PATOLOGIA GENERALE
•
•
•
DEFINIZIONI DI MALATTIA
Qualsiasi condizioni del corpo e della mente che diminuisce la probabilità
di sopravvivenza dell’individuo o della specie.
Assenza dello stato di salute.
Deviazione rilevabile della condizione omeostatica. Comporta un aumento
dell’entropia. E’ una condizione dinamica.
DEFINIZIONE DI SALUTE (OMS)
Stato di tenesse fisico, mentale e sociale completo e
non semplice assenza di malattia.
L’omeostasi è un disequilibrio controllato. L’azione di fattori patogeni intrinseci o estrinseci possono portare a:
•
adattamento/risposta difensiva Æ malattia
•
mancato adattamento Æ malattia, morte
Il mancato adattamento è anche una possibile descrizione dell’invecchiamento.
CRITERI DI CLASSIFICAZIONE DELLE MALATTIE
•
focale
•
diffusa
Topografico
•
disseminata
•
sistemica
•
generalizzata (da non confondersi con “sistemica”)
Funzionale
es. malattie psichiche, del metabolismo, ecc.
Eziologico
es. malattie virali, ambientali, professionali
Epidemiologico es. malattie stagionali, ricorrenti, ecc.
•
•
Sindrome (concorso): associazione di diversi fenomeni patologici che hanno in comune un nesso fisio-patologico, ovvero una sola
causa per anomalie di vari organi.
Stress: risultato non specifico di qualsiasi richiesta abnorme rivolta all’organismo.
Stimoli abnormi ed eterogenei portano ad una risposta unitaria, non specifica, della cellula in 3 fasi:
1. allerta
2. resistenza
3. esaurimento
Agenti stressanti ambientali
Condizioni patologiche
•
Processi flogistici
•
Shock termico
•
Febbre
•
Metalli pesanti
Neoplasie
•
Inibitori del metabolismo energetico •
•
Infezioni
•
Chemioterapici
•
Ischemia
•
Danno ossidativo
Tutti questi fattori portano alla denaturazione di proteine. Le HSP (Heat Shock Proteins) presenti si staccano dall’HSF (Heat
Shock Factor) monomerico, allo scopo di rinaturare le proteine. L’HSF può così trimerizzare e attivare nel nucleo la trascrizione delle
stress proteins. Il processo si verifica in pochi minuti: c’è un aumento misurabile delle HSP in mezz’ora. L’RNA delle HSP non
contiene introni. Le HSP sono altamente conservate (sono uguali nell’uomo e in Drosophila), quindi sono fondamentali per la
sopravvivenza.
Le HSP sono proteine chaperone che prevengono l’aggregazione delle proteine e ne ripristinano la giusta conformazione.
Esistono stress proteins costitutive e indotte. Quelle costitutive servono a garantire che le proteine siano prodotte correttamente.
Funzioni delle proteine chaperone:
impedire l’aggregazione e la precipitazione delle catene proteiche durante la traduzione e delle proteine con conformazione alterata;
aiutare le catene polipeptidiche ad assumere una conformazione corretta o a riacquistarla dopo un evento che ne ha alterato la
struttura;
hanno un ruolo nella degradazione delle proteine alterate strutturalmente.
Mutazioni delle proteine chaperone Æ chaperonopatie.
Fattori che possono disturbare il processo di ripiegamento di una proteina nascente:
1. mancata sintesi contemporanea di tutti i domini della proteina;
2. presenza di grande quantità di macromolecole;
3. esposizione di regioni che si ripiegano lentamente o idrofobiche.
Quando c’è una situazione di stress vengono sintetizzate le chaperonine inducibili. Tra le proteine da stress c’è l’ubiquitina (~
9 Da), che si lega alle proteine che devono essere degradate e le conduce al proteasoma dove vengono degradate.
Instabilità delle proteine nell’ambiente cellulare:
1. denaturazioni e modificazioni (ossidoriduzioni, glicosilazioni spontanee);
2. esposizione a tossici ambientali: radicali dell’O2, metalli pesanti, alcuni antibiotici;
3. mutazioni che causano l’assunzione di conformazioni anomale.
1
•
•
ADATTAMENTO
Capacità delle cellule di modulare alcune funzioni in caso di stress
Fisiologico
Patologico
Adattamento metabolico (es. digiuno, dopo rifampicina) Aumentata richiesta funzionale:
•
aumento del numero delle cellule Æ iperplasia
Adattamento strutturale
•
aumento delle dimensioni delle cellule Æ ipertrofia
•
entrambe
Diminuita richiesta funzionale:
•
riduzione del numero delle cellule Æ involuzione
•
riduzione delle dimensioni delle cellule Æ atrofia cellulare
•
entrambe
Quando lo stimolo termina si ha la restitutio ad integrum. Questi processi sono reversibili entro certi limiti.
