")
Prontuario Unico alla Dimissione (PUD)
e della visita specialistica
per la continuità di cura e l’appropriatezza
Aggiornamento giugno 2013
Nel PUD sono raccolti gli obiettivi di appropriatezza clinica ed economica per l’assistenza
farmaceutica, condivisi e sottoscritti in sede di accordo tra ASL Pavia e le Aziende
sanitarie pubbliche e private accreditate dell’ambito provinciale.
I contenuti del PUD sono sviluppati e verificati anche tramite osservazioni, proposte e
richieste motivate di modifica avanzate dai clinici. La verifica tecnica e l’aggiornamento
del PUD è affidata ad un tavolo tecnico permanente, multidisciplinare e
multiprofessionale, attivato dalla ASL di Pavia di concerto con le direzioni delle Aziende
sanitarie pubbliche e private accreditate e partecipato da Medici di Medicina Generale
(MMG) e da Pediatri di Libera Scelta (PLS). L’ordine del giorno del tavolo tecnico per il
PUD è di competenza della Direzione Sanitaria della ASL. La partecipazione ai lavori del
tavolo tecnico per il PUD è riservata ai professionisti della salute di volta in volta identificati
dalle rispettive Aziende in funzione degli argomenti discussi.
INDICE
PREMESSA
OBIETTIVI DI APPROPRIATEZZA CLINICA ED ECONOMICA
AMBITI DI APPLICAZIONE: DIMISSIONE, VISITA AMBULATORIALE
ADESIONE (COMPLIANCE) E PERSISTENZA ALLA CURA
ALLEGATI
1) Indicatori a livello di sistema provinciale, monitoraggi, verifiche e controlli in Medicina
Generale
2) Sintesi delle virtù terapeutiche dei farmaci in funzione della loro efficacia relativa e del loro
costo: Farmaci preferibili (costo-opportuni)
Farmaci di sintesi chimica
a) Statine e associazioni fisse contenenti una statina (aggiornamento maggio 2012)
b) ACE-Inibitori e Sartani
c) Inibitori di Pompa Protonica (IPP)
d) Antibiotici (in preparazione)
e) Eparine a Basso Peso Molecolare (EBPM) riassunto delle indicazioni registrate
Farmaci di sintesi biologica (biotecnologie)
f) Ormone della crescita
g) Fattori di crescita granulocitari
h) Eritropoietine
i) Nuovi antitrombotici
3) Osservazioni, proposte e richieste di modifica dei contenuti del PUD – per il tavolo tecnico
4) Bibliografia generale e citazioni inserite nel testo
Azienda Sanitaria Locale della provincia di Pavia
V.le Indipendenza, 3 – 27100 PAVIA +39 (0382)431341 Fax +39 (0382) 431271
Materiale di proprietà della ASL della Provincia di Pavia e della Regione Lombardia, utilizzabile nel rispetto della licenza Creative
Commons "Attribuisci e Condividi allo Stesso Modo 3.0" cioè con esplicita esclusione di modifica, commercializzazione o distribuzione
non autorizzata e previa comunicazione alla ASL (email: [email protected]) delle modalità di utilizzo.
PREMESSA
Secondo il Codice di Deontologia medica, il medico agisce secondo il principio di efficacia delle
cure nel rispetto dell’autonomia della persona tenendo conto dell’uso appropriato delle risorse
(articolo 6). L’articolo 13 ribadisce che le prescrizioni e i trattamenti devono essere ispirati ad
aggiornate e sperimentate acquisizioni scientifiche tenuto conto dell’uso appropriato delle risorse,
sempre perseguendo il beneficio del paziente secondo criteri di equità.
OBIETTIVI DI APPROPRIATEZZA CLINICA ED ECONOMICA
Gli obiettivi di appropriatezza clinica ed economica sono applicati da parte di tutti gli
operatori sanitari per quanto di competenza alla dimissione dal ricovero (ordinario, DH, Day
Surgery) e durante la visita ambulatoriale (prima visita, visita di controllo).
PRINCIPI ATTIVI PREFERIBILI (COSTO-OPPORTUNI):
farmaci per cui è dimostrabile maggiore sicurezza e/o maggiore efficacia terapeutica relativa e
minor costo per l’erario (ovvero con il miglior rapporto tra costo ed efficacia) alla luce delle
indicazioni normative e delle documentazioni scientifiche disponibili.
L’efficacia terapeutica relativa teorica è peraltro un riferimento di massima e mai puntuale che
deve sempre essere interpretato nel singolo paziente, dal momento che la risposta terapeutica nel
singolo è determinata anche da numerosi altri fattori genetici e fisiopatologici (variabilità biologica).
AREA TERAPEUTICA
Eparine a basso peso molecolare (EBPM)
(limitatamente alle indicazioni chirurgiche):
intero 1° ciclo alla dimissione nel rispetto delle indicazioni AIFA
Inibitori di pompa protonica (IPP):
principi attivi preferibili (Omeprazolo, Lansoprazolo, Pantoprazolo,
Esomeprazolo ai dosaggi minimi efficaci), in subordine altri principi attivi
(Rabeprazolo)
Statine e associazioni fisse contenenti una Statina:
principi attivi preferibili (Simvastatina, Pravastatina, Fluvastatina;
Atorvastatina - statina ad alta intensità costo-efficace - per sindrome
coronarica acuta), in subordine altri principi attivi
ACE-Inibitori o Sartani:
principi attivi preferibili per la prevenzione del rischio CV nei pazienti ipertesi
(ACE-Inibitori o Sartani equivalenti; Sartani per i pazienti per i quali sia stata
verificata intolleranza ad ACE-Inibitori), in subordine altri principi attivi
Antibioticoterapia:
possibilmente ciclo completo (vasta gamma di scelta tra i principi attivi)
Dimission
e
X
Visita
X
X
X
X
X
X
X
X
X
2
AMBITI DI APPLICAZIONE
Gli ambiti di applicazione sono due: dimissione e visita ambulatoriale.
ALLA DIMISSIONE il suggerimento prescrittivo oppure la prescrizione (e, laddove prevista, la
distribuzione diretta) avviene con le seguenti modalità:
• In caso di nuova prescrizione o modifica di terapia in atto, il medico specialista indica
(nella lettera di dimissione) oppure prescrive direttamente (su ricettario rosso regionale) in
linea di massima i farmaci contenenti principi attivi preferibili ovvero di minor costo a
parità di sicurezza ed efficacia o di maggior sicurezza ed efficacia a parità di costo, nel
rispetto delle indicazioni di immissione in commercio.
• La struttura consegna al paziente un quantitativo del farmaco di nuova prescrizione
sufficiente per garantire la copertura del dosaggio indicato per un periodo non inferiore ai
30 gg di terapia per le EPBM di indicazione chirurgica (Prot.per la gestione della
prevenzione della TVP….rev. 1 del 18 nov 2008 Fondaz IRCCS S.Matteo) e non superiore
ai 60 gg di terapia in tutti gli altri casi (L.405/2001) , e in ogni caso sufficiente a garantire la
copertura terapeutica fino alla prima visita del medico di Medicina Generale.(DGR n.VII
/10246 del 6 agosto 2002; Nota R.L. h1 2002 02598).
• Qualora sussistano controindicazioni specifiche ai principi attivi preferibili o indicazioni
specifiche per altri principi attivi, lo specialista indica (nella lettera di dimissione) oppure
prescrive direttamente (su ricettario rosso regionale) altri farmaci e ne dà informazione al
medico curante per garantire la correttezza del percorso di continuità assistenziale.
AL TERMINE DELLA VISITA SPECIALISTICA AMBULATORIALE il suggerimento prescrittivo
avviene con le seguenti modalità:
• In caso di nuova prescrizione o modifica di terapia in atto, il medico specialista indica (nel
referto della visita) oppure prescrive direttamente (su ricettario rosso regionale) in linea di
massima i farmaci contenenti principi attivi preferibili ovvero di minor costo a parità di
sicurezza ed efficacia o di maggior sicurezza ed efficacia a parità di costo, nel rispetto delle
indicazioni di immissione in commercio.
• Qualora sussistano controindicazioni specifiche ai principi attivi preferibili o indicazioni
specifiche per altri principi attivi, lo specialista indica (nel referto della visita) oppure
prescrive direttamente (su ricettario rosso regionale) altri farmaci e ne da’ informazione
(nel referto della visita) al medico curante per garantire la correttezza del percorso di
continuità assistenziale.
Le tipologie generali di pazienti interessati dagli obiettivi di appropriatezza raccolti nel PUD sono
due:
1) tutti i pazienti di nuova osservazione (naïve) che non presentino specifiche controindicazioni
a principi attivi preferibili (in tal caso vengono riportate in cartella clinica o nella lettera di
dimissione o nel referto di visita) e per i quali, a giudizio del clinico, sia indicato un trattamento
farmacologico con un farmaco appartenente alle categorie terapeutiche elencate nel PUD;
2) tutti i pazienti già in trattamento con farmaci appartenenti alle categorie terapeutiche elencate
nel PUD che, a giudizio del clinico, necessitino di sostituzione attiva con un altro farmaco
(della medesima o di altra categoria terapeutica) per uno dei seguenti motivi di inefficacia del
trattamento in corso:
a) non sono clinicamente controllati dalla terapia in atto, seppur inizialmente aderenti e poi
persistenti nell’uso della medesima;
b) non sono persistenti, sebbene inizialmente aderenti, e quindi non sono controllati e non
si presume possano divenire persistenti se non previa modifica della prescrizione.
La distinzione tra mancata adesione (compliance) e mancata persistenza alle prescrizioni di
medicinali è riportata nella pagina successiva. L’osservazione diretta della mancata persistenza ai
medicinali non è agevole nell’ambito ospedaliero, ed è difficile anche nell’ambito territoriale.
3
ADESIONE (COMPLIANCE) E PERSISTENZA ALLA CURA
Adesione alla cura (compliance): con questo termine si intende l'adesione alla terapia e alle prescrizioni
mediche da parte del paziente. Rispettare le prescrizioni nella posologia, nei tempi e nei modi di assunzione
di un farmaco è indispensabile per ottenere il massimo del risultato terapeutico. Una percentuale di malati
che oscilla fra il 30% e il 50% non aderisce alla posologia prescritta, rischiando di rendere vana l'azione
terapeutica. Tratto da Servizio di Epidemiologia e farmacologia Preventiva (www.sefap.it) Università di
Milano, dipartimento di Scienze farmacologiche.
Adesione ai medicinali: grado di volontaria cooperazione del paziente nell’assumere le medicine prescritte.
L’adesione riguarda i dosaggi, i tempi e la frequenza di assunzione. Tratto da PUBMED, Medication
Adherence (MeSH introdotto nel 2009; 1378 citazioni indicizzate “major”)
La non-adesione può essere classificata come parziale o completa: nel primo caso consiste nell’assunzione
di una dose considerevolmente minore, nel secondo nella mancata assunzione (talvolta denominata non1
persistenza) . La non-adesione aumenta col tempo dall’inizio del trattamento: la prevalenza di non-adesione
alle statine nei pazienti di età maggiore di 65 anni è stata stimata essere al 29%, 38%, 42%, e 56%
2
rispettivamente dopo sei, mesi, uno, due e cinque anni di trattamento . Gruppi differenti di pazienti
manifestano tassi differenti di non-adesione: entro due anni dall’inizio di un trattamento con statine, il 75%
dei pazienti in prevenzione primaria e il 40% dei pazienti con pregressa sindrome coronarica acuta non
3
aderisce più alla terapia . Molti pazienti infine assumono i medicinali ma non al dosaggio prescritto: ad
esempio, in uno studio si è notato che tra i pazienti che assumevano almeno il 20% dei medicinali loro
3
prescritti, circa 4 su 10 assumevano meno dell’80% di questi farmaci dopo un anno . Adattato con modifiche
da: Bell KJ, Kirby A, Hayen A, Irwig L, Glasziou P.: Monitoring adherence to drug treatment by using change
in cholesterol concentration: secondary analysis of trial data. BMJ. 2011 Jan 21;342:d12. doi:
10.1136/bmj.d12.
Persistenza ai medicinali: varie definizioni sono riportate in ambito specialistico. In sostanza e dal punto di
vista operativo, quando i pazienti inizialmente aderenti ai trattamenti terminano del tutto l’assunzione dei
medicinali prescritti, o li assumono saltuariamente intervallando più o meno ampi periodi di non assunzione
con periodi di assunzione, vengono considerati casi di non-persistenza.
Perché i pazienti possano avvantaggiarsi dei benefici osservati negli studi clinici, sia l’adesione iniziale che
la persistenza nel tempo ai trattamenti, soprattutto per le terapie croniche, devono essere aumentate nel
4
limite del possibile. Per poter vigilare ed intervenire sui pazienti meno aderenti e meno persistenti , è
necessario che i clinici possano diagnosticare in modo affidabile la non-adesione e la non-persistenza. Sono
disponibili metodi diretti, ad esempio l’osservazione clinica e la misurazione dei lipidi sierici durante le visite o
i ricoveri, e metodi indiretti quali questionari, conteggi di farmaci, controlli sulla frequenza di rinnovo delle
ricette, misure di marcatori fisiologici (es. il colesterolo) e sistemi di monitoraggio elettronico delle
5
prescrizioni . Adattato con modifiche da: Bell KJ et al, citato.
La identificazione della non persistenza negli assistiti della ASL di Pavia è possibile grazie
all’utilizzo della Banca Dati Assistito (BDA) e dei Ritorni Informativi Periodici Personalizzati (RIPP),
disponibili nella area interattiva del sito web aziendale, da anni utilizzati per il miglioramento
dell’appropriatezza d’uso dei farmaci nel contesto della Medicina Generale.
11
Ho PM, Bryson CL, Rumsfeld JS. Medication adherence: its importance in cardiovascular outcomes.
Circulation2009;119:3028-35.
2
Benner JS, Glynn RJ, Mogun H, Neumann PJ, Weinstein MC, Avorn J, et al. Long-term persistence in use of statin
therapy in elderly patients. JAMA2002;288:455-61.
3
Jackevicius CA, Mamdani M, Tu JV. Adherence with statin therapy in elderly patients with and without acute coronary
syndromes. JAMA2002;288:462-7.
4
Schedlbauer A, Schroeder K, Peters T, Fahey T. Interventions to improve adherence to lipid lowering medication.
Cochrane Database Syst Rev2004;4:CD004371.
5
Osterberg L, Blaschke T. Adherence to medication. N Engl J Med 2005;353:487-97.
4
Allegato 1
INDICATORI, MONITORAGGI, VERIFICHE E CONTROLLI
I seguenti indicatori, attualmente riferiti alla scheda “MMG consumi farmaceutici” disponibile nel
sito ASL per ogni MMG e PLS, verranno monitorati, discussi e verificati anche nell’ambito del
tavolo tecnico permanente per la il PUD:
INIBITORI DI POMPA PROTONICA (IPP)
Indicatore
9- Spesa, per 100 assistibili pesati, di farmaci 'IPP' (ATC A02BC e A02BD)
10 - % Pezzi di farmaco equivalente 'IPP' / Tot pezzi farmaci 'IPP'
STATINE
Indicatore
13 - Spesa, per 100 assistibili pesati, di 'Statine' (ATC A10AA)
14 - % Pezzi di farmaco equivalente 'STATINE' / Tot pezzi 'STATINE'
ACE INIBITORI E SARTANI
Indicatore
12 - Spesa, per 100 ass.bili pesati, di 'Sartani' (ATC C09C e C09D)
12a - % DDD sartani / tot. ATC 'C09'
ANTIBIOTICI
Indicatore
15 - Spesa, per 100 ass.bili pesati, di farmaci antimicrobici per uso sistemico
(ATC J)
15a - di cui per J01 - J02 - J04 (antimicrobici per uso sistemico)
15b - di cui per J05 (antivirali per uso sistemico)
15c - di cui per J06 (sieri ed immunoglobuline)
Nuovo possibile indicatore: % pezzi di antibiotico equivalente /Tot pezzi
5
Esempio di scheda MMG
Periodo: Gennaio - Dicembre 2010
MMG Distretto ASL
Scost.
Medico XY
ASL
1 - Media assistibili in carico nel periodo
1137 123669 466350
2 - Media assistibili pesati nel periodo
1653 180194 640964
3 - Numero di ricette per 100 assistibili pesati
545
659
657
-17
4 - Numero di ricette per 100 assistiti
1210
1349
1313
-8
5 - Spesa per 100 assistibili pesati
13790
16697 16343
-16
6 - % Spesa per farmaci equivalenti / Spesa farmaceutica complessiva
31
31
32
-1
7 - % Pezzi farmaco equivalente / Tot pezzi farmaci
56
56
57
-1
8 - Spesa, per 100 ass.bili pesati, di farmaci per l'apparato gastroint.
2430
2315
2347
4
(ATC A)
9 - Spesa, per 100 ass.bili pesati, di farmaci 'IPP' (ATC A02BC e
1111
1105
1031
8
A02BD)
10 - % Pezzi di farmaco equivalente 'IPP' / Tot pezzi farmaci 'IPP'
83
83
83
1
11 - Spesa, per 100 ass.bili pesati, di farmaci per il sistema cardiov.
5572
6083
6164
-10
(ATC C)
12 - Spesa, per 100 ass.bili pesati, di 'Sartani' (ATC C09C e C09D)
1691
1875
1732
-2
12a - % DDD sartani / tot. ATC 'C09'
34
37
34
-0
13 - Spesa, per 100 ass.bili pesati, di 'Statine' (ATC A10AA)
1315
1290
1412
-7
14 - % Pezzi di farmaco equivalente 'STATINE' / Tot pezzi 'STATINE'
51
46
45
6
15 - Spesa, per 100 ass.bili pesati, di farmaci antimicrobici uso sist.
576
897
861
-33
(ATC J)
15a - di cui per J01 - J02 - J04 (antimicrobici per uso sistemico)
564
812
771
-27
15b - di cui per J05 (antivirali per uso sistemico)
12
51
49
-75
15c - di cui per J06 (sieri ed immunoglobuline)
0
33
41 -100
16 - Spesa, per 100 ass.bili pesati, di farmaci antineopl. e immunomod.
1129
931
920
23
(ATC L)
16a - di cui antineoplastici (L01)
89
95
99
-11
16b - di cui terapia endocrina (L02)
665
609
592
12
16c - di cui immunostimolanti (L03)
77
29
35 124
16d - di cui immunosoppressori (L04)
299
199
194
54
17 - Spesa, per 100 assistibili pesati, di farmaci antinfiammatori (M01A)
160
187
180
-11
18 - % Pezzi 'Coxib' (ATC(M01AH) / Tot pezzi FANS (ATCM01A)
12
13
12
0
19 - Spesa, per 100 assistibili pesati, di farmaci del sistema nervoso
1330
2359
2157
-38
(ATC N)
19a - di cui oppioidi (N02A)
171
184
210
-19
19b - di cui inibitori sel. della serotonina-ricapt. (N06AB)
192
409
412
-53
20 - Spesa, per 100 assistibili pesati, di farmaci del sistema respiratorio
789
1266
1145
-31
(ATC R)
6
Allegato 2a
Sintesi delle virtù terapeutiche delle statine e di associazioni fisse contenti statine
in funzione della loro efficacia relativa e del loro costo per il SSN
STATINE DI PRIMA SCELTA
in assenza di specifiche controindicazioni
RIVISTA ALLA LUCE DELLA NOTA 13 AIFA 2012
Sei statine sono autorizzate alla commercializzazione in Italia: atorvastatina, fluvastatina,
lovastatina, pravastatina, rosuvastatina e simvastatina. L’ezetimibe da luglio 2011 è disponibile in
fascia A sia da solo che in associazione fissa con simvastatina. Per simvastatina, pravastatina,
fluvastatina e atorvastatina la copertura brevettuale è scaduta rispettivamente ad aprile 2007,
dicembre 2007, agosto 2008 e marzo 2012 e sono disponibili come farmaco equivalente: generico
o genericato (cioè con ritenzione del brand).
Le statine e l’associazione fissa simvavastina-ezetimibe differiscono tra loro riguardo ad efficacia
clinica (riduzione di mortalità globale, mortalità cardiovascolare, di rischio di IMA non fatale, di
angina, di ictus cerebri o di necessità di rivascolarizzazione: bypass, angioplastica o stenting), ad
efficacia biologica (riduzione di colesterolo LDL, C-LDL, aumento di colesterolo HDL, C-HDL) e a
costo.
Riguardo all’efficacia clinica, la seguente tabella sintetizza le conoscenze disponibili negli studi
clinici che hanno confrontato due statine tra loro in modo diretto (senza considerare cioè le
comparazioni indirette desumibili dagli studi in cui il controllo è il placebo)i,ii.
Efficacia clinica delle statine negli studi comparativi diretti (una statina vs un’altra statina)
Le statine:
In
prevenzione
primaria
In
prevenzione
secondaria
Sono equipotenti
sulla riduzione di
eventi CV
Non esistono studi
comparativi tra dosi
equipotenti di due o
più statine riguardo
al rischio di eventi
coronarici, di ictus o
di morte
mortalità
totale
mortalità
CV
Prava,
Simva,
Rosuva
Prava,
Simva
Atorva,
Prava,
Simva
Atorva,
Simva
eventi CV
ictus
Sono efficaci
nei diabetici
Atorva, Lova,
Prava,
Atorva, Sovrapponibile
Simva,
all’efficacia
Prava,
Rosuva
Simva, clinica nei non
Rosuva
diabetici
Atorva,
Prava, Simva
Riguardo alla efficacia biologica, le statine differiscono tra loro in termini di potenza
ipolipemizzante. Le diverse molecole determinano, infatti, diversi effetti su i valori di C-LDL:
1. “Statine di 1° livello” (nel linguaggio della No ta 13) ovvero quelle determinano una riduzione del
C-LDL fino a un massimo del 40% circa; rientrano in questo gruppo la simvastatina (10,20, 40
mg), la pravastatina, la fluvastatina e la lovastatina.
2. “Statine di 2° livello” ovvero quelle possono de terminare una riduzione del C-LDL superiore al
40% circa (indicate anche come statine ad alta intensità di azione); rientrano in questo gruppo
la simvastatina (80 mg), l’atorvastatina, la rosuvastatina e l’associazione fissa
simvastatina/ezetimibe.
Le dosi di statine (a bassa o ad alta intensità di azione) cui conseguono riduzioni percentuali di CLDL di comparabile entità sono riportate nella tabella seguente, ed i valori medi oltre che minimo e
massimo (che consentono di apprezzare l’intervallo di credibilità dell’effetto biologico ricercato)
delle statine sono riportati nel successivo grafico. Ogni riga riporta le dosi sostanzialmente
equipotenti di statine1.
