
GMP regolano per retroazione la loro stessa produzione agendo sui processi che trasformano IMP nei due
nucleotidi. Inoltre, poichè ATP è necessario per la produzione di GMP da IMP (necessario per le
fosforilazioni di GMP e GDP a dare rispettivamente GDP e GTP), e GTP lo è per quella di AMP da IMP (nella
prima tappa che porta da IMP a ATP è richiesta l’idrolisi di GTP), si ha un’ulteriore regolazione incrociata
che garantisce che la sintesi di un nucleotide diminuisca in carenza dell’altro.
Sintesi delle Pirimidine
La sintesi dei nucleotidi pirimidinici si distingue dalla sintesi di quelli purinici per diversi aspetti. Il PRPP,
pur essendo fondamentale, non è coinvolto nella reazione iniziale, e soprattutto, l’anello pirimidinico
viene sintetizzato separatamente e poi aggiunto al ribosio-5-P.
La reazione iniziale è la formazione di carbammil-fosfato a partire da glutammina, CO2 e due ATP (la
reazione è uguale alla prima tappa del ciclo dell’urea, ma avviene nel citosol e non nel mitocondrio). Due
condensazioni successive, la prima con aspartato, la seconda intramolecolare, portano rispettivamente
alla formazione di carbammil-aspartato e diidroorotato, una molecola ciclica.
Questo viene deidrogenato ad acido orotico che, reagendo con il PRPP, porta alla formazione di OMP. La
successiva decarbossilazione di OMP porta ad UMP. Questo è il ribonucleotide di partenza per formare gli
altri nucleotidi pirimidinici. L’attacco successivo di fosfati donati da ATP porta a UDP e UTP, dall’UTP si
forma CTP, dall’UDP si forma TMP.
Regolazione Allosterica della sintesi delle pirimidine nell’uomo. Il controllo della sintesi delle pirimidine si
esercita attraverso meccanismi di feedback negativo e positivo. L’enzima sede della regolazione è la
carbammil-fosfato sintetasi II, che catalizza la tappa iniziale del processo. UDP e UTP sono effettori
allosterici negativi, mentre PRPP e ATP sono effettori positivi. La sintesi di purine e pirimidine procede
comunque parallelamente, grazie all’esistenza di controlli incrociati che realizzano un coordinamento delle
due vie.
Enzime regolato
Carbammil-fosfato sintasi II
Effettore allosterico
UDP, UTP
PRPP, ATP
Effetto
inibizione da feedback
stimolante
In particolar modo il reciproco controllo si esercita sulla reazione che porta alla sintesi di PRPP, metabolica
fondamentale in entrambe le vie. ADP e GDP, ma anche TDP esercitano un effetto allosterico negativo
sulla ribosio-5-P pirofosfochinasi, rallentando la produzione di PRPP e quindi l’intero processo di sintesi di
nucleotidi.
La sintesi dei deossiribonucleotidi
Avviene a partire dai corrispondenti ribonucleotidi per azione dell’enzima ribonucleotide reduttasi.
La tioredoxina agisce da agente ossidante, tramite i gruppi –SH delle sue cisteine.
Catabolismo Intracellulare delle Purine
La degradazione intracellulare dei nucleotidi purinici avviene ad opera di nucleotidasi e fosforilasi che
rispettivamente trasformano i nucleotidi in nucleosidi e quest’ultimi in basi azotate e ribosio-1-P.
L’adenosina
è
trasformata
ad
inosina
ad
opera
dell’adenosina
deamminasi.
Guanina ed ipoxantina sono trasformate in xantina e questa in acido urico ad opera dell’enzima xantina
ossidasi. L’acido urico è il metabolita finale della degradazione delle purine, eliminato con le urine sotto
forma di cristalli, a causa della sua bassa solubilità.
Rilevanza biomedica del catabolismo delle purine
Salvataggio delle Purine. Le basi puriniche libere generate dalla degradazione dei nucleotidi vengono in
realtà per la maggior parte recuperate e riutilizzate per formare nuovi nucleotidi. Tale recupero è reso
possibile dagli enzimi adenina fosforibosiltransferasi (APRT) e ipoxantina-guanina fosforbosiltransferasi
(HGPRT), che catalizzano la reazione delle basi con il PRPP. AMP, IMP e GMP possono essere sintetizzati
anche senza ricorrere alla sintesi de novo. La sindrome di Lesh-Nyhan è una patologia dovuta all’assenza o
all’inattività dell’enzima per il recupero di guanina e ipoxantina. L’accumulo di questi composti e di PRPP
dà luogo a iperuricemia (con sintomi simili a quelli della gotta), ma anche a sintomi neurologici e ritardo
mentale.