Meccanismi alla base dell’ipertrofia:
•
stimoli meccanici;
•
stimoli nervosi;
•
fattori di crescita;
•
vengono indotti fattori trascrizionali e geni embrionali.
Meccanismi alla base dall’atrofia:
•
distruzione di proteine citosoliche: aumento dell’RNA codificante ubiquitina e proteasomi;
•
distruzione di costituenti cellulari Æ autofagia.
Autofagia:
•
nel reticolo endoplasmatico vengono prodotte vescicole dotate di una doppia membrana;
•
la vescicola avvolge l’organulo e si fonde con un lisosoma;
•
parte del materiale viene riutilizzata, parte rimane nel vacuolo e dà origine ai corpi residui, che si presentano elettrondensi (es.
lipofuscine, che rappresentano l’accumulo di fosfolipidi nei lisosomi);
•
è un processo ubiquitario, regolato nel corso dello sviluppo e in base alla nutrizione;
•
ruolo centrale della chinasi Tor: l’inattivazione di Tor ad opera della fosfatasi PP2A induce autofagia.
Autofagia e malattie:
•
eccessiva autofagia si associa a malattie neurodegenerative (es. Parkinson, cardiomiopatie);
•
deficit di autofagia si associa a tumori.
APOPTOSI
La morte cellulare si divide in 2 categorie:
apoptosi Æ morte cellulare programmata;
necrosi.
Esistono anche una serie di modalità intermedie.
Funzione dell’apoptosi:
•
nell’organismo viene mantenuta un’omeostasi cellulare;
•
il bilancio tra l’incremento (differenziamento da precursori e proliferazione) e il decremento (differenziamento e morte) della
popolazione cellulare è finemente regolato;
•
senza morte cellulare, una persona di 80 anni avrebbe 2 tonnellate di midollo osseo e linfonodi, e un intestino di 16 km.
Esempi di apoptosi:
•
morte cellulare durante embriogenesi (regressione vasi, ecc.);
•
sostituzione delle cellule dell’epitelio delle cripte intestinali;
•
regolazione della popolazione delle cellule B e T nel sangue; nelle malattie autoimmuni ci sono cellule del sangue che sono
iperreattive nei confronti di cellule self.
L’apoptosi è conservata nell’evoluzione: i geni che la regolano in C. elegans (studiati da Sydney Brenner), sono gli stessi che
la regolano nell’uomo.
Caratteristiche morfologiche della cellula apoptotica:
•
condensazione e frammentazione specifica della cromatina;
•
vacuolizzazione del citoplasma;
•
membrana intatta (non c’è rilascio di materiale all’esterno);
•
formazione di invaginazione e protrusioni della membrana;
•
formazione a partire dalle protrusioni di corpi apoptotici, che contengono organuli;
•
fagocitosi dei corpi apoptotici;
•
assenza di fenomeni infiammatori.
La cellula scompare senza indurre modificazioni nell’ambiente circostante. Se la cellula si rompesse, il contenuto si
spanderebbe nell’ambiente extracellulare e scatenerebbe fenomeni infiammatori.
2
Frammentazioni della cromatina:
•
il DNA genomico viene tagliato da endonucleasi specifiche in frammenti di lunghezza discreta;
•
questi frammenti sono lunghi 200 bp o multipli.
Il DNA è arrotolato sugli istoni. Tra un istone e l’altro il DNA è esposto e quindi può essere tagliato dall’endonucleasi. Il DNA
avvolto intorno ad un istone è lungo 200 bp. A volte l’endonucleasi manca un taglio e quindi ci sono filamenti di lunghezza multipla.
Nella cellula necrotica il DNA subisce una degradazione casuale.
Fagocitosi dei corpi apoptotici:
•
i macrofagi riconoscono i corpi apoptotici e li fagocitano;
•
il recettore della fosfatidilserina viene esposto sulla superficie esterna della membrana plasmatica dei corpi apoptotici;
•
molecole di superficie (TSP-1, CD36) o intracellulari (DOCK180) sono coinvolte nella fagocitosi.
Il passaggio dall’interno all’esterno della membrana del recettore della fosfatidilserina è catalizzato dalla flippasi.
VIE PER INDURRE L’APOPTOSI
Ligandi extracellulari
Æ
Via estrinseca
Attivazione di recettori specifici
Via intrinseca
Stress intra- ed extracellulari
Æ
(stress ossidativo, farmaci citotossici, radiazioni)
Æ
mitocondrio
caspasi
Æ
caspasi
Pathway recettore-mediato (death receptors)
Il ligando si lega al recettore: la porzione citosolica induce la reazione all’interno della cellula. Tra i recettori più importanti ci
sono il FAS, il CD95 e il TN-α. Ci sono anche dei recettori esca, che non hanno la componente intracellulare e non inducono nessuna
reazione.