7
Equipotenza: dosi di statine cui conseguono riduzioni % di C-LDL di comparabile
entità (efficacia biologica)*
% riduzione
attesa LDL
Atorva
statina
Fluva
statina
Lova
statina
Prava
statina
Rosuva
statina
Simva
statina
<=30%
30-40%
40-45%
45-50%
50-60%
> 60%
-10 mg
20 mg
40 mg
80 mg
--
40 mg
80 mg
-----
20 mg
40 o 80 mg
(80 mg n.c.)
----
20 mg
40 mg
80 mg
----
--5 o 10 mg
-20 mg
40 mg
10 mg
20 mg
40 mg
80 mg (2x40)
---
Simva
statinaEzetimibe
10-10 mg
20-10 mg
40-10 mg
*Stime basate sui risultati degli studi comparativi esistenti. In italico le dosi considerate “di 2° li vello” nella Nota 13 AIFA
2012. In grassetto le statine disponibili come equivalenti. N.C.: non commercializzata in Italia.
Riguardo al costo, nella tabella seguente è riportato il costo medio annuale in base al prezzo al
pubblico per unità posologica (calcolato per i farmaci coperti o meno da brevetto) per un
trattamento continuativo, che è l’obiettivo teorico per ciascun paziente. I farmaci sono ordinati in
funzione della riduzione attesa di colesterolo LDL desunta dall’analisi della linea guida ATP III
statunitense, degli studi pubblicati ed infine, in carenza di dati nelle analisi citate, nella
documentazione fornita dai produttori6.
Costo medio annuale per efficacia biologica delle statine e dell’associazione fissa
simvastatina-ezetimibe
Statina e dose (n° compresse)
Classe di riduzione Riduzione %
Costo medio
attesa di colesterolo media attesa di annuale in €
LDL
colesterolo LDL
Pravastatina 20 mg
24,0%
89,43
Lovastatina 20 mg
24,0%
202,58
< 30%
Fluvastatina 40 mg
25,0%
302,95
Simvastatina 10 mg
30,0%
62,05
Fluvastatina 80 mg
31,0%
169,73
Lovastatina 40 mg
31,0%
202,58
30-35%
Pravastatina 40 mg
34,0%
215,35
Simvastatina 20mg (28 cpr)
35,0%
105,85
Simvastatina 20mg (10 cpr)
35,0%
140,53
Sivastatina 10+ ezetimibe 10 mg *
36,0%
757,38
Atorvastatina 10 mg (30 cpr)
37,0%
85,78
35-40%
Atorvastatina 10 mg (10 cpr)
37,0%
262,80
Simvastatina 40 mg
41,0%
198,93
Simvastatina 20 +ezetimibe 10 mg *
42,0%
759,20
Atorvastatina 20 mg (30 cpr)
43,0%
147,83
40-45%
Atorvastatina 20 mg (10 cpr)
43,0%
459,90
Rosuvastatina 5 mg
45,0%
292,00
Simvastatina 80 mg (NB: 2 cpr da
46,0%
397,87
40)
Simvastatina 40 + ezetimibe 10 mg
49,2%
784,75
45-50%
*
Atorvastatina 40 mg
50,0%
182,50
Rosuvastatina 10 mg
52,0%
357,70
Rosuvastatina 20 mg
55,0%
540,20
> 50%
Atorvastatina 80 mg
57,0%
182,50
Rosuvastatina 40 mg
63,0%
562,10
6
Smith MEB, Lee NJ, Haney E, Carson S. Drug Class Review: HMG-CoA Reductase Inhibitors (Statins) and Fixed-dose
Combination Products Containing a Statin: Final Report Update 5. Drug Class Reviews. Portland (OR): Oregon Health &
Science University; 2009 Nov. e Ara R, Tumur I, Pandor A, Duenas A, Williams R, Wilkinson A, et al. Ezetimibe for the
treatment of hypercholesterolaemia: a systematic review and economic evaluation. Health Technol Assess 2008;12(21).
8
In grassetto le statine disponibili come equivalenti (genericate, cioè di marca, o generiche
pure). Evidenziate in giallo le statine equivalenti ormai disponibili per ciascuna classe di
riduzione attesa di colesterolo LDL.
L’efficacia biologica delle statine è stata indagata anche per la capacità di ridurre lo spessore
dell’intima-media carotidea, la rigidità vascolare ed altri marcatori di aterosclerosi subclinica, ma le
documentazioni a riguardo non sono sufficienti per una applicazione diffusa nella pratica clinica.
(*) Dalla Nota 13:
- l’ezetimibe in associazione ad una statina può determinare una ulteriore riduzione di LDL-C
indipendentemente dalla statina utilizzata; questa ulteriore riduzione è stata stimata non superiore
al 15%-22% (ed è praticamente la stessa qualunque sia la dose della statina associata)
- l’ezetimibe è utile e rimborsata dal SSN in due casi: in associazione (precostituita o
estemporanea) con statine quando queste da sole, a dosi ottimali, non consentono di raggiungere
gli obiettivi (target) terapeutici; in monoterapia nei pazienti intolleranti alle statine.
Per stimare la efficacia biologica della ezetimibe quando usata in associazione ad una statina è
stata quindi considerata una riduzione ulteriore pari al 20% (stima conservativa) della riduzione
attesa di colesterolo LDL ottenibile dalla sola statina associata (di corrispondente dosaggio).
Grafico aggiornato al marzo 2012.
9
ASL PAVIA – Otto anni di utilizzo di statine 2000-2009
In fugura è riportato la tendenza del numero di assistiti in terapia con statine, per principio attivo, osservata
nella Provincia di Pavia.
16.000
14.000
12.000
10.000
8.000
6.000
4.000
2.000
0
2001
2002
ATORVASTATINA
2003
2004
FLUVASTATINA
2005
LOVASTATINA
2006
PRAVASTATINA
2007
2008
2009
ROSUVASTATINA
2010
SIMVASTATINA
I due principi attivi con numero maggiore di terapie sono sempre Simvastatina ed Atorvastatina, anche se
negli ultimi anni è in forte ascesa la Rosuvastatina. Marginale infine il dato relativo ai rimanenti principi attivi.
Utilizzo di simvastatina-ezetimibe (ATC C10BA02) in provincia di Pavia
Anno 2011
tipo_soggetto
Prima prescrizione
Continuita terapeutica
Switc h da altre statine
di c ui
ATC
statina
C10BA02 SIMVASTATINA E EZETIMIBE
C10BA02 SIMVASTATINA E EZETIMIBE
C10AA01
C10AA02
C10AA03
C10AA04
C10AA05
C10AA07
soggetti
SIMVASTATINA
LOVASTATINA
PRAVASTATINA
FLUVASTATINA
ATORVASTATINA
ROSUVASTATINA
443
1.010
244
71
1
11
6
73
82
1.697
% su totale
soggetti
26,1%
59,5%
14,4%
29,1%
0,4%
4,5%
2,5%
29,9%
33,6%
E' stata presa in considerazione, per tutti i casi, la prima prescrizione di simva-ezetimibe del 2011:
• per prima prescrizione si intende la prescrizione di simva-ezetimibe senza che, in un periodo finestra di 3
mesi precedentemente, ci siano state prescrizioni di altre statine (C10AA)
• per continuità terapeutica si intende la continuità di terapia con simva-ezetimibe dal 2010
• per switch da altre statine, nel corso del 2011, nella tabella seguente è indicata la statina di partenza.
CATEGORIA BDA DI SOGGETTI IN TERAPIA CON SIMVASTATINA-EZETIMIBE
SCELTA DEL PRINCIPIO ATTIVO PER L’INIZIO DEL TRATTAMENTO
10
Le conoscenze disponibili riguardo ad efficacia clinica, efficacia biologica e al costo delle diverse
statine facilitano la scelta del trattamento farmacologico più appropriato, che andrà sempre
personalizzato in funzione delle caratteristiche cliniche e delle preferenze dei singoli pazienti:
a) EFFICACIA CLINICA: Le statine per cui è stata dimostrata efficacia clinica (riduzione di
mortalità globale e CV, riduzione di eventi CV) dovrebbero essere preferite rispetto alle
statine per cui è stata dimostrata solo l’efficacia biologica (la riduzione del C-LDL)
b) SICUREZZA: A parità di efficacia biologica, dovrebbero essere preferite le statine con più
favorevole rapporto tra efficacia e probabilità di eventi avversi indesiderati
c) COSTO: A parità di efficacia biologica, infine, dovrebbe essere preferita la prescrizione di
statine a minor costo di acquisto per il servizio sanitario e per il paziente.
In studi su terapia con statina a maggiore intensità vs statine a minore intensità di riduzione del
colesterolo LDL (C-LDL), l’ulteriore riduzione media del C-LDL a 1 anno è stata di 0,51 mmol/L.
Regimi più intensivi hanno prodotto una ulteriore riduzione del 15% (IC 95% 11-18; p<0.001) degli
eventi vascolari maggiori, ovvero riduzioni delle morti coronariche o degli infarti miocardici non
fatali del 13% (IC 95% 7-19; p<0.0001), delle rivascolarizzazioni coronariche del 19% (IC 95% 1524; p<0.0001), e dell’ictus ischemico del 16% (IC 95% 5-26; p=0.005). Per una riduzione del CLDL di 1.0 mmol/L queste ulteriori riduzioni del rischio sono risultate simili alle riduzioni
proporzionali negli studi su statine vs controlli.
Riduzione del rischio relativo per eventi coronarici maggior
La riduzione del Rischio
Relativo risulta indipendente dai
livelli iniziali di LDL-C e dalle
caratteristiche cliniche del
paziente
Ogni riduzione di LDL-C di 38
mg/dl si associa ad una
riduzione del Rischio Relativo
del 24%
Riduzione colesterolo LDL (mmol/l)
Più rilevante della riduzione del rischio relativa è la misura della riduzione del rischio assoluto, che,
a differenza della prima, dipende dalle condizioni effettive del singolo paziente, tra cui il livello di
rischio basale, determinato da numerosi altri fattori modificabili oltre al livello di C-LDL.
11
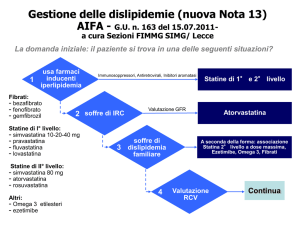
ALGORITMO PER LA PRESCRIZIONE NEI PAZIENTI IPERCOLESTEROLEMICI
(nelle note le specifiche prescrittive)
1) Il paziente assume immunosoppressori, antiretrovirali o inibitori delle aromatasi?iii
→ Qualsiasi statina in rapporto alla tolleranza individuale e all’interferenza con altri farmaci.
Se il paziente non assume immunosoppressori, antiretrovirali o inibitori delle aromatasi:
2) Il paziente ha una insufficienza renale cronica (IRC)?iv
→ Se trigliceridi > 500 mg/dl: omega-3 sostituti
→ Se C-LDL è > 130: I scelta Simvastatina+Ezetimibe, II scelta altre statine a minima escrezione
renale atorvastatina
Se il paziente non assume immunosoppressori, antiretrovirali o inibitori delle aromatasi e non ha
una IRC:
3) Il paziente ha una dislipidemie familiare?v
IPERCOLESTEROLEMIE AUTOSOMICHE DOMINANTI: Statine 2° livello a dose massima*
IPERCOLESTEROLEMIE AUTOSOMICHE RECESSIVE: Statine 2° livello a dose massima*
DISBETALIPOPROTEINEMIA: Statine 2° livello a dose m assima*
IPERLIPIDEMIA FAMILIARE COMBINATA: Statine 2° livel lo + omega 3
IPERCHILOMICRONEMIE e gravi IPERTRIGLICERIDEMIE: Omega 3 + fibrati
(* preferenzialmente Atorvastatina equivalente; in casi selezionati associate ad ezetimibe)
Se il paziente non assume immunosoppressori, antiretrovirali o inibitori delle aromatasi, non ha
una IRC e non ha alcuna dislipidemia familiare né ha altre cause di ipercolesterolemia II
(ipotiroidismo, ecc.):
4) Il paziente ha una ipercolesterolemia (poligenica)?
Prescrivere una terapia di prima scelta adatta al rischio cardiovascolare del paziente:
→ Se rischio basso: modifiche del comportamento alimentare e della attività fisica (non occorrono
farmaci)
→ Se rischio moderato: tendere a C-LDL < 130 usando statine di 1° livello
→ Se rischio alto: tendere a C-LDL < 100 usando statine di 1° livello
→ Se rischio molto alto: tendere a C-LDL < 70 usando Atorvastatina equivalente o altra statina
di 2° livello
Nei pazienti intolleranti alla dose considerata ottimale di statina, e nei pazienti non rispondenti
(cioè tolleranti alla dose considerata ottimale, ma che non lo raggiungono comunque l’obiettivo)
usare Atorvastatina equivalente o, se Atorvastatina verifica inefficace, altra statina di 2° livello.
Nei pazienti non sufficientemente rispondenti ad una statina di 2° livello aggiungere ezetimibe.
In tutti i casi precedenti:
laddove possibile preferire statine (di 1° o 2° li vello) a brevetto scaduto
se necessaria ezetimibe, evitare l’associazione fissa precostituita, più costosa e non più
efficace.
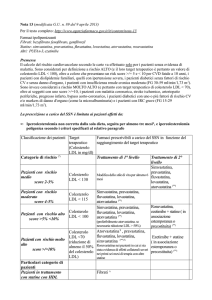
La determinazione del rischio necessita di particolare attenzione. Nella annotazione a
margine della tabella 1 nella stessa Nota 13 AIFA vengono elencati “I maggiori fattori individuali di
rischio considerati nella linea guida AHA/ACCe dell’ESC/EASD sono (secondo le indicazioni Adult
Treatment Panel III) per il trattamento della dislipidemia: età > 50 anni nei maschi e 60 nelle
femmine, abitudine al fumo, pressione arteriosa sistolica > 135 e diastolica > 85, o trattamento
antipertensivo in atto, bassi valori di colesterolo HDL (< di 40 mg/dl nei maschi e < di 50 mg/dl
nelle femmine), storia familiare di cardiopatia ischemica prematura in un familiare di 1° grado
(prima di 55 anni nei maschi e prima di 65 anni nelle femmine).”.
Preso alla lettera, questo elenco potrebbe indurre a considerazioni discutibili. Infatti, c’è fattore di
rischio e fattore di rischio, e i metodi per determinare il livello individuale di rischio variano a
seconda degli autori e dei contesti clinici da cui vengono ricavati i dati. La sommatoria dei singoli
fattori non equivale alla verifica dell’impatto di fattori multipli su dati sperimentali. E’
importante quindi tenere conto di conoscenze di alta qualità e per esiti clinici davvero importanti
per i pazienti e non limitarsi a “fare la conta” dei fattori.
12
Cosa dicono le fonti citate nell’elenco di fattori di rischio riportati nella Nota 13? Quella più recente,
della associazione statunitense di cardiologia: 2010 ACCF/AHA Guideline for Assessment of
Cardiovascular Risk in Asymptomatic Adults (Circulation 2010, 122:2748-2764 e disponibili sul sito
web della AHA), classifica le raccomandazioni cliniche in diverse aree, che vanno dalla anamnesi
familiare al calcolo del punteggio globale di rischio, a misurazioni dello spessore intimale carotideo
o della proteina C reattiva o del peptide natriuretico, al riscontro di diabete o di emoblobina
glicosilata HbA1c, all’ECG, alla microalbuminuria, a tecniche radiologiche avanzate, a tecniche
genetiche.
L’importanza dell’effetto clinico che si può ottenere applicando ciascuna di queste tecniche è
classificata, in questa linea guida, da I (benefici molto maggiori dei danni) a III (nessun beneficio o
danni maggiori dei benefici), mentre la qualità di queste conoscenze (cioè la stima di quanto
attendibili possano essere questi effetti clinici) è classificata da A (molti studi su popolazioni
diverse) a C (pochissimi studi su poche popolazioni e/o soltanto pareri di esperti).
In pratica, le raccomandazioni IA sarebbero di alta qualità per effetti clinici importanti, mentre
quelle IIIC sarebbero di molto bassa qualità e per effetti negativi o nulli.
Se si esamina il numero di raccomandazioni riportate da questa aggiornata linea guida, si ottiene il
grafico seguente.
Qualità delle conoscenze per importanza clinica
nella Linea Guida AHA 2010 sul rischio CV
Numero di
raccomandazioni
10
8
6
4
2
0
A
B
C
Qualità delle conoscenze
Importanza e valore dell'effetto clinico:
I
IIA
IIB
III
In sostanza: sul totale di 35 raccomandazioni, nessuna è di qualità A, mentre tra quelle di qualità B
solo tre raccomandazioni hanno un valore I di importanza e validità. E quali sono queste tre? Si
tratta di:
- fare l’anamnesi familiare specifica in tutti gli adulti asintomatici;
- calcolare un punteggio di rischio CV globale, con qualsiasi metodo, in tutti gli adulti asintomatici
che non siano già stati colpiti da un evento CV (in tal caso, sono già ad alto rischio)
- fare le due cose precedenti anche nelle femmine, non solo nei maschi.
La raccomandazione di considerare l’anamnesi familiare è desunta da due studi di un decennio fa
su coorti variabili da 5.000 a 25.000 pazienti. La raccomandazione del calcolo del rischio CV è
basata su circa 5.000 pazienti USA (Framingham), fino a oltre 200.000 pazienti europei (SCORE).
L’algoritmo italiano CUORE, inspiegabilmente scomparso nella nuova versione della Nota 13, è
stato costruito sulla osservazione di oltre 20.000 italiani fino al decesso o all’accertamento in vita.
CUORE dovrebbe essere lo strumento più sensibile e specifico per determinare il rischio CV in
pazienti italiani. L’alternativa potrebbe essere l’algoritmo europeo SCORE nella versione per paesi
a basso rischio CV, come l’Italia. In entrambi i casi si tratta di evitare di “fare la conta” dei fattori ma
13
di esaminare, dati alla mano, il probabile rischio futuro del paziente: un modo semplice ed efficace
per fare medicina personalizzata.
Prevenzione primaria nel paziente ad alto rischio cardiovascolare (secondo la Carta del RCV
> 20% a 10 anni)vi. Se dopo 90 giorni sono insufficienti le norme igieniche ben condotte:
• l’obiettivo cui tendere per il C-LDL è di 100 mg/dl
• se il paziente presenta C-LDL < 160 mg/dl è sufficiente Simvastatina fino a 40 mg o statina
equipotente. Se non si è raggiunto l’obiettivo passare a una statina ad elevata potenza a
brevetto scaduto7 o, se questa verificata inefficace, altra statina di 2° livello.
• se il paziente presenta C-LDL >160 mg/dlvii iniziare con Atorvastatina equivalente.
Prevenzione secondaria nel paziente con malattia coronarica cronica stabile o nel paziente
diabetico senza malattia cardiovascolare
• l’obiettivo cui tendere per il C-LDL è di 100 mg/dl
• se il paziente presenta C-LDL < 160 mg/dl è sufficiente Simvastatina fino a 40 mg o statina
equipotente. Se non si è raggiunto l’obiettivo passare a una statina ad elevata potenza a
brevetto scaduto o, se questa verificata inefficace, altra statina di 2° livello
• se il paziente presenta C-LDL >160 mg/ dlviii iniziare con una statina ad elevata potenza a
brevetto scaduto.
Tuttavia nei pazienti con diabete senza malattia cardiovascolare le documentazioni di efficacia
clinica sono meno evidenti e più discutibili.
Prevenzione secondaria nel paziente con malattia cronica stabile e affetto da diabete
oppure da insufficienza renale cronica
• l’obiettivo cui tendere per il C-LDL è di 80 mg/dl
• se il paziente presenta C-LDL < 130 mg/dl è sufficiente Simvastatina fino a 40 mg o statina
equipotente. Se non si è raggiunto l’obiettivo passare a una statina ad elevata potenza a
brevetto scaduto o, se questa verificata inefficace, altra statina di 2° livello.
• se il paziente presenta C-LDL >130 mg/ dlix iniziare con una statina ad elevata potenza a
brevetto scaduto.
L’obiettivo per il C-LDL va contemperato con la necessità di garantire il miglior grado possibile di
aderenza e persistenza al trattamento. Qualora il trattamento con statina a maggiore intensità non
venga tollerato o mantenuto dal paziente (registrazione nel follow up) è raccomandata la
prosecuzione con statina a minore intensità con l’obiettivo di mantenere il paziente il più vicino
possibile all’obiettivo lipidico di 100 mg/dl.
Prevenzione secondaria nel paziente con Sindrome Coronarica Acuta
• l’obiettivo cui tendere per il C-LDL è di 80 mg/dl (peraltro questo livello è sostenuto
dall’opinione di esperti e non da sicuri riscontri nella ricerca clinica)
• Iniziare con una statina ad elevata potenza a brevetto scaduto a dosaggio adeguatox,xi
(previa titolazione) indipendentemente dai valori basali di C-LDL
• La sostituzione nel tempo di una statina ad elevata potenza a brevetto scaduto con statina
equipotente non a brevetto scaduto (es. Rosuvastatina) è giustificata solo in pazienti ad
altissimo rischio cardiovascolare ovvero in pazienti con documentata intolleranza o inefficacia
della prima.
Le documentazioni di efficacia clinica sono al momento più robuste per Atorvastatina che per
Rosuvastatina o altre forme (comprese le associazioni fisse contenenti una statina).
7
Ad oggi solo Atorvastatina equivalente.
14
Ezetimibe (associazione precostituita: simvastatina + ezetimibe)
In attesa di nuovi trial che comprendano dei controlli veri (ad esempio IMPROVE IT, confronto tra
simvastatina 20 mg da sola e simvastatina 20 mg più ezetimibe 10 mg), l’utilizzo di simvastatina +
ezetimibe non può essere considerato alternativo all’utilizzo di statine di maggiore potenza tra
quelle indicate nella nuova Nota 13 come statine di “1° livello” (ad esempio simvastatina 40 mg) o
di “2° livello” (ad esempio atorvastatina 20 mg o r osuvastatina 10 mg), perché non è stato
dimostrato che l’uso di una bassa di simvastatina assieme ad ezetimibe sia clinicamente
più efficace né più tollerato rispetto a queste altre statine da sole: abbassa di più il
colesterolo, cioè è più efficace dal punto di vista biologico (dell’11% nei pazienti naive e del 22%
nei pazienti in precedenza trattati con statine), ma non riguardo agli esiti che contano per il
paziente ad esempio riduzione di eventi cardiovascolari.
