Rivista Italiana di Genetica e Immunologia Pediatrica - Italian Journal of Genetic and Pediatric Immunology
Anno III numero 1 - gennaio 2010 | direttore scientifico: Carmelo Salpietro - direttore responsabile: Giuseppe Micali
Home page
Norme editoriali | Stampa l'articolo
Motore di ricerca
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
Numeri precedenti
◀ Indietro pagina 4 Avanti ►
Genetica, clinica e diagnostica strumentale degli ipopituitarismi congeniti
Congenital hypopituitarisms: genetics, clinical aspects and instrumental diagnosis
1
2
3
E. David , A. Albani , A. Laganà
1
2
3
UOC di Radiodiagnostica Università degli Studi di Messina, UOC di Endocrinologia Università degli studi di Messina, UOC di Ginecologia ed Ostetricia Università
degli Studi di Messina
Abstract
Hypopituitarism is an endocrine syndrome characterized by reduced or absent
secretion of one or more adenohypophysial hormones, resulting in
dysfunction of the glands corresponding devices. The deficit of all hormones
constitute the framework of pan-hypopituitarism, deficiency of two or more
hormones is called partial hypopituitarism, while deficiency of single hormone
is called selective hypopituitarism. The pan-hypopituitarism is a rare
condition, but the partial hypopituitarism, especially iatrogenic, is more
frequent. There are various forms of congenital and acquired, organic and
functional.
The congenital hypopituitarism is caused by mutations of several genes that
encode for transcription factors. The phenotype varies depending on the
transcription factor involved: prop1 (deficit somatolattotropo hormone,
thyrotropic, gonadotropic and corticotropic times) (Fig. 1), POU1F1
(somatolattotropo and thyroid stimulating hormone deficiency, pituitary
hypoplasia) (Fig. 2), HESX1 (pituitary deficiencies variables, septo-optic
dysplasia) (Fig. 3), and less frequently LHX3 (somatolattotropo and
gonadotropic hormone deficiency, limitations in the rotation of the head and
neck) (Fig. 4) and LHX4 (deficit variables pituitary, ectopic the
neurohypophysis, abnormalities of the brain).
The clinical picture depends on the hormone deficient, the degree of
insufficiency of the gland and the age of onset. Prevalent in childhoodcongenital idiopathic forms, which occur mainly with delayed growth and
puberty and absence dell'adrenarca; TSH deficiency induces different clinical
pictures according to the age in which it occurs. Predominate form acquired in
adulthood, including macroedema pituitary, which gradually destroys the
pituitary GH secretion order compromised, then the gonadotropins, TSH and
ACTH then.
Riassunto
L’ipopituitarismo è una sindrome endocrina caratterizzata dalla ridotta o
assente secrezione di uno o più ormoni adenoipofisari, con conseguente
disfunzione delle ghiandole periferiche corrispondenti. Il deficit di tutti gli
ormoni configura il quadro di pan-ipopituitarismo, la carenza di due o più
ormoni è detta ipopituitarismo parziale, mentre il deficit di un solo ormone
viene definito ipopituitarismo selettivo. Il pan-ipopituitarismo è una condizione
rara, invece l’ipopituitarismo parziale, soprattutto quello iatrogeno, è più
frequente. Esistono varie forme congenite ed acquisite, organiche e funzionali.