L’attivazione della porzione citosolica delle proteine avviene legando il FADD, che adatta il recettore alla procaspasi. La
procaspasi contiene diverse caspasi. Legandosi al FADD viene tagliata e si formano le caspasi attive.
Le caspasi 8 e 10 sono attivatori di altre proteine della stessa famiglia, le caspasi 3, 6 e 7, che sono le caspasi effettrici. Le
caspasi effettrici attivano una serie di substrati coinvolti nell’apoptosi, come l’endonucleasi, substrati che fanno formare le bolle o
portare sulla superficie esterna della membrana il recettore della fosfatidilserina. Esiste anche una via di attivazione dalle caspasi 8 al
mitocondrio.
Pathway mitocondriale
L’attivazione avviene a livello mitocondriale: vengono alterate o regolate alcune proteine della famiglia delle BCL-2 (BAD, BAX,
BCL-2, BCL-XL). Alcune di queste promuovono l’apoptosi (BAD, BAX), altre la inibiscono (BCL-2, BCL-XL).
Apoptosoma: gruppo di proteine che contiene il citocromo c, proteina mitocondriale che si libera quando avviene l’apoptosi.
L’apoptosoma ha la stessa funzione delle caspasi attivatici: attiva le caspasi effettrici.
Il livello di ATP nella cellula è importante per il funzionamento dell’apoptosoma: se c’è ATP l’apoptosoma può funzionare, se
manca ATP la cellula muore per necrosi.
MEDIATORI DELL’APOPTOSI
Bcl-2
•
•
Famiglia di proteine caratterizzate da 4 BH (Bcl2 homology) domains (dominio: parte di una proteina dotata di una certa funzione).
Si dividono in:
•
antiapoptotici: Bcl2, Bcl-XL, Bcl-w, MCL1, DIVA, NR-13 (BH 1, 2, 4 domains);
•
proapoptotici: BAX, BAK, BOK (pochi BH domains) e Bad, Bik, Bid, Bim (solo BH3).
Caspasi
•
•
•
•
Proteasi sintetizzate come procaspasi e attivate da tagli proteolitici.
Agiscono su substrati specifici (altre caspasi e non), tagliando in corrispondenza dell’acido aspartico (Asp): cASPasi.
14 caspasi di mammifero conosciute.
Classificate in 3 gruppi:
caspasi (apoptotiche) iniziatrici: lungo prodominio (-2, -8, -9, -10);
caspasi (apoptotiche) effettrici o esecutive: corti prodomini (-3, -6, -7);
caspasi coinvolte nella maturazione delle citochine.
Substrati delle caspasi
•
•
•
•
ICAD (inibitore della DNAasi attivata dalle caspasi). CAD libero trasloca nel nucleo e frammenta il DNA genomico.
PARP (Poly (ADP-ribose) Polimerase): enzima che ripara il DNA Æ viene inattivato.
ROCK 1 (Rho-associated coiled-coil forming kinase I). Se tagliata, rimane attiva e fosforila permanentemente la MLC (miosine
light chian): avvengono modificazioni morfologiche della cellula.
Actina, lamina.
3
Inibitori
•
•
Proteine cellulari agiscono come inibitori dell’apoptosi a vari livelli:
•
FLIPs, simili alla procaspasi-8, ma prive di attività catalitica. Bloccano il death receptor;
•
Proteine Bcl-2: regolano la permeabilità della membrana mitocondriale.
Proteine virali e cellulari agiscono come inibitori dell’attività delle caspasi (IAPs):
•
NAIP (Neuronal Apoptosis Inhibitory Protein): protegge i neuroni da prematura apoptosi nel morbo di Parkinson;
•
Survivina;
•
P35 del Baculovirus: è un potente inibitore delle caspasi.
PATOLOGIE ASSOCIATE AD UN ECCESSO DI APOPTOSI
•
•
•
•
•
Malattie neurodegenerative:
Parkinson: neurotossine inducono stress ossidativo sul mitocondrio dei neuroni e produzione di Bax;
Alzheimer: APP e TAU possono indurre apoptosi.
Atrofie muscolari spinali: mutazione in Survival of Motor Neuron (SMN).
AIDS: HIV-1 induce l’apoptosi in CD4+ (helper) T cells / via mitocondriale.
Alcool
Stress ossidativo
PATOLOGIE ASSOCIATE AD UN DIFETTO DI APOPTOSI
Cancro:
•
da infezione virale;
•
da mutazione nel DNA genomico.