Riguardo alla efficacia biologica, ovvero alla capacità di ridurre il colesterolo LDL,
l’ezetimibe ha una efficacia aggiuntiva, seppure relativamente minore rispetto alle statine.
Riguardo alla efficacia clinica, ovvero alla capacità di ridurre mortalità o eventi CV (IMA,
ictus, ricoveri), l’ezetimibe è invece assai controversa. Negli studi clinici infatti
l’associazione simvastatina + ezetimibe è stata riscontrata esserexii,xiiixiv:
xv
•
efficace quanto la simvastatina da sola in soggetti con ipercolesterolemia familiare (ENHANCE) perché i
pazienti non avevano un IMT patologico al baseline (0,70 mm);
•
•
•
efficace quanto il placebo (e con 105 morti con l’associazione vs 100 col placebo, differenza statisticamente non
xvi
significativa ma che fa pensare) in soggetti con stenosi aortica (SEAS) , tranne che per la necessità di
ricorrere al by-pass Ao-Co, evento che si è ridotto in modo statisticamente significativo
nei pazienti trattati con eze-simva (p<0,02);
meno efficace di niacina + statina in obesi cardiopatici o con equivalenti di RCV (7
morti con l simvastatina + ezetimibe vs 1 con niacina + simvastatina) studiati su un end
point surrogato (ARBITER 6-HALTSxvii, studio in cui la mortalità non era un end point e
che è stato sospeso prematuramente dopo 14 mesi perché la niacina si è dimostrata
più efficace di ezetimibe nel ridurre l’IMT in obesi cardiopatici: l’associazione con la
statina era, per il 50% circa con simvastatina e per il 50 % circa con atorvastatina).
biologicamente meno efficace della simvastatina da sola (ma a dosaggio differente) in soggetti ad alto rischio
xviii
coronarico: nello studio VYCTOR , pubblicato benchè effettuato con solo 90 pazienti, gli Autori concludono per un
“beneficio della combinazione statine ed ezetimibe su un end point surrogato” che vedono solo loro. Trattasi di
uno studio molto piccolo, che ha dimostrato per la prima volta che l’associazione
ezetimibe-simvastatina, così come la simvastatina da sola e la pravastatina da sola,
riduce in modo statisticamente significativo l’IMT … non di più di queste statine da sole
… anche perché tutte e 3 le terapie portavano l’LDL a 48 mg/dl!
•
•
clnicamente contraddittoria, per una tendenza a maggiore mortalità rispetto al placebo che, benchè non sia
statisticamente differente né fosse un esito primario, solleva perplessità in nefropatici cronici non coronaropatici
xix
(SHARP)
xx
infine, la pubblicazione completa di questo studio SHARP ha chiarito :
o che il beneficio osservato (17% di riduzione di eventi aterosclerotici maggiori) potrebbe tranquillamente essere
ascritto alla simvastatina, più che all’aggiunta di ezetimibe;
o che una simile riduzione di colesterolo LDL può essere raggiunta con altri farmaci, ad esempio atorvastatina 20
mg, che nello studio 4D è stata usata, seppur senza raggiungere esiti clinici di interesse, in pazienti cinque volte
più a rischio dei pazienti esaminati in SHARP;
o che gli investigatori hanno cambiato gli end point dello studio durante la raccolta dati, arrivando a mescolare
esiti irrevocabili, come la morte o l’infarto cardiaco, con esiti, quali le rivascolarizzazioni, che possono essere
facilmente distorti dagli stessi ricercatori (i quali si rendono conto dei farmaci assunti, benchè in cieco, dai loro
pazienti);
o che se anche si volesse ritenere credibile tale mescola di end point, il risultato sarebbe assai deludente in
termini clinici: per prevenire un “evento aterosclerotico maggiore” (per 3/4 valorizzato come 1
rivascolarizzazione in meno) occorrerebbe trattare ben 48 pazienti per quasi 5 anni (1 evento prevenuto ogni
235 anni-paziente di trattamento; NNT attorno a 30 per la prevenzione secondaria e attorno
o
a 70 per la prevenzione primaria);
che nei pazienti in trattamento con ezetimibe si è osservato un numero maggiore di morti per cause totali e per
cancro, rispetto a coloro che erano trattati solo con simvastatina. Benchè questa differenza non sia
15
statisticamente significativa e la mortalità non fosse un esito primario, è disturbante che il trattamento “nuovo”
dia più morti del “vecchio” ed è opportuno che ai pazienti ciò venga riferito.
L’uso di simvastatina + ezetimibe dovrebbe quindi essere limitato a quei pochi casi in cui
l’obiettivo terapeutico non può essere raggiunto con le statine, ad esempio per una
ipercolesterolemia effettivamente resistente o per una intolleranza vera ad alte dosi di statine di
adeguata potenza, purchè tali casi siano stati adeguatamente presi in carico e ben documentati in
cartella clinica.
16
Allegato 2b
Sintesi delle virtù terapeutiche dei farmaci attivi sull’asse Renina-Angiotensina-Aldosterone
in funzione della loro efficacia relativa e del loro costo per il SSN
ACE-INIBITORI E SARTANI DI PRIMA SCELTA
in assenza di specifiche controindicazioni
Numerosi inibitori dell’enzima convertente l’angiotensina (ACE-inibitori) e inibitori recettoriali
dell’angiotensina (sartani) sono autorizzati alla commercializzazione in Italia. Per la maggior parte
degli ACE-inibitori la copertura brevettuale è scaduta, come per il sartano losartan. La scadenza
della copertura brevettuale per valsartan è prevista per il 2011 e per altri sartani nel 2012. Alcune
associazioni fisse (sartano + diuretico) hanno una copertura brevettuale più estesa nel tempo.
Gli ACE-inibitori ed i sartani non differiscono in modo clinicamente significativo tra loro riguardo ad
efficacia clinica (riduzione di mortalità globale, mortalità ed eventi cerebro-cardiovascolari)xxi e ad
efficacia biologica (riduzione della pressione arteriosa sistolica e diastolica) mentre differiscono
riguardo al costo.
Al momento attuale, le condizioni cliniche per le quali il Sistema Sanitario Nazionale garantisce la
rimborsabilità (oltre all’indicazione) di farmaci inibitori dell’angiotensina II, sono, oltre
all’ipertensione arteriosa, le seguenti:
1. Ipertensione arteriosa ed ipertrofia VS all’ECG (losartan)
2. Nefropatia diabetica e proteinuria (irbesartan)
3. Infarto miocardico recente (12 ore-10 giorni) in presenza di disfunzione VS (valsartan)
4. Insufficienza cardiaca cronica (in pazienti ≥ 60 anni), quando il trattamento con gli inibitori
dell’enzima di conversione dell’angiotensina non è considerato adatto (losartan)
5. Scompenso cardiaco e alterata funzione sistolica ventricolare sinistra (frazione di eiezione
ventricolare sinistra ≤ 40%). In aggiunta al trattamento con ACE-inibitori o quando gli ACEinibitori non siano tollerati (candesartan).
Nei pazienti con ipertensione arteriosa di qualsiasi grado o affetti da una delle condizioni sopra
elencate, quando è indicato un intervento farmacologico per la inibizione dell’asse reninaangiotensina-aldosterone, si raccomanda la prescrizione di ACE-inibitori come prima scelta,
riservando la prescrizione dei sartani:
• in sostituzione degli ACE-inibitori: nei pazienti con tosse prolungata non risolvibile (ovvero non
responsiva a trattamento sintomatico) o con anamnesi di angioedema;
• in aggiunta agli ACE-inibitori: nei pazienti con scompenso cardiaco e alterata funzione sistolica
ventricolare sinistra (frazione di eiezione ventricolare sinistra ≤ 40%). Quest’ultima
combinazione terapeutica va condotta sotto la sorveglianza degli Ambulatori specialistici
dedicati.
17
Allegato 2c
Sintesi delle virtù terapeutiche degli inibitori di pompa protonica (IPP)
in funzione della loro efficacia relativa e del loro costo per il SSN
INIBITORI DI POMPA PROTONICA (IPP) DI PRIMA SCELTA
in assenza di specifiche indicazioni o controindicazioni
Tre recenti valutazioni dell’utilizzo, dell’efficacia e della sicurezza comparative degli IPP sono state realizzate
8
9
10
in Canada , Francia e negli Stati Uniti . In tutti i casi è stata effettuata una analisi critica sistematica degli
studi disponibili e della ricerca clinica in corso. Questa sintesi è basata su questi rapporti e sulle note AIFA
n° 1 e 48.
Quattro delle cinque molecole distribuite in Italia sono disponibili come medicinale equivalente:
Dose
Nomi commerciali
* dosi clinicamente equivalenti negli studi clinici standard*
esomeprazolo
Nexium, Axagon, Lucen, Esopral ed equivalenti
20 mg
lansoprazolo
Lansox, Limpidex, Zoton ed equivalenti
15 mg
omeprazolo
Mepral, Omeprazen, Losec, Antra ed equivalenti
20 mg
pantoprazolo
Peptazol, Pantecta, Pantopan, Pantorc ed equivalenti
20 mg
rabeprazolo
Pariet
10 mg
Dose
doppia*
40 mg
30 mg
40 mg
40 mg
20 mg
11
Gli IPP hanno tre indicazioni principali :
- l’eradicazione dell’Helicobacter Pylori e il trattamento delle ulcere gastroduodenali;
- la prevenzione e il trattamento delle lesioni gastroduodenali da FANS nei pazienti a rischio;
- il trattamento della malattia da reflusso gastroesofageo (MRGE) e dell’esofagite da reflusso.
Le indicazioni da scheda tecnica e la posologia consigliata differiscono tra i vari IPP.
L’ESSENZIALE
Non esistono differenze tra IPP in termini di efficacia e di tolleranza.
Non esistono indicazioni preferenziali tra IPP per alcuna delle indicazioni cliniche, se usati a dosaggi
clinicamente equivalenti e per periodi di trattamento adeguati, mentre il costo di acquisto è differente tra le
varie molecole. La prescrizione appropriata deve sempre portare pari attenzione all’economicità di acquisto
e alla qualità della risposta, che può variare nel singolo paziente.
Molte prescrizioni sono clinicamente poco giustificate.
Per un consistente numero di pazienti la prescrizione di un IPP è inappropriata. Allo stato attuale delle
conoscenze sono considerabili clinicamente poco giustificate le prescrizioni di IPP per:
- dispepsia funzionale (tranne che in presenza di una MRGE studiata con endoscopia)
- la prevenzione di lesioni gastroduodenali da FANS utilizzati per brevi periodi in pazienti non a rischio
(adulti di età minore di 65 anni, senza anamnesi di ulcera né in trattamento cronico con antiaggreganti,
anticoagulanti o corticosteroidi)
Quali IPP preferire?
Per la terapia iniziale della maggioranza dei disturbi acido-correlati qualsiasi IPP va bene purchè utilizzato a
dosi clinicamente equivalenti (dose standard, dose doppia; dose mezza in alcuni casi).
Quale dose impiegare all’inizio?
Non è stata dimostrata la superiorità clinica di dosi doppie equivalenti rispetto a dosi standard equivalenti.
La dose standard è sufficiente per il trattamento iniziale di tutti i tipi di pazienti.
Quando non servono gli IPP?
Gli studi clinici suggeriscono che gli IPP non hanno efficacia nel migliorare i sintomi dell’asma, i sintomi
laringei o la tosse cronica che può essere (solo statisticamente, non clinicamente) associata alla MRGE.
8
Evidence for PPI Use in Gastroesophageal Reflux Disease, Dyspepsia and Peptic Ulcer Disease. March 2007
Canadian Agency for Drugs and Technologies in Health (CADTH, Canada). COMPUS - Optimal therapy report. 1(2).
9
Médicaments inhibiteurs de la pompe à protons chez l’adulte. Déc. 2009 Haute Authoritè de Santé (Francia).
10
Drug Class Review: Proton Pump Inhibitors - May 2009. DERP, Oregon Health & Science University (USA).
11
Tutti gli IPP sono inoltre indicati nel trattamento della sindrome di Zollinger-Ellison e in altre patologie rare a gestione
specialistica che non sono prese in considerazione in questo rapporto.
18
VOLUMI DI IPP PRESCRITTI IN LOMBARDIA NEL 2001-201012
VOLUMI DI IPP PRESCRITTI NELLA PROVINCIA DI PAVIA, ANNI 2002-2010
Volumi di IPP prescritti in Provincia di Pavia - Anni 2002-2010
18,0
16,0
Consumo SSN (in DDD / 1.000 ab. die)
14,0
12,0
omeprazolo
10,0
pantoprazolo
lansoprazolo
rabeprazolo
8,0
esomeprazolo
6,0
4,0
2,0
0,0
2002
2003
2004
2005
2006
2007
2008
2009
2010
12
L’uso degli Inibitori di Pompa Protonica in Lombardia: analisi di farmacoutilizzazione e di farmacovigilanza. Febbraio 2011. Disponibile
sul sito web del Centro Regionale di Farmacovigilanza. Olivia Leoni*, Valentino Conti*, Lucrezia Magistro**, Stefania Scotto*
*Centro Regionale di FarmacoVigilanza Regione Lombardia **Scuola di Specializzazione in Farmacologia Medica,
Università degli Studi di Milano.
19
SOVRATRATTAMENTO INAPPROPRIATO
13
14 15
- Reazioni avverse: aumentato rischio di fratture , di infezione da Clostridium difficile , , associazione con
16
17
polmonite (CAP) , possibile con cancro colorettale
- Comorbidità o interazioni negative: warfarin, fenitoina, clopidogrel (ampia discussione)
- Remissione ottenuta o risposta inadeguata (revisione annuale per riduzione dose, trattamento
18
intermittente, antiacidi, alginati)
19
- IPP NON INDICATO: IL PRINCIPALE PROBLEMA (7), spinto dal marketing. Per disturbi vaghi o modesti
20
e uso "diagnostico" è sufficiente un breve periodo, per evitare una cronicizzazione inappropriata .
- Inappropriata profilassi in ospedale. Quando è necessario completare sul territorio un ciclo di profilassi
iniziato in ospedale, va garantita una distribuzione diretta.
COME SOSPENDERE I TRATTAMENTI CON IPP
I problemi della sospensione sono:
- ricaduta, ricorrenza, rischio di sanguinamento (in funzione della patologia indicante): non vanno sospesi
nel Barrett e in alcune esofagiti complicate (stenosi, ulcere, emorragie)
21
- possibile rimbalzo ipersecretivo acido, studiato per 12 settimane su 120 volontari sani : otto settimane
di IPP inducono entro due settimane dalla sospensione ipersecrezione acida che dura fino a 4 settimane
La sospensione deve essere graduale solo nei dispeptici o nei pazienti con MRGE che si presume possano
ritornare sintomatici. Negli altri casi la sospensione non comporta significativi problemi.
IN SOSTANZA
Iniziare gli IPP solo se e quando clinicamente indicato
Usarli per il minor tempo possibile
In alcuni casi22 considerare gli anti-H2
13
U.S. Food and Drug Administration (FDA). FDA Drug Safety Communication. Possible increased risk of fractures of
the hip, wrist, and spine with the use of proton pump inhibitors. May 2010.
14
Howell MD et al. Iatrogenic gastric acid suppression and the risk of nosocomial Clostridium difficile infection. Arch
Intern Med 2010; 170: 784-790.
15
Linksky A. Proton pump inhibitors and risk for recurrent Clostridium difficile infection. Arch Intern Med 2010: 170: 772778.
16
Herzig SJ et al. Acid-suppressive medication use and the risk for hospital-acquired pneumonia. JAMA 2009; 301:
2120-2128.
17
Chubak J. Colorectal cancer risk in relation to use of acid suppressive medications. Pharmacoepidemiology and Drug
Safety. 2009: 18: 540-544
18
National Institute for Health and Clinical Excellence (NICE). Dyspepsia: Managing dyspepsia in adults in primary care.
Clinical Guideline CG17, August 2004.
19
Katz MH. Failing the acid test. Benefits of proton pump inhibitors may not justify the risks for many users. Arch Int Med
2010; 170: 747-748.
20
McColl K, Gillen D. Evidence that proton-pump inhibitor therapy induces the symptoms it is used to treat.
Gastroenterology 2009; 137: 20-39.
21
Reimer C et al. Proton-pump inhibitor therapy induces acid-related symptoms in healthy volunteers after withdrawal of
therapy. Gastroenterology 2009; 137: 80-87.
22
Smith AD et al. Dyspepsia on withdrawal of ranitidine in previously asymptomatic volunteers. Am J
Gastroenterology 1999; 94: 1209-1213.
20
Allegato 2d
Sintesi delle virtù terapeutiche degli antibiotici
in funzione della loro efficacia relativa e del loro costo per il SSN
ANTIBIOTICI DI PRIMA SCELTA
in assenza di specifiche indicazioni o controindicazioni
In preparazione
21
Allegato 2e
Sintesi delle virtù terapeutiche delle Eparine a Basso Peso Molecolare (EBMP)
in funzione della loro efficacia relativa e del loro costo per il SSN
EPARINE DI PRIMA SCELTA
in assenza di specifiche indicazioni o controindicazioni
La seguente tabella riporta tutte le EBPM commercializzate in Italia, ordinate per le seguenti
cinque macroaree terapeutiche:
1. PROFILASSI TVP (TROMBOSI VENOSA PROFONDA)
In area medica
In area chiurugica
2. TRATTAMENTO TVP (TROMBOSI VENOSA PROFONDA)
3. TRATTAMENTO TVS (TROMBOSI VENOSA SUPERFICIALE) (“flebite”)
4. TRATTAMENTO MALATTIA CORONARICA
IMA NON Q (UA/NSTEMI)
IMA Q (STEMI)
PREVENZIONE EVENTI ACUTI PTCA
5. PROFILASSI COAGULAZIONE IN EMODIALISI
Per ciascuna area terapeutica sono riportate in colonna le indicazioni registrate in Italia per
ciascuna EBPM, suddivisa per dosaggio (espresso in UI per le EBPM ovvero in mg per il
fundaparinux).
Per ciascuna area terapeutica sono riportate in riga il testo delle indicazioni autorizzative. L’esame
di questi testi consente di rispondere alla semplice domanda:
ESISTONO DIFFERENZE CLINICAMENTE RILEVANTI TRA LE DIVERSE EBPM
ALL’INTERNO DI CIASCUNA DELLE CINQUE MACROAREE TERAPEUTICHE?
La letteratura scientifica (disponibile a richiesta, in previsione di inserimento nel sito aziendale)
documenta la seguente risposta a questa domanda:
NON ESISTONO DIFFERENZE DI EFFICACIA / SICUREZZA DI IMPORTANZA CLINICA TRA
LE EBPM, ALL’INTERNO DELLE 5 MACROAREE DI INDICAZIONE CLINICA.
L’esperienza del medico (disponibile nelle cartelle cliniche) potrebbe documentare risposte
differenti. In tal caso la ASL sarebbe interessata a venire a conoscenza dei casi di differente
efficacia / sicurezza, in modo da poterne tener conto nell’aggiornamento del presente allegato e
documentarli a garanzia dell’uso appropriato nel territorio della provincia e, laddove necessario,
poter avanzare alla Regione e alla Agenzia Italiana del Farmaco eventuali richieste di revisione o
precisazione delle indicazioni registrate.
22
Fo
Fo
Fo
Fo
Fo
Fo
5-7,5-10 mg
Arixtra
2,5 mg
Arixtra
Re
Arixtra
Pa
1,5 mg
REVIPARINA
FONDAPARINU
X
FONDAPARINU
X
FONDAPARINU
X
Clivarina
Tutti
PARNAPARINA
Tutti
Fluxum
Fraxiparina,
Seleparina
NADROPARINA
2.850-3.800-5.7007.600-9.500
Clexane T
Farmaco
Fraxodi, Seledie 11.400-15.200-19.000
ENOXAPARINA
6.000-8.000-10.00030.000
NADROPARINA
ENOXAPARINA
2.000-4.000
DALTEPARINA
Clexane
5.000-7.500-10.000
Ivor
Tutti
3.500
Ivor
Fragmin
2.500
Ivor
Dosaggi: UI ovvero mg
BEMIPARINA
BEMIPARINA
BEMIPARINA
INDICAZIONI REGISTRATE IN ITALIA DELLE EPARINE A BASSO PESO MOLECOLARE
(EBPM)xxii
EPARINA A BASSO PESO
MOLECOLARE: principio attivo
*Indicazione sia in area chirurgica che
medica
PROFILASSI TVP (TROMBOSI VENOSA PROFONDA)
CHIRURGIA
Profilassi TVP in chirurgia generale o ortopedica
Profilassi TVP in chirurgia generale e ortopedica ed in pz.non
chirurgici allettati e a rischio TVP
Profilassi TVP in chirurgia generale e ortopedica e nei pz. a
rischio maggiore di TVP
Prevenzione delle tromboembolie in pz. sottoposti ad
interventi di chirurgia ortopedica
Prevenzione delle tromboembolie in pz. sottoposti ad
interventi di chirurgia generale
Prevenzione di Episodi Tromboembolici Venosi23 (TEV)
Prevenzione di Episodi Tromboembolici Venosi24 (TEV)
Da
Na
En
Be
Be
MEDICINA
Profilassi TVP in chirurgia generale e ortopedica ed in pz.non
chirurgici allettati e a rischio TVP
Profilassi TVP in chirurgia generale e ortopedica e nei pz. a
rischio maggiore di TVP
Prevenzione degli Episodi Tromboembolici Venosi (TEV) in
adulti di pertinenza medica considerati ad alto rischio di TEV
e che sono immobilizzati a causa di una patologia acuta quale
insufficienza cardiaca e/o disturbi respiratori acuti e/o infezioni
o pat
En
Pa
Re
TRATTAMENTO TVP (TROMBOSI VENOSA PROFONDA)
Trattamento TVP
Trattamento TVP con o senza Embolia Polmonare
Trattamento TVP con o senza EP in fase acuta
Trattamento TVP acuta
Trattamento della TVP e dell’Embolia Polmonare (EP) acuta
eccetto nei pazienti emodinamicamente instabili o che
richiedono trombolisi o embolectomia polmonare
Na
En
En
Na
Pa
Re
Be
Da
Fo
TRATTAMENTO TVS (TROMBOSI VENOSA SUPERFICIALE)
Trattamento di adulti con trombosi venosa superficiale
sintomatica spontanea acuta degli arti inferiori in assenza di
trombosi venosa profonda concomitante
Fo
Fo
23
Prevenzione di Episodi Tromboembolici Venosi (TEV) negli adulti sottoposti a chirurgia ortopedica maggiore degli arti
inferiori quali frattura dell’anca, chirurgia maggiore del ginocchio o chirurgia sostitutiva dell’anca.