L'ipopituitarismo congenito è causato da mutazioni di diversi geni che
codificano per i fattori di trascrizione. Il fenotipo varia a seconda del fattore di
trascrizione coinvolto: PROP1 (deficit degli ormoni somatolattotropo,
tireotropo, gonadotropo e a volte corticotropo) (fig. 1), POU1F1 (carenza degli
ormoni somatolattotropo e tireotropo, ipoplasia ipofisaria) (fig. 2), HESX1
(carenze ipofisarie variabili, displasia setto-ottica) (fig. 3) , e meno
frequentemente LHX3 (deficit degli ormoni somatolattotropo e gonadotropo,
limitazioni nella rotazione del capo e del collo) (fig. 4) e LHX4 (deficit ipofisari
variabili, ectopia della neuroipofisi, anomalie dell'encefalo). Il quadro clinico
dipende dall’ormone carente, dal grado di insufficienza della ghiandola e
dall’età di insorgenza. Nell’età evolutiva prevalgono le forme idiopatichecongenite, che si manifestano essenzialmente con ritardo della crescita e della
pubertà ed assenza dell’adrenarca; il deficit di TSH induce quadri clinici
diversi in base all’età in cui si presenta. Nell’età adulta prevalgono le forma
acquisite, tra cui il macroedema ipofisario, che distrugge gradualmente
l’ipofisi compromettendo nell’ordine la secrezione di GH, poi quella delle
gonadotropine, TSH ed infine ACTH.
Keywords: congenital hypopituitarism, etiology, diagnosis, treatment
Introduzione
L’ipopituitarismo è una sindrome endocrina caratterizzata dalla ridotta o assente
secrezione di uno o più ormoni adenoipofisari, con conseguente disfunzione delle
ghiandole periferiche corrispondenti. Il deficit di tutti gli ormoni configura il quadro di
pan-ipopituitarismo, la carenza di due o più ormoni è detta ipopituitarismo parziale,
mentre il deficit di un solo ormone viene definito ipopituitarismo selettivo. Il
pan-ipopituitarismo è una condizione rara, invece l’ipopituitarismo parziale,
soprattutto quello iatrogeno, è più frequente. Esistono varie forme congenite ed
acquisite, organiche e funzionali [1].
Epidemiologia
Uno studio italiano ha rilevato la prevalenza di deficit di GH di circa 9 casi su 1000
individui di una popolazione pediatrica [2]. I dati di screening del programma
regionale USA nel NordEst stimano la frequenza di carenza congenita di TSH di 1
caso ogni 29.000 nati vivi [3].
Mortalità/morbilità
I decessi a causa di carenze ormonali, di solito sono causati da insufficienza
surrenalica secondaria a deficit di ACTH. L'aumento della mortalità è dovuto ad
anomalie cardiovascolari, che sono correlate a deficit di GH: bambini ed adolescenti
con tale deficit, infatti, hanno dimostrato di avere alterazioni della funzionalità
cardiovascolare di grado importante [4, 5, 6]. Negli adulti con deficit di GH, il
trattamento con rhGH migliora i fattori di rischio cardiovascolare, ma non sono stati
segnalati studi a lungo termine che dimostrino la riduzione di incidenza di malattie
cardiovascolari in tali soggetti. [7-8].
Anatomia e Fisiologia
L’ipofisi è una ghiandola situata alla base del cranio, in un incavo dell’osso sfenoide
denominato sella turcica. E’ costituita per due terzi da una parte anteriore o
adenoipofisi, e per un terzo da una parte posteriore, o neuroipofisi.
I due lobi, anteriore e posteriore, sono distinti anatomicamente e funzionalmente ed
hanno una diversa origine embriologica.
La neuroipofisi origina dall'infundibulo (parte del diencefalo) ed è essenzialmente
costituita dai terminali di assoni originanti dalla porzione magnicellulare dei nuclei
SO e PV. Essa funge da deposito per la successiva liberazione di due nonapeptidi:
l’ormone antidiuretico (ADH) e l’ossitocina (OT).
L’ adenoipofisi deriva dalla tasca di Ratkhke, una estroflessione dell'ectoderma
stomodeale e, insieme all’ipotalamo, coordina le complesse funzioni regolatrici di
numerose altre ghiandole endocrine. [9]. La maggior parte dei casi di ipopituitarismo
dell’infanzia, in particolare quelli che si sviluppano dopo la nascita, sono il risultato di
patologie agenti principalmente sull'ipotalamo e sul peduncolo, e si traducono in un
difetto nella segnalazione neuroendocrina ipotalamo-ipofisaria. L’ipofisi in tal caso è
generalmente completamente funzionale, ma non è in grado di produrre livelli
normali di ormoni stimolanti, in quanto non riceve la stimolazione adeguata da parte
dei fattori di rilascio ipotalamici [10]. I bambini con ipopituitarismo congenito hanno
più probabilità di patologie primarie ipofisarie rispetto ai bambini con ipopituitarismo
acquisito, in cui le patologie dell’ipotalamo e del peduncolo sono molto più comuni.