Malattie autoimmuni:
•
diabete tipo I (mediatori infiammatori da T cells, glucosio/IL-1): l’organismo non riesce a distruggere le cellule autoreattivi, le
quali distruggono le isole di Langerhans;
•
encefalomieliti: antigeni contro mielina. Neuroni e oligodendrociti muoiono per apoptosi.
APOPTOSI E CANCRO
Molti dei meccanismi di controllo della proliferazione cellulare fanno capo all’apoptosi: eccessiva proliferazione o crescita in luoghi non
appropriati induce apoptosi nelle cellule interessate.
Mutazioni che causano overespressione di oncogeni pro-proliferativi come cMyc, E1A o E2F1, sensibilizzano le cellule verso l’apoptosi
e non sono sufficienti per la formazione del tumore.
Cellule tumorali possono proliferare se eludono i controlli apoptotici.
La resistenza delle cellule tumorali all’apoptosi è una caratteristica essenziale dello sviluppo del tumore ed è causata da
overespressione di geni anti-apoptotici o inattivazione di geni pro-apoptotici.
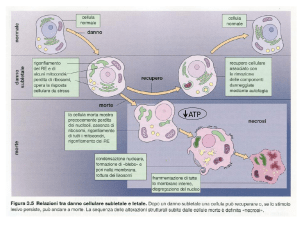
NECROSI
NECROSI:
•
morte cellulare patologica;
•
si instaura una condizione infiammatoria;
•
la necrosi, tuttavia, viene utilizzata anche in processi fisiologici.
NECROSI FISIOLOGICA:
rimpiazzamento degli enterociti intestinali: apoptosi e necrosi;
maturazione follicolare durante oogenesi: apoptosi e necrosi;
morte indotta da attivazione di linfociti T primari (regolazione negativa della risposta immunitaria).
Esperimenti di genetica indicano che la necrosi può sostituire l’apoptosi durante lo sviluppo embrionale.
Gli inneschi di apoptosi e necrosi usano molecole in comune.
LA CELLULA NECROTICA
•
Distribuzione dispersa della cromatina – degradazione del DNA.
•
Dilatazione degli organelli.
•
Degradazione delle membrane (membrane degli organelli e membrana plasmatica).
•
Materiale cellulare nel mezzo – no fagocitosi Æ INFIAMMAZIONE
Nell’apoptosi, il DNA viene tagliato in frammenti lunghi 200 bp o multipli; nella cellula necrotica, invece, avviene un taglio
casuale: le DNAasi tagliano a random.
STIMOLI PATOLOGICI CHE PORTANO ALLA NECROSI
Virus, batteri, protozoi.
Tossine batteriche.
Componenti del sistema immunitario: complemento, NK attivati, macrofagi peritoneali.
Deplezione di ossigeno, glucosio e altri fattori trofici (ischemia) – necrosi massiva (cellule endoteliali, neuroni, miocardiociti, cellule
renali, ecc.)
RECETTORI CELLULARI DEL PROGRAMMA DI NECROSI
4
Stimoli e recettori:
membri della famiglia dei recettori del TNF possono innescare la necrosi, oltre che l’apoptosi (es. linfociti T attivati muoiono per necrosi
via FAS e attraverso un meccanismo caspasi-indipendente);
excitotossine (NMDA, AMPA, Kainate) inducono necrosi in vari tipi di cellule via recettori del glutammato (NMDA receptor, ecc.);
ischemia, stress ossidativo e farmaci citotossici agiscono direttamente sui sistemi intracellulari.
Mediatori intracellulari:
•
ioni calcio;
•
kinasi indotte da stress (JNK, p38);
•
PARP: Poly(ADP-ribosio)polimerase.
Il recettore FAS può attivare sia la necrosi, sia l’apoptosi. La PARP nell’apoptosi viene degradata, mentre viene attivata nella necrosi.
Effettori mitocondriali:
proteine della famiglia Bcl-2: Bcl-XL, BNIP3. Geni anti-apoptotici favoriscono la necrosi bloccando Cyt C;
massiccia attivazione di PARP, che riduce drasticamente il livello di ATP;
ioni calcio inducono ROS (Reactive Oxygen Species)
Effettori finali:
•
Reactive Oxygen Species (ROS), come H2O2, vengono prodotte dal mitocondrio;
•
le ROS attivano proteasi, lipasi e nucleasi;
•
degradazione di proteine cellulari, membrane e DNA.
Proteasi:
calpaine;
catepsine;
ser-prot.
Le caspasi hanno substrati specifici. Le proteasi, invece, degradano le proteine che hanno una funzione all’interno della cellula.