24
Prevenzione degli Episodi Tromboembolici Venosi (TEV) negli adulti sottoposti a chirurgia addominale considerati ad
alto rischio di complicanze tromboemboliche, quali pazienti sottoposti a chirurgia addominale per patologie tumorali.
23
2,5 mg
5-7,5-10 mg
Arixtra
Arixtra
1,5 mg
Arixtra
REVIPARINA
FONDAPARINU
X
FONDAPARINU
X
FONDAPARINU
X
Tutti
Clivarina
PARNAPARINA
Tutti
Fluxum
Fraxiparina,
Seleparina
NADROPARINA
2.850-3.800-5.7007.600-9.500
Clexane T
Farmaco
Fraxodi, Seledie 11.400-15.200-19.000
ENOXAPARINA
6.000-8.000-10.00030.000
NADROPARINA
ENOXAPARINA
2.000-4.000
Clexane
DALTEPARINA
Tutti
Fragmin
Dosaggi: UI ovvero mg
BEMIPARINA
5.000-7.500-10.000
Ivor
BEMIPARINA
3.500
Ivor
BEMIPARINA
2.500
Ivor
EPARINA A BASSO PESO
MOLECOLARE: principio attivo
*Indicazione sia in area chirurgica che
medica
TRATTAMENTO MALATTIA CORONARICA
IMA NON Q (UA/NSTEMI)
Trattamento angina instabile e IMA non Q
Trattamento angina instabile e infarto miocardico non Q in
associazione ad ASA
Malattia coronarica instabile quale angina instabile e IMA non
Q in associazione ad ASA
Trattamento dell’angina instabile o dell’IMA25
Na
En
En
Da
Fo
IMA Q (STEMI)
26
Trattamento dell’IMA
Trattamento dell’IMA27
En
Fo
PREVENZIONE EVENTI ACUTI PTCA
Prevenzione degli eventi acuti in angioplastica coronarica
transluminale percutanea (PTCA)
Re
PROFILASSI COAGULAZIONE IN EMODIALISI
Prevenzione della coagulazione in corso di emodialisi
Prevenzione della coagulazione nella circolazione
extracorporea durante emodialisi
Profilassi della coagulazione extracorporea in emodialisi e
nell’emofiltrazione fino a 4h di durata
En
Be
Na
Be
Da
A seguito della Determina AIFA del 16 luglio 2013 pubblicata sulla GU n. 175 del 27-7-2013 avente per
oggetto "Modalita' e condizioni di impiego del medicinale «PHT Eparine»", le eparine possono essere
erogate direttamente dalle strutture pubbliche e private accreditate per la "profilassi della TVP e
continuazione della terapia iniziata in ospedale, sia dopo intervento ortopedico maggiore, che dopo
intervento di chirurgia generale maggiore”.
25
Trattamento dell’angina instabile o dell’IMA senza sopra-slivellamento del tratto ST (UA/NSTEMI) in adulti nei quali un
approccio invasivo urgente (PCI) (<120 minuti) non è indicato.
26
Trattamento dell’infarto acuto del miocardio con sovraslivellamento del segmento ST, inclusi pazienti in terapia medica
o da sottoporre a successivo intervento coronarico percutaneo.
27
Trattamento dell’IMA associato a sopra-slivellamento del tratto ST (STEMI) in adulti in terapia con trombolitici o che,
inizialmente, non sono deputati a ricevere altre forme di terapia di riperfusione.
24
EBPM per le indicazioni registrate e per la gestione perioperatoria del paziente
cronicamente anticoagulato (ACO): revisione della documentazione comparativa
disponibile
0
Valutazioni di tecnologia sanitaria (HTA)
0
Revisioni sistematiche e metanalisi
10
Studi clinici controllati randomizzati
6
Linee guida cliniche
Embolia polmonare DGR 78: nelle regioni italiane varia da 3.794 a 5.845 €
Tromboflebite delle vene profonde DRG 128: nelle regioni italiane varia da 2.087 a 3.763 €
25
Farmaci di sintesi biologica (biotecnologici)
I BIOSIMILARI
Introduzione
Negli anni 70-80, con l’avvento delle tecniche di ingegneria genetica, si è assistito alla diffusione di
farmaci biotecnologi, costituiti da proteine o glicoproteine, ottenuti mediante processi di estrazione
e purificazione a partire da substrati cellulari che hanno subito un procedimento di
ingegnerizzazione (inserzione del gene di interesse) o modifica (fusione cellulare, linee continue,
monoclonali) di varia entità. Tali farmaci hanno ampiamente migliorato il trattamento di diverse
condizioni patologiche come neoplasie, diabete, epatiti, sclerosi multipla, artrite reumatoide e
psoriasi. Nel 2010 i farmaci biologici disponibili erano circa 250 e diverse centinaia erano in via di
sviluppo. Tuttavia, a circa 30 anni di distanza dal primo farmaco biologico immesso sul mercato,
numerosi brevetti concernenti queste molecole sono scaduti o stanno per scadere, aprendo la
strada alla produzione di farmaci biosimilari, così come è successo per i generici di farmaci non
biologici. A differenza però di questi ultimi, vista la complessità sia di struttura sia di produzione dei
farmaci biologici, l’autorizzazione all’immissione in commercio (AIC) di un biosimilare deve seguire
procedure molto diverse da quelle necessarie per l’introduzione sul mercato di un generico.
Differenze tra farmaci biologici e farmaci di sintesi chimica
I farmaci biologici, costituiti, nella maggior parte dei casi, da molecole proteiche, presentano
importanti differenze rispetto ai farmaci convenzionali ottenuti tramite reazioni chimiche
standardizzate. Le differenze principali sono in termini di:
- peso molecolare: le molecole proteiche presentano un peso molecolare che varia da 10.000 a
200.000 Daltons, rispetto ai farmaci sintetizzati chimicamente che presentano un peso di circa
poche centinaia di Daltons;
- struttura: i farmaci proteici presentano una struttura tridimensionale altamente complessa. E’
possibile mimare la sequenza aminoacidica iniziale (struttura primaria), ma è molto difficile imitare
alla perfezione i “ripiegamenti” che ne costituiscono la struttura secondaria, terziaria e quaternaria;
- stabilità: le proteine sono molecole molto instabili e possono essere strutturalmente danneggiate
da fonti di calore, solventi organici, ossigeno, modificazione di equilibrio acido- base e altre fattori
che ne determinano la perdita completa o parziale dell’attività biologica;
- microeterogeneità: il prodotto proteico finale non dipende solo dalla codifica del DNA, ma anche
da modifiche legate alla cellula (glicosilazione, acetilazione, fosforilazione, proteolisi) e al
processo (ossidazione, deamidazione) o a modifiche intenzionali postraduzionali (es. aggiunta di
polietilenglicole). Ad esempio, in uno studio del 2005, analizzando con la spettrometria di massa l’
rh-GH (recombinant human growth hormone), è stata riscontratata una sostituzione aminoacidica
nel 2% dei prodotti finali (che può quindi portare a modifiche di struttura e quindi di antigenicità).
Inoltre, la complessità del farmaco biotecnologico è legata anche al processo produttivo,
caratterizzato da una certa variabilità: si può quindi affermare per questa classe di farmaci che “il
prodotto è il processo”. Le fasi principali di produzione sono:
1) clonazione della sequenza di DNA da codificare e inserimento di tale DNA in un vettore
plasmidico;
2) trasferimento del vettore plasmidico in “cellule ospiti”;
3) identificazione delle cellule che formano il prodotto in quantità e di qualità desiderate: tali cellule,
una volta selezionate, costituiranno una “banca cellulare” per le successive produzioni;
4) espansione del volume cellulare fino a migliaia di litri, in base alla quantità di prodotto
desiderata;
5) purificazione del prodotto proteico;
6) trasferimento del prodotto finale in una formulazione e device adatti per il trasporto, per la
conservazione e l’uso da parte del paziente.
Vista la complessità del processo, risulta chiaro che, se un secondo produttore volesse riprodurre il
medesimo farmaco, dovrebbe sicuramente seguire le tappe descritte sopra, ma non sarebbe in
grado di ottenere un prodotto identico senza avere accesso al materiale del prodotto di riferimento
26
(banca cellulare) e alle procedure operative. Nello specifico, il trasferimento nelle “cellule ospiti”
rappresenta un evento unico, che non può essere replicato in maniera identica e la
microeterogeneità del prodotto proteico dipende dal processo di produzione: queste variazioni
potrebbero avere un impatto sulla qualità, le caratteristiche farmacocinetiche/farmacodinamiche e
sull’efficacia clinica del prodotto.
Definizione di biosimilare e regolamentazione
Il farmaco biosimilare (definito anche follow-on biologicals, follow-on protein products) è un
farmaco biologico che presenta caratteristiche di similarità in termini di qualità, sicurezza ed
efficacia rispetto ad prodotto di riferimento già in commercio. Esso può essere commercializzato
alla scadenza del brevetto del farmaco di riferimento e, vista la complessità di produzione,
l’autorizzazione all’immissione in commercio deve essere strettamente regolamentata.
La differenza tra generico convenzionale e biosimilare viene definito nell’Articolo 10 (4) della
Direttiva 2001/83/CE modificata dalla Direttiva 2004/27/CE:
“ Medicinale generico: un medicinale che ha la stessa composizione qualitatitiva e quantitativa di
sostanze attive e la stessa forma farmaceutica del medicinale di riferimento nonché una
bioequivalenza con il medicinale di riferimento dimostrata da studi appropriati di biodisponibilità”
“Quando un medicinale biologico simile a un medicinale biologico di riferimento non soddisfa le
condizioni della definizione del medicinale generico, a causa in particolare di differenze attinenti
alle materie prime o di differenze nei processi di fabbricazione del medicinale biologico di
riferimento, devono essere forniti i risultati delle appropriate prove precliniche o delle
sperimentazioni cliniche relative a dette condizioni”.
Bisogna tenere in considerazione che anche una minima differenza strutturale del prodotto
proteico può avere un impatto clinico significativo. Le differenze nella struttura primaria proteica
non sono compatibili con il concetto di biosimilarità, secondo le linee guida EMA che vedremo in
seguito. Per le proteine glicosilate, le differenze nel pattern di glicosilazione possono influenzare
significativamente l’interazione tra receptor-binding protein e proteina e le caratteristiche
farmacocinetiche. In termini di sicurezza, l’immunogenicità (tipica della maggior parte dei farmaci
biologici), non dipende solo da fattori legati al paziente, ma anche dalla qualità del prodotto
proteico: ad esempio la presenza/assenza di glicosilazione così come la presenza di impurità o
aggregati possono essere associati alla produzione di anticorpi. Quindi, biofarmaci prodotti da
aziende diverse non possono essere considerati a priori equivalenti dal punto di vista del rischio
immunogenico. Ricordiamo il caso di qualche anno fa che, dopo l’approvazione di una nuova
formulazione di eritropoietina, si è assistito ad un aumento di casi di aplasia pura della serie rossa.
Un importante prerequisito per lo sviluppo dei farmaci biosimilari è stato il progresso delle tecniche
analitiche negli ultimi 20 anni. Le tecniche disponibili una decina di anni fa infatti non erano in
grado di studiare le complesse strutture delle molecole proteiche. Le tecniche oggi disponibili sono
invece in grado di studiare la struttura secondaria, terziaria e quaternaria delle proteine, fornendo
dati in termini di qualità e caratteristiche dei biofarmaci. In realtà, la eventuali differenze del
prodotto finale non sono completamente evidenziabili dai metodi analitici, per cui risulta
fondamentale, per la qualità del prodotto, un buon controllo sul processo produttivo. E’ di
fondamentale importanza che qualsiasi regolamentazione per l’approvazione dei farmaci
biosimilari trovi il giusto equilibrio tra l’ingresso nel commercio di tali farmaci, la loro competitività
sul mercato e la sicurezza per il paziente. Bisogna infine ricordare che i farmaci biosimilari, benché
portino benefici dal punto di vista economico, per definizione non portano alcun progresso
scientifico, in quanto il medicinale di riferimento è già disponibile sul mercato con provata efficacia
e sicurezza da molti anni.
L’EMA (European Medicines Agency) è stata la prima a pubblicare delle Linee Guida sui farmaci
biosimilari nell’ottobre 2005 (ultima revisione 31 maggio 2012). Esse prevedono, oltre alle
informazioni sulla qualità che si rendono necessarie per l’AIC (Autorizzazione Immissione in
Commercio) di qualsiasi farmaco, l’obbligo di condurre studi comparativi preclinici e clinici che
confrontino il biosimilare con il farmaco biologico di riferimento, definiti come comparability
exercise o esercizio di comparabilità. Tali studi hanno l’obiettivo di dimostrare come il farmaco
biosimilare abbia un profilo di efficacia e sicurezza sovrapponibile a quello del farmaco di
riferimento.
27
Nello specifico le linee guida si dividono in:
- Linee guida generali per i farmaci biosimilari che comprendono:
1) Linee guida sulla qualità:
- il principo attivo del biosimilare in studio deve essere simile in termini molecolari e biologici al
principio attivo del prodotto di riferimento; inoltre la forma farmaceutica, la potenza e la via di
somministrazione devono essere le stesse del prodotto di riferimento (se non lo sono occorreranno
dati aggiuntivi);
- devono essere valutate e considerate le caratteristiche della molecola e del processo di
produzione con esercizio di comparabilità (il range di accettabilità delle differenze dovrà sempre
essere stabilito a priori): è fondamentale comparare e definire le caratteristiche chimico-fisiche
(composizione, proprietà fisiche, impurità), attività biologica e purezza. Bisogna sottolineare che
quando si confronta la stabilità di due molecole proteiche prodotte da due aziende diverse, si
possono riscontrare delle differenze
che possono dipendere dalle diverse formulazioni:
rimuovendo tali “interferenze” (eccipienti, ad esempio) con metodi specifici, si possono ottenere
delle conclusioni più verosimili in termini di equivalenza fisico-chimica (come dimostrato in uno
studio del 2010 su due formulazioni di rhGH).
2) Linee guida sulle valutazioni precliniche:
- studi in vitro: per stabilire il grado di comparabilità in termini di reattività;
- studi in vivo su animali allo scopo di valutare gli effetti farmacodinamici/attività rilevanti per
l’applicazione clinica e la tossicità non clinica compreso il titolo anticorpale e la reattività crociata.
Non sono richiesti invece studi sulla tossicità a livello riproduttivo, sulla mutagenicità e
carcinogenicità, a meno che non sia specificatamente indicato;
3) Linee guida sulle valutazioni cliniche: i requisiti dipendono dalle conoscenze esistenti sul
farmaco biologico di riferimento e dalle indicazioni terapeutiche richieste. Lo studio di comparabilità
in termini clinici inizia con studi di farmacocinetica e farmacodinamica, seguiti da studi di efficacia e
sicurezza. Nello specifico:
- studi di farmacocinetica: è necessario comparare le caratteristiche farmacocinetiche (clearance,
emivita, assorbimento e biodisponibilità) del biosimilare e del farmaco di riferimento;
- studi di farmacodinamica: gli effetti farmacodinamici del biosimilare e del farmaco di riferimento
devono essere comparati in soggetti dove possono essere osservate le maggiori differenze;
- studi di efficacia e di sicurezza per dimostrare che il biosimilare e il farmaco di riferimento sono
comparabili dal punto di vista dell’efficacia (equivalenza più che “non inferiorità”) e della sicurezza.
Esse dovrebbero essere valutate per ciascuna delle indicazioni terapeutiche, anche se è possibile
in alcuni casi estrapolarle dal prodotto di riferimento. Nello specifico, per quanto riguarda la
sicurezza, sono richiesti dati di sicurezza pre-marketing su un numero di pazienti sufficiente per
identificare il tipo, la gravità e la frequenza degli eventi avversi nel biosimilare e nel prodotto di
riferimento e dovrà essere eseguito un monitoraggio post-marketing (al momento della
commercializzazione la ditta produttrice dovrà presentare un programma di farmacovigilanza in
accordo con la legislazione EU). Uno dei punti cruciali è l’immunogenicità, caratteristica di tutti i
farmaci biotecnologici e non solo dei biosimilari. In generale, le principali conseguenze cliniche
dell’immunogenicità sono reazioni allergiche, aumento di efficacia, perdita di efficacia e
neutralizzazione di proteine endogene (come nel caso della aplasia cellulare della serie rossa
dopo somministrazione di Epoetina alfa (Eprex®), in cui la risposta anticorpo mediata indotta dal
farmaco ha neutralizzato anche l’EPO endogena). I fattori di rischio per l’immunogenicità sono
legati alla proteine e al paziente.
Fattori di rischio legati alle proteine:
- struttura (glicosilazione, variazione della sequenza primaria rispetto alla endogena);
- contaminanti (provenienti dalla cellula ospite): a volte si formano nella parte iniziale del processo
di produzione e la loro presenza può essere ridotta migliorando la tecnica di purificazione, in altri
casi si liberano a seguito di cambiamenti introdotti nella catena di produzione o a valle del
processo;
- formulazione ed eccipienti: possono modificare la conformazione e il potenziale immunogeno,
quindi la formulazione dovrà essere in grado di mantenere la conformazione originaria della
proteina sintetizzata. In particolare alcune tecniche come la liofilizzazione possono indurre
l’ossidazione e l’aggregazione delle proteine aumentandone il potenziale immunogenico.
Fattori di rischio legati al paziente
28
- via di somministrazione: le modalità di somministrazione intramuscolare e sottocutanea sono
maggiormente immunogene rispetto alla via endovenosa e al trattamento topico;
- dose e durata del trattamento: le terapia a breve termine sembrano essere meno immunogene
rispetto a quelle a lungo termine e i prodotti somministrati in maniera intermittente sono associati
ad una maggiore probabilità di sviluppare risposta immune rispetto a quelli somministrati in
maniera continuativa;
- immunodepressione: legata a minore probabilità di sviluppare anticorpi rispetto ai soggetti
immunocompetenti;
- assenza congenita di proteine endogene: soggetti con deficit di ormone della crescita o di fattore
VIII potrebbero non avere la normale tolleranza verso i prodotti biotecnologici ed essere più rapidi
a sviluppare anticorpi in quanto il loro sistema immunologico non ha avuto precedenti contatti con
tale tipo di proteina;
- infezioni croniche: i pazienti con tali infezioni sono più predisposti a sviluppare risposte immuni;
- terapie concomitanti: alcuni farmaci possono aumentare o ridurre il rischio di una risposta
immune a una proteina terapeutica;
- precedente esposizione a proteina simile: tale evenienza può essere causa di una risposta
immune a seguito di presensibilizzazione.
Quindi, dal punto di vista regolatorio, nella valutazione della tossicità di un nuovo prodotto
biotecnologico, è richiesta anche negli studi pre-clinici la misurazione del titolo anticorpale, la
valutazione dei possibili meccanismi che la determinano e le possibili conseguenze cliniche.
Ricordiamo però che l’immunogenicità negli umani non è prevedibile solo con test in vitro o con
animali, per cui sono fondamentali gli studi clinici.
Ritornando ai biosimilari, nella valutazione degli aspetti clinici e non clinici, l’immunogenicità deve
essere valutata seguendo:
- una strategia di rilevazione/identificazione degli anticorpi: la strategia per la rilevazione degli
anticorpi prevede diverse tipologie di tecniche analitiche (saggi di screening, di conferma, di
specificità degli anticorpi e di valutazione dell’esistenza del fenomeno della neutralizzazione);
dovrebbero essere utilizzati test validati e abbastanza sensibili al fine di rilevare anche un basso
titolo anticorpale o anticorpi con bassa affinità di legame; la periodicità e la tempistica applicata
per la rilevazione degli anticorpi dovrebbe essere giustificata dal produttore.
- caratterizzazione della risposta immune osservata: è importante raccogliere dati sulla
caratterizzazione della risposta anticorpale per tutte le indicazioni di un determinato prodotto; in
particolare, per i prodotti di uso cronico, è necessario conoscere l’evoluzione e la persistenza della
risposta immune osservata ( per i prodotti di uso cronico è previsto che tale valutazione venga
effettuata con studi preregistrativi della durata di almeno un anno; la mancanza di questi dati dovrà
essere motivata).
- valutazione della correlazione tra gli anticorpi prodotti e la farmacocinetica/farmacodinamica: il
confronto della risposta clinica prima e dopo l’induzione di anticorpi può fornire informazioni sulla
correlazione tra lo sviluppo di anticorpi e la risposta clinica stessa;
- esercizio di comparabilità in termini di risposta anticorpale: è richiesto il confronto tra il biosimilare
e il prodotto di riferimento in termini di risposta anticorpale. In assenza di questi dati o in caso di
ipotetico rischio per la sicurezza derivante da studi pre clinici e clinici, si rende necessario ottenere
ulteriori informazioni sulle conseguenze di una potenziale risposta immune non desiderata. Per i
farmaci a uso prolungato sono richiesti dati per un follow up di un anno;
- piano di gestione del rischio: quando si programma uno studio su un nuovo farmaco
biotecnologico, deve essere presentato all’EMA un piano di gestione del rischio che prevede la
valutazione dell’immunogenicità. I dati richiesti nella fase di sviluppo clinico dipenderanno dalla
frequenza dell’evento avverso, dal potenziale immunogeno della proteina e della rarità della
condizione clinica di base.
Oltre alle linee guida generali, l’EMA ha approvato delle linee guida specifiche per le diverse
classi di farmaci:
- Linee guida per le eritropoietine ricombinanti (prima stesura marzo 2006, sostituite aprile 2010);
- Linee guida per l’ormone della crescita (febbraio 2006);
- Linee guida per i G-CSF (febbraio 2006);
- Linee guida per le insuline ricombinanti (febbraio 2006, revisionate luglio 2011);
- Linee guida per gli interferoni α ricombinanti (giugno 2009);
29
- Linee guida per le eparine a basso peso molecolare (aprile 2009, revisionate luglio 2011);
- Linee guida per gli anticorpi monoclonali (giugno 2012).
Risultano in bozza le linee guida per l’interferone β ricombinante e per l’ormone follicolo
stimolante.