Gli ormoni rilasciati dall’ipofisi anteriore sono:
• Ormone adrenocorticotropo (ACTH) : stimola le ghiandole surrenali a liberare il
cortisolo che aiuta a mantenere i valori normali della pressione arteriosa e della
glicemia;
• Ormone antidiuretico (ADH) : controlla la perdita di acqua da parte dei reni
• Ormone follicolo stimolante (FSH) : Controlla la funzione sessuale e la fertilità nei
maschi e nelle femmine
• L'ormone della crescita (GH) : stimola la crescita dei tessuti e delle ossa
• Ormone luteinizzante (LH) : controlla la funzione sessuale e la fertilità nei maschi e
nelle femmine
Gli ormoni rilasciati dall’ipofisi posteriore sono:
• L'ossitocina: stimola la contrazione dell'utero durante il travaglio ed la produzione
di latte
• Prolattina: stimola lo sviluppo della ghiandola mammaria e la produzione di latte
• Ormone stimolante la tiroide (TSH) : stimola la tiroide a rilasciare gli ormoni tiroidei
e ne mantiene il trofismo.
Eziopatogenesi
Un elevato numero di patologie infiammatorie, granulomatose o neoplastiche, così
come traumatismi o insulti da radiazioni interessanti la regione ipotalamo-ipofisaria
possono esserne all’origine. Inoltre anche alcune anomalie genetiche possono
determinare deficit isolati o multipli di ormoni ipofisari.
Cause organiche:
Ipopituitaristno acquisito
- Neoplastiche: adenoma ipofisano, craniofaringioma, glioma, ependimoma,
meningioma, germinoma, gangliocitoma, amartoma, pinealoma, metastasi (es. da
carcinoma della mammella, polmone e colon), Linfomi e leucemie
- Lesioni iatrogene: chirurgia sellare e parasellare, radioterapia nella regione del
collo o della testa, chemioterapia, interruzione della terapia con glucocorticoidi
- cause vascolari: apoplessia ipoflsarla, sìndrome di Sheehan, arteriti, aneurisma
della carotide interna
- infezioni: meningite, encefalite, tubercolosi, sifilide terziaria, micosi, toxoplasmosi
- Patologie infiltrative/infiammatorie: sarcoidosi, istiocitosi X, emocromatosi,
amiloidosi, ipofisite linfocitaria, Ipofisite granulomatosa
- Traumi cranici
- Idiopatico (processo autoimmune)
Ipopituitarismo congenito
- Displasia ipofisariaa (aplasia, ipoplasia, ectopia)
- Sindromi della linea mediana (es. displasia setto-ottica)
- Deficit ormonali su base genetica: Mutazioni di fattori di trascrizione ipofisari
(Prop-1, Pil-1, Hexsl), sindrome di Kallmann, deficit isolato di GH, sindrome di
Laurence-Moon-Biedl, Sindrome di Prader-Willi
- Sella vuota primaria
- Tumore intracranico congenito
- Encefalocele
- Traumi da parto (emorragia intracranica, asfissia)
- Idiopatico
Cause funzionali
- Psichiche: anoressia nervosa, depressione, stress, nanismo psico-sociale
- Nutrizionali/metaboliche: malnutrizione, malassorbimento, calo ponderale,
magrezza, esercizio fisico intenso
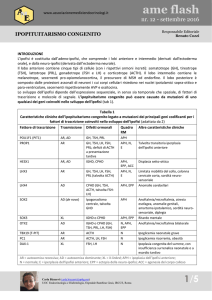
Genetica
L'ipopituitarismo congenito è causato da mutazioni di diversi geni che codificano per
i fattori di trascrizione. Il fenotipo varia a seconda del fattore di trascrizione coinvolto:
PROP1 (deficit degli ormoni somatolattotropo, tireotropo, gonadotropo e a volte
corticotropo) (fig. 1), POU1F1 (carenza degli ormoni somatolattotropo e tireotropo,