CONSEGUENZE DELLA NECROSI: INFIAMMAZIONE
•
L’organismo necessita di uno stimolo infiammatorio per attivare il sistema immunitario e far fronte a condizioni patologiche come
infezioni, traumi, cellule trasformate, ecc.
•
La morte cellulare per necrosi crea questo stato infiammatorio rilasciando nel mezzo mediatori solubili (prima o dopo la distruzione
della cellula).
•
HSP70, calreticinina, oligonucleosomi e carboidrati sono noti per la loro capacità di attivare le Antigen Presenting Cells (APC).
•
Molti virus esprimono inibitori delle caspasi. La necrosi rappresenta una via alternativa per indurre la morte cellulare (significato
evolutivo).
APOPTOSI O NECROSI?
•
Il bilanciamento tra i fattori pro-apoptotici (anti-necrotici) e pro-necrotici (anti-apoptotici) determinano se una cellula morirà per
apoptosi o per necrosi.
•
Una stessa specie cellulare può passare da apoptosi a necrosi, se il suddetto bilanciamento cambia (es. glucatione in U937).
•
Specie cellulari diverse hanno sistemi regolativi diversi (es. NMDA su NMDAR porta a necrosi o ad apoptosi a seconda del tipo
cellulare, della presenza di inibitori o della diversa espressione dei recettori).
•
L’organismo “decide” se, in quella particolare condizione patologica o fisiologica, quel tipo di cellula deve morire per necrosi o per
apoptosi:
•
le condizioni patologiche prediligono la necrosi perché necessitano di una condizione infiammatoria per far fronte alla
patologia;
•
in condizioni fisiologiche prevale l’apoptosi.
PATOLOGIE ASSOCIATE A NECROSI
•
Infezioni
•
Morbo di Alzheimer.
•
Morbo di Creuzfeldt-Jakob.
•
Epilessia.
•
Malattie infiammatorie.
•
Ischemie.
MODULAZIONE DELLA MORTE CELLULARE COME TERAPIA
In molte patologie, la modulazione della necrosi e dell’apoptosi viene considerata come un possibile approccio terapeutico:
•
aumento della morte cellulare (tumori);
•
inibizione della morte cellulare (ischemia).
Ischemia:
•
inibitori delle caspasi sono già utilizzati in danni ischemici del miocardio e del cervello per ridurre il fenomeno apoptotico;
•
in aree ischemiche, il blocco della necrosi favorirebbe una riduzione della morte del tessuto;
•
SB203580 è un inibitore della stress kinase p38, utilizzato con successo in modelli di ischemia cerebrale;
•
UU126 (inibitore di MEK/ERK) protegge l’ippocampo da ischemia cerebrale;
•
3-aminobenzamide (inibitore di PARP) previene danni ischemici al miocardio e al cervello.
5
RADICALI LIBERI
I radicali liberi sono molecole o frammenti di molecole che hanno uno o più elettroni spaiati negli orbitali esterni. Sottraggono
elettroni ad altre molecole per completare il loro ottetto.
Molecole bersaglio:
•
acidi nucleici;
•
proteine;
•
lipidi delle membrane biologiche.
Danno origine a stress ossidativo.
Fattori che influenzano l’iperproduzione di radicali liberi:
•
diete sbilanciate, prive di verdure;
•
fumo;
•
alcool;
•
inquinamento;
•
raggi solari;
•
esercizio fisico intenso.
In condizioni fisiologiche, il 3% dell’O2 a livello mitocondriale non entra in catena respiratoria, ma forma radicali liberi, che
vengono neutralizzati dalle difese antiradicaliche. Alla formazione dei radicali liberi concorrono diverse molecole, tra cui il citocromo b
2+
ed enzimi citoplasmatici, come la xantina ossidasi. Anche la presenza di Fe e Cu catalizza la formazione di radicali liberi.
I radicali liberi più frequenti sono:
•
O2•: si forma a livello della catena respiratoria, oppure per azione degli enzimi ossidasi.
•
H2O2: prodotto dalla superossidodismutasi a partire da O2• e trasformato in acqua dalla catalasi.
•
OH-: il glucatione difende da questo radicale libero passando alternativamente dalla forma ossidata a quella ridotta.
•
NO.
Le vitamine C, E ed A hanno azione antiradicalica.
Se i meccanismi di protezione sono insufficienti, la cellula viene danneggiata: si può arrivare ad un danno tale da uccidere la
cellula. Le proteine con gruppi sulfidrilici (SH) sono molto sensibili ai radicali liberi, che formano un cross-legame tra i gruppi SH.
I doppi legami dei lipidi polinsaturi sono suscettibili all’azione dei radicali liberi, che formano perossidi (radicali liberi di
natura lipidica) innescando una reazione autocatalitica a catena.