Gotta. Quando la concentrazione dell’acido urico e dei suoi sali urati (iperuricemia) raggiunge livelli
elevati, il composto, scarsamente solubile, precipita sotto forma di cristalli che si depositano nei tessuti
molli e nelle articolazioni, formando depositi detti tofi. Lo stato patologico dovuto a tali depositi viene
definita gotta, caratterizzata da attacchi ricorrenti di natura artritica (artrite gottosa), alterazioni renali e
urolitiasi. Esistono forme primitive e secondarie di iperuricemia. Tra le prime si annoverano quelle
associate a diminuita attività dell’enzima che media il salvataggio delle purine (HGPRT) oppure
all’aumentata attività della PRPP sintetasi. In entrambi i casi si ha produzione ed escrezione in eccesso di
nucleotidi purinici, con conseguente iperuricemia. Farmaci in grado di bloccare l’enzima chiave della
produzione di acido urico, la xantina ossidasi, sono efficaci nel trattamento della gotta. L’allopurinolo è un
analogo dell’ipoxantina, che si lega all’enzima in modo irreversibile, bloccandone quindi l’attività.
Deficit di ADA. Un’altra patologia associata dovuta alla mancanza o inattività di un enzima del
metabolismo purinico e il deficit di Adenina Deamminasi (ADA), che si associa ad una immunodeficienza
severa combinata (SCID), caratterizzata da carenza di linfociti B e T. I bambini affetti da tale patologia sono
fortemente immunodepressi e soggetti a infezioni di varia natura. Spesso denominati “bambini bolla”,
perchè obbligati a vivere in ambienti sterili ed isolati. La malattia è stata una delle prime patologie ad
essere trattata con successo tramite terapia genica e cellulare.
Farmaci chemioterapici. Molti farmaci chemioterapici agiscono bloccando o alterando il metabolismo
purifico o pirimidinico. Le cellule tumorali infatti, duplicandosi ad alta velocità, necessitano di una efficace
sintesi di nucleotidi per la replicazione del DNA.
Questo tipo di farmaci antitumorali ne blocca la produzione e quindi determina la morte delle cellule in
attiva crescita.
Poiché anche le cellule normali si duplicano, si hanno effetti collaterali, spesso considerevoli, soprattutto a
carico dei distretti cellulari che si rinnovano con maggiore velocità (capelli, linfociti, etc).
Uno di questi farmaci è il 5-fluoro-uracile. All’interno dell’organismo è convertito a 5-F-dUMP, che
compete con il dUMP per il sito attivo dell’enzima timidilato sintasi. Legandosi all’enzima in modo
irreversibile ne blocca l’attività. La mancata produzione di TMP, rende le cellule incapaci di replicare il
proprio DNA.
Il metotrexato determina lo stesso tipo di effetto finale, agendo tuttavia in modo diverso. Blocca la
diidrofolato redattasi, necessaria per la produzione di tetraidrofolato, coenzima essenziale per la sintesi di
dTMP.
Catabolismo Pirimidine
La degradazione dei nucleotidi pirimidinici è inizialmente simile a quella dei nucleotidi purinici con enzimi
specifici che defosforilano e deamminano rispettivamente il nucleotide ed il nucleoside.
Le basi pirimidiniche seguono un percorso catabolico diverso da quello delle purine, in quanto non danno
luogo ad acido urico, ma a metaboliti altamente solubili ( β -alanina e β -amminobutirrato) ed in parte
utilizzabili da altre vie metaboliche.
Riepilogo
Basi azotate (adenina e guanina = purine, timina, citosina e gracile= pirimidine) coniugate allo zucchero
ribosio formano i nucleosidi. L’attacco di gruppi fosfato al ribosio dei nucleosidi forma i nucleotidi (mono,
di e trifosfati), Sono molecole fondamentali per la sintesi degli acidi nucleici, quali fonte di energia chimica
(ATP), quali molecole segnale (AMP) e componenti di altri composti (NAD+, FAD+).
I nucleotidi non sono essenziali e l’organismo umano può sintetizzarli, anche se attraverso vie metaboliche
complesse.
La sintesi delle purine ha come precursore fondamentale il PRPP, cui vengono aggiunti progressivamente
atomi di carbonio ed azoto a costruire l’anello purinico.
La sintesi delle pirimidine vede invece la costruzione separata dell’anello della base azotata, che viene
successivamente coniugato al PRPP.
La degradazione delle purine dà luogo ad acido urico, composto scarsamente solubile, il cui accumulo si
associa a stati patologici diversi (gotta, sindrome di Lesh Nyhan). Quella delle pirimidine produce invece
composti solubili e, in parte, utilizzabili da altri metabolismi.