I farmaci biosimilari ad oggi approvati dall’EMA sono elencati nella tabella che segue:
INN
Name
Company
Somatropin Omnitrope
Sandoz
Somatropin Valtropin
Biopartners
Epoetin
alfa
Epoetin
alfa
Epoetin
alfa
Epoetin
zeta
Epoetin
zeta
Binocrit
Sandoz
Epoetin alfa
Hexal
Sandoz
(Hexal)
Abseamed
Medice
Retacrit
Hospira
Silapro
Stada
Filgrastim TevaGrastim
Filgrastim
Filgrastim
Ratiopharm
Filgrastim Biograstim
Teva
Ratiopharm
CT
Arzneimittel
Filgrastim Filgrastim Hexal Hexal
Filgrastim Zarzio
Sandoz
Filgrastim Nivestim
Hospira
CHMP
opinion
January
Genetropin (Pfizer)
2006
February
Humatrope (Lilly)
2006
Eprex/Erypro
June 2007
(JnJ/Amgen)
Eprex/Erypro
June 2007
(JnJ/Amgen)
Eprex/Erypro
June 2007
(JnJ/Amgen)
Eprex/Erypro
October
(JnJ/Amgen)
2007
Eprex/Erypro
October
(JnJ/Amgen)
2007
February
Neupogen (Amgen)
2008
February
Neupogen (Amgen)
2008
February
Neupogen (Amgen)
2008
October
Neupogen (Amgen)
2008
October
Neupogen (Amgen)
2008
Neupogen (Amgen) March 2010
Reference
EU approval
April 2006
April 2006
August 2007
August 2007
August 2007
December
2007
December
2007
September
2008
September
2008
September
2008
February
2009
February
2009
June 2010
Non tutti i biosimilari presentati all’EMA sono stati autorizzati e in particolare nel luglio 2011 un
farmaco è stato rifiutato e altri 3 sono stati ritirati. E’ il caso ad esempio del biosimilare della
classe degli interferoni-2a, Alpheon®, comparato con Roferon-A (farmaco di riferimento).
Innanzitutto erano state rilevate delle differenze tra i 2 medicinali in termini di impurità; inoltre la
CHMP (Committee for Medicinal Products for Human Use) aveva stabilito che il processo in uso
per la produzione del farmaco non era stato adeguatamente validato. Infine, negli studi clinici,
Alpheon era risultato equivalente a Roferon-A nel trattamento dell’epatite C ma con una
percentuale più elevata di ripresa di malattia e di eventi avversi.
Biosimilari e problemi “aperti”
Benchè la regolamentazione dei biosimilari sia in atto in molti Paesi, rimangono ancora dei
problemi aperti, fonte di discussione.
1) Prodotto di riferimento: deve essere un farmaco in uso e autorizzato dalla Comunità Europea,
mentre dati relativi a farmaci in uso fuori dalla Comunità Europea possono solo fornire informazioni
di supporto; in questo modo le autorità regolatorie hanno pieno accesso ai dati del prodotto di
riferimento per l’esercizio di comparabilità. Così facendo, se in tutti i paesi vigesse la stessa regola,
il farmaco biosimilare autorizzato in Paesi diversi potrebbe differire in termini di formulazione,
produzione e potenza poichè l’azienda produttrice dovrebbe condurre studi di comparabilità con
medicinali di riferimento autorizzati in quel paese;
30
2) Foglietto informativo: per i generici di farmaci convenzionali, il foglietto illustrativo è identico a
quello del prodotto di riferimento. Questo non può essere un approccio corretto per il biosimilare: in
particolare è necessario differenziare i dati di efficacia e sicurezza che sono stati ottenuti con studi
clinici con il farmaco biosimilare per determinate indicazioni da quelli estrapolati, per altre
indicazioni, dal farmaco di riferimento. Questa distinzione non è presente per alcuni biosimilari
autorizzati in Europa;
3) Farmacovigilanza: se la farmacovigilanza è fondamentale per tutti i nuovi farmaci immessi sul
mercato, ancora di più lo è per i biosimilari per cui la strada dell’autorizzazione in commercio è
molto più rapida. Alcuni autori ritengono che il sistema di farmacovigilanza basato sulla
segnalazione spontanea degli operatori sanitari può non essere sufficiente (rischio di “sotto
segnalazioni”), per cui dovrebbe essere ideato un metodo più appropriato. Inoltre, per il
monitoraggio post marketing del farmaco è fondamentale la tracciabilità, per cui i biosimilari
dovrebbero avere nomi commerciali e non solo essere nominati secondo il sistema INN
(International non-proprietary name)
4) Interscambialità: mentre i generici dei farmaci convenzionali sono considerati (entro certi limiti)
interscambiabili, per i biosimilari l’interscambiabilità dovrebbe essere dimostrata da studi scientifici
di comparabilità soprattutto in termini di sicurezza ed in particolare di immunogenicità. L’EMA
afferma che la scelta di trattare un soggetto con il prodotto di riferimento o con il biosimilare
dovrebbe essere di pertinenza del professionista medico con provata esperienza.
31
Allegato 2f
Alla luce della normativa e delle indicazioni regolatorie vigenti:
1) I farmaci di sintesi biologica (originatori o biosimilari):
- non possono essere considerati sostituibili dal farmacista all’atto della dispenzazione;
- non possono essere considerati intercambiabili dal medico all’atto della prescrizione.
2) I farmaci biosimilari:
- consistono in una alternativa terapeutica aggiuntiva a disposizione del medico e del
paziente;
- possono essere una alternativa preferibile, quando presentino:
a) efficacia terapeutica relativa comparabile ai prodotti originatori e
b) minor costo per il SSN e per il cittadino
Sintesi delle virtù terapeutiche dei farmaci con ormone somatotropo
in funzione della loro efficacia relativa e del loro costo per il SSN
ORMONE DELLA CRESCITA
in assenza di specifiche indicazioni o controindicazioni
La somatotropina o ormone della crescita è impiegata in clinica come trattamento sostitutivo nei
casi di carenza congenita o acquisita dell’ormone ipofisario. La prima somatotropina ricombinante
umana è stata approvata nel 1985 con il nome di Sumatrem®, seguita in breve tempo
dall’approvazione da parte dell’FDA di altri sette ormoni della crescita ricombinanti.
A seguito della scadenza del brevetto di Genotropin® (GH umano ricombinante registrato e
protetto da brevetto Pfizer), nel 2003 è stato presentato alla FDA un dossier registrativo di un GH
ricombinante umano biosimile, Omnitrope® (Sandoz), completo di dati di sicurezza ed efficacia in
parte originali ed in parte desunti dal dossier registrativo di Genotropin®. Gli studi chimico-fisici
dimostravano un alto grado di equivalenza tra Omnitrope® e Genotropin®, con identica struttura
primaria, secondaria e terziaria della proteina terapeutica e livelli simili di impurezza. I dati di
farmacologia e tossicologia pre-clinica erano favorevoli in diverse specie e i dati di farmacocinetica
e farmacodinamica umana (fase I) dimostravano stretta equivalenza tra il biosimile ed il suo
originatore. La valutazione di efficacia e sicurezza clinica si era basata su studi clinici di fase III,
condotti su popolazioni pediatriche trattate per carenza di GH, con brevi o lunghi periodi di
osservazione. La valutazione dei parametri clinici “altezza” e “tasso di crescita” ha
confermato il profilo di efficacia di Omnitrope®, mentre l’osservazione di 51 pazienti pediatrici per
24 mesi ha confermato lo scarso potere immunogeno del biosimile, dal momento che nessuno dei
pazienti aveva sviluppato anticorpi durante il trattamento. Sulla base dei risultati di questi studi il 12
Aprile 2006 la Commissione Europea ha espresso il parere che Omnitrope® possiede un profilo
simile a quello di Genotropin® per quanto riguarda qualità di produzione, efficacia clinica e
sicurezza e ha concesso a Sandoz
l’autorizzazione all’immissione in commercio di Omnitrope® con l’indicazione “deficit di GH in
pazienti pediatrici”. L’approvazione della seconda indicazione richiesta e cioè “deficit di GH in
pazienti adulti” è stata ottenuta invece sulla base di dati desunti dalla letteratura scientifica
internazionale e dalla valutazione del dossier registrativo di Genotropin® Pfizer (EMEA/H/C/607).
L’osservazione post-marketing di efficacia e tollerabilità a lungo termine di Omnitrope® è stata
valutata in 89 bambini con deficit dell’ormone della crescita in uno studio randomizzato e
controllato con Genotropin®, che prevedeva il trattamento farmacologico con GH ricombinante per
69 mesi (Romer et al., 2009a). Omnitrope® ha dimostrato efficacia e tollerabilità comparabile a
Genotropin® nei 7 anni di follow-up.
Le linee guida EMA per la somatotropina prevedono:
1)Studi non clinici che comprendono:
- studi di farmacodinamica: in vitro, per evidenziare qualsiasi differenza in termini di reattività tra il
biosimilare e il farmaco di riferimento; in vivo, utilizzando un modello murino per comparare
l’attività farmacodinamica dei due farmaci;
- studi tossicologici: si richiede almeno uno studio di tossicità a dose ripetuta della durata di
almeno 4 settimane in una specie animale come il topo, dando particolare importanza alla risposta
32
immunogenica. Come già accennato in precedenza per le linee guida generali, non sono richiesti
studi di sicurezza farmacologica, tossicità riproduttiva, mutagenicità e carcinogenicità.
2) Studi clinici che comprendono:
- studi di farmacocinetica: le proprietà farmacocinetiche del biosimilare e del farmaco di riferimento
devono essere valutate in uno studio clinico in crossover utilizzando la via di somministrazione
sottocutanea. Sono ritenuti appropriati dei volontari sani, ma è richiesta la soppressione della
produzione di GH endogeno utilizzando un analogo della somatostatina. I parametri da
considerare sono la AUC, la Cmax e l’emivita. Il margine di comparabilità deve essere definito a
priori;
- studi di farmacodinamica: la farmacodinamica dovrebbe essere valutata come parte dello studio
comparativo di farmacocinetica. Il marker farmacodinamico da considerare è il valore di IGF1, in
aggiunta si può usare anche IGFBP3. Però, poiché non esiste una chiara relazione lineare tra i
livelli di IGF1 e la risposta clinica in termini di crescita, IGF1 non può essere considerato un marker
surrogato di efficacia negli studi clinici;
- studi di efficacia: l’efficacia clinica deve essere dimostrata in almeno un RCT adeguatamente
potenziato di confronto tra il biosimilare e il farmaco di riferimento, condotti in doppio cieco. Gli
studi dovrebbero avere come popolazione target soggetti con deficit di GH naive per la terapia e in
età prepubere (per evitare l’interferenza dello spurt puberale) e dovrebbero fornire indicazioni su
come misurare l’altezza. Il primo marker di efficacia è la modifica della velocità di crescita e delle
SD della velocità di crescita rispetto al basale. La durata dello studio deve essere minimo di 6
mesi. La crescita deve essere valutata per un periodo minimo di 6 mesi e massimo di 18 mesi
prima di iniziare lo studio;
- studi di sicurezza: dati di sicurezza derivati da RCT di efficacia. I dati dell’immunogenicità si
ottengono dai pazienti arruolati in trial di efficacia dosando gli anticorpi ogni 3 mesi con metodi
altamente specifici e sensibili. In aggiunta, si possono ottenere campioni per dosare IGF1,
IGFBP3, insulina e glicemia a digiuno;
- piano di farmacovigilanza: include un dettaglio di farmacovigilanza su come impostare le
valutazioni post marketing sulla base dei rischi individuati negli studi effettuati.
Biosimilari e Ormone della Crescita
La somatotropina (rh-GH) è un polipeptide di 22 kD non glicosilato prodotto da E. coli, immessa in
commercio in USA e in Europa nel 1987 sotto il nome commerciale di Humatrope®.
Successivamente è stata autorizzata sul mercato sotto il nome commerciale di Genotropin® in
Europa nel 1987 e in USA nel 1995. La versione biosimilare di Genotropin®, commercializzata con
il nome commerciale di Omnitrope® (Sandoz), è stato il primo biosimilare approvato in Europa nel
dicembre 2006; è stato invece approvato negli USA nel maggio 2006, non come biosimilare ma
sotto la sezione 505 (b) del FD&C Act (Federal Food and Cosmetics Act). Un altro biosimilare di
Humatrope, Valtropin® (LG Life Sciences, Biopartners), non in commercio in Italia, è stato
autorizzato dall’EMA nell’aprile 2006 e successivamente dalla FDA nell’aprile 2007.
Dati di letteratura: studi di farmacocinetica e farmacodinamica
Stanhope R. et al. Bioequivalence studies of Omnitrope, the first biosimilar/rhGH follow on protein.
J Clin Pharmacol, 2010.
Sono stati condotti due studi di fase I, randomizzati, crossover, in doppio cieco, su 24 volontari
sani con soppressione farmacologica della produzione endogena di GH. Sono state paragonate 3
formulazioni differenti di rhGH: Omnitrope® liofilizzato, Omnitrope® liquido e Genotropin® (polvere
liofilizzata). Sono stati valutati i parametri farmacocinetici (AUC, Cmax, Tmax ed T1/2) e
farmacodinamici (livelli di IGF1, IGFBP3 e acidi grassi non esterificati) dopo una singola
somministrazione sottocutanea del farmaco alla dose di 5 mg. Le tre formulazioni sono risultate
equivalenti in termini farmacocinetici e farmacodinamici. In particolare Omnitrope® liofilizzato è
risultato equivalente a Genotropin® e Omnitrope® liquido.
Fuhr U et al. Bioequivalence between novel ready-to-use liquid formulations of the recombinant
human GH Omnitrope and the original lyophilized formulations for reconstitution of Omnitrope and
Genotropin. Eur J End 2010
Sono stati condotti 2 studi randomizzati e in doppio cieco su 36 volontari sani con soppressione
farmacologica della produzione di GH endogena. Scopo degli studi è stata quella di comparare in
termini di farmacocinetica, farmacodinamica, sicurezza e tolleranza locale una nuova formulazione
di Omnitrope® liquida con la formulazione di Omnitrope® in polvere e di Genotropin® in polvere,
33
dopo una singola dose di 5 mg sc. Lo studio ha concluso per la bioequivalenza, in termini di
farmacocinetica, farmacodinamica e di sicurezza delle 3 formulazioni.
Dati della letteratura: studi clinici
Romer T et al. Seven years of safety and efficacy of the recombinant human growth hormone
Omnitrope in the treatment of Growth hormone deficient children. 2009, Hormone Res.
E’ stato condotto uno studio clinico di fase III su 89 bambini in età prepubere con deficit di GH.
Obiettivo dello studio era comparare efficacia e sicurezza di Omnitrope® con Genotropin® e
valutare l’efficacia e la sicurezza di Omnitrope® in un periodo di trattamento di 7 anni. Lo studio
consisteva di 3 fasi:
Fase 1: 44 pazienti ricevevano Omnitrope® liofilizzato e 45 pazienti Genotropin® per 9 mesi alla
dose di 0.03 mg/kg/die.
Fase 2: i pazienti che avevano ricevuto Omnitrope® hanno proseguito lo stesso farmaco per 6
mesi, mentre i pazienti che avevano ricevuto Genotropin® hanno proseguito con Omnitrope®
liquido per 6 mesi.
Fase 3: i pazienti di entrambi i gruppi ricevevano Omnitrope® liquido per un periodo di 69 mesi.
Risultati: sono stati valutati 4 parametri auxologici (altezza, altezza in SD, velocità di crescita e
velocità di crescita in SD) e dosaggio di IGF1 e IGFBP3. Tali parametri erano comparabili tra i due
gruppi di pazienti e confermavano l’efficacia e la sicurezza di Omnitrope® per un periodo di
trattamento di 7 anni.
In una fase dello studio di pre marketing, il 60% circa dei pazienti che avevano ricevuto una
formulazione liofilizzata di Omnitrope® (poi non commercializzata), avevano sviluppato anticorpi
anti GH. Questa caratteristica era stata attribuita alla presenza di un livello non accettabile di
proteine derivate dalle “cellule ospiti”; la presenza di questi anticorpi non alterava però le
caratteristiche di efficacia e sicurezza di Omnitrope®. Il farmaco ovviamente non è stato
commercializzato e sono stati apportati dei miglioramenti nel processo di produzione in termini di
purificazione alle successive formulazioni.
Lopez –Siguero J. Long term safety and efficacy of the recombinant human growth hormone
Omnitrope® in the treatment of Spanish growth hormone deficient children: results of phase III
study. Adv ther 2011.
E’ stato condotto uno studio di fase III per dimostrare l’efficacia e la sicurezza del trattamento con
Omnitrope® a lungo termine. Sono stati arruolati 70 bambini prepuberi spagnoli (naive per la
terapia) con deficit di GH isolato, trattati con Omnitrope® sottocute alla dose di 0.03 mg/kg/die. I
risultati hanno dimostrato, dopo 4 anni di terapia, buoni parametri di crescita in termini di altezza,
altezza in SDS, velocità di crescita e velocità di crescita in SDS. Inoltre è stato riportato un
aumento significativo dei valori di IGF1 e IGFBP3. Tale studio ha concluso per l’efficacia, sicurezza
e buona tolleranza del trattamento con Omnitrope® in bambini con deficit di GH.
Peterkova V. a randomized, double-bind study to assess the efficacy and safety of valtropin, a
biosimilar growth hormone, in children with growth hormone deficiency. Horm Res, 2007.
Scopo di questo studio è stato quello di comparare Valtropin® con Humatrope® nel trattamento di
bambini in età prepubere con deficit di GH isolato. 98 e 49 bambini sono stati trattati
rispettivamente con Valtropin® e Humatrope® per un anno. I pazienti sono stati valutati con
misurazioni in altezza e velocità di crescita ogni 3 mesi; inoltre sono stati dosati IGF1, IGFBP3 e
anticorpi anti GH. I risultati hanno evidenziato: velocità di crescita pari a 11.3 +/- 3 cm/anno per
Valtropin® e 10.5 +/- 2.8 cm/anno per Humatrope® con una differenza tra i due trattamenti di 0.09
cm/anno (IC: - 0.71-0.90). Inoltre la velocità di crescita era aumentata per entrambi i trattamenti
senza avanzamento dell’età ossea così come i valori di IGF1 e IGFBP3. Non sono state segnalate
differenze significative in termini di eventi avversi e sono stati riscontrati anticorpi anti GH in 3
pazienti trattati con Valtropin® (3.1 %) e in 1 paziente trattato con Humatrope® (2.0%); in
particolare in questi pazienti non è stato evidenziato un ridotto pattern di crescita rispetto agli altri
pazienti dello studio. Lo studio ha dimostrato equivalenza di Valtropin® in termini di efficacia e
sicurezza con Humatrope®.
Per quanto riguarda le indicazioni d’uso, in Europa Omnitrope® presenta tutte le indicazioni
cliniche del prodotto di riferimento (Genotropin®), benché gli studi di fase III siano stai condotti
solo su pazienti in età pediatrica con deficit di GH. Le altre indicazioni (deficit di GH in pazienti
adulti, piccoli per età gestazionale, insufficienza renale cronica, sindrome di Turner e sindrome di
Prader Willi) sono state estrapolate dal prodotto di riferimento. Contrariamente alla situazione
europea, in Canada l’indicazione d’uso è stata estrapolata solo per il deficit di GH nel paziente
34
adulto e in USA, oltre che per il paziente adulto, anche per i nati piccoli per età gestazionale, per le
basse stature idiopatiche e per la sindrome di Prader Willi.
L' Omnitrope® è ancora un "farmaco sottoposto a monitoraggio intensivo” all’interno della Rete
Nazionale di Farmacovigilanza. Lo scopo di questo monitoraggio è quello di raccogliere in maniera
esaustiva tutte le informazioni relative alla sicurezza dei farmaci di nuova introduzione in
commercio, dei farmaci per cui sussistono delle particolari problematiche di farmacovigilanza, o
per i quali sia stata approvata una modifica sostanziale delle condizioni di impiego.
Anche per la formulazione biosimilare di rh-GH rimangono dei problemi aperti di cui vale la pena
discutere. Un primo punto riguarda gli studi necessari per l’approvazione del biosimilare: a
differenza di quanto succede per il prodotto di riferimento, non sono richiesti studi di sicurezza
farmacologica, tossicità per la riproduzione, mutagenicità e carcinogenicità. Inoltre, per quanto
riguarda l’efficacia e la sicurezza, è richiesto un solo studio di breve durata (12 mesi) e soprattutto
non per tutte le indicazioni cliniche del prodotto, che possono essere estrapolate dal prodotto di
riferimento (in realtà Omnitrope sarà monitorato in uno studio di farmacovigilanza in fase IV -studio
PATRO-sui bambini nati piccoli per età gestazionale). Vista però la bassa frequenza degli eventi
avversi in corso di terapia con GH, solo uno studio a lungo termine potrà evidenziare gli eventi
avversi. Inoltre i database esistenti non contengono dati sufficienti per dirci se la terapia con GH
può aumentare il rischio di sviluppo tumorale: vale comunque la pena indagare questo possibile
quanto raro evento avverso per tutte le formulazioni di GH, biosimilari compresi. Infine punto
cruciale rimane quello della interscambiabilità: diversamente da quanto succede in Europa, in USA
il New Biologics Price Competition and Innovation ACT 2009-2010 ha permesso alla FDA di
considerare interscambiabili biosimilari e prodotto di riferimento.
Bibliografia principale
1) Saenger P. Biosimilar growth hormone. Indian J Pediatr. 2012; 79:92-98
2)Ahmed I et al. Biosimilars: impact of biologic product life cycle and European experience on the regulatory trajectory in
the United States. Clin Ther. 2012; 34:2
3) Kresse GB. Biosimilars- Science, status and strategic perspective. Eur J Pharm Bioph. 2009; 479-486
4) Cauchy M et al. Excipient exchange in the comparison of preparartions of the same biologic made by different
manufacturing process: an esploratory study with recombinant human growth hormone. Biological. 2010; 637-643
5) Lopez –Siguero J. Long term safety and efficacy of the recombinant human growth hormone Omnitrope® in the
treatment of Spanish growth hormone deficient children: results of phase III study. Adv ther 2011.
6) Romer T et al. Seven years of safety and efficacy of the recombinant human growth hormone Omnitrope in the
treatment of Growth hormone deficient children. Hormone Res. 2009; 72: 359-369
7) Fuhr U et al. Bioequivalence between novel ready-to-use liquid formulations of the recombinant human GH Omnitrope
and the original lyophilized formulations for reconstitution of Omnitrope and Genotropin. Eur J End. 2010; 152: 10511058
8) Stanhope R. et al. Bioequivalence studies of Omnitrope, the first biosimilar/rhGH follow on protein. J Clin
Pharmacol.2010
9) EMA- Guideline on similar biological medicinal products containing biotechnology-derived proteins as active
substance: quality issue (revision 1). 31 May 2012
10) EMA- Guideline on similar biological medicinal products containing biotechnology-derived proteins as active
substance: non clinical and clinical issues. October 2005
ORMONE DEL LOBO ANTERIORE DELL’IPOFISI, ORMONE DELLA CRESCITA H01AC01
Principio
attivo
SOMATR
OPINA
Biosimilare
Omnitrope
Titolare
AIC
In commercio
in Italia
Sandoz
GmbH
Marzo 2007
Originatore
Genotropin
(Pfizer)
Modalità
erogative/prescrittive
Classe A-PHT, erogazione
diretta ospedaliera
(rendicontazione File F tip 6)
oppure ricetta SSN con
indicazione PHT
35
Somatropina biosimilare:
Humatrope
Dosaggi in commercio:
Humatrope 1 cart 24 mg
Humatrope 1 cart 12 mg
Humatrope 1 cart 6 mg
Indicazioni terapeutiche:
Pazienti pediatrici:
Humatrope è indicato per il trattamento a lungo termine dei
bambini con deficit di statura dovuto ad inadeguata secrezione
dell'ormone della crescita endogeno.