I RL interagiscono con le timine del DNA genomico e mitocondriale, provocando una rottura dell’elica. Se gli enzimi riparatori
non funzionano si possono avere mutazioni. I RL possono inoltre convertire la deossiguanosina in idrodeossiguanosina.
DANNO DA RIPERFUSIONE
Se una coronaria è occlusa da un trombo, il tessuto miocardico a valle di essa diventa ischemico. Quando si rivascolarizza, il
danno diventa più esteso di quello provocato dall’ischemia. Questo fenomeno è detto danno da riperfusione.
Ischemia
ATP Æ ADP Æ AMP Æ Adenosina Æ Inosina Æ Ipoxantina
Attivazione di
proteasi citosoliche
Æ
Conversione proteolitca
della xantina deidrogenasi in
Se non c’è O2, la xantina ossidasi è inattiva.
Riperfusione
O2 Æ ipoxantina + xantina ossidasi + O2
↓
urato + H2O2 + O2• (radicale superossido)
↓
Fe3+ (catalizzatore)
↓
O2 + OH- + •OH (radicale ossidrile)
↓
Danno
↓
PMN (polimorfonucleati)
↓
Radicali liberi↓
↓
Danno
6
Æ
xantina
ossidasi
I radicali liberi sono uno dei meccanismi antimicrobici più efficienti. I globuli bianchi usano mieloperossidanti per uccidere i
microbi invasori.
O2
↓
O2 • -
NADPH ossidasi
superossido
↓
H2O2
-
-
-
-
Cl , Br , I , Scn
↓
HOX
•
•
•
perossidasi
Acido ipoanico
Terapia O2 Æ eccesso di O2
PMN, macrofagi Æ infiammazione
PMN, xantina ossidasi Æ danno da riperfusione
MECCANISMO DI SMALTIMENTO DEI RADICALI LIBERI
ENDOGENI
Sistemi enzimatici:
•
superossidodismutasi;
•
catalasi;
•
glucatione perossidasi;
•
denaturasi;
•
acido lipoico.
ESOGENI
Dieta:
•
antiossidanti;
•
grassi polinsaturi.
Molecole chelanti i metalli:
•
albumina;
•
ferritina;
•
transferrina;
•
celluloplasmina.
INVECCHIAMENTO
Progressiva perdita di efficienza dei meccanismi di conservazione dell’omeostasi biologica.
Teorie evoluzionistiche
•
•
•
Pleiotropia antagonistica: geni vantaggiosi in giovane età sono deleteri in età avanzata.
Disposable soma: le cellule somatiche sono mantenute solo per assicurare la riproduzione della specie.
Accumulo di mutazioni: non c’è selezione contro quelle mutazioni che compromettono la capacità di adattamento e di
sopravvivenza nella vecchiaia.
Teorie sistemiche
•
•
•
Immulogica.
Neuroendocrina.
Rate of living: il potenziale metabolico di ogni organismo è prefissato.
Teorie cellulari
•
•
Radicali liberi: il metabolismo ossidativo produce RL che danneggiano proteine e DNA (anche mt).
Wear & tear: accumulo di danni per alterazioni posttraduzionali (glicossidazione e infiammazione).
Teorie molecolari
•
•
•
Restrizione dei codoni: l’accuratezza della traduzione degli mRNA è compromessa.
Catastrofe molecolare.
Genetica.
7
Principali teorie dell’invecchiamento
•
Errori macromolecolari (crosslinking/glicossidazione)
•
Radicali liberi
•
Danno mitocondriale
•
Teoria genomica
ERRORI MACROMOLECOLARI
Crosslinking
•
•
Calore + glucosio: crosslinking di aa.
Diabete + A.G.E. Æ invecchiamento precoce.
Glucosio
•
•
+
Gruppo
amminico proteico
Æ
Base di Schiff
Æ
Prodotto di Amadori
Æ
Prodotto con
legame trasversale
Le molecole di collagene crosslinkano in modo irreversibile Æ i vasi e il cristallino diventano più rigidi.
Il proteasoma non è più in grado di eliminare le proteine anomale.
RADICALI LIBERI
•
•
•
I tessuti postmitotici (cervello, muscolo, miocardio) possono essere i bersagli critici del ROS nell’invecchiamento.
Un drammatico aumento della mortalità correlata all’età coinvolge malattie del cuore e del sistema nervoso.
La restrizione calorica riduce la produzione di radicali liberi nei mitocondri.
DANNO MITOCONDRIALE
•
•
•
•
La riparazione del DNA mt non è efficiente.
Il DNA mitocondriale codifica per 13 subunità proteiche, 22 tRNA e 2 rRNA.
La delezione di ogni singolo tRNA mt compromette la sintesi proteica mitocondriale.