ipoplasia ipofisaria) (fig. 2), HESX1 (carenze ipofisarie variabili, displasia settoottica) (fig. 3) , e meno frequentemente LHX3 (deficit degli ormoni somatolattotropo
e gonadotropo, limitazioni nella rotazione del capo e del collo) (fig. 4) e LHX4 (deficit
ipofisari variabili, ectopia della neuroipofisi, anomalie dell'encefalo) (fig. 5). E'
fondamentale il monitoraggio a lungo termine poiché i pazienti sviluppano nuove
carenze (per esempio un esordio tardivo di deficit dell'ormone corticotropo in pazienti
con mutazioni di PROP1). I modelli di trasmissione variano con i fattori e la
mutazione coinvolti (trasmissione recessiva per PROP1 e LHX3, dominante per
LHX4, autosomica recessiva per PUO1F1 e HESX1). Il trattamento è equivalente a
quello dei pazienti senza deficit ipofisari, se intrapreso immediatamente dopo la
conferma della diagnosi e seguito da un follow-up specifico [11].
Clinica
Il quadro clinico dipende dall’ormone carente, dal grado di insufficienza e dall’età di
insorgenza. Nell’età evolutiva prevalgono le forme idiopatiche-congenite, che si
manifestano essenzialmente con ritardo della crescita e della pubertà ed assenza
dell’adrenarca; il deficit di TSH induce quadri clinici diversi in base all’età in cui si
presenta. Nell’età adulta prevalgono le forma acquisite, tra cui il macroedema
ipofisario, che distrugge gradualmente l’ipofisi compromettendo nell’ordine la
secrezione di GH, poi quella delle gonadotropine, TSH ed infine ACTH [1].
Nell’adulto i primi sintomi evidenti sono quelli da carenza di gonadotropine, mentre il
deficit di ACTH si manifesta solo nei casi gravi ed è spesso parziale. [1].
Ipopituitarismo congenito
Ipopituitarismo deve essere sospettato in bambini con difetti della linea mediana o
atrofia ottica (indicativi di una displasia setto-ottica) [12-13] e nei ragazzi con
micropene (indicativi di una carenza di gonadotropina) [14-15]. Un'altra
presentazione è la disidratazione ipernatremica a causa di diabete insipido;
l’associazione di diabete insipido e deficit di cortisolo può però compensare il
bilancio idrico, e mascherare quindi l’iniziale condizione [16]. I sintomi
dell'ipotiroidismo includono affaticamento, secchezza della cute, e stipsi a causa
della carenza dell’ormone stimolante la tiroide (TSH), e la concomitante carenza di
ACTH e cortisolo causa nausea, vomito e malessere.
Ipopituitarismo acquisito
Il quadro clinico è simile a quello dei bambini affetti da ipopituitarismo congenito.
Bisogna valutare la funzione dell'ipofisi prima e dopo il trattamento in bambini con
craniofaringiomi o altri tumori dell'ipofisi o dell'ipotalamo. Lo stesso vale per i
bambini che hanno ricevuto irradiazione cranica (ad esempio, prima del trapianto di
midollo osseo o per i tumori del cranio). I bambini senza un insulto ipotalamico noto
o ipofisario, con ipopituitarismo, si presentano spesso con disturbi della crescita a
causa del deficit di GH. Gli adolescenti possono presentare pubertà assente o
parziale. Le ragazze possono presentare amenorrea primaria o secondaria. Diabete
insipido, poliuria e polidipsia, possono essere i sintomi di presentazione. Raramente,
i pazienti con deficit di ACTH possono presentarsi con iponatriemia [16].