Humatrope è indicato anche per il trattamento della bassa
statura nelle bambine con Sindrome di Turner, confermata
dall'analisi cromosomica.
Humatrope è indicato inoltre per il trattamento del ritardo di
crescita nei bambini prepuberi con insufficienza renale cronica.
Humatrope è indicato inoltre per il trattamento del disturbo di
crescita (altezza attuale SDS < -2.5 ed altezza corretta in base
alla statura dei genitori SDS < -1) in bambini di bassa statura
nati piccoli per l'età gestazionale (SGA), con peso e/o lunghezza
alla nascita inferiore a –2 SD, che non hanno presentato un
recupero della crescita (velocità di crescita SDS < 0 durante
l'ultimo anno) entro l'età di 4 anni od oltre.
Humatrope è indicato anche per il trattamento di pazienti
con deficit staturale associato ad un'alterata funzione del gene
SHOX, confermata dall'analisi del DNA.
Pazienti adulti
Humatrope è indicato per la terapia sostitutiva negli adulti
con deficit marcato dell'ormone della crescita.
I pazienti con grave deficit dell'ormone della crescita nell'età
adulta sono definiti come pazienti con patologia ipofisariaipotalamica nota e con almeno un deficit noto di un ormone
ipofisario, che non sia prolattina. Questi pazienti devono
sottoporsi ad un singolo test dinamico al fine di diagnosticare od
escludere un deficit della crescita.
Nei pazienti con deficit isolato di ormone della crescita ad
insorgenza in età pediatrica (senza evidenza di patologia
ipofisaria-ipotalamica o che non siano stati sottoposti ad
irradiazione cranica) si raccomanda l'esecuzione di due test
dinamici, ad eccezione di quelli con un basso valore di IGF-1 (<
-2SDS) per i quali può essere effettuato un singolo test. Il limite
di riferimento del test dinamico impiegato deve essere preciso.
36
Somatropina: Genotropin
Dosaggi in commercio:
Genotropin Mini 4tbf 1,2 mg
Genotropin Mini 4tbf 1,4 mg
Genotropin Mini 4tbf 1,6 mg
Genotropin Mini 4tbf 1,8 mg
Genotropin Mini 4tbf 2 mg
Genotropin Mini 7tbf 0,2
mg
Genotropin Mini 7tbf 0,4 mg
Genotropin Mini 7tbf 0,8 mg
Genotropin Mini 7tbf 1 mg
Genotropin Pen 1tbf 12 mg
Genotropin Kabipen 1 tbf
5,3 mg
Indicazioni terapeutiche:
Nei bambini:
1. Disturbi della crescita dovuti a insufficiente increzione di
ormone somatotropo (deficit di ormone della crescita, GHD)
e disturbi della crescita associati a Sindrome di Turner o a
insufficienza renale cronica.
2. Disturbi della crescita (altezza attuale < -2,5 SDS e altezza
corretta in base alla statura dei genitori < - 1 SDS) in
bambini di bassa statura nati piccoli per l’età gestazionale
(SGA), con peso e/o lunghezza alla nascita inferiore a - 2
SD, che non hanno presentato recupero di crescita (HV < 0
SDS durante l’ultimo anno) entro l’età di 4 anni od oltre.
3. Sindrome di Prader-Willi per il miglioramento della crescita e
della composizione corporea. La diagnosi di Sindrome di
Prader-Willi deve essere confermata da appropriati test
genetici.
Negli adulti
Trattamento sostitutivo nei pazienti adulti con marcato deficit
di ormone della crescita che viene gestito differentemente in
funzione dell’età di insorgenza:
In caso di insorgenza in età adulta: pazienti che hanno un
grave deficit di ormone della crescita associato a deficit ormonali
multipli come conseguenza di una patologia ipotalamica o
ipofisaria nota, e che hanno almeno un deficit di un ormone
ipofisario, ad eccezione della prolattina. Questi pazienti devono
essere sottoposti ad un appropriato test dinamico di secrezione
per la diagnosi o per l’esclusione del deficit di ormone della
crescita. In caso di insorgenza in età infantile: pazienti carenti di
ormone della crescita in età infantile per cause congenite,
genetiche, acquisite, o idiopatiche.
In caso di insorgenza in età infantile: devono essere
rivalutati per quanto riguarda la capacità secretoria dell’ormone
della crescita al completamento della crescita longitudinale. Nei
pazienti con elevata probabilità di GHD persistente, ad esempio
per una causa congenita o GHD secondario ad una malattia o
ad un danno ipotalamo-ipofisario, livelli di IGF-I con SDS< -2 in
assenza di trattamento con ormone della crescita per almeno 4
settimane, devono essere considerati segno evidente di totale
GHD.
Per tutti gli altri pazienti saranno necessari il
dosaggio dei livelli di IGF-I e un test di stimolazione
dell’ormone della crescita.
37
Allegato 2g
Alla luce della normativa e delle indicazioni regolatorie vigenti:
1) I farmaci di sintesi biologica (originatori o biosimilari):
- non possono essere considerati sostituibili dal farmacista all’atto della dispenzazione;
- non possono essere considerati intercambiabili dal medico all’atto della prescrizione.
2) I farmaci biosimilari:
- consistono in una alternativa terapeutica aggiuntiva a disposizione del medico e del
paziente;
- possono essere una alternativa preferibile, quando presentino:
c) efficacia terapeutica relativa comparabile ai prodotti originatori e
d) minor costo per il SSN e per il cittadino
Sintesi delle virtù terapeutiche dei farmaci con G-CSF
in funzione della loro efficacia relativa e del loro costo per il SSN
FATTORI DI CRESCITA GRANULOCITARI
in assenza di specifiche indicazioni o controindicazioni
Il fattore stimolante le colonie di granulociti umano (filgrastim o GCSF) è una glicoproteina che
regola la produzione e la liberazione dal midollo osseo di granulociti neutrofili funzionali. Filgrastim
(r-metHuG-CSF) è impiegato per stimolare la produzione di globuli bianchi nelle situazioni di
neutropenia in pazienti in corso di chemioterapia, in pazienti sottoposti a mieloablazione prima di
un trapianto del midollo emopoietico e per trattare la neutropenia persistente in pazienti affetti da
infezione avanzata da virus dell’immunodeficienza umana (HIV), allo scopo di ridurre il rischio di
infezioni opportunistiche quando altri trattamenti risultano inadeguati. Il primo G-CSF ricombinante
è stato immesso in commercio nel 1991 con il nome di Neupogen®. Nel Novembre 2008 sei
formulazioni biosimili di filgrastim hanno ricevuto parere positivo dall’EMA (Ratiogastrim® e
Filgastim Ratiopharm® della Ratiopharm; Filgrastim Hexal® della Hexal; Biograstim® della CT
Arzneimittel; Zarzio® della Sandoz e Tevagastrim® della Teva) che ha rilasciato l’autorizzazione
all’immissione in commercio, valida in tutta l’Unione Europea. In Italia sono state approvate per il
commercio nel Novembre 2009 le formulazioni Zarzio® Sandoz e Ratiogastrim® Ratiopharm e
Tevagrastim® Teva Generics.
G.U. n. 270 del 18 novembre 2010 - Determinazione AIFA 2 novembre 2010 avente per oggetto
“Aggiornamento del Piano terapeutico AIFA per prescrizione di fattori di crescita granulocitari (ex
Nota 30 e 30bis)”.
L’aggiornamento riguarda l’inserimento dei biosimilari tra i principi attivi prescrivibili nel PT (nuovo
PT AIFA fattori di crescita granulocitari). Il PT AIFA aggiornato è in vigore dal 19 novembre 2010.
Si ricorda che nel Piano Terapeutico il testo della prima condizione clinica «Trattamento della
neutropenia febbrile da chemioterapia (filgrastim, lenograstim, pegfilgrastim)» deve intendersi
condizione clinica comprensiva anche della profilassi, mantenendo in tal modo a carico del SSN
anche il trattamento preventivo (profilassi) della neutropenia febbrile da chemioterapia.
38
Filgrastim biosimilare:
Ratiograstim
Dosaggi in commercio:
Ratiograstim 30 MUI (300
mcg/0,5 ml) soluzione
iniettabile in siringa
preriempita
Indicazioni terapeutiche:
1. Riduzione della durata della neutropenia e dell'incidenza di
neutropenia febbrile in pazienti trattati con chemioterapia
citotossica standard per affezioni maligne (con l'eccezione
della leucemia mieloide cronica e delle sindromi
mielodisplastiche) e per la riduzione della durata della
neutropenia in pazienti sottoposti a terapia mieloablativa
seguita da trapianto di midollo osseo considerati a maggior
rischio di neutropenia grave prolungata. La sicurezza e
l'efficacia di filgrastim sono simili negli adulti e nei bambini
trattati con chemioterapia citotossica.
2. Mobilizzazione delle cellule progenitrici del sangue periferico
(PBPC).
3. In pazienti, bambini o adulti, con neutropenia grave
congenita, ciclica o idiopatica, con una conta assoluta di
neutrofili (CAN) ≤0,5 x 109/l, e una storia di infezioni gravi o
ricorrenti, una somministrazione a lungo termine di
Ratiograstim è indicata per incrementare la conta dei
neutrofili e per ridurre l'incidenza e la durata delle
complicanze correlate all'infezione.
4. Trattamento della neutropenia persistente (CAN minore o
uguale a 1,0 x 109/l) in pazienti con infezione da HIV
avanzata, per ridurre il rischio di infezioni batteriche quando
non siano appropriate altre opzioni per controllare la
neutropenia.
Indicazioni terapeutiche:
Filgrastim biosimilare:
Tevagrastim
Dosaggi in commercio:
Tevagrastim 30 MUI/0,5 ml
soluzione iniettabile in
siringa preriempita
Tevagrastim 48 MUI/0,8 ml
soluzione iniettabile
1. Riduzione della durata della neutropenia e dell'incidenza di
neutropenia febbrile in pazienti trattati con chemioterapia
citotossica standard per affezioni maligne (con l'eccezione
della leucemia mieloide cronica e delle sindromi
mielodisplastiche) e per la riduzione della durata della
neutropenia in pazienti sottoposti a terapia mieloablativa
seguita da trapianto di midollo osseo considerati a maggior
rischio di neutropenia grave prolungata. La sicurezza e
l'efficacia del filgrastim sono simili negli adulti e nei bambini
trattati con chemioterapia citotossica.
2. Mobilizzazione delle cellule progenitrici del sangue periferico
(PBPC).
3. In pazienti, bambini o adulti, con neutropenia grave
congenita, ciclica o idiopatica, con una conta assoluta di
neutrofili (CAN) ≤ 0,5 x 109/l, e una storia di infezioni gravi o
ricorrenti, una somministrazione a lungo termine di
Tevagrastim è indicata per incrementare la conta dei
neutrofili e per ridurre l'incidenza e la durata delle
complicanze correlate all'infezione.
4. Trattamento della neutropenia persistente (CAN minore o
uguale a 1,0 x 109/l) in pazienti con infezione da HIV
avanzata, per ridurre il rischio di infezioni batteriche quando
non siano appropriate altre opzioni per controllare la
neutropenia.
39
Filgrastim biosimilare:
Zarzio
Dosaggi in commercio:
Zarzio 48 MU/0,5 ml
soluzione iniettabile in
siringa preriempita
Zarzio 30 MU/0,5 ml
soluzione iniettabile in
siringa preriempita
Filgrastim: Granulokine
Dosaggi in commercio:
Granulokine 30 siringa
preriempita 0,5 ml 30 MU
Granulokine 30 flaconcino
iniettabile 1 ml 30 MU
Indicazioni terapeutiche:
1. Riduzione della durata della neutropenia e dell'incidenza di
neutropenia febbrile nei pazienti trattati con chemioterapia
citotossica standard per patologie maligne (con l'eccezione
della leucemia mieloide cronica e delle sindromi
mielodisplastiche) e riduzione della durata della neutropenia
nei pazienti sottoposti a terapia mieloablativa seguita da
trapianto di midollo osseo considerati ad alto rischio di
neutropenia grave prolungata.
La sicurezza e l'efficacia del filgrastim sono simili negli adulti
e nei bambini sottoposti a chemioterapia citotossica.
2. Mobilizzazione delle cellule progenitrici del sangue periferico
(PBPC).
3. Nei bambini e negli adulti con grave neutropenia congenita,
ciclica o idiopatica, con una conta assoluta di neutrofili (ANC)
≤ 0,5 x 109/l e una storia di infezioni gravi o ricorrenti, la
somministrazione a lungo termine del filgrastim è indicata per
incrementare la conta dei neutrofili e ridurre l'incidenza e la
durata degli eventi correlati alle infezioni.
4. Trattamento della neutropenia persistente (ANC ≤ 1,0 x 109/l)
nei pazienti con infezione avanzata da HIV, per ridurre il
rischio di infezioni batteriche quando altre opzioni
terapeutiche siano inadeguate.
Indicazioni terapeutiche:
1. Ridurre la durata della neutropenia e l'incidenza della
neutropenia febbrile in pazienti trattati con chemioterapia
citotossica standard per affezioni maligne (con l'eccezione
della leucemia mieloide cronica e delle sindromi
mielodisplastiche) e nel ridurre la durata della neutropenia in
pazienti sottoposti a terapia mieloablativa seguita da
trapianto di midollo osseo considerati a maggior rischio di
neutropenia severa prolungata. La sicurezza e l'efficacia di
Granulokine sono simili negli adulti e nei bambini trattati con
chemioterapia citotossica.
2. Mobilizzazione delle cellule progenitrici del sangue periferico
(PBPC).
3. In pazienti, bambini o adulti, con neutropenia grave
congenita, ciclica o idiopatica, con una CAN (conta assoluta
dei neutrofili) < 0,5 x 109/l e una storia di infezioni gravi o
ricorrenti, una somministrazione a lungo termine di
Granulokine è indicata per incrementare la conta dei
neutrofili e per ridurre l'incidenza e la durata delle
complicanze infettive.
4. Trattamento della neutropenia persistente (CAN uguale o
minore di 1,0 x 109/l) in pazienti con infezione da HIV
avanzata, per ridurre il rischio di infezioni batteriche quando
non siano appropriate altre opzioni per controllare la
neutropenia.
40
41
Allegato 2h
Alla luce della normativa e delle indicazioni regolatorie vigenti:
1) I farmaci di sintesi biologica (originatori o biosimilari):
- non possono essere considerati sostituibili dal farmacista all’atto della dispenzazione;
- non possono essere considerati intercambiabili dal medico all’atto della prescrizione.
2) I farmaci biosimilari:
- consistono in una alternativa terapeutica aggiuntiva a disposizione del medico e del
paziente;
- possono essere una alternativa preferibile, quando presentino:
e) efficacia terapeutica relativa comparabile ai prodotti originatori e
f) minor costo per il SSN e per il cittadino
Sintesi delle virtù terapeutiche dei farmaci con ESA
in funzione della loro efficacia relativa e del loro costo per il SSN
ERITROPOIETINE
in assenza di specifiche indicazioni o controindicazioni
L’eritropoietina umana ricombinante fu purificata nel 1985 dopo la clonazione del gene EPO
umano, e immessa in commercio già dal 1988 per il trattamento dell’anemia secondaria a malattie
renali e, successivamente, per la terapia di sostegno dei pazienti oncologici con anemia
secondaria a trattamento chemioterapico. In Europa, la copertura brevettuale di EPO alfa,
commercializzata da Janssen Cilag con il nome Eprex®/Erypo®, è scaduta nel 2004.
Attualmente sono stati prodotti due tipi diversi di EPO alfa biosimili, ottenute secondo due diversi
standard operativi di sintesi e purificazione, dotati di struttura aminoacidica identica ma con
composizione glicosilica diversa: uno mantiene il nome di EPO alfa e l’altro è stato denominato
EPO zeta.
EPO alfa biosimile è in commercio con il nome di Binocrit® (Sandoz), Abseamed® (Medice
Arzneimittel): Sandoz è azienda sussidiaria di Novartis, mentre Medice Arzneimittel è un “licensing
partner” di Sandoz.
EPO zeta biosimile è invece commercializzata con il nome di Retacrit® (Hospira). Poichè i
procedimenti di produzione per le due molecole sono diversi, anche le rispettive proteine
terapeutiche non possono essere considerate identiche. Pertanto è stato necessario per le ditte
farmaceutiche presentare alla Commissione Europea dossier registrativi specifici: uno per il
biosimile EPO alfa, l’altro per il biosimile EPO zeta.
Durata
di azione
Breve
Breve
Breve
Breve
Breve
Breve
Breve
Breve
Lunga
Ultralunga
INN (nome internazionale) Nome commerciale
Epoetin alfa
Epoetin alfa
Epoetin alfa
Epoetin alfa
Epoetin zeta
Epoetin zeta
Epoetin beta
Epoetin teta
Darbepoetin alfa
MetossiPEG-Epoetin beta
Eprex
Binocrit
Epoetin alfa Hexal
Abseamed
Retacrit
Silapro
Neorecormon
Eporatio
Aranesp
Mircera
Protezione
commerciale
originatore
biosimilare
biosimilare
biosimilare
biosimilare
biosimilare
originatore
originatore
originatore
originatore
Via
ev, sc
ev
ev
ev
ev, sc
ev, sc
ev, sc
ev, sc
sc
ev, sc
42
NEFROLOGIA
1. Trattamento dell'anemia sintomatica associata a insufficienza renale
cronica (IRC) in pazienti adulti e pediatrici: trattamento dell'anemia
associata a insufficienza renale cronica in pazienti adulti e pediatrici
emodializzati e in pazienti adulti sottoposti a dialisi peritoneale.
Trattamento dell'anemia grave, di origine renale, accompagnata da
sintomi clinici, in pazienti adulti con insufficienza renale non ancora
dializzati.
1. Trattamento dell'anemia sintomatica associata ad insufficienza renale
cronica (IRC) in pazienti adulti e pediatrici.
1. Trattamento dell'anemia sintomatica associata ad insufficienza renale
cronica (IRC) in pazienti adulti.
NEONATOLOGIA
2. Prevenzione dell'anemia dei neonati prematuri con un peso alla
nascita compreso tra 750 e 1500 g e con un periodo di gestazione
inferiore a 3-4 settimane.
ONCOEMATOLOGIA
3. Trattamento dell'anemia e riduzione del fabbisogno trasfusionale in
pazienti adulti in trattamento chemioterapico per tumori solidi, linfoma
maligno o mieloma multiplo e a rischio di trasfusione, come indicato
dallo stato generale del paziente (situazione cardiovascolare, anemia
preesistente all'inizio della chemioterapia).
3. Trattamento dell'anemia sintomatica in pazienti adulti con tumore non
mieloide sottoposti a chemioterapia.
CHIRURGIA
4. Aumentare la produzione di sangue autologo nei pazienti facenti
parte di un programma di predonazione autologa. L'impiego per tale
indicazione deve essere valutato in rapporto al noto rischio di eventi
tromboembolici. Il trattamento deve essere effettuato solo in pazienti
non sideropenici con anemia moderata (emoglobina (Hb) 10-13 g/dl
(6,2-8,1 mmol/l)), quando le tecniche di risparmio di sangue non siano
disponibili o siano insufficienti e l'intervento programmato di chirurgia
elettiva maggiore richieda un elevato quantitativo di sangue (4 o più
unità di sangue per le donne, 5 o più unità per gli uomini).
4. Ridurre l'esposizione a trasfusioni di sangue allogenico in pazienti
adulti non sideropenici, ritenuti ad alto rischio di complicanze
trasfusionali, prima di un intervento elettivo di chirurgia ortopedica
maggiore. Limitare l'uso ai pazienti con anemia moderata (Hb 10-13
g/dl) non facenti parte di un programma di predonazione autologa e per
i quali si preveda una perdita ematica di 900-1800 ml.
Eporatio
Breve
X
X
Darb
Lunga
Ultra
lunga
X
X
X
X
X
X
X
X
X
X
X
X
X
X
PEG
Mircera
θ
Aranesp
Durata di azione
β
Neorecormon
Protezione
commerciale
originatore
biosimilare
biosimilare
biosimilare
biosimilare
biosimilare
originatore
originatore
originatore
originatore
Z
Retacrit, Silapro
Nome
commerciale
Eprex
Binocrit
Epoetin alfa Hexal
Abseamed
Retacrit
Silapro
Neorecormon
Eporatio
Aranesp
Mircera
α
Binocrit, Epoetin alfa Hexal,
Abseamed
INN
(nome internazionale)
Epoetin alfa
Epoetin alfa
Epoetin alfa
Epoetin alfa
Epoetin zeta
Epoetin zeta
Epoetin beta
Epoetin teta
Darbepoetin alfa
MetossiPEG-Epoetin beta
α
Eprex
ERITROPOIETINE - Indicazioni registrative
X
X
X
X
43
1. Utilizzo nel trattamento dell’anemia (Hgb < 10 g/dl o riduzione
dell’emoglobina > 2 g/dl durante un qualsiasi periodo di 4 settimane di
trattamento) nei pazienti che ricevono ribavirina in combinazione con
interferone standard o peghilato per il trattamento dell’infezione cronica da HCV
e che presentano risposta virologica alla terapia.
2. In pazienti HIV pluritrattati con anemia (Hgb < 8,5 g/dl) nei quali l’uso dei
farmaci anemizzanti è l’unica alternativa terapeutica.
3. Sindromi mielodisplastiche (MDS)
4. Anemia refrattaria (AR), con sideroblasti (RARS) e senza sideroblasti (RA)
X
X
X
X
X
X
X
X
X
44
Eporatio
Neorecormon
Retacrit
Binocrit
Eprex
Estensione di indicazione relative ad usi consolidati sulla base di
evidenze scientifiche presenti in letteratura L. 648/96
Allegato 2i
Sintesi delle virtù terapeutiche dei Nuovi Antitrombotici
in funzione della loro efficacia relativa e del loro costo per il SSN
RIVAROXABAN (Xarelto), APIXABAN (Eliquis), DABIGATRAN, (Pradaxa)
in assenza di specifiche indicazioni o controindicazioni
Le seguenti tabelle riportano i nuovi antitrombotici commercializzati in Italia, i rispettivi meccanismi
di azione e le indicazioni registrative.