Un deficit di proteine mitocondriali aumenta la generazione di radicali liberi.
1.
2.
3.
Declino nel trasposto di elettroni Æ riduzione della sintesi di ATP.
Aumentata sintesi di radicali liberi.
Induzione di morte cellulare per rilascio del citocromo c, che entra nel citoplasma provocando l’apoptosi.
SENESCENZA REPLICATIVA – TEORIA GENOMICA
Una cellula normale si può replicare al massimo 30-40 volte, poi raggiunge la senescenza replicativa: la cellula non muore,
ma non risponde più a stimoli proliferativi. Gli organismi vivono più a lungo se le loro cellule sono capaci di più divisioni cellulari.
Significato della senescenza replicativa:
•
le cellule neoplastiche richiedono più 4-6 mutazioni per assumere un fenotipo maligno;
•
contenere la proliferazione (vale a dire un numero minore di duplicazioni del DNA) riduce l’incidenza di mutazioni.
SINDROMI DA INVECCHIAMENTO PRECOCE
•
•
•
•
•
Sindrome di Verner (AR): wrn codifica una DNA elicasi Æ aumento delle mutazioni (soprattutto delezioni).
Atassia telangectasica (AR): atm, una protein chinasi della famiglia delle PI-3 chinasi, media la risposta a rotture nella doppia
elica.
Progeria di Hutchinson-Gilford (AD): difetto molecolare non identificato.
Sindrome di Down.
Sindrome di Cockayne.
Gli animali modificati geneticamente per essere più piccoli della norma vivono più a lungo.
•
•
In C. elegans, la mutazione di age-1 aumenta l’attività della SOD e della catalasi Æ aumentata resistenza allo stress ossidativo.
In C. elegans, la mutazione di clk-1, coinvolto nella sintesi del coenzima Q (componente della catena di trasporto di elettroni
mitocondriale) rallenta lo sviluppo e ritarda l’invecchiamento, probabilmente come risultato di un rallentato metabolismo.
Questi link genetici tra invecchiamento e danno ossidativo sono validi in animali in cui la maggior parte delle cellule è in fase
postmitotica Æ un topo k/o per SOD e glucatione perossidasi non ha invecchiamento precoce.
Topi k/o per p66shc: durata della vita aumentata del 30%. p66shc è una molecola che traduce il segnale dopo stress ossidativo.
Il fattore trascrizionale DAF-16 controlla l’espressione di vari geni, alcuni dei quali implicati nella longevità. Tra questi ci sono
geni anti-aging, tra cui gli enzimi antiossidanti HSP, e geni pro-aging. Questi ultimi hanno a che fare con la riproduzione.
TELOMERI
I telomeri costituiscono l’estremità dei cromosomi e sono formati da sequenze ripetute. Hanno la funzione di proteggere i
cromosomi dai danni ed evitare che si chiudano su se stessi o crosslinkino.
8
Ogni volta che una cellula si divide, perde circa 350 basi di telomeri. Nelle cellule staminali è presente la telomerasi, che
mantiene invariata la lunghezza dei telomeri, rigenerandoli dopo ogni replicazione. La telomerasi si trova nelle cellule embrionali,
staminali e nel 90% dei tumori maligni dell’uomo.
•
•
Cellula senza telomerasi Æ
Cellula con telomerasi Æ
senescenza replicativa.
NO senescenza replicativa.
Nella progeria, i telomeri alla nascita sono molto corti.
EPIGENETICA E INVECCHIAMENTO
Le variazioni epigenetiche sono variazioni dell’espressione genica non implicanti mutazioni del DNA.
Nelle cellule vecchie è stata osservata un’ipometilazione generalizzata accompagnata da ipermetilazione selettiva,
soprattutto a carico dei geni controllori del mantenimento dell’integrità del genoma (caretaker), come p53.
Nell’invecchiamento sono importanti tanto i fattori genetici, tanto i fattori ambientali.
Ci sono cellule che non crescono perché sono quiescenti: esiste quindi la possibilità di confrontare la quiescenza con la
senescenza. La quiescenza è reversibile.
La quiescenza è determinata dall’inibizione da contatto. L’eterocromatina è presente solo nelle cellule vecchie, ma non nelle
cellule quiescenti.
EZIOLOGIA DELLE MALATTIE
Eziologia delle malattie:
•
cause intrinseche;
•
cause estrinseche.
Nella risposta a stimoli estrinseci esiste comunque una variabilità individuale.
•
•
Malattia congenita: presente alla nascita.
Malattia ereditaria: dovuta al patrimonio genetico, ma non necessariamente presente alla nascita (es. Corea di Huntington).