Laboratorio
La diagnosi di Ipopituitarismo si basa sulla determinazione dei livelli sierici delle
tropine ipofisarie che sono inappropiatamente bassi-normali o comunque non
elevati, e degli ormoni delle ghiandole bersaglio, che sono ridotti.
In alcuni casi tuttavia, per una corretta diagnosi, è necessario far seguire ai test
basali, dei test dinamici della funzionalità ipofisaria. Il deficit di GH ad esempio, non
può essere diagnosticato con i soli test statici, in quanto i livelli basali di GH sono
spesso indosabili anche in soggetti normali e, mentre un valore ampiamente normale
di IGF 1 esclude la diagnosi di GHD, un valore basso non è sufficiente .
Analogamente, per la diagnosi di iposurrenalismo secondario, al dosaggio dell ‘
ACTH basale e della cortisolemia del mattino, farà seguito il test di stimolo con
ACTH oppure il test all’ipoglicemia insulinica (ITT).
Per identificare la causa di Ipopituitarismo è inoltre necessaria la valutazione
morfologica della regione ipotalamo-ipofisaria tramite RMN con gadolinio; in
presenza di lesioni espansive il quadro diagnostico è infine completato dalla visita
oculistica (fundus oculi, campimetria, acuità visiva) [1].
Diagnostica per Immagini
La Risonanza Magnetica con mezzo di contrasto (gadolinio), la quale evidenzia
eventuali anomalie quali sella vuota, agenesia, effetto massa, emorragia,
rappresenta in assoluto la tecnica più accurata per uno studio completo della
ghiandola pituitaria.Risulta chiaro come, per poter riconoscere queste anomalie, tra
le più svariate, sia d’obbligo una conoscenza precisa del normale, nel contesto della
ghiandola pituitaria.Subordinata a questa, quando la RM non è disponibile o
effettuabile, è invece la TC. Reperti più tipici sono in ogni caso:ghiandola piccola per
età, riempimento della sella con liquido cerebrospinale (sella vuota), ectopia.
In RMN, la ghiandola pituitaria anteriore, nelle immagini T1 pesate, appare scura e
di uguale intensità della materia grigia, mentre la ghiandola pituitaria posteriore
appare bianca e si fa riferimento, radiologicamente, come il "punto luminoso"
posteriore. Questa differenza può riflettere uno strato di grasso dentro/intorno alla
ghiandola pituitaria posteriore. In alcuni casi di ipopituitarismo congenito, questa
"macchia" è completamente assente e correla bene con la presenza clinica di
diabete insipido (DI). Negli altri casi di ipopituitarismo congenito, la "macchia" si
trova in una posizione anatomica ectopica nella via del normale sviluppo embrionale
dell'ipofisi posteriore, oppure è assente. Tali reperti radiologici sono a volte utili per
accertare la natura congenita di ipopituitarismo. Nelle forme acquisite di
ipopituitarismo, in cui DI sviluppa spontaneamente o dopo l'intervento, la "macchia"
è in genere assente [17].
Vi sono infine indagini di secondo livello, il cui utilizzo può essere giustificato
relativamente al singolo caso e comunque complementarmente alla RM o alla TC:
Rx Cranio, che mostra eventuali grossolane alterazioni della sella turcica.
Angiografia, per eventuali alterazioni su base vascolare, di cui l’aplasia della
carotide interna, come dimostrano in merito numerose pubblicazioni scientifiche, è il
maggiore esempio, sebbene anche in questo caso la RMN assuma un ruolo di
assoluto rilievo.