P.A., nome commerciale (anno 1° commercializzazione ), meccanismo di azione
Rivaroxaban, Xarelto (2012)
Rivaroxaban è un inibitore diretto e altamente selettivo del fattore Xa, con biodisponibilità orale.
L’inibizione del Fattore Xa interrompe le vie intrinseca ed estrinseca della cascata della coagulazione e
inibisce sia la formazione di trombina, sia lo sviluppo di trombi. Rivaroxaban non inibisce la trombina
(Fattore II attivato) e non è stato dimostrato alcun effetto sulle piastrine.
Apixaban, Eliquis (2009)
Apixaban è un potente inibitore orale, reversibile, diretto e altamente selettivo del sito attivo del
fattore Xa. Non ha bisogno dell’antitrombina III per esercitare l’attività antitrombotica. Apixaban inibisce il
fattore Xa libero e legato al coagulo, e l’attività della protrombinasi. Apixaban non ha effetti diretti
sull’aggregazione piastrinica, ma inibisce indirettamente l’aggregazione piastrinica indotta dalla trombina.
Con l’inibizione del fattore Xa, apixaban previene la generazione della trombina e lo sviluppo del trombo. Gli
studi preclinici di apixaban nei modelli animali hanno dimostrato efficacia antitrombotica nella prevenzione
della trombosi arteriosa e venosa a dosi che preservavano l’emostasi.
Dabigatran, Pradaxa (2008)
Dabigatran etexilato è un profarmaco di piccole dimensioni molecolari che non esercita alcuna attività
farmacologica. Dopo somministrazione orale, dabigatran etexilato è rapidamente assorbito e convertito in
dabigatran mediante idrolisi catalizzata da esterasi nel plasma e nel fegato. Dabigatran è un potente
inibitore diretto, competitivo, reversibile della trombina ed è il principio attivo principale che si ritrova nel
plasma.
45
P.A., nome
commerciale
(anno 1° comm.)
Rivaroxaban,
Xarelto (2012)
Apixaban, Eliquis
(2009)
Dabigatran,
Pradaxa (2008)
Fascia e
prescrittore
Indicazione
FASCIA A :
SU PRESCRIZIONE
SPECIALISTICA
ORTOPEDICO,
EMATOLOGO,
ANESTESISTA,
MED INTERNA
FASCIA A :
SU PRESCRIZIONE
SPECIALISTICA
ORTOPEDICO,
EMATOLOGO,
ANESTESISTA,
MED INTERNA
Prevenzione del tromboembolismo venoso (TEV) nei pazienti
adulti sottoposti a interventi di sostituzione elettiva di anca o di
ginocchio
Prevenzione degli eventi tromboembolici venosi (TEV) nei
pazienti adulti sottoposti a intervento chirurgico di sostituzione
elettiva dell’anca o del ginocchio.
FASCIA A :
75 MG: Prevenzione primaria di episodi tromboembolici in
SU PRESCRIZIONE pazienti adulti sottoposti a chirurgia sostitutiva elettiva totale
SPECIALISTICA
dell’anca o del ginocchio.
ORTOPEDICO ED
EMATOLOGO
110MG:
- Prevenzione primaria di episodi tromboembolici in pazienti adulti
sottoposti a chirurgia sostitutiva elettiva totale dell’anca o del
ginocchio.
FASCIA A: SU
PIANO
TERAPEUTICO DI:
CARDIOLOGO,
INTERNISTA,
NEUROLOGO,
GERIATRA,
EMATOLOGO
- Prevenzione di ictus e embolia sistemica in pazienti adulti con
fibrillazione atriale non valvolare con uno o più dei seguenti fattori
di rischio:
• Precedente ictus, attacco ischemico transitorio o embolia sistemica
(ES)
• Frazione di eiezione del ventricolo sinistro < 40%
• Insufficienza cardiaca sintomatica, ≥ Classe 2 della classificazione
della New York Heart Association (NYHA)
• Età ≥ 75 anni
• Età ≥ 65 anni associata con una delle seguenti condizioni: diabete
mellito, coronaropatia o ipertensione.
150MG: Prevenzione di ictus e embolia sistemica in pazienti adulti
con fibrillazione atriale non valvolare con uno o più dei seguenti
fattori di rischio:
• Precedente ictus, attacco ischemico transitorio o embolia sistemica
(ES)
• Frazione di eiezione del ventricolo sinistro < 40%
• Insufficienza cardiaca sintomatica, ≥ Classe 2 della classificazione
della New York Heart Association (NYHA)
• Età ≥ 75 anni
• Età ≥ 65 anni associata con una delle seguenti condizioni: diabete
mellito, coronaropatia o ipertensione.
Sintesi di Documentazione di Efficacia correlate:
1) DABIGATRAN O WARFARIN IN PAZIENTI CON FIBRILLAZIONE ATRIALE – dicembre
2009
46
47
Allegato 3
OSSERVAZIONI, PROPOSTE E RICHIESTE DI MODIFICA DEI CONTENUTI DEL PUD
Sezione DIMISSIONE
(SUGGERIMENTO PRESCRITTIVO E DISTRIBUZIONE DIRETTA)
Osservazioni, proposte e richieste di modifica:
Sezione VISITA AMBULATORIALE
(SUGGERIMENTO PRESCRITTIVO)
Osservazioni, proposte e richieste di modifica:
Sezione ALLEGATI
Sintesi delle virtù terapeutiche dei farmaci in funzione della loro efficacia relativa e del loro costo per il SSN
STATINE ED ALTRI FARMACI ANTIDISLIPIDEMICI
ACE-INIBITORI E SARTANI
INIBITORI DI POMPA PROTONICA
ANTIBIOTICI
EPARINE A BASSO PESO MOLECOLARE (EBPM)
ORMONE DELLA CRESCITA
FATTORI DI CRESCITA GRANULOCITARI
ERITROPOIETINE
Osservazioni, proposte e richieste di modifica:
Dott./sig._____________________________________________________________
UO/Struttura _________________________________________________________
Azienda _____________________________________________________________
Data ___/___ /___
48
Allegato 4
BIBLIOGRAFIA E CITAZIONI INSERITE NEL TESTO
BIBLIOGRAFIA STATINE
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
World Health Organization. Regional Office for Europe. Appropriateness in health care services. Report on a WHO
workshop, Koblenz, Germany; 23–25 March 2000.
Hicks NR. Some observations on attempts to measure appropriateness of care. Br Med J 1994; 309: 730–733.
Britten N, Jenkis L, Barber N, Bradley C, Stevenson F. Developing a measure for the appropriateness of prescribing
in general practice. Qual Saf Health Care 2003; 12: 246-50.
Federazione Nazionale degli Ordini dei Medici Chirurghi e degli Odontoiatri. Codice di Deontologia Medica. 16
Dicembre 2006.
European Cardiovascular Disease Statistics 2008; British Heart Foundation, Health Promotion Research Group,
Department of Public Health, University of Oxford.
Quarta Task Force congiunta della Società Europea di Cardiologia e di altre società sulla prevenzione delle malattie
cardiovascolari nella pratica clinica. Linee guida europee sulla prevenzione delle malattie cardiovascolari nella
pratica clinica (traduzione italiana). G Ital Cardiol 2008; 9: 11-59.
Gruppo di lavoro OsMed. L’uso dei farmaci in Italia. Rapporto nazionale anno 2009. Roma: Il Pensiero Scientifico
Editore, 2010.
Nota 13 AIFA, GU no. 72 del 27 marzo 2007 – Serie Generale.
Grundy SM, Cleeman JI, Bairey Merz CN, et al., for the Coordinating Committee of the National Cholesterol
Education Program. Implications of recent clinical trials for the National Cholesterol Education Program Adult
Treatment Panel III guidelines. Circulation 2004;110:227-239.
Regione Puglia, Deliberazione della Giunta Regionale no.1384 del 22/07/2008. Interventi in materia farmaceutica in
attuazione dell'art. 3, C. 26 della legge regionale 31.12.2007, n. 10.
Regione Campania, Deliberazione della Giunta Regionale no.1883 del 26/11/2008. Piano di contenimento della
spesa farmaceutica, Promozione dell'appropriatezza diagnostica e terapeutica nelle dislipidemie e dei farmaci
equivalenti nella classe C10AA degli inibitori dell' HGM-CoA Reduttasi.
Decreto del Presidente in qualità di Commissario ad acta; 2 luglio 2009, n. 45. Promozione dell’appropriatezza
diagnostica e terapeutica nelle dislipidemie e dei farmaci equivalenti nelle classi C10AA e C10BA degli inibitori del
HGM-coa Reduttasi singoli o in associazione. Boll. Uff. Reg. Lazio 2009; supplemento ordinario no. 135 al no.29.
Regione Toscana, Deliberazione della Giunta Regionale no. 986 del 22/11/2010. Allegato A. Linee di indirizzo sulla
terapia farmacologica della ipercolesterolemia.
Neaton JD, Wentworth D. Serum Cholesterol, Blood Pressure, Cigarette Smoking, and Death From Coronary Heart
Disease Overall Findings and Differences by Age for 316099 White Men. Arch Intern Med. 1992;152:56-64.
Baigent C, Keech A, Kearney PM, Blackwell L, Buck G, Pollicino C, Kirby A, Sourjina T, Peto R, Collins R,
Simes R; Cholesterol Treatment Trialists' (CTT) Collaborators. Efficacy and safety of cholesterol-lowering
treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins.
Lancet. 2005; 366:1267-78.
Cholesterol Treatment Trialists’ (CTT) Collaboration, Baigent C, Blackwell L, Emberson J, Holland LE, Reith
C, Bhala N, Peto R, Barnes EH, Keech A, Simes J, Collins R. Efficacy and safety of more intensive lowering
of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 2010;
376:1670-81.
Mahley RW, Bersot T. Terapia farmacologica dell’ipercolesterolemia e delle dislipidemie. In Goodman &
Gilman ed. Le basi farmacologiche della terapia; 11ma edizione, 2006.
Smith MEB, Lee NJ, Haney E, Carson S. Drug Class Review: HMG-CoA Reductase Inhibitors (Statins) and
Fixed-dose Combination Products Containing a Statin: Final Report Update 5. Drug Class Reviews. Portland
(OR): Oregon Health & Science University; 2009 Nov.
National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of
High Blood Cholesterol in Adults (Adult Treatment Panel, III). Third Report of the National Cholesterol
Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol
in Adults (Adult Treatment Panel III) final report. Circulation 2002; 106:3143.
Task Force per la Diagnosi e il Trattamento delle Sindromi Coronariche Acute Senza Sopraslivellamento del
Tratto ST della Società Europea di Cardiologia. Linee guida per la diagnosi e il trattamento delle sindromi
coronariche acute senza sopraslivellamento del tratto ST (traduzione italiana). G Ital Cardiol 2007; 8: 599675.
Task Force per il Trattamento dell’Infarto Miocardico Acuto con Sopraslivellamento del Tratto ST della
Società Europea di Cardiologia. Trattamento dell’infarto miocardico acuto nei pazienti con
sopraslivellamento persistente del tratto ST alla presentazione (traduzione italiana). G Ital Cardiol 2009; 10:
450-489.
Cannon CP, Braunwald E, McCabe CH, Rader DJ, Rouleau JL, Belder R, et al. Intensive versus moderate
lipid lowering with statins after acute coronary syndromes. N Engl J Med. 2004;350:1495–1504.
Lablanche JM, Leone A, Merkely B, Morais J, Alonso J, Santini M, Eha J, Demil N, Licour M, Tardif JC, for
the CENTAURUS investigators. Comparison of the efficacy of rosuvastatin versus atorvastatin in reducing
apolipoprotein B/apolipoprotein A-1 ratio in patients with acute coronary syndrome: Results of the
CENTAURUS study. Arch Cardiovasc Dis 2010
Jones PH, Davidson MH, Stein EA et al. Comparison of the efficacy and safety of rosuvastatin versus
atorvastatin, simvastatin, and pravastatin across doses (STELLAR Trial). Am J Cardiol 2003; 92:152–160.
49
25. Nicholls SJ, Brandrup-Wognsen G, Palmer M, Carter PJ. Meta-analysis of Comparative Efficacy of
Increasing Dose of Atorvastatin Versus Rosuvastatin Versus Simvastatin on Lowering Levels of
Atherogenic Lipids (from VOYAGER). Am J Cardiol 2010; 105: 69-76.
26. Pasternak RC, Smith SC Jr, Bairey-Merz CN, Grundy SM, Cleeman JI, Lenfant C. ACC/AHA/NHBLI clinical
advisory on the use and safety of statins. Circulation 2002;106:1024-8.
27. Thompson PD, Clarkson P, Karas RH. Statin-Associated Myopathy. JAMA 2003; 289:1681-1690.
28. Athyros VG, Tziomalos K, Gossios TD, Griva T, Anagnostis P, Kargiotis K,Pagourelias ED, Theocharidou E,
Karagiannis A, Mikhailidis DP. Safety and efficacy of long-term statin treatment for cardiovascular events in
patients with coronary heart disease and abnormal liver tests in the Greek Atorvastatin and Coronary Heart
Disease Evaluation (GREACE) Study: a post-hoc analysis. Lancet 2010;376:1916-22.
29. Colivicchi F, Uguccioni M, Ragonese M et al: Cardiovascular risk factor control among diabetic patients
attending community-based diabetics care clinics in Italy. Diab Res Clin Prac. 2007; 75: 176-183.
30. Filippi A, D'Ambrosio G, Giustini SE, Pecchioli S, Mazzaglia G, Cricelli C. Pharmacological treatment after
acute myocardial infarction from 2001 to 2006: a survey in Italian primary care. J Cardiovasc Med
(Hagerstown). 2009;10:714-8.
31. Colivicchi F, Bassi A, Santini M, Caltagirone C. Discontinuation of statin therapy and clinical outcome after
ischemic stroke. Stroke 2007;38:2652-2657.
32. Deambrosis P, Saramin C, Terrazzani G, et al: Evaluation of the prescription and utilization patterns of
statins in an Italian local health unit during the period 1994-2003. Eur J Clin Pharmacol 2007;63:197-203.
33. Simpson RJ, Mendys P. The effects of adherence and persistence on clinical outcomes in patients treated
with statins: A systematic review. J Clin Lipidol 2010; 4:462-471
34. Moon JC, Bogle RC. Switching statins. BMJ 2006;332:1344–5.
35. Atar D, Carmena R, Clemmensen P, Laflamme AK, Wassmann S,Lansberg P, Hobbs R. Clinical review:
impact of statin substitution policies on patient outcomes. Ann Med 2009; 41:242-256.
36. Aronow HD, Hess G, Hill J, Kuznik A, Liu LZ. Switching from atorvastatin to simvastatin in patients at high
cardiovascular risk: effects on low-density lipoprotein cholesterol. Am J Ther 2010; 17:167-175.
37. Athyros VG, Tziomalos K, Karagiannis A, Mikhailidis DP. To switch (statins) or not to switch? That is the
question. Expert Opin Pharmacother. 2010;11:2943-6.
38. Thiebaud P, Patel BV, Nichol MB, Berenbeim DM. The effect of switching on compliance and persistence:
The case of statin treatment. Am J Manag Care 2005; 11:670-674.
39. Chapman RH, Benner JS, Girase P, Benigno M, Axelsen K, Liu LZ, Nichol MB. Generic and therapeutic statin
switches and disruptions in therapy. Curr Med Res Opin. 2009; 25:1247-60.
40. Thomas M, Mann J. Increased thrombotic vascular events after change of statin. Lancet 1998; 352: 1830–
1831.
41. Phillips B, Roberts C, Rudolph AE, Morant S, Aziz F, O’Regan CP. Switching statins: the impact on patient
outcomes. Br J Cardiol 2007; 14: 280-285.
42. Butler R, Wainwright J. Cholesterol lowering in patients with CHD and metabolic syndrome [Letter]. Lancet.
2007;369:27.
43. Colivicchi F, Tubaro M, Santini M. Clinical implications of switching from intensive to moderate statin
therapy after acute coronary syndromes. Int J Cardiol. 2010 Jul 30. [Epub ahead of print]
44. Catapano AL. Perspectives on low-density lipoprotein cholesterol goal achievement. Curr Med Res Opin.
2009;25:431–447.
45. McCormack T, Harvey P, Gaunt R, Allgar V, Chipperfield R, Robinson P. Incremental cholesterol reduction
with ezetimibe/simvastatin, atorvastatin and rosuvastatin in UK General Practice (IN-PRACTICE):
randomised controlled trial of achievement of Joint British Societies (JBS-2) cholesterol targets. Int J Clin
Pract. 2010;64:1052–1061.
46. Willey VJ, Reinhold JA, Willey KH, Kelly BL, Cziraky MJ.: Clinical and economic outcomes in patients
switched to simvastatin in a community-based family medicine practice. Int J Clin Pract. 2010
Aug;64(9):1235-8.
47. Rublee DA, Burke JP.: LDL-C goal attainment in patients who remain on atorvastatin or switch to equivalent or nonequivalent doses of simvastatin: a retrospective matched cohort study in clinical practice. Postgrad Med. 2010
Mar;122(2):16-24.
48. Insull W Jr, Ghali JK, Hassman DR, Y As JW, Gandhi SK, Miller E; SOLAR Study Group: Achieving low-density
lipoprotein cholesterol goals in high-risk patients in managed care: comparison of rosuvastatin, atorvastatin, and
simvastatin in the SOLAR trial. Mayo Clin Proc. 2007 May;82(5):543-50. Erratum in: Mayo Clin Proc. 2007
Jul;82(7):890. Comment in: Mayo Clin Proc. 2007 May;82(5):539-42.
49. Harley CR, Gandhi SK, Anoka N, Bullano MF, McKenney JM.: Understanding practice patterns and low-density
lipoprotein cholesterol goal attainment implications of switching patients from simvastatin in a health plan setting. Am
J Manag Care. 2007 Dec;13 Suppl 10:S276-81.
50. Health Economics and Outcomes, i3 Innovus, 12125 Technology Drive, Minnesota, MN 55244, USA.
51. Usher-Smith JA, Ramsbottom T, Pearmain H, Kirby M.: Evaluation of the cost savings and clinical outcomes of
switching patients from atorvastatin to simvastatin and losartan to candesartan in a Primary Care setting. Int J Clin
Pract. 2007 Jan;61(1):15-23.
52. Usher-Smith JA, Ramsbottom T, Pearmain H, Kirby M.: Evaluation of the clinical outcomes of switching patients
from atorvastatin to simvastatin and losartan to candesartan in a primary care setting: 2 years on. Int J Clin Pract.
2008 Mar;62(3):480-4.
53. Analisi “I FARMACI EQUIVALENTI – ATTUALITÀ E PROSPETTIVE”. Marzo 2010. SEXTANTFARMA
54. National Institute for Health and Clinical Excellence (NICE). Clinical guideline 67. Lipid modification: cardiovascular
risk assessment and the modification of blood lipids for the primary and secondary prevention of cardiovascular
disease (2008). La linea guida è accompagnata da alcuni algoritmi aggiornati al marzo 2010.
50
th
55. All Wales Medicines Strategy Group - Statin Template Guidance. Approved by AWMSG 11 December 2008. Use
of statins in the primary and secondary prevention of cardiovascular disease. Aggiornato al dicembre 2009.
56. Haute Autorité de Santé (Paris): Place des hypolipémiants dans le traitement des hypercholestérolémies et la
prévention cardio- vasculaire. Septembre 2010.
57. NICE – TA 94 Statins for the prevention of cardiovascular events. Issue date: January 2006 - Review date:
November 2008 (confermato 2009 senza modifiche).
58. Brugts JJ, Yetgin T, Hoeks SE, Gotto AM, Shepherd J, Westendorp RGJ de Craen AGM, Knop RH, Nakamura H,
Ridker P, van Domburg R, Deckers JW: The benefits of statins in people without established cardiovascular disease
but with cardiovascular risk factors: meta-analysis of randomised controlled trials. BMJ 2009;338:b2376
doi:10.1136/bmj.b2376.
59. Cannon CP, Braunwald E, McCabe CH, et al. Intensive versus moderate lipid lowering with statins after acute
coronary syndromes. N Engl J Med. Apr 8 2004;350(15):1495-1504. Finanziato da Bristol-Meyers Squibb e Sanyko.
60. Pedersen TR, Faergeman O, Kastelein JJP, et al. High-dose atorvastatin vs usual-dose simvastatin for secondary
prevention after myocardial infarction: the IDEAL study: a randomized controlled trial. JAMA. Nov 16
2005;294(19):2437-2445. Finanziato da Pfizer.
61. LaRosa JC, Grundy SM, Waters DD, et al. Intensive lipid lowering with atorvastatin in patients with stable coronary
disease. The New England journal of medicine. 2005;352(14):1425-1435.
62. Sharma M, Ansari MT, Abou-Setta AM, Soares-Weiser K, Ooi TC, Sears M, Yazdi F, Tsertsvadze A, Moher D.:
Systematic Review: Comparative Effectiveness and Harms of Combinations of Lipid-Modifying Agents and HighDose Statin Monotherapy. Ann Intern Med. 2009 Aug 31.
63. Kastelein JJ, Akdim F, Stroes ES, Zwinderman AH, Bots ML, Stalenhoef AF, et al; ENHANCE Investigators.
Simvastatin with or without ezetimibe in familial hypercholesterolemia. N Engl J Med. 2008;358:1431-43. [PMID:
18376000]
64. Sharp Collaborative Group: Study of Heart and Renal Protection (SHARP): randomized trial to assess the effects of
lowering low-density lipoprotein cholesterol among 9,438 patients with chronic kidney disease. Am Heart J. 2010
Nov;160(5):785-794.e10. Epub 2010 Sep 18.
65. Melloni C, Shah BR, Ou FS, Roe MT, Smith SC Jr, Pollack CV Jr, Ohman M, Gibler WB, Peterson ED, Alexander
KP.: Lipid-lowering intensification and low-density lipoprotein cholesterol achievement from hospital admission to 1year follow-up after an acute coronary syndrome event: results from the Medications ApplIed aNd SusTAINed Over
Time (MAINTAIN) registry. Am Heart J. 2010 Dec;160(6):1121-9, 1129.e1.