Malattie dello sviluppo:
•
errori di morfogenesi (malformazioni);
•
anomalie cromosomiche;
•
single gene disease;
•
malattie poligeniche.
Malformazione: errore di morfogenesi. Le malformazioni possono essere dovute a:
•
cause sconosciute;
•
agenti chimici (talidomide Æ blocco della formazione vascolare Æ focomelia);
•
infezioni (rosolia nel primo trimestre);
•
radiazioni ionizzanti.
Nel corso dello sviluppo fetale ci sono momenti di particolare sensibilità:
•
aborto: entro i primi 15 giorni;
•
SNC: 2a-5a settimana;
•
cuore: 2a-6a settimana;
•
estremità: 3a-7a settimana;
•
occhi: 3a-7a settimana.
Il periodo più a rischio è il primo trimestre.
Deformazione: risultato di un fattore meccanico (es. mancanza di liquido amniotico).
La sequenza del DNA può cambiare, ma ciò non è sempre negativo perché questo meccanismo è alla base dell’evoluzione. I
processi evolutivi sono caratterizzati dalla selezione di mutazioni casuali della sequenza degli acidi nucleici, che conferiscono alle
specie nuove caratteristiche favorevoli alle condizioni ambientali.
Mutazione: alterazione permanente del DNA:
•
mutazioni geniche in regioni codificanti o non codificanti;
•
disordini citogenetici.
Molte mutazioni possono essere silenti.
MUTAZIONI PUNTIFORMI
•
•
•
Missenso: sostituzione di un amminoacido con un altro.
Non senso: sostituzione di un amminoacido con un segnale di stop. Si ottiene una proteina più corta non funzionante o instabile.
Frame shift: sfasamento del corpo di lettura dovuto ad un’inserzione o a una delezione che porta all’inserimento di amminoacidi
che non sono quelli della proteina nativa.
9
Effetti:
1. perdita di funzione completa (null);
2. mutazioni costitutivamente attive (es. RAS: serve a tradurre il segnale dalla membrana al nucleo; nei tumori il RAS è sempre attivo,
quindi la cellula riceve il segnale di crescita anche in assenza di un fattore di crescita);
3. acquisizione di nuove funzioni (gain of function).
DISORDINI CITOGENETICI
Anomalie numeriche
Euploidia (multipli di N):
•
mancata divisione meiotica Æ gamete diploide;
•
fecondazione di una cellula uovo con 2 spermatozoi.
Cardiociti ed epatociti possono essere fisiologicamente poliploidi.
Aneuploidia (non multipli di N):
•
non disgiunzione meiotica (molti casi di sindrome di Down);
•
anafase ritardata (alcuni casi di sindrome di Turner).
Anomalie strutturali
•
•
•
•
Delezioni
Traslocazioni
Amplificazioni
Inversioni:
•
paracentriche (dalla stessa parte del centromero);
•
pericentriche (da parti opposte del centromero).
Delezioni terminali o interstiziali
•
•
•
•
Sindrome cri du chat: delezione del segmento distale di 5p.
Sindrome di Wolf-Hirschorn (delezione di 4p).
Rb (retinoblastoma) e Wilms (tumore).
Sindrome di Di George/sindrome velo-cardio-facciale: delezione 22q11 Æ difetto di sviluppo delle strutture del III e IV arco
branchiale:
•
dismorfie facciali;
•
alterazioni del palato;
•
cardiopatie;
•
aplasia timica;
•
ipoplasia delle parotidi.
Traslocazioni
•
Leucemia mieloide cronica (un frammento del cromosoma 22 migra sul cromosoma 9 dando origine ad un cromosoma
chimerico detto cromosoma Philadelphia): abl è un oncogene che viene attivato con la traslocazione Æ proliferazione dei blasti
mieloide.
Inversione
•
Emofilia: mancato funzionamento del fattore VIII della coagulazione.
Amplificazione
•
Neuroblastoma (amplificazione di regioni del cromosoma 2): vengono prodotte grandi quantità del gene Mic, che stimola la
proliferazione cellulare.
TRISOMIA 21 – SINDROME DI DOWN
•
•
•
95%: mancata disgiunzione meiotica;
4%: traslocazione robertsoniana di q21 su un cromosoma acrocentrico (22 o 14);
1%: mosaicismo.
L’incidenza della sindrome di Down aumenta con l’età della madre (1/50 sopra i 45 anni), perché le cellule uovo non sono
soggette a ricambio, quindi con il tempo aumentano le probabilità che subiscano mutazioni.
Normalmente gli spermatozoi alterati non arrivano a fecondare la cellula uovo, quindi la causa è quasi sempre materna.
Gli affetti da sindrome di Down sono soggetti ad Alzheimer precoce e sono esenti da aterosclerosi.
10