Rx polso e ginocchio, che evidenziano il ritardo o l’assenza di nuclei epifisiari
permettendo di determinare l’età ossea comparativamente a quella cronologica.[18]
Terapia
La terapia è essenzialmente una terapia medica sostitutiva. Tuttavia quando
l'ipopituitarismo è dovuto a un tumore ipofisario il trattamento specifico deve essere
rivolto non solo al deficit ormonale ma anche al tumore. Con la sola eccezione del
prolattinoma la chirurgia costituisce oggi la prima scelta di trattamento per la
maggior parte degli adenomi. Se il tumore è piccolo i più preferiscono la rimozione
trans-sfenoidale della neoplasia. Può essere utilizzata anche l'irradiazione ad alto
voltaggio dell'ipofisi. In presenza di tumori più grandi e di estensione soprasellare, la
resezione totale della neoplasia può non essere attuabile, né per via transsfenoidale né trans-frontale, e può essere giustificata l'irradiazione aggiuntiva ad alto
voltaggio. Sia il trattamento chirurgico sia l'irradiazione possono essere seguiti dalla
perdita di altre funzioni ormonali ipofisarie. I pazienti irradiati possono perdere la
funzione endocrina lentamente, nel corso di anni, e sviluppare problemi visivi legati
alla fibrosi del chiasma ottico. Di conseguenza, lo stato ormonale post-trattamento
deve essere valutato a intervalli ravvicinati, preferibilmente a 3 e a 6 mesi e poi una
volta l'anno. Questa valutazione deve comprendere lo studio della funzionalità
ipofisaria, la valutazione radiologica della sella turcica ed un esame del campo
visivo. Per quanto concerne i prolattinomi, la maggior parte degli endocrinologi
considera la cabergolina come trattamento di prima scelta, indipendentemente dalle
loro dimensioni. Vi sono tuttavia alcune prove che soggetti con macroadenomi > 2
cm con livelli circolanti di prolattina estremamente elevati richiedano il trattamento
chirurgico o la terapia radiante in aggiunta alla terapia medica. Nell'apoplessia
ipofisaria, è giustificato il trattamento chirurgico immediato se compaiono
improvvisamente disturbi del campo visivo o paralisi dell'oculomotore, oppure se la
sonnolenza progredisce verso il coma a causa di fenomeni compressivi
sull'ipotalamo. Sebbene in alcuni casi il trattamento medico con corticosteroidi ad
alte dosi e la terapia di supporto generale possano essere sufficienti, di regola va
eseguita prontamente la decompressione trans-sfenoidale del tumore, che risulta
spesso emorragico.
Referenze
[1] Principi di Terapia Endocrina e Metabolica. Fabbrizio Monaco. Società Editrice
Universo – SEU Roma. Cap. II – Malattie dell’adenoipofisi o Ipofisi anteriore – Livia
Santarelli, Fabrizio Monaco.
[2] Migliaretti G, Aimaretti G, Borraccino A, Bellone J, Vannelli S, Angeli A, et al.
Incidence and prevalence rate estimation of GH treatment exposure in Piedmont
pediatric population in the years 2002-2004: Data from the GH Registry.
[3] Hanna CE, Krainz PL, Skeels MR, Miyahira RS, Sesser DE, LaFranchi SH.
Detection of congenital hypopituitary hypothyroidism: ten-year experience in the
Northwest Regional Screening Program. J Pediatr. 1986 Dec;109 (6) :959-64.
[4] Rosén T, Bengtsson BA. Premature mortality due to cardiovascular disease in
hypopituitarism. Lancet. 1990 Aug 4;336 (8710) :285-8.
[5] Twickler TB, Wilmink HW, Schreuder PC, Cabezas MC, van Dam PS,
Koppeschaar HP, et al. Growth hormone (GH) treatment decreases postprandial
remnant-like particle cholesterol concentration and improves endothelial function in
adult-onset GH deficiency.
Departments of Internal Medicine, University Medical Center Utrecht, 3508 GA
Utrecht, The Netherlands. J Clin Endocrinol Metab. 2000 Dec;85 (12) :4683-9.
[6] Hoffman RP. Growth hormone (GH) treatment does not restore endothelial
function in children with GH deficiency. J Pediatr Endocrinol Metab. 2008 Apr;21 (4)
:323-8.
Home page
[7] Lanes R, Soros A, Flores K, Gunczler P, Carrillo E, Bandel J. Endothelial
function, carotid artery intima-media thickness, epicardial adipose tissue, and left
ventricular mass and function in growth hormone-deficient adolescents: apparent
effects of growth hormone treatment on these parameters. J Clin Endocrinol Metab.