66. Meaney A, Ceballos G, Asbun J, Solache G, Mendoza E, Vela A, Meaney E.: The VYtorin on Carotid intima-media
thickness and overall arterial rigidity (VYCTOR) study. J Clin Pharmacol. 2009 Jul;49(7):838-47. Epub 2009 May 14.
67. Meaney Villines TC, Stanek EJ, Devine PJ, Turco M, Miller M, Weissman NJ, Griffen L, Taylor AJ.: The ARBITER 6HALTS Trial (Arterial Biology for the Investigation of the Treatment Effects of Reducing Cholesterol 6-HDL and LDL
Treatment Strategies in Atherosclerosis): final results and the impact of medication adherence, dose, and treatment
duration. J Am Coll Cardiol. 2010 Jun 15;55(24):2721-6
68. Istituto Superiore di Sanità: Progetto CUORE - Carta del rischio cardiovascolare. Disponibile all’indirizzo:
http://www.cuore.iss.it/valutazione/carte.asp
51
BIBLIOGRAFIA SARTANI
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
Dahlof B, Devereux RB, Kjeldsen SE, Julius S, Beevers G, de Faire U, Fyhrquist F, Ibsen H, Kristiansson K,
Lederballe-Pedersen O, Lindholm LH, Nieminen MS, Omvik P, Oparil S, Wedel H. Cardiovascular morbidity and
mortality in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomised trial against
atenolol. Lancet. 2002;359(9311):995-1003.
Brenner BM, Cooper ME, de Zeeuw D, Keane WF, Mitch WE, Parving HH, Remuzzi G, Snapinn SM, Zhang Z,
Shahinfar S. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and
nephropathy. N Engl J Med. 2001;345(12):861-869.
Pitt B, Poole-Wilson PA, Segal R, Martinez FA, Dickstein K, Camm AJ, Konstam MA, Riegger G, Klinger GH, Neaton
J, Sharma D, Thiyagarajan B. Effect of losartan compared with captopril on mortality in patients with symptomatic
heart failure: randomised trial—the Losartan Heart Failure Survival Study ELITE II. Lancet. 2000;355(9215):15821587.
Dickstein K, Kjekshus J. Effects of losartan and captopril on mortality and morbidity in high-risk patients after acute
myocardial infarction: the OPTIMAAL randomised trial. Optimal Trial in Myocardial Infarction with Angiotensin II
Antagonist Losartan. Lancet. 2002;360(9335):752-760.
Lewis EJ, Hunsicker LG, Clarke WR, Berl T, Pohl MA, Lewis JB, Ritz E, Atkins RC, Rohde R, Raz I. Renoprotective
effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J
Med. 2001;345(12):851-860.
Pfeffer MA, McMurray JJ, Velazquez EJ, Rouleau JL, Kober L, Maggioni AP, Solomon SD, Swedberg K, Van de
Werf F, White H, Leimberger JD, Henis M, Edwards S, Zelenkofske S, Sellers MA, Califf RM. Valsartan, captopril, or
both in myocardial infarction complicated by heart failure, left ventricular dysfunction, or both. N Engl J Med.
2003;349(20):1893-1906.
Cohn JN, Tognoni G. A randomized trial of the angiotensin-receptor blocker valsartan in chronic heart failure. N Engl
J Med. 2001;345(23):1667-1675.
McMurray JJ, Ostergren J, Swedberg K, Granger CB, Held P, Michelson EL, Olofsson B, Yusuf S, Pfeffer MA.
Effects of candesartan in patients with chronic heart failure and reduced left-ventricular systolic function taking
angiotensin-converting-enzyme inhibitors: the CHARM-Added trial. Lancet. 2003;362(9386):767-771.
Granger CB, McMurray JJ, Yusuf S, Held P, Michelson EL, Olofsson B, Ostergren J, Pfeffer MA, Swedberg K.
Effects of candesartan in patients with chronic heart failure and reduced left-ventricular systolic function intolerant to
angiotensin-converting-enzyme inhibitors: the CHARM-Alternative trial. Lancet. 2003;362(9386):772-776.
Yusuf S, Teo KK, Pogue J, Dyal L, Copland I, Schumacher H, Dagenais G, Sleight P, Anderson C. Telmisartan,
ramipril, or both in patients at high risk for vascular events. N Engl J Med. 2008;358(15):1547-1559.
Norris S, Weinstein J, Peterson K, Thakurta S, McDonagh M, Helfand M: Drug Class Review - Direct Renin
Inhibitors, Angiotensin Converting Enzyme Inhibitors, and Angiotensin II Receptor Blockers. Final Report January
2010. Copyright © 2009 by Oregon Health & Science University Portland, Oregon 97239.
Baker WL, Coleman CI, Kluger J, Reinhart KM, Talati R, Quercia R, Phung OJ, White CM. Systematic Review:
Comparative Effectiveness of Angiotensin-Converting Enzyme Inhibitors or Angiotensin II–Receptor Blockersfor
Ischemic Heart Disease. Ann Intern Med, 2009; 151:861-71
Matchar DB, McCrory DC, Orlando LA, Patel MR, Patel UD, Patwardhan MB, Powers, B, Samsa GP, Gray RN.
Systematic review: comparative effectiveness of angiotensin-converting enzyme inhibitors and angiotensin II
receptor blockers for treating essential hypertension. Ann Intern Med. 2008 Jan 1;148(1):16-29.
The ONTARGET Investigators; Yusuf S, Teo KK, Pogue J, et al. Telmisartan, ramipril, or both in patients at high risk
for vascular events. N Engl J Med 2008;358:1547-1559.
52
CITAZIONI INSERITE NEL TESTO
i
Smith MEB, Lee NJ, Haney E, Carson S. Drug Class Review: HMG-CoA Reductase Inhibitors (Statins) and
Fixed-dose Combination Products Containing a Statin: Final Report Update 5. Drug Class Reviews. Portland
(OR): Oregon Health & Science University; 2009 Nov.
ii
Brugts JJ, Yetgin T, Hoeks SE, Gotto AM, Shepherd J, Westendorp RGJ de Craen AGM, Knop RH,
Nakamura H, Ridker P, van Domburg R, Deckers JW: The benefits of statins in people without established
cardiovascular disease but with cardiovascular risk factors: meta-analysis of randomised controlled trials.
BMJ 2009;338:b2376 doi:10.1136/bmj.b2376.
iii
Dalla Nota 13 AIFA: Un incremento del colesterolo totale e delle frazioni a basso peso molecolare (LDL e
VLVL), dei TG e dell’apolipoproteina B sono stati riscontrati nel 60-80% dei pazienti sottoposti a trapianto di
cuore e che ricevono una terapia immunosoppressiva standard comprensiva di steroidi, ciclosporina e
azatioprina nel 45% dei pazienti sottoposti a trapianto di fegato e in una percentuale di pazienti sottoposti a
trapianto di rene che a seconda delle varie casistiche considerate arriva fino al 60%. Numerosi studi
effettuati su campioni di popolazione di adeguata numerosità hanno consentito di dimostrare la correlazione
tra iperlipidemia e lo sviluppo di aterosclerosi e conseguentemente di malattia cardiovascolare l’iperlipidemia
indotta dai farmaci immunosoppressivi, inoltre, accelera lo sviluppo della cosiddetta GVC (graft coronary
vasculopathy), una forma di aterosclerosi coronarica accelerata che rappresenta la più comune causa di
morte tardiva post-trapianto di cuore e che si riscontra in questi pazienti con un’incidenza annua pari al 10%
Alla luce di questi dati nella pratica clinica l’utilizzo di farmaci ipolipemizzanti nei pazienti sottoposti a
trapianto di organo solido si è reso indispensabile laddove l’utilizzo di un regime dietetico controllato a basso
contenuto di colesterolo e la riduzione di eventuali ulteriori fattori di rischio cardiovascolare modificabili non
sia stata sufficiente per mantenere i valori di colesterolemia entro i limiti consigliati e laddove non sia
proponibile l’utilizzo di uno schema alternativo di terapia antirigetto.
Nei pazienti con infezione da HIV, a seguito dell’introduzione della HAART (terapia antiretrovirale di
combinazione ad alta efficacia), è frequente l’insorgenza di dislipidemia indotta dai farmaci antiretrovirali che,
nel tempo, può contribuire ad un aumento dell’incidenza di eventi cardio-vascolari, sviluppabili anche in
giovane età.
Da studi di coorte prospettici, se pur non tutti concordi, emerge un rischio relativo di eventi ischemici
vascolari pari a circa 1.25 per anno con incremento progressivo e proporzionale alla durata di esposizione
alla terapia antiretrovirale. La prevalenza di dislipidemia nei pazienti HIV positivi è variabile in rapporto al tipo
di terapia antiretrovirale, comunque è intorno al 25% per la colesterolemia e oltre il 30% per
l’ipertrigliceridemia (indotta in particolare dall’interferone).
Alla luce di questi dati, nella pratica clinica l’utilizzo di farmaci ipolipemizzanti nei pazienti con infezione da
HIV in trattamento antiretrovirale si è reso necessario, laddove la riduzione dei fattori di rischio
cardiovascolare “modificabili” non si riveli sufficiente a mantenere i valori di colesterolemia e trigliceridemia
entro i limiti presenti nel box e laddove, per motivi clinici e/o virologici, non sia sostituibile la terapia
antiretrovirale in atto. In questi casi si possono utilizzare statine di 2° livello in eventuale assoc iazione con gli
omega 3.
iv
Dalla Nota 13 AIFA: Il danno aterosclerotico nei pazienti con insufficienza renale cronica (IRC), a parità
di livello dei fattori di rischio, è superiore a quello che si osserva nella popolazione generale; le malattie
cardiovascolari sono infatti la principale causa di morte dei pazienti con IRC. Per tale motivo è
necessario, in questi pazienti, un controllo particolarmente accurato dei fattori di rischio delle malattie
cardiovascolari, tra cui la dislipidemia.
Le statine sembrano efficaci nella prevenzione di eventi vascolari in pazienti vasculopatici con IRC e sono
in grado di ridurre la proteinuria e di rallentare la progressione della malattia renale. Per pazienti adulti
con IRC in stadio 3-4 (GFR < 60ml/min, ma non ancora in trattamento sostitutivo della funzione renale),
così come per coloro che pur con una GFR > 60 ml/min presentino segni di malattia renale in atto
(proteinuria dosabile), va considerato un trattamento farmacologico ipocolesterolemizzante, nel caso di
insuccesso della correzione dello stile di vita, con l’obiettivo di raggiungere un TT per LDL-col almeno <
100 mg/dL; secondo alcuni autorevoli enti internazionali, il TT può essere fissato a < 70-80 mg/dL (specie
in presenza di condizioni che aumentano ulteriormente il rischio, come una storia clinica di eventi
cardiovascolari accertati o diabete mellito).
Se i livelli della trigliceridemia sono ≥ 500 mg/dL, va considerato un trattamento con fibrati, tenendo conto
dell’esigenza di adeguare il dosaggio di questi farmaci, escreti per via renale, alla funzione renale
residua.
Nei pazienti con IRC in stadio 5 (GFR < 15 ml/min o in trattamento sostitutivo della funzione renale) le
evidenze attuali, desunte dai pochi studi di intervento pubblicati, non sono favorevoli al trattamento della
dislipidemia. Il recentissimo risultato dello studio AURORA, che valutava l’effetto di rosuvastatina in una
popolazione di pazienti con IRC allo stadio finale, ha dimostrato che, a fronte di una riduzione del LDL-C,
53
il trattamento con statina non era associato ad una riduzione dell’end-point combinato di IMA, stroke e
morte cardiovascolare.
v
Dalla Nota 13 AIFA: Dislipidemie familiari. Le dislipidemie familiari sono malattie su base genetica
caratterizzate da elevati livelli di alcune frazioni lipidiche plasmatiche e, spesso da una grave e precoce
insorgenza di malattie CV.
Le dislipidemie erano classicamente distinte secondo la classificazione di Frederickson, basata
sull’individuazione delle frazioni lipoproteiche aumentate; questa classificazione è oggi in parte superata da
una classificazione genotipica, basata sull’identificazione delle alterazioni geniche responsabili.
Ad oggi non sono tuttavia definiti criteri internazionali consolidati per la diagnosi molecolare di tutte le
principali dislipidemie familiari e l’applicabilità clinica pratica di tali criteri è comunque limitata: il loro
riconoscimento va quindi effettuato impiegando algoritmi diagnostici che si basano sulla combinazione di
criteri biochimici, clinici ed anamnestici. E’ essenziale per la diagnosi di dislipidemia familiare escludere
preliminarmente tutte le forme di iperlipidemia secondaria o da farmaci le principali delle quali sono elencate
nelle già ricordate tabelle III e IV.
Tra le dislipidemia familiari che più frequentemente si associano ad un rischio aumentato di cardiopatia
ischemia prematura, vanno ricordate le ipercolesterolemie familiari autosomiche dominanti (ADH1, ADH2,
ADH3; geni affetti rispettivamente LDLR, APOBPCSK9), l’iperlipidemia familiare combinata (FCH; gene
affetto non conosciuto), la disbetalipoproteinemia (gene affetto APOE) e le gravi iperchilomicronemie /
ipertrigliceridemie (Geni affetti LPL, APOC2, APOA5, GPIHBP1, LMF1, l’ipercolesterolemia da difetto della
proteina ARH (gene affetto LDLRAP1) e la sitosterolemia (gene affetto ABCG5/ABCG8) come indicate nel
box. In tutti questi pazienti l’obiettivo primario della terapia è di portare la colesterolemia a valori più bassi
possibile.
Criteri clinici per la diagnosi clinica dell’ipercolesterolemia familiare ai fini dell’appropriatezza prescrittiva dei
medicinali in Nota 13 sono i seguenti:
- Ipercolesterolemia familiare monogenica, o FH
Malattia genetica (con prevalenza nel nostro Paese intorno ad 1:500) frequentemente dovuta a mutazioni del
gene che codifica il recettore delle LDL. Benché una diagnosi certa sia ottenibile solamente mediante
metodiche di analisi molecolare, questa dislipidemia, nella pratica clinica, può essere diagnosticata con
ragionevole certezza mediante un complesso di criteri biochimici, clinici ed anamnestici. I cardini di questi
criteri, sostanzialmente condivisi da tutti gli algoritmi diagnostici proposti, includono:
- colesterolemia LDL superiore a 190 mg/dL più
- trasmissione verticale della malattia, documentata dalla presenza di analoga alterazione biochimica nei
familiari del probando.
In assenza di informazioni sul profilo lipidico dei familiari il sospetto è molto forte se insieme alla
colesterolemia LDL superiore a 190 mg/dL si osservano:
- presenza di xantomatosi tendinea nel probando oppure
- un’anamnesi positiva per cardiopatia ischemica precoce (prima dei 55 anni negli uomini, prima dei 60 nelle
donne) nel probando o nei familiari di I e II grado (nonni, genitori, fratelli) o la presenza di grave
ipercolesterolemia in figli in età prepubere.
Dati recenti suggeriscono che un appropriato trattamento dei pazienti con ipercolesterolemia familiare
conduce ad un sostanziale abbattimento del loro eccesso di rischio cardiovascolare.
- Iperlipidemia combinata familiare, o FCH
Questa malattia (con prevalenza nel nostro Paese intorno ad 1-2:100) è caratterizzata da una importante
variabilità fenotipica ed è collegata a numerose variazioni genetiche, con meccanismi fisiopatologici
apparentemente legati ad un’iperproduzione di apo B-100, e quindi delle VLDL.
I criteri diagnostici sui quali è presente un consenso sono:
- colesterolemia LDL superiore a 160 mg/dl e/o trigliceridemia superiore a 200 mg/dl più
- documentazione nei membri della stessa famiglia (I e II grado) di più casi di ipercolesterolemia e/o
ipertrigliceridemia (fenotipi multipli), spesso con variabilità fenotipica nel tempo (passaggio da
ipercolesterolemia ad ipertrigliceridemia, o a forme miste).
In assenza di documentazione sui familiari, la dislipidemia familiare è fortemente sospetta in presenza
anamnestica o clinica o strumentale di arteriosclerosi precoce.
È indispensabile per la validità della diagnosi di iperlipidemia combinata familiare escludere le famiglie in cui
siano presenti unicamente ipercolesterolemia o ipertrigliceridemia.
- Disbetalipoproteinemia familiare
Patologia molto rara (con prevalenza nel nostro Paese intorno ad 1:10.000) che si manifesta in soggetti
omozigoti per l’isoforma E2 dell’apolipoproteina E. La patologia si manifesta in realtà solamente in una
piccola percentuale dei pazienti E2/E2, per motivi non ancora ben noti.
I criteri diagnostici includono:
- valori sia di colesterolemia che di trigliceridemia intorno ai 400-500 mg/dl più
- presenza di larga banda beta, da fusione delle bande VLDL ed LDL, alla elettroforesi delle lipoproteine.
La presenza di uno di questi fattori aumenta la validità della diagnosi:
54
- xantomi tuberosi,
- xantomi striati palmari (strie giallastre nelle pieghe interdigitali o sulla superficie palmare delle mani, da
considerare molto specifici).
vi
Istituto Superiore di Sanità: Progetto CUORE - Carta del rischio cardiovascolare. Disponibile all’indirizzo:
http://www.cuore.iss.it/valutazione/carte.asp
vii
Documentazione in cartella: colesterolo basale (prima dell’applicazione di stili di vita salubri), descrizione
degli interventi sugli stili di vita (con privilegio per la attività fisica), colesterolo a tre mesi.
viii
Documentazione in cartella: colesterolo prima della prescrizione alla dimissione, descrizione degli
interventi sugli stili di vita (con privilegio per la attività fisica) raccomandati oltre alla terapia farmacologica.
ix
Documentazione in cartella: C-LDL prima della prescrizione alla dimissione, descrizione degli interventi
sugli stili di vita (con privilegio per la attività fisica) raccomandati oltre alla terapia farmacologica.
x
Cannon CP, Braunwald E, McCabe CH, et al. Intensive versus moderate lipid lowering with statins after
acute coronary syndromes. N Engl J Med. Apr 8 2004;350(15):1495-1504. Finanziato da Bristol-Meyers
Squibb e Sanyko.
xi
LaRosa JC, Grundy SM, Waters DD, et al. Intensive lipid lowering with atorvastatin in patients with stable
coronary disease. The New England journal of medicine. 2005;352(14):1425-1435.
xii
Sharma M, et al: Systematic Review: Comparative Effectiveness and Harms of Combinations of LipidModifying Agents and High-Dose Statin Monotherapy. Ann Intern Med. 2009 Aug 31.
xiii
Ara R, et al: Ezetimibe for the treatment of hypercholesterolaemia: a systematic review and economic
evaluation. Health Technol Assess. 2008 May;12(21):iii, xi-xiii, 1-212.
xiv
A. Donzelli, Pillola di Buona Pratica Clinica 2011 (in press).
xv
Kastelein JJ, et al: ENHANCE Investigators. Simvastatin with or without ezetimibe in familial
hypercholesterolemia. N Engl J Med. 2008;358:1431-43. [PMID: 18376000].
xvi
Rossebø AB, et al: SEAS Investigators: Intensive lipid lowering with simvastatin and ezetimibe in aortic
stenosis. N Engl J Med. 2008 Sep 25;359(13):1343-56. Epub 2008 Sep 2.
xvii
Villines TC, et. al: The ARBITER 6-HALTS Trial (Arterial Biology for the Investigation of the Treatment
Effects of Reducing Cholesterol 6-HDL and LDL Treatment Strategies in Atherosclerosis): final results and
the impact of medication adherence, dose, and treatment duration. J Am Coll Cardiol. 2010 Jun
15;55(24):2721-6.
xviii
Meaney A, et al: The VYtorin on Carotid intima-media thickness and overall arterial rigidity (VYCTOR)
study. J Clin Pharmacol. 2009 Jul;49(7):838-47. Epub 2009 May 14.
xix
Sharp Collaborative Group: Study of Heart and Renal Protection (SHARP): randomized trial to assess the
effects of lowering low-density lipoprotein cholesterol among 9,438 patients with chronic kidney disease. Am
Heart J. 2010 Nov;160(5):785-794.e10. Epub 2010 Sep 18.
xx
Baigent C et al. (SHARP Investigators): The effects of lowering LDLc with simvastatin plus ezetimibe in
patients with chronic kidney disease (Study of Heart and Renal Protection): a randomised placebo-controlled
trial. Lancet 2011; 377:2181.
xxi
Cosa risulta dal confronto tra i farmaci (estratto dal PDT ASL Pavia “Ipertensione arteriosa 2010”):
a) Ipertensione – Monoterapia – Mortalità: l'effetto sulla mortalità è stato riportato solo da 8 studi per
un totale di 3.492 pazienti. Nessuno di questi studi era stato progettato per misurare la mortalità per
qualsiasi causa come esito clinico primario. Nello studio di maggiore durata (12 mesi di
osservazione per 369 pazienti) non sono accaduti decessi nel gruppo trattato con Valsartan o nel
gruppo di confronto trattato con Ramipril. Negli studi di breve durata il tasso di mortalità variava tra
lo 0% e il 2%, con una maggioranza di studi che non riportavano decessi. In nessuno degli studi è
stata riscontrata una differenza significativa tra Sartano e ACE- inibitore riguardo alla mortalità.
b) Ipertensione, scompenso cardiaco e malattia cardiovascolare – Mortalità ed esiti compositi che
comprendono la mortalità: Monoterapia: Sartani non inferiori ad ACE-Inibitori (2 studi); Sartani simili ad ACEInibitori per la mortalità o per gli esiti primari compositi che comprendono la mortalità (5 studi); il primo studo
ELITE riportava un vantaggio del Losartan rispetto al Captopril per la morte cardiaca improvvisa. Terapia di
combinazione: Sartani simili ad ACE-Inibitori (3 studi).
xxii
Nome del principio attivo, formulazione, nome del prodotto e indicazione registrata sono stati desunti da
CODIFA (maggio 2011). Le indicazioni registrate sono state ordinate in cinque aree omogenee. Per
ciascuna area le indicazioni registrate delle EBPM sono state reperite nella seguente documentazione:
Nadroparina: Fraxiparina ®, Fraxodi ® - RCP (Masson - CODIFA)
Nadroparina: Seleparina ®, Seledie ® - RCP (Masson - CODIFA)
Dalteparina: Fragmin ® - RCP (Masson - CODIFA)
Reviparina: Clivarina ® - RCP (Masson - CODIFA)
Parnaparina: Fluxum ® - RCP (Masson - CODIFA)
Bepiparina: Ivor ® - RCP (Masson - CODIFA) RCP: Riassunto delle Caratteristiche del Prodotto
55
")