2005 Jul;90 (7) :3978-82. Epub 2005 May 3.
[8] O'Neal D, Hew FL, Sikaris K, Ward G, Alford F, Best JD. Low density lipoprotein
particle size in hypopituitary adults receiving conventional hormone replacement
therapy. J Clin Endocrinol Metab. 1996 Jul;81 (7) :2448-54.
[9] McGraw-Hill - New York – 1995 Williams Textbook of Wilson. Endocrinologia.
Chapter 6.The anterior pituitary. Thorner MO, Vance ML, Horvath E, et al. Saunders.
Philadelphia. 1992.
[10] Pediatric Endocrinology. Rosenfeld RG. Disorders of growth hormone and
insulin-like growth factor secretion and action. Sperling MA. Saunders. Philadelphia.
1996.
[11] Castinetti F, Reynaud R, Saveanu A, Quentien MH, Albarel F, Barlier A, et al.
Clinical and genetic aspects of combined pituitary hormone deficiencies. Ann
Endocrinol (Paris). 2008 Feb;69 (1) :7-17. Epub 2008 Mar 4.
[12] Matthai SM, Smith CS. Pituitary hypoplasia associated with a single central
maxillary incisor. J Pediatr Endocrinol Metab. 1996 Sep-Oct;9 (5) :543-4.
[13] Willnow S, Kiess W, Butenandt O, Dorr HG, Enders A, Strasser-Vogel B, et al.
Endocrine disorders in septo-optic dysplasia (De Morsier syndrome) --evaluation and
follow up of 18 patients. Eur J Pediatr. 1996 Mar;155 (3) :179-84.
[14] Burgner DP, Kinmond S, Wallace AM, Young DG, Forest MG, Donaldson MD
Male pseudohermaphroditism secondary to panhypopituitarism.. Arch Dis Child.
1996 Aug;75 (2) :153-5.
[15] Setian N, Aguiar CH, Galvão JA, Crivellaro CE, Dichtchekenian V, Damiani D.
Rathke's cleft cyst as a cause of growth hormone deficiency and micropenis. Childs
Nerv Syst. 1999 May;15 (5) :271-3.
[16] Rajaratnam S, Seshadri MS, Chandy MJ, Rajshekhar V. Hydrocortisone dose
and postoperative diabetes insipidus in patients undergoing transsphenoidal pituitary
surgery: a prospective randomized controlled study. Br J Neurosurg. 2003 Oct;17 (5)
:437-42.
[17] Root AW, Martinez CR. Magnetic resonance imaging in patients with
hypopituitarism. Trends Endocrinol Metab. 1992 Oct;3 (8) :283-7.
[18] Dutta P, Bhansali A, Singh P, Rajput R, Khandelwal N, Bhadada S. Congenital
hypopituitarism:clinico-radiological correlation. J Pediatr Endocrinol Metab 2009
Oct;22 (10) :921-8
[19] Manuale Merck Malattie endocrine e metaboliche. Malattie dell'Ipofisi. Malattie
dell'Ipofisi Anteriore. Iposecrezione Degli Ormoni Dell'ipofisi Anteriore. Terapia.
[20] Slosman FX.Atlante di Risonanza Magnetica del Cervello.Inforadiologie.ch.2005-2011
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
◀ Indietro pagina 4 Avanti ►
Scarica l'articolo: pagina 4.pdf
Sommario 20 pagine
Direttore scientifico
Trimestrale di divulgazione scientifica dell'Associazione Pediatrica di Immunologia e Genetica
Legge 7 marzo 2001, n. 62 - Registro della Stampa Tribunale di Messina n. 3/09 - 11 maggio 2009
Carmelo Salpietro - Direttore responsabile
Giuseppe Micali - Segreteria redazione
Basilia Piraino - Piera Vicchio
Direzione-Redazione: UOC Genetica e Immunologia Pediatrica - AOU Policlicnico Messina